


Abstract
Cross-talk between competitive endogenous RNAs (ceRNAs) through shared miRNAs represents a novel layer of gene regulation that plays important roles in the physiology and development of cancers. However, a global view of their system-level properties across various types of cancers is still unknown. Here, we constructed the mRNA related ceRNA–ceRNA interaction landscape across 20 cancer types by systematically analyzing molecular profiles of 5203 tumors and miRNA regulations. Our study highlights the conserved features shared by pan-cancer and higher similarity within similar origin cell type. Moreover, a core ceRNA network was identified. Function analysis identified a common theme of cancer hallmarks, however they exhibit phenotype-specific connectivity patterns. Besides, we found a marked rewiring in the ceRNA program between various cancers, and further revealed conserved and rewired network ceRNA hubs in each cancer, which were tensely competitive interactions to constitute conserved and cancer-specific modules. By providing mechanistic linkage between known cancer miRNAs, their mediated ceRNA–ceRNA interactions, and the associations with known cancer hallmarks, the inferred cancer ceRNA–ceRNA interaction landscape will serve as a powerful public resource for further biological discoveries of tumorigenesis.
INTRODUCTION
MicroRNAs (miRNA) are an abundant class of small, non-coding RNAs (∼22 nt long), which negatively regulate gene expression at the level of messenger RNAs (mRNAs) stability and translation inhibition (1). Dysregulation of miRNA activity has been shown to play an important role in tumor initiation and progression (2–4). In addition to the conventional miRNA/mRNA function, recent studies have shown that the interaction of the miRNA seed region with mRNA is not unidirectional, but that the pool of mRNAs can crosstalk through their ability to compete for miRNA binding (5,6). These competitive endogenous RNAs (ceRNAs) act as molecular sponges for a miRNA through their miRNA binding sites (also referred to as miRNA response elements, MRE), thereby de-repressing all target genes of the respective miRNA. Yet few such modulators of miRNA activity have been characterized and both the extent and relevance of their role in cancers are poorly understood.
More recently, comprehensive multidimensional molecular profiles of large tumor populations generated by research consortia such as The Cancer Genome Atlas (TCGA) have enabled integrated analysis of molecular alterations associated with individual human cancer types (7,8). And some ceRNAs were revealed in multiple types of cancer (9,10). PTEN is a critical tumor suppressor gene which is frequently altered in multiple human cancers. Three recent studies have identified and successfully validated protein-coding transcripts as PTEN ceRNAs in prostate cancer (10), glioblastoma (11) and melanoma (9). In addition, examples are already emerging of non-coding RNAs as competitive platforms for miRNAs, such as lincRNA-p21(12), lincMD1(13) and linc-RoR (14). When the analysis was significantly extended beyond the binary ceRNA associations described in these studies, the identified ceRNA interactions were found to be important part of the miRNA-mediated interactions. However, these studies demonstrated that previously uncharacterized transcripts could be functionalized, partly through the identification of their ceRNA interactors, and presented a framework for the prediction and validation of ceRNA interactions. Sumazin et al. investigated the ability of coding genes to act as ceRNAs in human glioblastoma and identified a broad network of sponge interactions as mediators of crosstalk between different regulatory pathways (11). In addition, Paci et al. had performed a computational analysis and identified a sponge interaction network between long non-coding RNAs and mRNAs in human breast cancer (15). An increasing number of researchers have attempted to identify the ceRNA interactions in specific cancer. However, previous studies had focused on the properties of individual ceRNA interactions in a specific cancer, but had lacked a global view of their system-level properties across cancers. Moreover, individual miRNA can target hundreds or thousands of mRNAs on the basis of sequence complementarity, only a substantial fraction of these predicted interactions may depend on cell type and context (16,17). This suggests that the ceRNA interactions may be cancer-specific, systematic studies that evaluate ceRNA cross-talks across multiple cancer types are needed to explore.
With the discovery of ceRNAs, many of genes with known ceRNA interactors identified so far have been implicated in human disease. For example, PTEN is a potent tumor suppressor gene that is frequently disrupted in multiple cancers and governs multiple cellular processes, including survival, proliferation and energy metabolism (18). In addition, HULC is the most upregulated gene in hepatic celluler cancer (HCC) and has been shown to regulate several genes involved in liver cancer (19). linc-MD1 expression in primary muscle cells has been shown to result in partial mitigation of the correct timing of the differentiation program (13). These suggest that ceRNA crosstalk is not only of fundamental importance in physiological conditions, but is also crucially relevant in various cancers. However, cancer encompasses more than 100 related diseases (20), making it crucial to understand the commonalities and differences of ceRNA interactions among various types and subtypes (21). TCGA was founded to address these needs, and its large datasets are providing unprecedented opportunities for us to systematically analyze the ceRNA networks across cancers.
Here, we performed a systematic analysis of 5203 tumors from 20 cancer types to investigate the mRNA-related ceRNA cross-talks (Supplementary Figure S1). Besides the conserved topological features across cancers, our study highlights a marked rewiring in the ceRNA program between various types of cancers. By applying functional enrichment we have identified specific ceRNAs associated with hallmarks of cancers. Through in-depth analyses of the structure of the pan-cancer ceRNA networks, we identified the conserved and differential hubs, which further constituted the conserved ceRNA modules and cancer-specific ceRNA modules. These analyses and validations demonstrate how the cancer-associated ceRNA regulatory network can be used to accelerate discovery of ceRNA-based biomarkers and potentially therapeutics. To extend the impact and usage of mRNA related ceRNA–ceRNA landscape across cancers, Pan-ceRNADB (http://www.bio-bigdata.com/pan-cernadb/ or http://www.bio-bigdata.net/pan-cernadb/), a convenient and available resource is further constructed for biomedical scientists. Systematic construction and analysis of mRNA-related ceRNA regulatory network across multiple cancers, can help to elucidate the commonalities and differences in mechanisms of cancers.
MATERIALS AND METHODS
Genome-wide protein-coding gene expression profiles across cancers
All protein-coding gene expression datasets were obtained from the TCGA. In total, 20 types of cancers were analyzed in the current study (Supplementary Table S1). Normalized TCGA level 3 Agilent microarray mRNA expression profiles were used for glioblastoma multiforme (GBM) and ovarian serous cystadenocarcinoma (OV) . Genes with missing values in >30% samples were removed and then we imputed the other missing values by the k-nearest neighbor method. For the remaining cancer types, mapped and gene-level summarized (RPKM) RNA-seq datasets were used (22). To filter genes not expressed across most samples in RNA-seq datasets, we removed genes with RPKM expression values of 0 in all of the samples. To allow log transformation, gene RPKM expression values of 0 were set to 0.05 in the given samples. The microarray and RNA-seq mRNA expression values were log2 transformed for all subsequent analysis.
Ago CLIP-supported miRNA–target interactions
Recently, several studies have reported that the use of cross-linking and Argonaute (Ago) immunoprecipitation coupled with high-throughput sequencing (CLIP-Seq) could identify endogenous genome-wide interaction maps for miRNAs (23,24). In this study, the AGO-CLIP altas were generated by compiling available AGO-CLIP data from starBase V2.0(25), and compared with coding sequence (CDS) and 5′ UTR regions, 3′ UTR regions are overlap with more CLIP-Seq peak clusters (Supplementary Table S2). To investigate human miRNA–target regulatory relationships, miRNA target sites are predicted by five prediction programs and directly downloaded from their corresponding websites, including TargetScan (26), miRanda (27), Pictar (28), PITA (29) and RNA22(30). Especially, the RNA22 algorithm also predicted the target sites in CDS and 5′ UTR. Then, all CLIP-Seq peak clusters were intersected with these predicted miRNA target sites. In total, we characterized ∼420 000 interactions between 386 conserved miRNAs and 13 802 protein-coding genes. Most of these miRNA interactions are located at the 3′ UTR regions, and about 8.5 and 3.35% interactions were in the CDS and 5′ UTR regions, respectively.
