


Abstract
Single-molecule manipulation technologies have been extensively applied to studies of the structures and interactions of DNA and proteins. An important aspect of such studies is to obtain the dynamics of interactions; however the initial binding is often difficult to obtain due to large mechanical perturbation during solution introduction. Here, we report a simple disturbance-free rapid solution exchange method for magnetic tweezers single-molecule manipulation experiments, which is achieved by tethering the molecules inside microwells (typical dimensions–diameter (D): 40–50 μm, height (H): 100 μm; H:D∼2:1). Our simulations and experiments show that the flow speed can be reduced by several orders of magnitude near the bottom of the microwells from that in the flow chamber, effectively eliminating the flow disturbance to molecules tethered in the microwells. We demonstrate a wide scope of applications of this method by measuring the force dependent DNA structural transitions in response to solution condition change, and polymerization dynamics of RecA on ssDNA/SSB-coated ssDNA/dsDNA of various tether lengths under constant forces, as well as the dynamics of vinculin binding to α-catenin at a constant force (< 5 pN) applied to the α-catenin protein.
INTRODUCTION
Over the past decades, single molecule manipulation methods have emerged as versatile and sensitive tools to investigate properties of DNA, proteins and their interactions. Due to their capability of applying forces to molecules and measuring molecular deformation at a nanometer resolution, they have become powerful tools to address important questions in the emerging field of mechanobiology.
Optical tweezers and magnetic tweezers are two widely applied technologies in single-molecule manipulation studies. Most of previous studies using these two technologies were conducted in the absence of flow to avoid mechanical perturbation that impairs high resolution single-molecule measurements (1). As a result, data during solution exchange often could not be recorded, causing loss of important information regarding the initial conformation and binding dynamics in response to solution condition changes.
Several methods have been developed to probe the initial binding for optical tweezers by quickly moving the trapped bead and its attached molecule from one solution condition to another in laminar flow, and the molecular binding was monitored by fluorescence imaging (2,3). More recently, additional method has been developed to avoid the influence from the flow drag in the laminar flow by further moving the bead attached molecule from the laminar flow into a flow-free harbor which is connected to the laminar flow through a thin neck (4,5). These rapid solution exchange approaches have enabled optical tweezers to probe initial binding dynamics of molecules in response to solution condition changes.
However, the above bead-moving and laminar flow based rapid solution exchange methods cannot be applied to typical magnetic tweezers experiments, as magnetic tweezers apply forces to a paramagnetic bead attached to molecules immobilized on coverslip surfaces. Solution flow inevitably introduces drag force to the tethered bead, resulting in loss of measurement accuracy during flow exchange. High-speed solution exchange makes things worse, as it would result in large drag force to the bead that can cause large conformational perturbation to the molecule and cause tether breakage. The scope of the applications of magnetic tweezers would be greatly broadened if a rapid disturbance-free solution exchange method could be developed.
In this work, we report a simple method to achieve rapid disturbance-free solution exchange for magnetic tweezers by tethering molecules at the bottom of microwells (typical dimensions tested in our experiments—diameter (D): 40–50 μm; height (H): 100 μm) in a microwell array. Our results show that complete solution exchange can take place in the order of seconds, making it possible to obtain early binding dynamics upon solution exchange at constant forces. To demonstrate the wide scope of applications of this method, we measured the force dependent DNA structural transitions in response to solution condition changes, and polymerization dynamics of RecA on ssDNA/SSB-coated ssDNA/dsDNA of various lengths, as well as dynamics of vinculin D1 domain binding to transiently exposed α-catenin M1 domain under a low force, which were technically challenging in previous magnetic tweezers studies.
MATERIALS AND METHODS
Fabrication of the microwell array
The microwells were fabricated following the procedures described in (6). Positive mould consisting of 40 μm-diameter, 100 μm-height microwells were constructed using UV light lithography on a 4 inch silicone wafer with SU8 photo-resist. The wafer was passivized by silanization and 10:1 PDMS mixture was casted on the SU8 wafer to form a pillar-like negative mould. The PDMS mould was mounted on a glass slide with the top of the pillar facing the slide surface. Then UV curable polymer (OFN-134) was added to the side of the mould and was allowed to completely fill the gap between the PDMS mould and the slide. The slide was then submerged in DI water cured under 200 W UV for 8 min. Then the PDMS mould was carefully lifted from the assembly, leaving the cured polymer microwell membrane on the slide for storage.
Flow channel design
A typical flow channel was assembled by sticking two pieces of coverslips together by parafilm with a chamber volume of ∼ 20 ul (22 mm (L) × 5 mm (W) × 0.2 mm (H)) (Figure 1A&B). The bottom coverslip (32 mm × 22 mm) was functionalized with chemicals such as streptavidin for attaching biotin-labeled molecules of interest. The fabricated array of polymer microwells was stuck to the center of the bottom coverslip inside the flow channel. The channel was placed on an in-house built magnetic tweezers that can apply a wide range of constant forces (7). One entry of the channel was connected to a computer-controlled pump that can draw solution from the channel to a syringe, while the other entry was connected with a tube for flowing in new solution (Figure 1C). Note that a flow speed in channel can be roughly estimated by withdraw speed (unit of volume) dividing the channel size (W × H, unit of area). A preparation protocol of flow channel with microwell array is included in SI-text S1.
Figure 1.
