

Abstract
Ocular coloboma is a common eye malformation resulting from incomplete fusion of the optic fissure during development. Coloboma is often associated with microphthalmia and/or contralateral anophthalmia. Coloboma shows extensive locus heterogeneity associated with causative mutations identified in genes encoding developmental transcription factors or components of signaling pathways. We report an ultra-rare, heterozygous frameshift mutation in FZD5 (p.Ala219Glufs*49) that was identified independently in two branches of a large family with autosomal dominant non-syndromic coloboma. FZD5 has a single-coding exon and consequently a transcript with this frameshift variant is not a canonical substrate for nonsense-mediated decay. FZD5 encodes a transmembrane receptor with a conserved extracellular cysteine rich domain for ligand binding. The frameshift mutation results in the production of a truncated protein, which retains the Wingless-type MMTV integration site family member-ligand-binding domain, but lacks the transmembrane domain. The truncated protein was secreted from cells, and behaved as a dominant-negative FZD5 receptor, antagonizing both canonical and non-canonical WNT signaling. Expression of the resultant mutant protein caused coloboma and microphthalmia in zebrafish, and disruption of the apical junction of the retinal neural epithelium in mouse, mimicking the phenotype of Fz5/Fz8 compound conditional knockout mutants. Our studies have revealed a conserved role of Wnt–Frizzled (FZD) signaling in ocular development and directly implicate WNT–FZD signaling both in normal closure of the human optic fissure and pathogenesis of coloboma.
Introduction
Ocular coloboma (OC) is a developmental structural defect caused by the abnormal persistence of the optic fissure in post-embryonic life. In combination with microphthalmia (small eyes) and anophthalmia (absent eyes), OC represents a spectrum of malformations that account for an estimated 10–15% of pediatric blindness (1). Transcription factors and signaling pathways play crucial roles in optic-cup morphogenesis and fissure closure (2,3). Accordingly, human genetic studies together with vertebrate models have implicated bone morphogenetic protein (4–7), Hedgehog (Hh) (8), retinoic acid (9–11) and Hippo (12) pathways in the pathogenesis of these ocular malformations (8,13–15). Defects in components of Wnt signaling have been attributed to syndromic and non-syndromic ocular diseases, including Norrie disease (16,17), osteoporosis-pseudoglioma syndrome (18) and familial exudative vitreoretinopathy (16,17,19–21), but not as a cause of abnormal ocular morphogenesis.
A growing body of evidence from several vertebrate models indicates that Wnt signaling is indispensable in optic-field development and ocular morphogenesis. In the Wnt pathway, non-canonical (β-catenin-independent) signaling interacts with canonical (β-catenin-dependent) signaling to control presumptive retinal versus forebrain fates (22). Loss of non-canonical ligands, Wnt5 and Wnt11, causes failure of eye field segregation (22), whereas inactivation of β-catenin prior to optic vesicle differentiation causes anophthalmia (23). At later stages of development, the canonical pathway also contributes to optic-cup morphogenesis, with overexpression of the Wnt inhibitor Dkk1 leading to abnormal lens formation and coloboma (24,25). Furthermore, the loss of the secreted Frizzled (FZD)-related proteins (known modulators of Wnt signaling) causes defects in optic-cup patterning (26).
The Wnt receptor Fzd5 mediates both canonical and non-canonical signaling (22,27). During eye field specification, Fzd5 is specifically expressed in evaginating eye precursors (28,29). In zebrafish, Wnt11–Fzd5 signaling promotes eye field specification using the non-canonical pathway (22). In Xenopus, Fzd5 acting via the canonical pathway controls the neural potential of retinal progenitors through regulation of Sox2 (30). Mouse Fzd5−/− mutants display extreme optic-cup invagination defects with failure to induce lens formation (31), whereas conditional Fzd5 mutants (Supplementary Material, Fig. S1) exhibit both microphthalmia and coloboma with disrupted retinal epithelial apical junctions (27,32) implicating Fzd5 in mammalian ocular morphogenesis and early neurogenesis. Additionally, mouse knockout mutants of Lrp6, encoding Fzd co-receptor presumed to be in the canonical Wnt signaling pathway (33), demonstrate ocular phenotypes similar to those observed in the conditional Fzd5 mutants. We therefore hypothesized that mutations in FZD5 may be involved in the development of human congenital ocular malformations. In this study, an unbiased genetic screen, and a candidate gene approach were used with both identifying the same single novel frameshift mutation in FZD5 in a large, extended family in which non-syndromic OC segregated as an autosomal dominant disorder. Functional analysis of the mutant protein, using zebrafish, mouse retinal explants and co-culture assays, strongly suggests a dominant-negative effect on WNT signaling, which is likely responsible for optic fissure closure defects. The present study, therefore, directly implicates WNT–FZD signaling in the pathogenesis of human coloboma.