Genes associated with cancer hallmarks
A list of Gene Ontology (GO) terms that were related to the hallmarks of cancer were obtained from a previous study (31). The genes annotated to these hallmark-associated GO terms were obtained from MsigDB V4.0, which is a collection of annotated gene sets for use with GSEA software (32).
Collection of cancer-associated miRNAs
Several database systems have proposed to provide a comprehensive resource of miRNA dysregulation in various human diseases. We collected the cancer-associated miRNAs from six databases, including HMDD (33), miR2Disease (34), miREnvironment (35), OncomiRDB (36), PhenomiR 2.0(37) and SM2miR (38). And all these cancer-related miRNAs were manually corresponded to each cancer.
Construction of the ceRNA networks in individual cancer type
Having got the Ago CLIP-supported miRNA–mRNA regulatory data, we mainly followed two principles listed below to identify ceRNA pairs in each cancer. A central tenet of our hypothesis is that trans-regulatory ceRNA crosstalk increased with the high miRNA regulatory similarity between mRNAs and their strong co-expression in specific cancer (Supplementary Figure S1). Firstly, a hypergeometric test is used to compute the significance of shared miRNAs for each possible gene pairs (detailed description can be found in Supplementary Method). Moreover, the number of shared miRNAs is required to at least three. All P-values were subject to false discovery rate (FDR) correction and mRNA pairs with FDR < 0.01 were considered as candidate ceRNA interaction pairs.
Depending on the total number of functional miR-binding sites that they share with a target, ceRNA modulators can decrease the number of free miR molecules available to repress other target genes. This indicated that the expression of ceRNA pairs was positively correlated with each other. To identify the active ceRNA pairs in a specific cancer, we computed the Pearson correlation coefficient (R) of each candidate ceRNA pairs identified above. All the candidate ceRNA pairs with R > 0 and P-adjusted < 0.05 were identified as ceRNA–ceRNA interactions.
After assembling all identified ceRNA pairs, we generated the mRNA-related ceRNA network for each cancer type. A node represents a mRNA, and two nodes are connected if they are co-regulated by miRNAs and positively co-expressed in this cancer.
Topological measurements of the ceRNA networks
For each ceRNA in a network, degree is defined as the number of edges incident to it. On the one hand, it is reported that the hub genes with higher degrees in biological networks are more likely to be essential. On the other hand, experimental studies have demonstrated that nearly 10% of the nodes in a network are essential. Thus in this work, we selected the top 10% of genes with the highest degrees in the ceRNA network as the hub genes. In addition, we also analyzed the results using another two commonly used thresholds (15 and 20%).
To systematical analysis of the hub across cancers, we split the hubs into three groups: (i) cancer-specific hubs, that is the ceRNAs were only hubs in only one cancer ceRNA network; (ii) differential hubs, the ceRNAs that are hubs in more than one ceRNA network but their neighborhoods change between different cancer networks; and (iii) common ceRNA hubs, these ceRNAs are hubs in more than one cancer and their interacting ceRNAs are similar in different cancers. To define the first category, ceRNAs that were ranked in top 10% in at least one network, but were not ranked in top 10% in any other cancers were selected. To identify the other two types of hubs, we calculated the similarity of their interacting ceRNAs between pairs of ceRNA networks. For each pair of ceRNA networks, we calculated the Simpson index. If the Simpson index of a ceRNA is higher than 0.8 in at least one pair of ceRNA networks, the hub ceRNA was grouped into the common hubs. Otherwise, we added the ceRNA to the secondary category.
Construction of hallmark associated ceRNA networks in each cancer
The hallmark-associated ceRNA networks were constructed by mapping the hallmark genes to the ceRNA network and then the edges linked by two hallmark genes were extracted. Then we test whether the number of edges is statistically significant larger than the random cases. Instead of the real ceRNA networks, 1000 random degree-conserved networks were chosen as control, and the number of edges comprised by hallmark genes in each random network was counted. The P-value is the fraction of the number of edges, which is larger than that in the real one.
Network visualization and comparison analysis
The ceRNA networks were visualized by Network Workbench (39) and Cytoscape 3.0.2(40) and topology analysis was performed by the package of ‘igraph’ in R language. To estimate the similarity of two cancer ceRNA networks, we calculated the number of edges that are present in both networks (common ceRNA interactions) and Simpson index was used. In addition, a hypergeometric test was used to test that if two ceRNA networks significantly shared the common ceRNA interactions.
Identification of ceRNA network modules
First, we used the clique percolation clustering method to identify ceRNA modules in each cancer, which are defined as cliques. Cliques are all of complete subgraphs that are not parts of larger complete subgraphs. This procedure is performed using CFinder (41), which is a fast program for locating and visualizing overlapping.
To identify the conserved ceRNA modules across cancers, the hallmark associated ceRNA networks in each cancer were merged. And then we obtained the hallmark associated subnetwork of the common hubs. And then the cliques were identified from the subnetwork. The conserved ceRNA modules were defined as the community with genes from 10 to 50. Similarly, the cancer specific ceRNA modules were identified from the cancer specific hub subnetworks.
RESULTS
The miR program-mediated mRNA-related ceRNA–ceRNA regulatory landscape in pan-cancer
To evaluate both the range and potential tumorigenic role of this class of miR-mediated interactions, we explored a two-stage analysis method and constructed the ceRNA–ceRNA regulatory landscape across diverse types of cancers. In the first step, we identified nearly 7328 genes participating in 521 621 pairwise miR program-mediated RNA–RNA interactions at a FDR < 0.01. These candidate ceRNA pairs were regulated by at least three common miRNAs. We then constructed cancer-specific ceRNA interactomes by filtering the global ceRNA interactome according to specific cancer expressomes. As a result, cancer-specific ceRNA interactome covered more than half of the candidate ceRNA interactome, implicated with 11.9–84.9% genes and 0.27–22.3% of the candidate ceRNA interactions (Supplementary Figures S2 and S3). Moreover, to provide a convenient and available resource about mRNA related ceRNA–ceRNA landscape across cancers for biomedical scientists, Pan-ceRNADB, a free and web-accessible database, is further constructed (http://www.bio-bigdata.com/pan-cernadb/ or http://www.bio-bigdata.net/pan-cernadb/).
Together, all these ceRNA–ceRNA interactions constitute a large and previously uncharacterized ceRNA networks across diverse types of cancers. We modeled the network graphically, with genes represented as nodes and their miRNA-mediated interactions as undirected edges. In this work, we mainly constructed the ceRNA networks of multiple cancers by using the dataset of TCGA. In order to test the robust of the ceRNA networks in a specific cancer, we also analyzed multiple independent datasets obtained from Gene Expression Omnibus. As a result, these ceRNA interactions show high co-expression in dependent datasets (Supplementary Figure S4). Thus, a conclusion can be drawn that the ceRNA interactions are stable across the ceRNA networks constructed using different datasets. Moreover, benchmark analyses have indicated that such a two-step approach is valuable to identify the association of competition among mRNAs (Supplementary Text S1). All these results validate the stability of the ceRNA networks across cancers and prove those can be used to understand the biological mechanism of cancers.