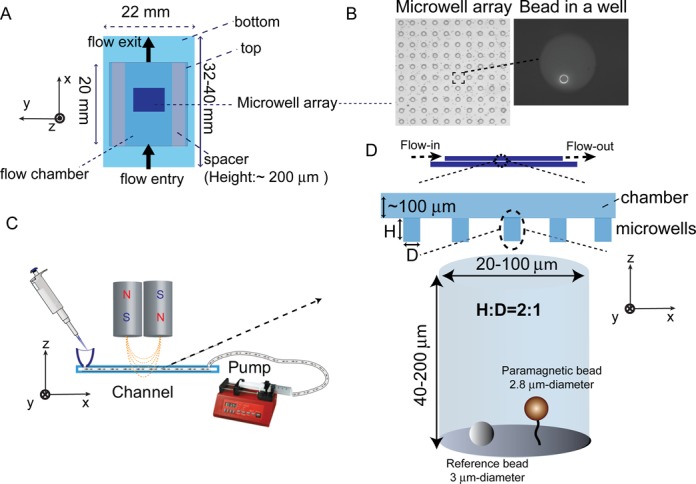
Microwell array and flow channel design. (A) Schematic of flow channel with a middle area covered by a thin microwell array. z- denotes the force direction, x–y plane denotes the focal plane. (B) A part of microwell array (D: 40 μm; H: 100 μm) covered area imaged using a 20X objective (left panel) and a DNA tethered bead at the bottom of a microwell imaged using a 100X objective (right panel). (C) A sketch of the instrument design. Solutions are added to the flow channel entry at the left, while a pump withdraws the solution from the exit at the right. Force is applied to tethers by a pair of magnets placed above the channel. (D) Sketches of the cross-section of the flow channel (top panel) and a tethered molecule at the bottom of a microwell (bottom panel). A reference bead stuck to the coverslip surface used to eliminate spatial drift is also shown.
During the experiments, the molecules of interest were tethered between the bottom coverslip surface and a paramagnetic bead (2.8 μm-diameter Dynal bead (Invitrogen) in our case) inside the microwells (Figure 1D). More details of the magnetic tweezers used in this study and its applications can be found in our previous publications (7,8).
Flow dynamics simulation method
To evaluate flow dynamics inside the experimental chamber, we used SOLIDWORKS Flow Simulation package to solve the Navier-Stokes equation for the setup. In these calculations we modeled the flow of buffer solution (density 1 g/cm3, dynamic viscosity 10−3 Pa s) in the central part of the chamber (dimensions: 5 × 5 × 0.2 mm) containing a single well with various dimensions (diameter 20–100 μm, height 40–200 μm). To estimate the upper bound of the flow velocity at the bottom of the well (where protein/DNA stretching is performed) in the simulation we considered the extreme case when the buffer solution is pumped into the chamber at a very high speed (100 μl/s, ∼16 cm/s). The simulated flow dynamics within the microwells are shown in Figure 2 and Supplementary Figure S1.
Figure 2.
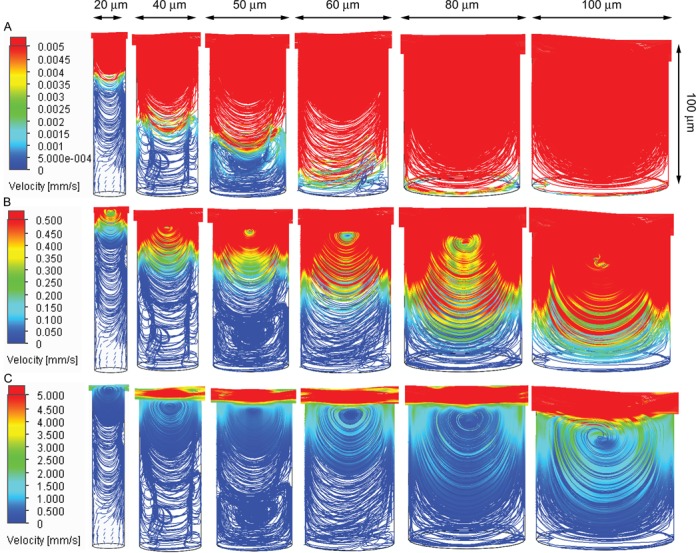
Simulated flow dynamics inside the microwells. (A–C). Flow dynamics inside microwells with a fixed height of 100 μm and various diameters from 20 μm to 100 μm (from left to right), with different flow speed scale bars. In the simulations, the flow speed in the center of the channel is set at 100 μl/s (∼16 cm/s). The details of the simulations are described in the methods section.
DNA and proteins
The end-labeled 576 bp dsDNA, 576 nt ssDNA, 600 bp end-closed GC-rich DNA and 48,502 bp λ-DNA are generated as previously described (9–12). The SSB and RecA proteins are generous gifts from K. Muniyappa. The α-catenin and vinculin proteins are prepared as previously described (13,14).
RESULTS
Simulations of the flow dynamics in the microwell
We first searched for dimensions of the microwells such that it could simultaneously achieve disturbance-free and rapid exchange between the solution in the microwell and that in the channel. The former requires high height-to-diameter (H:D) aspect ratio while the latter requires low height of the microwell (Figure 1D). The flow dynamics in the microwells in the presence of flow with constant velocity in the channel was simulated using Flow Simulation package from SOLIDWORKS (Methods). We found that microwells with diameters of 20–100 μm with an aspect ratio of H:D ∼2:1 could meet our need.