Results
A frameshift mutation in FZD5 causes autosomal dominant coloboma
Whole-exome sequencing (WES) was performed as part of the rare disease component of UK10K (www.uk10k.org) in five members of a large family with autosomal dominant OC (Family 3483; Fig. 1A–C). The affected individuals IV:6, V:1, VI:2 and VI:5 shared only one ultra-rare variant (not present in exome aggregation consortium (ExAC), EVS, 1000G, UK10K internal databases); a frameshift mutation in FZD5 (c.656delCinsAG; p.Ala219Glufs*49, hereafter referred to as A219Xfs*49). This variant was then shown to co-segregate with the disease in all affected individuals available for testing with one exception, IV:7 (Fig. 1A). IV:7 has bilateral coloboma, but is ‘married-in’ to the family being unrelated to the affected individuals VI:2, V:1, IV:1, IV:4 and IV:6 (his wife). He has no prior family history of eye malformations and no other plausible causative variants could be identified in his exome sequence data. Two unaffected individuals (III:2 and V:8) also carried the mutation and were considered as non-penetrant. Targeted re-sequencing of FZD5 in an additional 380 unrelated coloboma patients (COL) from the MRC Human Genetics Unit Cohort as part of UK10K revealed no other potentially pathogenic variants.
Figure 1.

A FZD5 frameshift mutation identified in a family with autosomal dominant coloboma. (A) Six- and three-generation family pedigrees of Family 3483 and Family 111, respectively. The dotted line links these independently ascertained pedigrees carrying the same mutation on an identical haplotype. This link is plausible based on the history obtained from both Mennonite families, with the likely linking individual (Family 3483 II:4) having emigrated from Europe to North America. For Family 3483, ocular images from the affected individuals are shown adjacent to the cognate pedigree symbol. Coloboma patient numbers indicate individuals whose exomes were sequenced. Otherwise, Sanger sequencing was used for segregation analysis, which reveals high (0.8) but incomplete penetrance, as indicated by two obligate carriers that are unaffected. The pedigree key is in the top left corner. (B) Representative images showing eye malformations in affected individuals from Family 111. The LOD score for the combined pedigree is shown below the family tree. (C) Chromatopherogram of the frameshift FZD5 mutation (c.656delCinsAG). (D) Schematic of the human FZD5 gene with hg19 coordinates on chromosome 2. This gene is transcribed in the antisense direction relative to the genomic coordinate numbering. The position of the cDNA mutation is indicated in the ORF, which is entirely contained in the second exon. Below are diagrammatic representations of the WT and ‘mutant’ FZD5 peptides. The WNT-binding domain (dark blue box) is common to both, and the seven transmembrane domains (orange boxes) are present only in the WT protein of 585 residues. The mutation results in the truncation at Ala219 (substituted to Glu) with an aberrant extension of 48 residues (red box), resulting in a protein of 266 amino acids.
Concurrently, FZD5 was screened as a candidate gene, based on mouse studies (27,32), in 172 unrelated individuals with coloboma from cohorts at National Eye Institute (NEI), USA and University of Alberta (U of A), Canada. These studies revealed the identical A219Xfs*49 mutation in all four affected individuals from family 111, where each exhibited bilateral coloboma and related phenotypes (e.g., microphthalmia, and cataract) (Fig. 1A and B).