Common features of ceRNA interactomes
Analysis of the topological features of the ceRNA networks across cancers, some common topological features of the ceRNA network were revealed (Figure 1A). Firstly, the examination of the degree distribution of these ceRNA networks reveals a power law distribution, showing that the ceRNA network is scale free, similar as most types of biological networks. In each cancer ceRNA network, most genes had few interacting partners, while a small subset of genes, denoted hubs, had many interacting partners each (Figure 1B and Supplementary Figure S5). We also found that the overall regulatory effect on a node depends on many variables, including the number of ceRNA neighbors and the number of miRNAs they share with its neighbors. In general, nodes in larger highly connected graphs will have more neighbors and will thus be more strongly regulated by other ceRNAs. Analysis of the ceRNA networks show that highly connected ceRNAs are more co-expressed with their neighbors than others (Figure 1C and Supplementary Figure S6). In additon, co-expression of ceRNAs in the network increases with the number of common miRNAs (Figure 1D and Supplementary Figure S7). Next, we analyzed the modular structure of the ceRNA networks. Here, we defined a ceRNA module as a clique that is a maximal complete subgraph. All modules in the ceRNA networks are identified using CFinder (41). Figure 1E and Supplementary Figure S8 show the number of modules corresponding to each k-value, and the cumulative fraction of ceRNAs contained in modules. With an increase in the value of k, there is a sharp decrease in the number of modules. In total, about 30–75% ceRNAs are involved in at least one module. We interpreted this feature as a consequence that ceRNAs implement specific regulations as small clusters rather than as individual or big modules.
Figure 1.
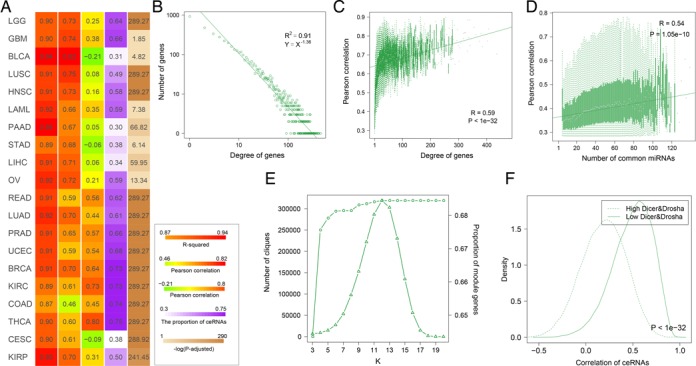
The global topological features of ceRNA–ceRNA interaction networks. (A) A global view of the topological features for 20 cancers. The first column represents the goodness of fit of degree distribution. And the second and the third columns represent the correlation coefficient. The fourth indicates the proportion of ceRNAs in cliques. The fifth column is the −log10(p) of ranksum test. The numbers were colored based on the adjacent color map. The details of these results for UCEC are shown in (B–F). (B) The distribution of the degree of ceRNAs. (C) ceRNAs with high degree are strongly coexpressed. (D) The correlation between expression of genes and the total expression of their ceRNAs is plotted as a function of the number of its ceRNA regulators. (E) Number of cliques at different k-values and cumulative ratios of ceRNAs in cliques with k-values are not bigger than k. The left y-axis represents number of cliques under different k-values, corresponding to the triangle line. The right y-axis represents cumulative ratios of ceRNAs in cliques, corresponding to the dot line. (F) The ceRNA–ceRNA interactions were likely to co-express in Dicer and Drosha low expressed groups.
The expression levels of miRNAs had shown to be critical for cross-regulation of ceRNAs in theoretical model (42), we further explore the role of miRNA concentrations on ceRNA cross-regulation across each cancer. Dicer and Drosha are two central regulators of miRNA maturation, we divided the cancer samples based on the Dicer and Drosha expression. As a result, ceRNA pairs were strongly co-expressed in Dicer/Drosha-low expressed groups (Supplementary Figures S9 and 10). Specifically, the co-expression were more obvious in Dicer and Drosha low expressed groups (Figure 1F and Supplementary Figure S11). We presumed that if miRNA molecules are more abundant, cross-regulation is unlikely to occur as most genes are fully repressed by the abundant miRNAs. This may be an important supporting evidence that the majority of the co-expressed genes in the network are ceRNA pairs. Another common feature of the cancer ceRNA interactomes was that ceRNA interactions participate in distal regulation between genes within and across chromosomes. Moreover, analysis of the pathway genes in KEGG shows that these ceRNA interactions mediate crosstalk between numerous pathways.
Network level analysis highlights a core ceRNA networks across cancers
Although the pan-cancer ceRNA networks share several common features, viewing the ceRNA network across cancers, our study highlights a marked rewiring in the ceRNA program between different cancers, documented by its ‘on/off’ switch from cancer to cancer and vice-versa. We found that ∼34.56% ceRNA regulations were occurred only in one cancer (Figure 2A) and only 3.78% ceRNA–ceRNA interactions were conserved in more than 10 cancers. The low conservation of ceRNA regulations may be explained in part by the cancer-specific expression of genes. Within any particular tissue or cell, only a subset of genes is expressed active (43) and thus only a subset of ceRNA interactions can work.
Figure 2.
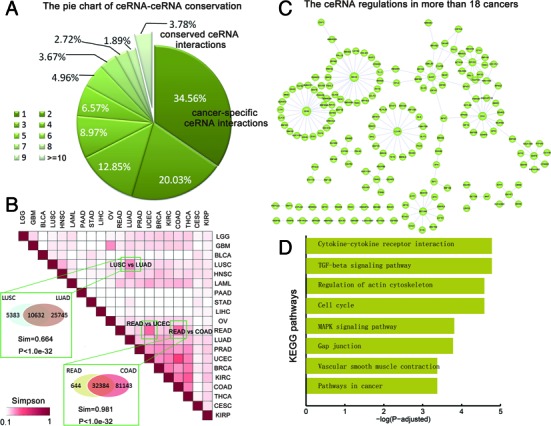
The network level comparision of ceRNA–ceRNA interaction networks across cancers. (A) The pie chart shows the proportion of ceRNA interactions presented in different number of cancers. The majority of the ceRNA interactions are cancer specific. (B) The simpson index matrix shows the similarity between each pair of ceRNA–ceRNA networks. Some pairs of cancers with same origin were specifically shown. (C) The core neuron ceRNA–ceRNA network that presented in more than 18 cancers. (D) The KEGG pathways enriched by the genes in the core ceRNA network.
Although most of the ceRNA regulations were cancer-specific, the cancers with similar tissue-of-origin share common ceRNAs (Figure 2B). Based on the Simpson index, we found that each cancer network varies in similarity to other cancer networks and reveal both known and new relationships among these cancers. Importantly, the adenocarcinomas, such as kidney renal clear cell carcinoma (KIRC), breast invasive carcinoma (BRCA), prostate adenocarcinoma (PRAD) and lung adenocarcinoma (LUAD), show greater similarities with each other than other cancers (Figure 2B). For instance, as expected, LUAD and lung squamous cell carcinoma (LUSC) are two types of lung cancers, we found that the similarity of their ceRNA networks were higher than those with other cancers. Approximately 66.4% ceRNA interactions in LUSC also worked in LUAD, which was significantly higher than expected (P < 1.0E-32). Another example is the colon cancer (COAD) and rectal cancer (READ). The 98.1% ceRNA interactions in the READ were shared in COAD, which was significantly higher than expected (sim = 0.981, P < 1.0E-32). To assess whether there is a common core of ceRNA regulatory interactions to maintain the architecture of ceRNA networks across cancers, we focused on the ceRNA interactions occurred in more than 90% cancers. This analysis revealed that most of the conserved ceRNA regulations formed a large connected component (Figure 2C). These findings indicate that the conserved ‘neuronal’ ceRNA network may maintain the ceRNA network architecture across cancers. Testing for KEGG pathway enrichments we found that these ceRNAs were highly enriched for basic cellular processes common to mutiple type of cancers (Figure 2D), such as cytokine–cytokine receptor interaction, cell cycle and mitogen-activated protein kinase (MAPK) signaling pathway. These observations suggest that the represented cancers might have a common feature of aberrant immune system function and cell cycle.