As shown in Figure 2, for microwells with diameters of 20–100 μm and a fixed height of 100 μm, when a high-speed flow of 100 μl/s (∼16 cm/s) was applied in the channel, flow vortices are developed at the upper level of the microwells, while the flow speeds are reduced significantly near microwell bottom. For example, for microwells with diameter of 40–50 μm and height of 100 μm (H:D ∼ 2:1), the flow speed is drastically reduced by ∼106-folds (< 0.1 μm/s) within 20 μm from the bottom. Hence the flow perturbation on the tethered molecules (with extension < 20 μm for most of single-molecule experiments) at the bottom is negligible. In addition, the simulated flow dynamics of microwells with 40–200 μm heights at a fixed H:D ratio of 2:1 (Supplementary Figure S1) consistently shows drastically reduced flow speed near bottom of microwells. Further, we estimated the complete solution exchange takes a time scale of several seconds for these microwells (SI-text S2, Supplementary Table S1).
Experimental validation of disturbance elimination in the microwell
According to the above simulations, the flow velocity is dramatically reduced to a negligible level near the bottom of the microwells in the presence of high-speed flow in the channel. We tested this prediction by monitoring the position of a 2.8-μm-diameter paramagnetic bead attached to one end of single dsDNA tethers before, during and after a flow of 60 μl/s (∼10 cm/s) was applied at constant forces generated by the magnets. Figure 3A show results obtained from single dsDNA tethers with various contour lengths (top: 576 bp; middle: ∼3.5 kbp, bottom: ∼48.5 kbp), where the flow in channel has negligible perturbation to the extensions of tethered molecules for all the molecules, consistent with the simulation results. As such, the designed microwell array is suitable for magnetic tweezers single-molecule manipulation experiments with disturbance-free rapid solution exchange.
Figure 3.
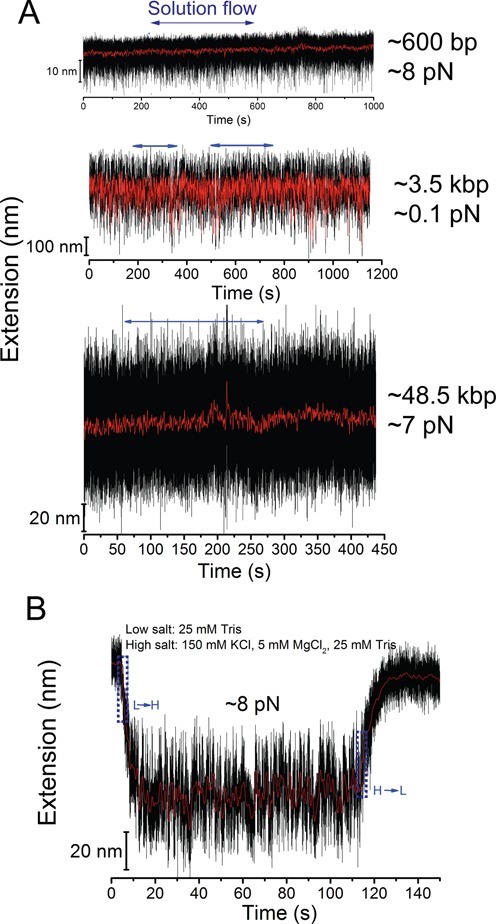
Experimental test of the performance of the disturbance-elimination method. (A) Representative extension time traces of dsDNA molecules with three different contour lengths (top—576 bp; middle—∼3.5 kbp; bottom—∼48.5 kbp) at the bottom of a microwell (H: 100 μm; D: 40 μm) before, during and after introduction of a flow with speed of ∼10 cm/s. The horizontal blue arrows indicate the period of flow. The results show that the extension fluctuation is not affected by the flow. (B) Representative extension time traces of a 576 nt ssDNA switched between a low salt concentration (20 mM Tris without other salts) and a higher salt concentration (20 mM Tris with 100 mM NaCl and 5 mM MgCl2), which indicates a time scale of several seconds needed for complete solution exchange between fluids in the microwell and in the channel. The blue dash boxes indicate the periods of solution exchange.
Quantification of the solution exchange time scale
In order to quantify the exchange rate between solution in the microwells and that in the channel, we measured the characteristic relaxation time of the ssDNA extension change in response to salt concentration change. Being a highly charged flexible polymer, the force-extension curve of ssDNA has been known highly sensitive to salt concentration (9,15). At the same force, an ssDNA has a longer extension in lower salt concentration due to electric repelling and shorter extension in higher salt concentration due to electric screening. Utilizing this property, we examined the extension change time course of single 576 nt ssDNA tethers during switching between 20 mM Tris without other salts and 20 mM Tris with 100 mM NaCl, 5 mM MgCl2 (Figure 3B). The results show that it took ∼5 s (salt concentration increase) to ∼10 s (salt concentration decrease) for the ssDNA to be relaxed to a new steady extension level upon solution exchange in the channel, indicating that the microwell is capable of rapid complete solution exchange within several seconds during high speed solution introduction in the channel, consistent with our theoretical estimation (SI-text 2 and Supplementary Table S1). The slightly longer time scale involved in the salt-decrease induced ssDNA extension elongation than ssDNA extension reduction during salt increase can be explained by a longer time needed for decreasing than increasing salt concentration (SI-text S3).
Interconversions between force-dependent DNA structures induced by salt concentration change
In this and following sections, we demonstrate the applications of this disturbance-free, rapid solution exchange method by probing the force dependent DNA structural transitions in response to solution change, and the polymerization dynamics of RecA on ssDNA/SSB-coated ssDNA/λ-DNA, as well as binding of vinculin D1 domain to transiently exposed α-catenin M1 domain under a low force (∼2 pN).