Haplotype analysis using five microsatellite markers flanking the FZD5 gene suggested a recent common ancestry between Family 3483 and Family 111 (Supplementary Material, Fig. S2). Based on the information provided by family 3483 that individual II:4 had emigrated to North America, this female represented a plausible genetic link with Family 111. In addition, both families are of Mennonite ancestry and originated from the same region in Europe. For the purpose of calculating the two-point LOD score, we designated II:4 in Family 3483 as the maternal great-grandmother of individual I:2 in Family 111, which is the closest possible link based on information from Family 111. This was a conservative approach, as it would generate a minimum possible LOD score associated with co-segregation of the disease and the mutation in the combined family. The linkage analysis was performed using the R package paramlink. Co-segregation of the FZD5 mutation with coloboma in the extended pedigree gave a two-point LOD score (θ = 0) ranging from 3.9 to 4.2 using penetrance values between 0.1 and 1. It was not possible to obtain an accurate estimate of the penetrance for this mutation as we were not able to examine or genotype all apparently unaffected individuals in both branches of the family. However, on the basis of the genotypes, we can safely conclude a relatively high, but incomplete penetrance of the disease mutation.
FZD5 has a single-coding exon with a 5′ non-coding exon. As such the A219Xfs*49 mutant transcript is not predicted to be a substrate for nonsense-mediated decay. The A219Xfs*49 mutation is thus likely to result in production of a truncated FZD5 protein with an intact highly conserved ligand-binding domain [extracellular cysteine rich domain (CRD)], but lacking the seven transmembrane domains (Fig. 1D) (Supplementary Material, Fig. S3).
Within the NEI and U of A cohorts, one additional rare missense variant was identified (c. 290A>T; p.Asp97Val (D97V); Supplementary Material, Table S1 and Fig. S4A and B). This variant is of uncertain significance as this variant was not present in the unaffected mother or brother and the father was deceased (Supplementary Material, Fig. S4A). This variant did not significantly change the FZD5 protein level or its membrane localization by in vitro transfection assay (Supplementary Material, Fig. S4C). Atomic non-local environment assessment predicted that the D97V variant would perturb local interactions (Supplementary Material, Fig. S4D). Super-Topflash (STF) reporter assays indicated a slight, but consistent increase of Wnt9b-stimulated canonical Wnt activity by D97V mutation (Supplementary Material, Fig. S4E) suggesting a gain of function.
Altered expression of FZD5 A219Xfs*49 in zebrafish results in microphthalmia and coloboma
To elucidate the functional relevance of the human FZD5 A219Xfs*49 mutation, zebrafish were used as a model system. Concordant with observations in mouse Fzd5 mutants (27,32), zebrafish fzd5 morphants exhibited coloboma and microphthalmia phenotypes (Fig. 2A–D). In addition, overexpression of the FZD5-A219Xfs*49 mutant in zebrafish embryos also resulted in coloboma and microphthalmia (Fig. 2E–J). Surprisingly, these phenotypes were more prevalent when the wild-type (WT) FZD5 protein was overexpressed (Fig. 2K–L). We noted that the eye size was similar when either WT or FZD5-A219Xfs*49 mutant was overexpressed (Supplementary Material, Fig. S5). These observations suggest that precise FZD5 and/or Wnt signaling dosage is critical for ocular development.
Figure 2.