Differential network analysis reveals conserved and rewired network hubs in each cancer type
Differential gene expression profiling studies have led to the identification of several cancer associated genes. However, evidences have shown that the oncogenic alterations in coding regions can modify the gene functions without affecting their own expression profiles (44). Next, we attempted at providing insights into how to identify cancer-related genes via the ceRNA network topological features. Comparing the degree distribution across cancers, we found that these most of the ceRNA networks were characterized by nodes with highly variable degrees, from genes with a few connections to ‘hubs’ with hundreds of links (Figure 3A). For example, the ceRNA network of GBM presents an increased connectivity with respect to the brain lower grade glioma (LGG). A Kolmogorov–Smirnov test showed that the GBM network was characterized by a gene degree which is stochastically increased with respect to the LGG network (P < 7.98E-7). The significant changes of ceRNA network connectivity in different stages of cancer indicate that ceRNAs with strongly altered connections can have a role in cancer biology and motivate us on a connectivity-based scoring measure for the identification of putative cancer associated genes.
Figure 3.
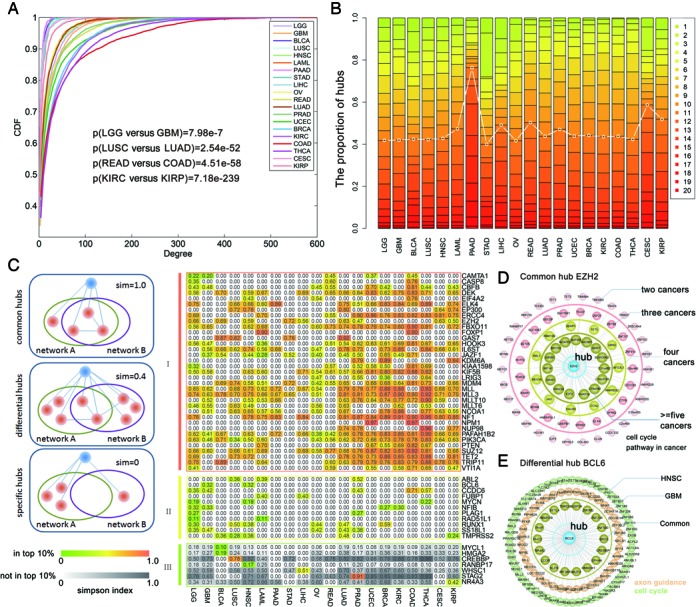
The conserved and rewired network hubs in each cancer type. (A) Cumulative distribution functions of the ceRNA degree in each cancer. (B) The number of hubs distribute in 1–20 cancers in which the hubs can occur. (C) The conserved and rewired network hubs. Shown are the simpson indexes of different types of hubs from the ceRNA networks. Columns of each heatmap correspond to one of the cancers, and the rows represent a cancer associated hub. The ith row and jth column show the average similarity of the ceRNA partners across all other cancer networks. The upper color map corresponds to the hubs being present in the top 10% of an inferred network. The bottom color map is for hubs that are not in the top 10%. (D) An example of common hub EZH2. ceRNAs ocurred in different numbers of cancers were grouped by circle. (E) An example of differential hub BCL6.
Since hub nodes have been found to play important roles in many networks, we identified hub ceRNAs in each network. Generally, these ceRNA hubs retained their high degree across other cancers (Figure 3B and Supplementary Figures S12 and 13). To systematically assess the extent to which these hub ceRNAs were shared among the different cancer-specific ceRNA networks, differential network analysis was performed and these hubs were grouped into three categories (see ‘Materials and Methods’ section): common hubs, differential hubs and cancer-specific hubs. As a result, we identified 769 common hubs across these cancers which constituted the largest proportion of the hub ceRNAs. In addition, ∼35 common hubs were included in Cancer Gene Cencus database, including EZH2, CASP8 and EP300 (Figure 3C). For instance, the ceRNA EZH2 was in the top 10% of hubs in nine ceRNA networks. We found that 15 interacting partners of EZH2 are represented in more than five ceRNA networks (Figure 3D). These results indicated that these ceRNAs might influence the cancerous state in different cancers through the same mechanisms. Functional enrichment analysis of these ceRNAs indicated that EZH2 may regulate cell cycle in cancers. In addition, there are 360 differential hubs, 11 of which were also known cancer genes (Figure 3C). To examine whether these differential ceRNA hubs were targeting different cancer signaling pathways in each cancer type, we associated each ceRNA hubs with a cancer signaling pathway by using the enrichment analysis. One interesting example was BCL6, which was in the top 10% of hubs in the GBM and head and neck squamous cell carcinoma (HNSC) networks (Figure 3E). Functional enrichment of the ceRNA partners of BCL6 in GBM, we identified the most significant pathway is ‘axon guidance’ (P-adjusted = 0.012). However, the partners of BCL6 in HNSC network was found to be mostly associated with ‘cell cycle’ (P-adjusted = 0.018). Overall, these results suggest that the ceRNAs may selectively regulate differential pathways in any specific cancer. Finally, 223 ceRNAs were identified as hubs exclusively in the ceRNA network of a specific cancer (Figure 3C). Several of these cancer-specific ceRNA hubs have specific roles in the type of cancer that they were uniquely identified with. For instance, the bladder urothelial carcinoma (BLCA) specific hub-HMGA2, has been demonstrated to be upregulated in bladder cancer at both the transcriptional and translational levels compared with normal bladder tissue, and HMGA2 protein is a potential prognostic marker for predicting tumor recurrence and progression (45). In addition, the LUSC-specific hub ceRNA-CREBBP had been found frequently mutated in LUSC (46). Overall, these results suggest that several of the cancer-specific ceRNA hubs have known roles in the relevant cancer type, while some ceRNA hubs are also associated with multiple disease states. A more detailed study of these ceRNAs coupled with further functional validation studies can reveal previously uncharacterized pathways for these cancer types.
miRNA-mediated ceRNA regulations control broad cancer-related hallmarks
Although the biology of cancer is extremely complex, the complexity of cancer can be reduced and represented by a few cancer hallmarks that enable tumor growth and metastasis dissemination (47). These hallmarks provide a framework for understanding the remarkable diversity of cancers. Next, we focused on the ceRNA regulations in the context of cancer hallmarks. According to one of the recent studies, there are 2954 genes related to cancer hallmarks. Firstly, analysis of the miRNA regulation of these hallmark genes shows that hallmark genes were regulated by more miRNAs than other genes (Supplementary Figure S14, P = 1.48E-31), suggesting that hallmark genes are more likely to be precise expressed and under the strictly regulatory control of miRNAs and ceRNAs. Functional enrichment analysis reveals that the cancer-related ceRNA networks enriched at least one hallmark of cancers. About 20% hallmark genes were involved in ceRNA regulations, which was significantly larger than randomly chosen genes (Figure 4A, right panel, P < 1.0E-3). On the other hand, the ceRNA networks cover most genes of the hallmark-related functions (range from 18.75 to 80%, Figure 4A, top panel). Another interesting observation is that all the 20 ceRNA networks are enriched in the function of ‘regulation of cell proliferation’, highlighting its roles in the development of pan-cancers.
Figure 4.
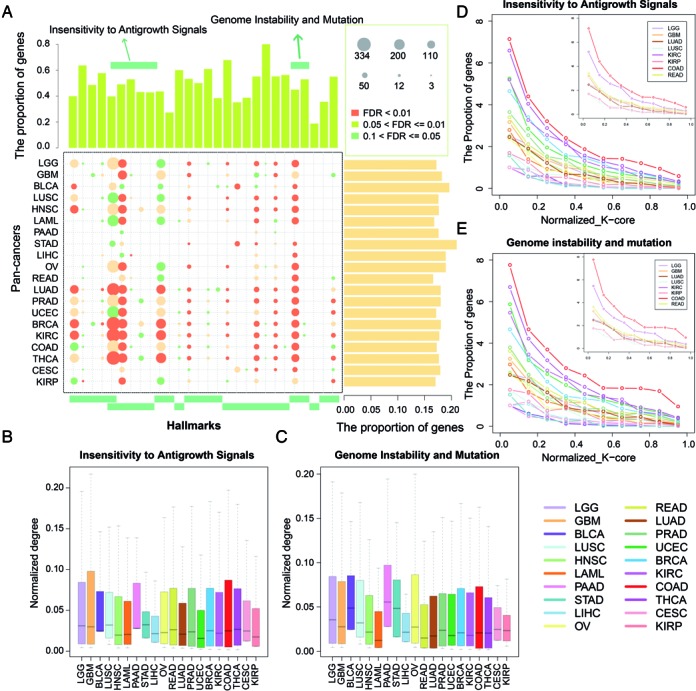
The ceRNA networks control broad cancer associated hallmarks. (A) The summary bubble-bar plot show the functional enrichment results of the ceRNA networks across the cancers. The top bars show the percentage of ceRNAs annotated in each term. And the bars on the right show the percentage of ceRNAs annotated in the cancer hallmarks. The bubble size indicates the number of genes in each term, and different color corresponds to different FDRs. The darker of the color, the smaller of the FDR. (B and C) The normalized degree of ceRNAs annotated in the two hallmarks. (D and E) Relationships between ceRNA layers and frequency of ceRNAs implicated in two hallmarks identified in each layer. Increasing layer numbers correspond to regions of increasing densities in the network. The layers of each network were normalized to [0–1] and the frequencies were summarized in each interval.