Tensile force of ∼ 65 pN can induce a so-called DNA overstretching transition for torsion-unconstrained DNA, elongating the contour length of B-DNA by about ∼ 1.7 fold (16,17). Later studies have revealed three possible overstretched DNA structures (18–34): 1) peeled ssDNA—a DNA strand peeling apart from the other at open ends or nicks to produce a ssDNA under tension while the other coils; 2) DNA bubble—two DNA stands separating from inside to produce two antiparallel ssDNA sharing tension; and 3) S-DNA—a new form of elongated, base-paired DNA.
The relative stability of different DNA structures is highly sensitive to environmental factors that influence DNA basepair stability such as sequence, temperature and salt concentration (31–35). Therefore, their inter-conversions can be induced by changing these factors while maintaining a constant force applied to the DNA, as demonstrated in a recent study through changing salt concentrations (36). Surprisingly, in that study, the NaCl concentration decrease induced B-to-S transition took > 40 s to finish, which was > 10 times slower than that observed in previously reported force-induced B-to-S transition at a constant NaCl concentration. We reason that this slow salt decrease induced B-to-S transition was caused by the slow solution exchange (> 5 minutes) that was needed to avoid flow perturbation in that study. To test it, we used the disturbance-free rapid solution exchange method to revisit the dynamics of inter-conversions between B-DNA and S-DNA induced by increasing/decreasing NaCl concentrations under a constant force.
Figure 4A shows the extension change of a 600 bp GC-rich torsion-unconstrained, end-closed DNA, being held at a constant force of ∼ 60 pN at 23o, induced by decreasing/increasing NaCl concentration between 250 mM and 0 mM in 20 mM Tris (pH 7.4). We observed inter-conversion between the B-DNA at 250 mM with a shorter extension and an overstretched DNA structure at 0 mM NaCl with an elongated extension by ∼ 1.6 fold. Due to the use of the end-closed (therefore peeling is pre-excluded) GC-rich DNA, the overstretched form is likely the S-DNA, which could be mixed with some small fraction of internally melted DNA (32,35).
Figure 4.
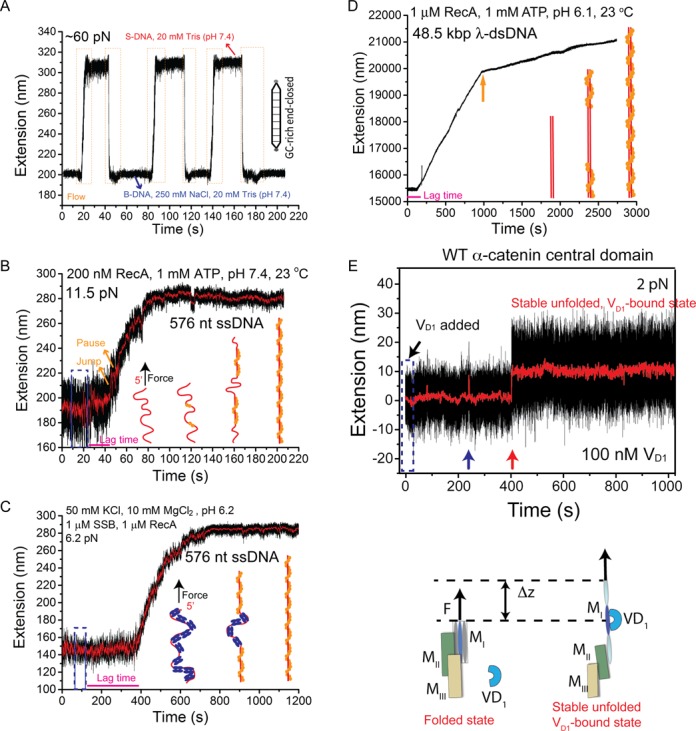
Representative applications. (A) Interconversions of a ∼ 600 bp end-closed GC-rich dsDNA between the B-form and the overstretched S-form DNA structures induced by salt concentration change at a constant force of ∼ 60 pN. During cycles of switching between 250 mM and 0 mM NaCl in 20 mM Tris (pH 7.4), the DNA extension correspondingly switched between a shorter level (B-DNA) in 250 mM NaCl and a longer level (S-DNA) in 0 mM NaCl. The orange dash boxes indicate the time windows during rapid solution exchange. (B) A typical extension time trace of a 576 nt ssDNA before, during and after 200 nM RecA (with 1 mM ATP, 1x ATP regeneration system, 50 mM NaCl, 10 mM MgCl2, 20 mM Tris pH 7.4) was introduced at a constant force of ∼ 11 pN. (C) A typical extension time trace of a 576 nt SSB-coated ssDNA before, during and after solution containing 1 μM RecA and 1 μM SSB (with 1 mM ATP, 1x ATP regeneration system, 50 mM NaCl, 10 mM MgCl2, 20 mM MES pH 6.2) was introduced at a force of ∼ 6.5 pN. For B&C, schematics of the RecA polymerization process are shown in figure panels. (D) A representative extension time trace of a 48.5 kbp λ-DNA before, during and after 1 μM RecA (with 1 mM ATP, 1x ATP regeneration system, 50 mM NaCl, 10 mM MgCl2, 20 mM MES pH 6.1) was introduced at a constant force of ∼ 10 pN. The solution was introduced within 10 s at the beginning of the time trace. The spike at ∼ 240 s is due to transient diffusion of a polystyrene bead into the view area that affected the bead imaging. (E) A representative time trace of the extension of an α-catenin MI domain tether during and after rapid introduction of 100 nM vinculin D1 solution at a constant low force of ∼ 2 pN. The short-lived spikes in 0–400 s are transient unfolding of the α-catenin MI domain. A vinculin D1 molecule binds to transiently exposed α-catenin MI domain at ∼ 400 s, stabilizing it in the unfolded state, as illustrated in the bottom panel. Blue dash boxes in panels B–E indicate the flow periods.