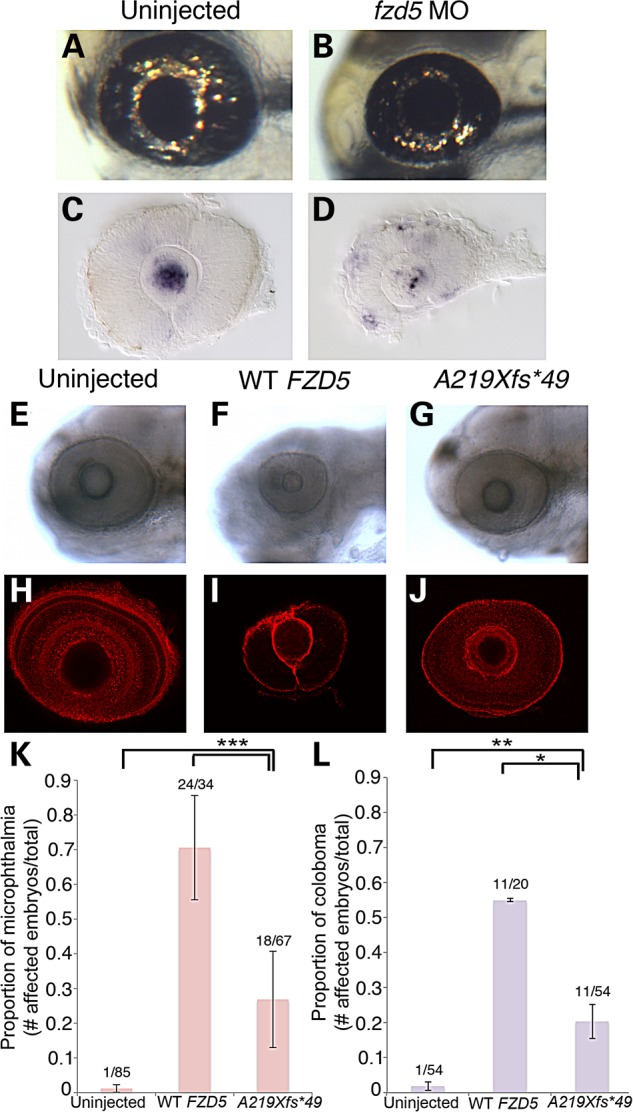
Morpholino knockdown and overexpression of FZD5 causes microphthalmia and coloboma in zebrafish. (A and B) Representative images of live embryos at 3 dpf, either uninjected (A) or injected with 1.2 pmol of fzd5 translation blocking morpholino (MO; B). (C and D) In situ hybridization for GFP was performed at 28 hpf in Tg[TOP:dGFP] embryos to assess levels of canonical Wnt signaling in uninjected (C; n = 26/26 eyes) or fzd5 morphants (D; n = 23/25 eyes). Retinal GFP expression was increased in morphants, while lens expression was decreased (D compare with C), suggesting a tissue-specific function for fzd5 in Wnt signaling. (E–L) Embryos were injected at the one-cell stage with either 200 pg human WT FZD5 mRNA or A219Xfs*49 FZD5 mRNA and imaged to analyze eye size and prevalence of coloboma. Injection of WT FZD5 caused higher incidence of microphthalmia (K, ***P < 0.0002) and coloboma (L, **P = 0.016; *P = 0.008) compared with injection of A219Xfs*49 FZD5 mRNA. All images represent majority of observed phenotypes in each injection group. (E–G) Live images of larvae at 3 dpf; (H–J) eyes labeled with β-laminin antibody at 3 dpf. (K and L) Quantification of ocular phenotypes seen in E–J. (E–L) The number of embryos analyzed for microphthalmia: uninjected (n = 85), WT FZD5 (n = 34), A219Xfs*49 (n = 67), two experimental replicates. The number of embryos analyzed for coloboma: uninjected (n = 54), WT FZD5 (n = 20), A219Xfs*49 (n = 54), two experimental replicates.
FZD5 A219Xfs*49 is a secreted protein that binds to Wnt, but is incapable of mediating Wnt signaling
To further understand the functional consequences of the human FZD5 A219Xfs*49 mutation, we examined mutant protein expression and localization in vitro. Transfection of A219Xfs*49 cDNA construct into Human embryonic kidney (HEK)293 cells produced a truncated FZD5 protein as predicted, containing the entire ligand-binding domain, but not the transmembrane domains. Under non-reducing conditions, the mutant FZD5 A219Xfs*49 protein shows multiple bands in the cell extracts, including one of ∼50 kDa and several ∼21 kDa (Fig. 3A). With the addition of β-mercaptoethanol, the truncated FZD5 protein primarily migrated at a lower-molecular-weight in the ECM fraction (Fig. 3A). Live cell surface immunofluorescence analysis confirmed that truncated FZD5 did not localize to the outer cell membrane (in contrast to the full-length FZD5) and instead displayed punctate and/or irregular extracellular staining (Fig. 3B and Supplementary Material, Fig. S6). As predicted, the A219Xfs*49 truncated FZD5 protein abrogated the ability to mediate both canonical (Fig. 3C, integrated T Cell Factor (TCF)-dependent reporter) and non-canonical WNT signaling activities (Fig. 3D, pulldown assay of Wnt5a stimulated guanosine triphosphate (GTP)-RhoA). An engineered secreted FZD5-CRD protein (sCRD, fused with human Ig-Fc fragment) had an effect similar to the A219Xfs*49 FZD5 mutant (Fig. 3C and D), suggesting that the secretion of the latter is critical for its abnormal function. To examine whether A219Xfs*49 FZD5 binds to Wnt, Co-immunoprecipitation (co-IP) experiments were conducted using cell extracts transfected with Wnt3a-myc, WNT7A-HA, FZD5 and FZD5 A219Xfs*49 constructs in different combinations (34,35). We detected binding of FZD5 A219Xfs*49 to WNT7A as well as FZD5 (Supplementary Material, Fig. S7), suggesting that a competition may exist between A219Xfs*49 FZD5 mutant and WT for WNT utilization.