An overall view of the functional profiles of ceRNA networks can also reveal some cancer-specific functions. For example, compared to LGG, we found that the ceRNAs in GBM were enriched in the process of ‘cellular response to hypoxia’. In addition, most of the adenocarcinoma (including LUAD, PRAD and BRCA) were also enriched in this process (Figure 4A), suggesting the tumor hypoxia is might be a classical feature of these cancers. As a tumor grows, it rapidly outgrows its blood supply, leaving portions of the tumor with regions where the oxygen concentration is significantly lower than in healthy tissues. In order to support continuous growth and proliferation in challenging hypoxic environments, cancer cells may active these ceRNA interactions for responsing to hypoxia. Next, we examined whether the genes enriched in the same hallmarks exhibit different connectivity patterns. The connections number of each ceRNA (node degree) was scaled to a value between 0 and 1 by dividing each node degree by the largest degree in a ceRNA network. We found that the ceRNAs enriched in the same hallmarks show varied degree across cancers (Figure 4B and C). In addition, by peeling each ceRNA network, we found that the ceRNAs with similar functions localized in different layers of the networks. For instance, ceRNAs associated with ‘insensitivity to antigrowth signals’ were with higher degree in the BRCA ceRNA network and OV (Figure 4B). However, these ceRNAs were localized to the more dense layers in BRCA (Figure 4D). These results suggest that ‘insensitivity to antigrowth signals’ associated ceRNAs play key roles in the development of BRCA. Another instance is the ‘Genome Instability and Mutation’ associated ceRNAs, that were with higher degree in COAD than those of READ. These ceRNAs were also more likely to localize in the more dense layers of the COAD ceRNA networks (Figure 4E). This structural property of hallmark genes in the context of pan-cancer ceRNA networks may be used to identify additional genes involved in specific cancer based on their phenotype-specific connectivity patterns.
Investigating the pan-cancer ceRNA networks reveals several conserved and cancer-specific ceRNA modules
We then mapped all hallmark genes to nodes in the ceRNA networks across cancers, and then extracted the connected components as hallmark ceRNA networks (HCNs, Supplementary Dataset S1). As shown in Supplementary Figure S15, except LIHC, other HCNs are much denser than expected by chance (1000 random degree-conserved ceRNA networks are chosen as control). These results demonstrate that cancer hallmark genes tend to be connected in the ceRNA networks. Next, we were interested in identifying the conserved and specific ceRNA modules. Modules in the ceRNA networks represent groups of functionally related genes dedicated to specific biological processes. Investigating the pan-cancer ceRNA networks reveals several conserved and cancer-specific modules of particular interest (see ‘Materials and Methods’ section). In total, 20 conserved ceRNA modules were identified (Supplementary Table S3). Functional analysis of these conserved modules revealed that the majority of these ceRNAs were involved in multiple cancer associated hallmarks. For instance, a conserved hub module consisted of 86 pairs of ceRNA interactions among 14 ceRNAs (Figure 5A). Among these ceRNAs, some have been demonstrated to play key roles in multiple cancers, such as HIF1A, MAPK6 and RASA1. Hypoxia-inducible factors (HIF) play pivotal roles in the regulation of cellular utilization of oxygen and are essential transcriptional regulators of angiogenesis in solid tumor (48). Previous studies have demonstrated that MAPK signaling facilitates HIF activation through p300/CBP (49). Here, we observed that 14 miRNAs mediated the regulation between HIF1A and MAPK6 in 10 cancers. Several miRNAs had been demonstrated to be dysregulated in many cancers, including miR-107, miR-17 and let-7 family. These results suggest another miRNA mediated pathways to activate the HIF signaling in cancers. Another example is the conserved module consisted 45 interactions among 10 ceRNAs (Figure 5B), including the cancer-associated genes, ITGA2, PCDHAC1 and SOS1. The ceRNA ITGA2 encodes the alpha subunit of a transmembrane receptor for collagens and related proteins. And PCDHAC1 is a member of the protocadherin alpha gene cluster. It had been demonstrated that integrin- and cadherin-mediated signals in cooperation then oppose each other to lead the keratinocyte into cell-cycle exit, growth arrest and onset of terminal differentiation (50). In our current study, we observed that ITGA2 and PCDHAC1 were co-regulated by two miRNAs, miR-17–5p and miR-20a-5p. The current knowledge summarized herein highlights the critical regulation the integrin and cadherin network in cooperation with miRNAs.
Figure 5.
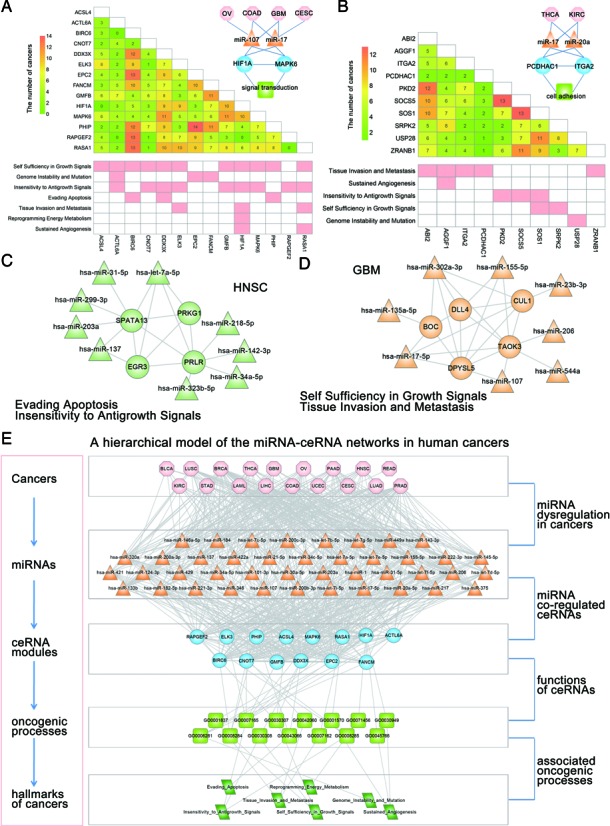
The conserved and specific ceRNA modules across the cancers. (A and B) Two conserved ceRNA modules across multiple cancers. The number in the matrix indicates the number of cancers the ceRNA interaction occurred. (C and D) Two examples of cancer-specific ceRNA modules. The cancer-associated miRNAs were also shown in the modules. (E) Summary of the hierarchical model to systematically understand the miRNA–ceRNA networks in human cancers. This model uses the module in (A) as an example. The model is laid out hierarchically with (from the top down) cancers, miRNAs, ceRNA modules, annotated GO biological process terms for the ceRNAs and finally the GO terms associated with hallmarks of cancers.