Such DNA structural interconversion induced by decreasing/increasing NaCl concentration was highly reproducible, which took place immediately upon solution exchange and completed within ∼ 3 s (during salt decrease) and ∼ 0.3 s (during salt increase). Given that it takes ∼ 5 s for complete solution exchange in our system, the result indicates that the actual transition dynamics are faster than the observed time scales. The longer time scale involved in the salt-decrease induced DNA elongation transition than that in the reverse transition during salt increase can be explained by a longer time needed for decreasing than increasing salt concentration (supplementary SI-text S3) and/or due to that the critical salt concentration for the transition might not be in the middle of the two concentrations we tested.
The polymerization dynamics of RecA on ssDNA
RecA is a crucial recombinase in bacteria that plays a critical role in homologous recombinational DNA damage repair. It nucleates and polymerizes on ssDNA, forming a rigid extended helical nucleoprotein filament (9,10,37–40). RecA binds to ssDNA with a high affinity of ∼ 100 nM and polymerizes with high cooperativity in a preferential 5′-to-3′ direction (37). RecA filament can also polymerize on one ssDNA strand inside a double-stranded DNA (10,37). Due to a high nucleation barrier and slowed-down polymerization rate inside dsDNA, its polymerization was able to be measured from 2 to 20 monomers/s on micrometer sized DNA (37,38,41,42). In contrast to dsDNA, due to decreased nucleation barrier and faster polymerization rate, the dynamics of RecA polymerization on ssDNA has been difficult to be probed in previous single-ssDNA stretching experiments. To our knowledge, there was only one single-ssDNA stretching experiment reporting the polymerization dynamics of RecA, which was achieved using a long ssDNA (7.3 kb) that allowed a longer time of observation (40). Here we show that the RecA nucleation and polymerization dynamics can be conveniently recorded on much shorter (∼ 600 bp) ssDNA (therefore better signal-to-noise ratio in extension measurements) using the microwell based disturbance-free rapid solution exchange method.
Figure 4B shows a typical time trace of the extension of a 576 nt ssDNA under a constant force of ∼ 11.5 pN, before, during and after 200 nM RecA was introduced with a flow speed of ∼7 mm/s, in the standard RecA reaction buffered solution containing 50 mM NaCl, 10 mM MgCl2, 1 mM ATP, 1x ATP regeneration system and 20 mM Tris-HCl (pH 7.4), at 23°C. The polymerization of RecA began several seconds after the complete solution exchange of RecA solution. We reason that the several seconds lag time (indicated by the magenta line) corresponds to the time for RecA nucleation, as it has been known that RecA polymerization requires a nucleation step (37). The elongation of the ssDNA extension is due to formation of the rigid RecA nucleoprotein filament (bending persistence of RecA filament is ∼ 1000 nm (40)).
The progressive elongation shows an interesting jump-pause-like dynamics of RecA polymerization (indicated by orange arrows). We reason that the jump corresponds to a cooperative polymerization from a newly nucleated RecA patch till the canonical polymerizing end (the 3′ end) (37) reached the end of ssDNA, or reached the 5′ end of an existing downstream RecA filament. Each pause can be explained to be the time for formation of a new nucleation site. Overall, the results are consistent with previously reported cooperative nucleation and polymerization of RecA on ssDNA (37).
It took about 60 s to reach a steady elongated extension with a total extension increase of ∼ 80 nm. It has been known that in RecA-ssDNA nucleoprotein filament, a RecA monomer occupies 3 nt of ssDNA. Therefore, our result of fully polymerization on the 576 nt ssDNA reveals an overall polymerization rate of 3 ± 1 monomer s−1 (calculated from three independent experiments) in 200 nM RecA solution under our solution condition. Given that multiple nucleation sites might exist in the 576 nt ssDNA, the observed overall polymerization rate might be greater than that of a single filament.
Dynamics of RecA loading onto SSB coated ssDNA
In bacteria, newly generated ssDNA fragment is immediately bound by SSB to prevent the formation of secondary structure or ssDNA degradation. The nucleoprotein array formed on ssDNA by SSB on one hand facilitates RecA loading onto ssDNA by removal of secondary structure, on the other hand suppresses the RecA filament formation due to the energy cost to remove the SSB on ssDNA (37). The dynamics and kinetics of RecA loading onto SSB coated ssDNA therefore are physiologically important. Recently, the RecA loading onto SSB coated ssDNA was probed using combination of optical traps and fluorescent dynamic imaging in the presence of laminar flow, which revealed a bi-directional (5′-to-3′ and 3′-to-5′) loading processes of RecA onto SSB-coated ssDNA (2). Here, we demonstrate fluorescence-label-free measurements of the dynamics of RecA loading onto SSB-ssDNA nucleoprotein array using the disturbance-free rapid solution exchange method with magnetic tweezers.