Besides the conserved ceRNA modules in multiple cancers, we should not overlook the importance of cancer-specific ceRNA modules. In total, we identified 35 cancer specific ceRNA modules (Supplementary Table S4). A series of experimental studies corroborate the role of PRLR in cancer biology (51). High level of PRLR expression has demonstrated to be an independent negative prognostic factor for overall survival in patients with HNSC. The PRLR mainly activated Stats and promoted the growth of human cancer cells by regulation Bcl-XL (52). In addition to this classical PRLR-dependent signal transduction pathway, here we found that PRLR may alter the activity of EGR3 through miRNA regulations (such as let-7a-5p), which is an immediate-early growth response gene which is induced by mitogenic stimulation (Figure 5C). Another example is the GBM specific ceRNA module, including the gene DPYSL5, which is also known as CRMP5(53). CRMP5 has been shown to highly express in the developing brain and in adult brain neurogenesis areas. The expression of CRMP5 in GBM activities Notch signaling pathway to promote proliferation and poor survival. However, the underlying mechanisms are still unknown. We found that CRMP5 can regulate the transmembrane Notch ligand-DLL4(54) through competing for the miRNA-miR-155–5p (Figure 5D). miR-155 has been demonstrated to play crucial roles in GBM. All these observations suggest that targeting CRMP5–DLL4 interaction mediated by miRNAs may be a promising strategy for future glioblastoma treatment.
A cancer hallmark network framework for understanding the ceRNA regulations
miRNAs are a class of small RNAs functioning as negative regulators of gene expression at post-transcriptional level. And the dysregulation of miRNAs has been demonstrated to play critical roles in cancer through regulating the genes inappropriately. In addition, recent and our current studies have suggested that miRNAs could act as a regulatory language and mediated the RNA–RNA interactions. Here, we proposed a hierarchical model to systematically understand the miRNA–ceRNA networks in human cancers (Figure 5E). Evidences have demonstrated that cancers develop from the accumulation of mutations and epigenetic changes (55), such as changes in DNA copy number, translocation as well as gene fusions. As a result, all of such events may induce altered expression of mRNAs. Changes of the genes impact the MREs and further alter the capacity of a proper mRNA to attach or titrate miRNAs. Consequently, the ceRNA network or modules are perturbed, inducing the cancer associated hallmark dysregulation. All these processes may contribute to the development and progression of cancers. Given the complex nature of biological systems, these regulatory systems often need to function in a coordinated fashion in order to produce appropriate physiological responses to both internal and external stimuli. Therefore, exploring the interaction and crosstalk between the regulatory systems is important for understanding the function of both cells and more complex systems. Taken together, the identification of the ceRNA networks herein expands the theory of dynamic and complexity of the miRNA-gene regulatory network and provides more challenge for development of miRNA-based cancer therapy.
DISCUSSION
In this study, we performed a systematic analysis of the mRNA-related ceRNA–ceRNA interaction landscape across 20 major types of cancer. Importantly, the global topological features of these cancer ceRNA networks are conserved. For example, the degree distributions of all mRNA-related ceRNA networks follow a power-law distribution. Comparison of the ceRNA networks across cancers, we showed that only a small proportion of the ceRNA interactions are conserved. The mRNA-related ceRNA interactions varied greatly from one cancer type to another. In addition, our study revealed that the cancers with similar tissue of origin show higher network similarity. Furthermore, the analysis of the ceRNA networks across tumor types revealed a core subnetworks, including genes involved in cell cycle. And our analysis also provides a hub-based view to elucidate the common and specific ceRNA modules across cancers. Hubs are topologically centered in the ceRNA network, having maximal informational links with other ceRNAs. Despite the majority of the hub ceRNAs share the common interacting partners, we found that some hubs rewired the partners in differential cancers (such as, BCL6). One possible explanation for this finding is that the hubs active different pathways in distinct cancers by selectively regulating these ceRNAs. Finally, conserved and specific ceRNA modules were analyzed, motivating us to propose a hierarchical model to systematically understand the miRNA–ceRNA networks in human cancers.
MiRNAs have been shown to regulate PTEN and thus contribute to cell transformation mediated by aberrant activation of the PI3K/AKT pathway (9). Much attention of recent studies is paid to the ceRNAs of PTEN, and some studies have identified and verified several of these ceRNAs. We also investigated the PTEN associated ceRNA subnetworks across cancers. A ceRNA sub-network of PTEN was constructed across 20 types of cancer and 285 interactions were included, including GBM. Recently, Sumazin et al. have validated a substantial set of miR-mediated PTEN modulators in multiple cell lines (11), which were significantly overlapped with our PTEN ceRNA subnetwork (P = 4.56E-27) and some of the known ceRNAs were included, such as CNOT6L, CCDC6, ZEB2, KLF6 and LRCH1 (Supplementary Table S5). siRNA silencing of these miR-mediated PTEN regulators had been shown to be sufficient to downregulate PTEN in a 3′ UTR-dependent manner and to increase tumor cell growth rates. Moreover, we found that ∼31.93% ceRNAs of PTEN were supported by other literatures or computational methods (Supplementary Table S5). In addition, many other novel RNAs were unveiled to modulate the PTEN RNA levels, supplementing the ceRNA networks of PTEN across cancers. Next, we compared the expression patterns and function correlations between the novel ceRNAs and those known ones. As a result, we observed that these novel ceRNAs showed similar expression patterns with PTEN as the known ones (Supplementary Figure S16). They also showed significantly higher function similarity with the known ceRNAs (Supplementary Figure S17, P < 0.001). When we explored whether these ceRNAs can be regulated by PTEN, we observed that that the expression of 67.02% ceRNAs were upregulated and 42.1% of the ceRNAs were upregulated above 1.5-fold change in PTEN overexpressed U87 cell line (Supplementary Figure S18). The proportion is slightly higher than that of the known ceRNAs of PTEN. Because the known ceRNAs of PTEN are incomplete and mainly focus on GBM, these novel ceRNAs provided here could provide more ceRNA candidate for PTEN across cancers. These results indicate the complex regulatory mechanism of PTEN tumor suppressor gene. Totally, the recently identified PTEN-centered ceRNA networks contribute to increase even further the relevance of PTEN in human cancer, and bring a deeper understanding of the molecular alterations that are at the basis of human cancer.
In the past several years, significant efforts have been made in determining biologically relevant miRNA–target interactions using high-throughput experimental approaches. The use of CLIP-Seq could identify endogenous genome-wide interaction maps for animal miRNAs. On the other hand, miRNA target identification is challenging owing to the imperfect nature of base pairing between an miRNA and its target, and the rules of targeting are not completely understood. Currently, several miRNA–target prediction algorithms, including TargetScan, miRanda, RNA22 and PITA, have been used to identify miRNA regulations. Moreover, the CLIP-Seq atlas allowed us to integrate experimentally defined miRNA–mRNA interaction with prediction programs to create more accurate prediction of miRNA regulations and also reduce the size of the search space for miRNA target sites (6,56,57). It is complementary between sequencing method and in silico prediction strategies. We observed that the combination of the computational approach with the experimental approach refined the computational predictions by more than 30-fold, similar as the result of previous study (56). Therefore, further development and optimization of these prediction algorithms based on CLIP-seq data will improve subsequent predictions of ceRNA interactions, and harnessing these experimental techniques will provide further insight into ceRNA regulation beyond that which is possible with in silico target predictions. In addition, although this analysis is mainly limited to the 3′ UTRs of protein-coding transcripts, we believed that it is still useful for the identification and analysis of putative ceRNA crosstalk across different cancers. As the understanding of miRNA regulatory mechanism widens, it can be expected that the identification of miRNA interactions as well as the ceRNA interactions will become progressively more accurate.