In the experiments, a fully covered SSB nucleoprotein array was formed on ssDNA by incubating the ssDNA tether with 1 μM SSB (in standard RecA reaction solution at pH 6.2) at ∼ 6 pN. Then, a mixture of 1 μM SSB and 1 μM RecA was introduced. The extension of ssDNA was recorded before, during and after the introduction of the protein solution (Figure 4C). After a lag phase of ∼ 200 s (indicated by magenta line) following the protein introduction, the extension began to gradually increase without apparent pause-jump-like dynamics observed in the absence of SSB. It took ∼ 400 s till RecA fully covered the ssDNA. From the extension time traces, a loading rate of ∼ 0.5 monomer s−1 was estimated, which is consistent with recent reported speed of 0.1–1 monomer s−1 by fluorescent dynamic imaging using optical tweezers (2).
Multiple independent experiments revealed that the lag time before RecA loading onto SSB-coated ssDNA is over ∼10-folds longer than that of loading onto naked ssDNA. Further, the growing rate of RecA filament on SSB-coated ssDNA is ∼ 10 folds slower than that on the naked ssDNA. These results indicate that SSB imposes a large energy barrier for both RecA nucleation and polymerization on ssDNA. Moreover, the lack of apparent jump-pause-like nucleation and polymerization dynamics of on SSB coated ssDNA indicates that the growing dynamics is likely restricted by removing the SSB from ssDNA, which reduces the level of polymerization cooperativity. Overall, these results demonstrated that the disturbance-free solution exchange method provides magnetic tweezers with new capability to investigate the dynamics of complex interplay between ssDNA processing proteins.
The polymerization dynamics of RecA on long double-stranded λ-DNA
To demonstrate that the disturbance-free rapid solution exchange method is also suitable for longer tethers, we used RecA polymerization dynamics on long λ-DNA (48,502 bp, ∼ 16 μm) as an example. As shown in Figure 4D, after RecA solution (1 μM) was introduced, there was a lag phase of over 200 s before RecA polymerization started. This long lag phase likely corresponds to nucleation of RecA inside dsDNA. After the nucleation step, RecA began to polymerize on the dsDNA, indicated by progressive extension increase of the dsDNA with a rapid growth phase followed by a slowly growing phase after a transition point (indicated by orange arrow in Figure 4E). These observations are consistent with previous observations (37,38,41,42). Together, these results demonstrate that this method is capable to probe dynamics of DNA and DNA-protein interactions over a wide range of length scales.
Dynamics of conformational change and interactions of mechanosensing proteins
Fine-tuning transmission of mechanical load from cell to extracellular matrix (ECM) and from cell to cell is essential for cell survival, growth and migration (43). Recent studies have uncovered a set of mechanosensitive proteins that are a part of the linkage between the cytoskeleton and cell adhesions (43). They are responsible for sensing mechanical force and regulating cell-ECM and cell-cell adhesions. Such proteins are often activated at low forces (e.g. ∼ 5 pN for talin and α-catenin) (13,14), leading to a difficulty in single-molecule studies of the activation of these proteins. This is because the drag force during solution introduction can easily exceed 5 pN that may cause uncontrolled activation of such proteins. Here, we demonstrate the application of the disturbance-free rapid solution exchange method to investigate the dynamics of vinculin binding to α-catenin that plays crucial mechansensing function at cell-cell junction, at a constant force (< 5 pN) applied to α-catenin.
Figure 4E shows a typical time trace of a tether of α-catenin central domain composed of a vinculin binding domain MI followed by the so-called modulation domains MII and MIII (13), under a constant force of ∼ 2 pN after rapid introduction (∼10 cm/s) of 100 nM vinculin D1 domain solution in the channel. The vinculin D1 is known binding to a single α-helix masked inside the folded α-catenin MI domain, and its binding requires unfolding of the MI domain at forces > 5 pN (13) (illustration in Figure 4E). At 2 pN, the α-catenin MI domain is predominated in the folded conformation, with a very low unfolding probability of only 10% (13). Such rare unfolding events are indicated by the spikes in the extension (blue arrow in Figure 4E).
An interesting question is whether such transiently unfolded conformation could expose the vinculin-binding site for the vinculin D1 binding. Our experiments showed that the vinculin D1 indeed could bind to the transiently exposed vinculin binding site at 2 pN, indicated by the stable elongated extension after binding occurred (Figure 4E, red arrow). The unfolded conformation of the α-catenin MI was locked by a stably bound vinculin D1 domain, indicated by the stable extension remained at the unfolded level for more than 10 minutes. Note that the top panel of Figure 4E is adapted from our previous publication (13). This example also demonstrates the advantage of the method; otherwise, unwanted unfolding of the α-catenin MI would occur due to drag force during rapid introduction of vinculin D1 solution.
DISCUSSION
In this work, we report a simple yet useful method that allows disturbance-free rapid solution exchange for magnetic tweezers single-molecule manipulation studies. We show that the flow speed can be dramatically reduced to a negligible level near the bottom of a microwell in an array while maintaining a high-speed flow in the channel, which enables complete solution exchange between the microwell and channel within 5–10 s. To our knowledge, this is the first implementation of disturbance-free rapid solution exchange that is suitable for magnetic tweezers experiments.
We have used several examples to demonstrate the wide scope of its applications to obtain the dynamics of: (i) DNA structural changes in response to salt concentration change under constant force, (ii) RecA polymerization on naked ssDNA/SSB-coated ssDNA/long dsDNA upon RecA solution was introduced and (iii) vinculin D1 domain binding to a transiently exposed α-catenin M1 domain under a low force of ∼ 2 pN. The dynamics of molecular structural changes or interactions revealed in these examples would be difficult to be obtained without implementation of the disturbance-free rapid solution exchange method. Thus, we believe that this method will make magnetic tweezers a more versatile single-molecule manipulation tool.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Dr Virgile Viasnoff's research group, the Nano-Micro-fabrication Core in Mechanobiology Institute and Dr Mark B H Breese in Department of Physics, National University of Singapore for the assistance in microwells preparation.
FUNDING
Singapore Ministry of Education Academic Research Fund Tier 3 [MOE2012-T3-1-001] and Tier 2 [MOE 2013-T2-1-154]; National Research Foundation of Singapore through the Mechanobiology Institute at National University of Singapore. Funding for open access charge: Singapore Ministry of Education Academic Research Fund Tier 3 [MOE2012-T3-1-001].
Author contributions: J.Y., M.Y. and S.L. conceived the study and designed the experiments; S.L., M.Y. and J.C. performed experiments, analyzed and interpreted the data; A.E. performed the simulation; S.A. provided technological support. S.L., M.Y., J.C., A.E. and J.Y. wrote the manuscript.
Conflict of interest statement. None declared.
REFERENCES
- 1.Neuman K.C., Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods. 2008;5:491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell J.C., Plank J.L., Dombrowski C.C., Kowalczykowski S.C. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature. 2012;491:274–278. doi: 10.1038/nature11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Candelli A., Wuite G.J.L., Peterman E.J.G. Combining optical trapping, fluorescence microscopy and micro-fluidics for single molecule studies of DNA-protein interactions. Phys. Chem. Chem. Phys. 2011;13:7263–7272. doi: 10.1039/c0cp02844d. [DOI] [PubMed] [Google Scholar]
- 4.Omar M.A., Miskovsky P., Bano G. Proof-of-principle for simple microshelter-assisted buffer exchange in laser tweezers: interaction of hypericin with single cells. Lab Chip. 2014;14:1579–1584. doi: 10.1039/c3lc51199e. [DOI] [PubMed] [Google Scholar]
- 5.Forget A.L., Dombrowski C.C., Amitani I., Kowalczykowski S.C. Exploring protein-DNA interactions in 3D using in situ construction, manipulation and visualization of individual DNA dumbbells with optical traps, microfluidics and fluorescence microscopy. Nat. Protoc. 2013;8:525–538. doi: 10.1038/nprot.2013.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masters T., Engl W., Weng Z.L., Arasi B., Gauthier N., Viasnoff V. Easy fabrication of thin membranes with through holes. application to protein patterning. PloS One. 2012;7:e44261. doi: 10.1371/journal.pone.0044261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H., Fu H., Zhu X., Cong P., Nakamura F., Yan J. Improved high-force magnetic tweezers for stretching and refolding of proteins and short DNA. Biophys. J. 2011;100:517–523. doi: 10.1016/j.bpj.2010.12.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le S., Chen H., Cong P., Lin J., Droge P., Yan J. Mechanosensing of DNA bending in a single specific protein-DNA complex. Sci. Rep. 2013;3:3508. doi: 10.1038/srep03508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu H., Le S., Chen H., Muniyappa K., Yan J. Force and ATP hydrolysis dependent regulation of RecA nucleoprotein filament by single-stranded DNA binding protein. Nucleic Acids Res. 2013;41:924–932. doi: 10.1093/nar/gks1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu H., Le S., Muniyappa K., Yan J. Dynamics and regulation of RecA polymerization and de-polymerization on double-stranded DNA. PloS One. 2013;8:e66712. doi: 10.1371/journal.pone.0066712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le S., Chen H., Zhang X., Chen J., Patil K.N., Muniyappa K., Yan J. Mechanical force antagonizes the inhibitory effects of RecX on RecA filament formation in Mycobacterium tuberculosis. Nucleic Acids Res. 2014;42:11992–11999. doi: 10.1093/nar/gku899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J., Le S., Basu A., Chazin W.J., Yan J. Mechanochemical regulations of RPA's binding to ssDNA. Sci. Rep. 2015;5:9296. doi: 10.1038/srep09296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao M.X., Qiu W., Liu R.C., Efremov A.K., Cong P.W., Seddiki R., Payre M., Lim C.T., Ladoux B., Mege R.M., et al. Force-dependent conformational switch of alpha-catenin controls vinculin binding. Nat. Commun. 2014;5:4525. doi: 10.1038/ncomms5525. [DOI] [PubMed] [Google Scholar]
- 14.Yao M.X., Goult B.T., Chen H., Cong P.W., Sheetz M.P., Yan J. Mechanical activation of vinculin binding to talin locks talin in an unfolded conformation. Sci. Rep. 2014;4:4610. doi: 10.1038/srep04610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McIntosh D.B., Saleh O.A. Salt species-dependent electrostatic effects on ssDNA elasticity. Macromolecules. 2011;44:2328–2333. [Google Scholar]
- 16.Smith S.B., Cui Y.J., Bustamante C. Overstretching B-DNA: The elastic response of individual double-stranded and single-stranded DNA molecules. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 17.Cluzel P., Lebrun A., Heller C., Lavery R., Viovy J.L., Chatenay D., Caron F. DNA: An extensible molecule. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 18.Leger J.F., Romano G., Sarkar A., Robert J., Bourdieu L., Chatenay D., Marko J.F. Structural transitions of a twisted and stretched DNA molecule. Phys. Rev. Lett. 1999;83:1066–1069. [Google Scholar]
- 19.Rief M., Clausen-Schaumann H., Gaub H.E. Sequence-dependent mechanics of single DNA molecules. Nat. Struct. Biol. 1999;6:346–349. doi: 10.1038/7582. [DOI] [PubMed] [Google Scholar]