In our current study, the ceRNA–ceRNA interactions were identified in each cancer by considering the similarity of miRNA regulation and expression in specific cancer. The co-expression of mRNAs may reflect a variety of scenarios. For example, the genes may co-localize within the genome or may be co-regulated by the same transcription factors. To exclude these scenarios, we calculated the proporion of co-localized and co-regulated mRNA pairs (details in Supplementary Methods). As a result, there are only 0.13–19.59% pairs of ceRNAs were co-localization or co-regulated by TFs across 20 types of cancer (Supplementary Table S6). Analyzing the ceRNA networks after filtering those co-localized or co-regulated pairs, we obtained the similar topological and functional landscapes of ceRNAs networks across human cancers (Supplementary Text S2). These results provide further evidences that the expression correlations in the ceRNA networks were mainly mediated by miRNAs. On the other hand, with the increasement of miRNA expression, DNA methylation and DNA copy number available for the same tumors, integration of these information may provide further evidence that the two correlated genes are competitively binding the same miRNAs. The multivariate linear model could measure the expression association between a miRNA and a mRNA, that also factors in variation (noise) in mRNA expression induced by changes in DNA copy number and promoter methylation at the mRNA gene locus. Motivated by the recent studies (58,59), the association of miRNA and mRNA were obtained in 10 types of cancers and an miRNA–mRNA pair was considered as associated if the FDR is under 0.05 (Supplementary Text S3). And then integrated with the CLIP-seq supported target sites in the text, we obtained the cancer specific miRNA–mRNA regulations. First, we explored whether the integration can improve the prediction accuracy of miRNA–mRNA interaction in our study. Using the experimentally validated miRNA–targets obtained from TarBase (60), miRTarBase (61) and miRecords (62), we found that integration of miRNA expression datasets can refine miRNA interaction in specific conditions by calculating two indexs, accuracy and F-score (Supplementary Figure S19). These results suggest that integration of the miRNA expression information can refine the miRNA-regulations in specific cancer. Next, we performed the same procedure and reconstructed the ceRNA–ceRNA networks in each cancer. To explore whether the ceRNA–ceRNA network is indeed mediated by miRNAs, for each ceRNA interaction we computed the difference between the Pearson and partial correlation coefficients and defined it sensitivity correlation (details in Supplementary Methods). About 77.78–100% of the ceRNA pairs were with significantly higher sensitivity correlation than random conditions (FDR < 0.05). Moreover, we re-introduced PTEN in U87 glioma cell line and observed that the expression of ∼67.56% ceRNAs were upregulated and 37.84% of the ceRNAs were upregulated with above 1.5-fold change (Supplementary Figure S18). Furthermore, we observed that more than 24.32 and 43.24% of the ceRNAs with downregulated expression in another two public PTEN knockdown experimental datasets (GSE68869 and GSE54269). These results provide further evidences that majority of the co-expressed genes in the networks are ceRNA pairs. Analyzing the ceRNA networks, we obtained the similar topological and functional landscapes of ceRNAs networks across human cancers (Supplementary Text S3). The ceRNA networks also show scale-free and modular structures and the ceRNA pairs were strongly co-expressed in Dicer/Drosha-low expressed groups. In addition, we found that in the Dicer/Drosha-low expressed groups, the overall expression levels of miRNA are indeed lower (Supplementary Figure S20). These results further evidence that the structures of the ceRNA networks and most of the results obtained in our study are robust. Considering the potential need of biomedical scientists, we constructed Pan-ceRNADB to store these two types of ceRNA–ceRNA interactions identified in our study: the level 1 datasets store the ceRNA–ceRNA interactions without integration of the miRNA expression, DNA methylation and DNA copy number; the level 2 datasets store the intergrated results.
In summary, we presented the ceRNA–ceRNA interaction landscape across human major cancers and showed the importance at various aspects. A bird's eye view of the functional ceRNA networks of large sample sets encompassing multiple tumor lineages may help to suggest potential unexpected targets that are applicable to cancer subsets or across cancers. Our study opens new avenues for leveraging publicly available genomic data to study the functions and mechanisms of ceRNAs across human cancers.
Supplementary Material
Acknowledgments
The authors gratefully thank the TCGA Research Network for providing data for this work.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National High Technology Research and Development Program of China [863 Program, 2014AA021102]; National Program on Key Basic Research Project [973 Program, 2014CB910504]; National Natural Science Foundation of China [91439117, 61473106, 61203264, 81371324, 81171122]; Natural Science Foundation of Heilongjiang Province [ZD201208, QC2015020]; China Postdoctoral Science Foundation [2014T70364, 2015M571436, LBH-Z14134]; WeihanYu Youth Science Fund Project of Harbin Medical University; Specialized Research Fund for the Doctoral Program of Higher Education of China [20132307110008]. Funding for open access charge: National High Technology Research and Development Program of China [863 Program, 2014AA021102]; National Program on Key Basic Research Project [973 Program, 2014CB910504]; National Natural Science Foundation of China [91439117, 61473106, 61203264, 81371324, 81171122]; Natural Science Foundation of Heilongjiang Province [ZD201208, QC2015020]; China Postdoctoral Science Foundation [2014T70364, 2015M571436, LBH-Z14134]; WeihanYu Youth Science Fund Project of Harbin Medical University; Specialized Research Fund for the Doctoral Program of Higher Education of China [20132307110008].
Conflict of interest statement. None declared.
REFERENCES
- 1.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 2.Kloosterman W.P., Plasterk R.H. The diverse functions of microRNAs in animal development and disease. Dev. Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 4.Li Y., Xu J., Chen H., Bai J., Li S., Zhao Z., Shao T., Jiang T., Ren H., Kang C., et al. Comprehensive analysis of the functional microRNA-mRNA regulatory network identifies miRNA signatures associated with glioma malignant progression. Nucleic Acids Res. 2013;41:e203. doi: 10.1093/nar/gkt1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language. Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tay Y., Rinn J., Pandolfi P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Omberg L., Ellrott K., Yuan Y., Kandoth C., Wong C., Kellen M.R., Friend S.H., Stuart J., Liang H., Margolin A.A. Enabling transparent and collaborative computational analysis of 12 tumor types within The Cancer Genome Atlas. Nat. Genet. 2013;45:1121–1126. doi: 10.1038/ng.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karreth F.A., Tay Y., Perna D., Ala U., Tan S.M., Rust A.G., DeNicola G., Webster K.A., Weiss D., Perez-Mancera P.A., et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147:382–395. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tay Y., Kats L., Salmena L., Weiss D., Tan S.M., Ala U., Karreth F., Poliseno L., Provero P., Di Cunto F., et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sumazin P., Yang X., Chiu H.S., Chung W.J., Iyer A., Llobet-Navas D., Rajbhandari P., Bansal M., Guarnieri P., Silva J., et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147:370–381. doi: 10.1016/j.cell.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoon J.H., Abdelmohsen K., Srikantan S., Yang X., Martindale J.L., De S., Huarte M., Zhan M., Becker K.G., Gorospe M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell. 2012;47:648–655. doi: 10.1016/j.molcel.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cesana M., Cacchiarelli D., Legnini I., Santini T., Sthandier O., Chinappi M., Tramontano A., Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y., Xu Z., Jiang J., Xu C., Kang J., Xiao L., Wu M., Xiong J., Guo X., Liu H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell. 2013;25:69–80. doi: 10.1016/j.devcel.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Paci P., Colombo T., Farina L. Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC Syst. Biol. 2014;8:83. doi: 10.1186/1752-0509-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erhard F., Haas J., Lieber D., Malterer G., Jaskiewicz L., Zavolan M., Dolken L., Zimmer R. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014;24:906–919. doi: 10.1101/gr.166702.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bossel Ben-Moshe N., Avraham R., Kedmi M., Zeisel A., Yitzhaky A., Yarden Y., Domany E. Context-specific microRNA analysis: identification of functional microRNAs and their mRNA targets. Nucleic Acids Res. 2012;40:10614–10627. doi: 10.1093/nar/gks841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalhoub N., Baker S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J., Liu X., Wu H., Ni P., Gu Z., Qiao Y., Chen N., Sun F., Fan Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38:5366–5383. doi: 10.1093/nar/gkq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 21.Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A., et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 23.Helwak A., Kudla G., Dudnakova T., Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C., Darnell R.B. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011;29:607–614. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 27.Betel D., Koppal A., Agius P., Sander C., Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krek A., Grun D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M., et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 29.Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 30.Miranda K.C., Huynh T., Tay Y., Ang Y.S., Tam W.L., Thomson A.M., Lim B., Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 31.Plaisier C.L., Pan M., Baliga N.S. A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome Res. 2012;22:2302–2314. doi: 10.1101/gr.133991.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y., Qiu C., Tu J., Geng B., Yang J., Jiang T., Cui Q. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42:D1070–1074. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Q., Wang Y., Hao Y., Juan L., Teng M., Zhang X., Li M., Wang G., Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–D104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Q., Qiu C., Yang J., Wu Q., Cui Q. miREnvironment database: providing a bridge for microRNAs, environmental factors and phenotypes. Bioinformatics. 2011;27:3329–3330. doi: 10.1093/bioinformatics/btr556. [DOI] [PubMed] [Google Scholar]
- 36.Wang D., Gu J., Wang T., Ding Z. OncomiRDB: a database for the experimentally verified oncogenic and tumor-suppressive microRNAs. Bioinformatics. 2014;30:2237–2238. doi: 10.1093/bioinformatics/btu155. [DOI] [PubMed] [Google Scholar]
- 37.Ruepp A., Kowarsch A., Theis F. PhenomiR: microRNAs in human diseases and biological processes. Methods Mol. Biol. 2012;822:249–260. doi: 10.1007/978-1-61779-427-8_17. [DOI] [PubMed] [Google Scholar]
- 38.Liu X., Wang S., Meng F., Wang J., Zhang Y., Dai E., Yu X., Li X., Jiang W. SM2miR: a database of the experimentally validated small molecules’ effects on microRNA expression. Bioinformatics. 2013;29:409–411. doi: 10.1093/bioinformatics/bts698. [DOI] [PubMed] [Google Scholar]
- 39.Team NWB. Indiana University; Northeastern University and University of Michigan; 2006. Network Workbench Tool. [Google Scholar]
- 40.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palla G., Derenyi I., Farkas I., Vicsek T. Uncovering the overlapping community structure of complex networks in nature and society. Nature. 2005;435:814–818. doi: 10.1038/nature03607. [DOI] [PubMed] [Google Scholar]
- 42.Ala U., Karreth F.A., Bosia C., Pagnani A., Taulli R., Leopold V., Tay Y., Provero P., Zecchina R., Pandolfi P.P. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc. Natl. Acad. Sci. U.S.A. 2013;110:7154–7159. doi: 10.1073/pnas.1222509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barshir R., Shwartz O., Smoly I.Y., Yeger-Lotem E. Comparative analysis of human tissue interactomes reveals factors leading to tissue-specific manifestation of hereditary diseases. PLoS Comput. Biol. 2014;10:e1003632. doi: 10.1371/journal.pcbi.1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anglani R., Creanza T.M., Liuzzi V.C., Piepoli A., Panza A., Andriulli A., Ancona N. Loss of connectivity in cancer co-expression networks. PLoS One. 2014;9:e87075. doi: 10.1371/journal.pone.0087075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang G.L., Zhang L.H., Bo J.J., Hou K.L., Cai X., Chen Y.Y., Li H., Liu D.M., Huang Y.R. Overexpression of HMGA2 in bladder cancer and its association with clinicopathologic features and prognosis HMGA2 as a prognostic marker of bladder cancer. Eur. J. Surg. Oncol. 2011;37:265–271. doi: 10.1016/j.ejso.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Kishimoto M., Kohno T., Okudela K., Otsuka A., Sasaki H., Tanabe C., Sakiyama T., Hirama C., Kitabayashi I., Minna J.D., et al. Mutations and deletions of the CBP gene in human lung cancer. Clin. Cancer Res. 2005;11:512–519. [PubMed] [Google Scholar]
- 47.Wang E., Zaman N., McGee S., Milanese J., Masoudi-Nejad A., O'Connor-McCourt M. Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin. Cancer Biol. 2015;30:4–12. doi: 10.1016/j.semcancer.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Matsuo Y., Ding Q., Desaki R., Maemura K., Mataki Y., Shinchi H., Natsugoe S., Takao S. Hypoxia inducible factor-1 alpha plays a pivotal role in hepatic metastasis of pancreatic cancer: an immunohistochemical study. J. Hepatobiliary Pancreat. Sci. 2014;21:105–112. doi: 10.1002/jhbp.6. [DOI] [PubMed] [Google Scholar]
- 49.Sang N., Stiehl D.P., Bohensky J., Leshchinsky I., Srinivas V., Caro J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J. Biol. Chem. 2003;278:14013–14019. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weber G.F., Bjerke M.A., DeSimone D.W. Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 2011;124:1183–1193. doi: 10.1242/jcs.064618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bauernhofer T., Pichler M., Wieckowski E., Stanson J., Aigelsreiter A., Griesbacher A., Groselj-Strele A., Linecker A., Samonigg H., Langner C., et al. Prolactin receptor is a negative prognostic factor in patients with squamous cell carcinoma of the head and neck. Br. J. Cancer. 2011;104:1641–1648. doi: 10.1038/bjc.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dagvadorj A., Kirken R.A., Leiby B., Karras J., Nevalainen M.T. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin. Cancer Res. 2008;14:1317–1324. doi: 10.1158/1078-0432.CCR-07-2024. [DOI] [PubMed] [Google Scholar]
- 53.Veyrac A., Reibel S., Sacquet J., Mutin M., Camdessanche J.P., Kolattukudy P., Honnorat J., Jourdan F. CRMP5 regulates generation and survival of newborn neurons in olfactory and hippocampal neurogenic areas of the adult mouse brain. PLoS One. 2011;6:e23721. doi: 10.1371/journal.pone.0023721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El Hindy N., Keyvani K., Pagenstecher A., Dammann P., Sandalcioglu I.E., Sure U., Zhu Y. Implications of Dll4-Notch signaling activation in primary glioblastoma multiforme. Neuro. Oncol. 2013;15:1366–1378. doi: 10.1093/neuonc/not071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sadikovic B., Al-Romaih K., Squire J.A., Zielenska M. Cause and consequences of genetic and epigenetic alterations in human cancer. Curr. Genomics. 2008;9:394–408. doi: 10.2174/138920208785699580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schug J., McKenna L.B., Walton G., Hand N., Mukherjee S., Essuman K., Shi Z., Gao Y., Markley K., Nakagawa M., et al. Dynamic recruitment of microRNAs to their mRNA targets in the regenerating liver. BMC Genomics. 2013;14:264. doi: 10.1186/1471-2164-14-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang J.H., Li J.H., Shao P., Zhou H., Chen Y.Q., Qu L.H. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–D209. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang D., Sun Y., Hu L., Zheng H., Ji P., Pecot C.V., Zhao Y., Reynolds S., Cheng H., Rupaimoole R., et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell. 2013;23:186–199. doi: 10.1016/j.ccr.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacobsen A., Silber J., Harinath G., Huse J.T., Schultz N., Sander C. Analysis of microRNA-target interactions across diverse cancer types. Nat. Struct. Mol. Biol. 2013;20:1325–1332. doi: 10.1038/nsmb.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vlachos I.S., Paraskevopoulou M.D., Karagkouni D., Georgakilas G., Vergoulis T., Kanellos I., Anastasopoulos I.L., Maniou S., Karathanou K., Kalfakakou D., et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43:D153–D159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsu S.D., Tseng Y.T., Shrestha S., Lin Y.L., Khaleel A., Chou C.H., Chu C.F., Huang H.Y., Lin C.M., Ho S.Y., et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42:D78–D85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao F., Zuo Z., Cai G., Kang S., Gao X., Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.