- 20.Williams M.C., Wenner J.R., Rouzina L., Bloomfield V.A. Effect of on on the overstretching transition of double-stranded DNA: Evidence of force-induced DNA melting. Biophys. J. 2001;80:874–881. doi: 10.1016/S0006-3495(01)76066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouzina I., Bloomfield V.A. Force-induced melting of the DNA double helix. 2. Effect of solution conditions. Biophys. J. 2001;80:894–900. doi: 10.1016/S0006-3495(01)76068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rouzina I., Bloomfield V.A. Force-induced melting of the DNA double helix - 1. Thermodynamic analysis. Biophys. J. 2001;80:882–893. doi: 10.1016/S0006-3495(01)76067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rouzina I., Williams M.C., Wenner J., Bloomfield V.A. DNA melting by mechanical force. Biophys. J. 2001;80:338a–339a. doi: 10.1016/S0006-3495(01)76066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenner J.R., Williams M.C., Rouzina I., Bloomfield V.A. Salt dependence of the elasticity and overstretching transition of single DNA molecules. Biophys. J. 2002;82:3160–3169. doi: 10.1016/S0006-3495(02)75658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cocco S., Yan J., Leger J.F., Chatenay D., Marko J.F. Overstretching and force-driven strand separation of double-helix DNA. Phys. Rev. E. 2004;70:011910. doi: 10.1103/PhysRevE.70.011910. [DOI] [PubMed] [Google Scholar]
- 26.Calderon C.P., Chen W.H., Lin K.J., Harris N.C., Kiang C.H. Quantifying DNA melting transitions using single-molecule force spectroscopy. J. Phys-Condens. Mat. 2009;21:34114. doi: 10.1088/0953-8984/21/3/034114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Mameren J., Gross P., Farge G., Hooijman P., Modesti M., Falkenberg M., Wuite G.J.L., Peterman E.J.G. Unraveling the structure of DNA during overstretching by using multicolor, single-molecule fluorescence imaging. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18231–18236. doi: 10.1073/pnas.0904322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gross P., Laurens N., Oddershede L.B., Bockelmann U., Peterman E.J.G., Wuite G.J.L. Quantifying how DNA stretches, melts and changes twist under tension. Nat. Phys. 2011;7:731–736. [Google Scholar]
- 29.Paik D.H., Perkins T.T. Overstretching DNA at 65 pN does not require peeling from free ends or nicks. J. Am. Chem. Soc. 2011;133:3221–3219. doi: 10.1021/ja108952v. [DOI] [PubMed] [Google Scholar]
- 30.Bongini L., Melli L., Lombardi V., Bianco P. Transient kinetics measured with force steps discriminate between double-stranded DNA elongation and melting and define the reaction energetics. Nucleic Acids Res. 2014;42:3436–3449. doi: 10.1093/nar/gkt1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu H., Chen H., Zhang X., Qu Y., Marko J.F., Yan J. Transition dynamics and selection of the distinct S-DNA and strand unpeeling modes of double helix overstretching. Nucleic Acids Res. 2011;39:3473–3481. doi: 10.1093/nar/gkq1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X., Chen H., Le S., Rouzina I., Doyle P.S., Yan J. Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching by single-molecule calorimetry. Proc. Natl. Acad. Sci. U.S.A. 2013;110:3865–3870. doi: 10.1073/pnas.1213740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X.H., Chen H., Fu H.X., Doyle P.S., Yan J. Two distinct overstretched DNA structures revealed by single-molecule thermodynamics measurements. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8103–8108. doi: 10.1073/pnas.1109824109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu H.X., Chen H., Marko J.F., Yan J. Two distinct overstretched DNA states. Biophys. J. 2011;100:176–176. [Google Scholar]
- 35.King G.A., Gross P., Bockelmann U., Modesti M., Wuite G.J.L., Peterman E.J.G. Revealing the competition between peeled-Ssdna, melting bubbles and S-DNA during DNA overstretching using fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 2013;110:3859–3864. doi: 10.1073/pnas.1213676110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X., Qu Y., Chen H., Rouzina I., Zhang S., Doyle P.S., Yan J. Interconversion between three overstretched DNA structures. J. Am. Chem. Soc. 2014;136:16073–16080. doi: 10.1021/ja5090805. [DOI] [PubMed] [Google Scholar]
- 37.Cox M.M. Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 2007;42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- 38.Galletto R., Amitani I., Baskin R.J., Kowalczykowski S.C. Direct observation of individual RecA filaments assembling on single DNA molecules. Nature. 2006;443:875–878. doi: 10.1038/nature05197. [DOI] [PubMed] [Google Scholar]
- 39.Merrin J., Kumar P., Libchaber A. Effects of pressure and temperature on the binding of RecA protein to single-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 2011;108:19913–19918. doi: 10.1073/pnas.1112646108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Loenhout M.T., van der Heijden T., Kanaar R., Wyman C., Dekker C. Dynamics of RecA filaments on single-stranded DNA. Nucleic Acids Res. 2009;37:4089–4099. doi: 10.1093/nar/gkp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shivashankar G.V., Feingold M., Krichevsky O., Libchaber A. RecA polymerization on double-stranded DNA by using single-molecule manipulation: The role of ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7916–7921. doi: 10.1073/pnas.96.14.7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Heijden T., van Noort J., van Leest H., Kanaar R., Wyman C., Dekker N., Dekker C. Torque-limited RecA polymerization on dsDNA. Nucleic Acids Res. 2005;33:2099–2105. doi: 10.1093/nar/gki512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogel V., Sheetz M. Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell. Biol. 2006;7:265–275. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.