Figure 3.

FZD5-A219Xfs*49 is incapable of mediating Wnt signaling. (A) Immunoblot analysis of subcellular fractions from transfected HEK293 cells. FZD5 A219Xfs*49mutant protein is detected primarily in ECM fraction. sCRD is expressed in both the culture medium and ECM. CE, cell extract. (B) Live cell immunofluorescence detection. Immunofluorescence staining was conducted to detect FZD5 proteins expression in transfected cells on coated coverslips (see the ‘Materials and Methods’ section). WT FZD5 is primarily present on the cell surface (left panel), while the majority of A219Xfs*49 mutant protein is detected extracellularly (middle panel), presumptively in ECM (doted staining). Negative control with vector transfection is shown in the right panel. (C) Wnt9b-induced canonical Wnt signaling in STF reporter cell line. Cells were transfected with 0.5 µg Wnt9b plasmid combined with 0.25 µg other plasmids. Like sCRD, FZD5 A219Xfs*49 mutant protein is not able to mediate Wnt9b-induced canonical Wnt signaling. The rightmost bar represents Wnt9b-induced canonical Wnt activity by WT FZD5, which is significantly different from all other forms of FZD5. (D) Representative image for active-Rho pulldown assays for non-canonical Wnt signaling. HEK293 cells were transfected with FZD5 WT, A219Xfs*49 and sCRD plasmids, treated with Wnt5a recombinant protein conditioned medium. Active GTP-RhoA assays strictly followed the manufacturer instructions (Cytoskeleton, Inc., Cat. no. Bk-030) (for details see the ‘Materials and Methods’ section). Wnt5a-enhanced formation of GTP-RhoA is obtained in the presence of FZD5, but not A219Xfs*49 mutant or sCRD. The signal intensities were quantified by NIH ImageJ from three immunoblots for three independent experiments, and analyzed by the Microsoft Excel (Student's t-test, ***P < 0.001).
FZD5 A219Xfs*49 antagonizes both canonical and non-canonical Wnt signaling
Given the abnormal function of truncated FZD5 A219Xfs*49, as indicated by its aberrant localization at the plasma membrane, and that A219Xfs*49 was associated with a dominant mode of inheritance in Family 3483 and Family 111, we reasoned that FZD5 A219Xfs*49 may act as a secreted FZD-related protein (36). This acquired secretory function may act non-cell autonomously and antagonize WNT–FZD5 activity expressed from the WT allele. To test this hypothesis, a co-culture assay was developed in which constructs A219Xfs*49 and Wnt9b plus FZD5 were, respectively, transfected into HEK293 and STF cells containing a built-in TCF luciferase reporter. Measurement of luciferase activity (schematically illustrated in Fig. 4A, left panel) revealed dose-dependent, non-cell-autonomous inhibition of FZD5-mediated canonical WNT activity with co-cultured A219Xfs*49 expressing cells (Fig. 4A, middle panel). Moreover, the inhibition was reversed in a dose-dependent manner by increasing FZD5 expression (Fig. 4A, right panel). Similar results were obtained in a Wnt5a/FZD5-induced RhoA activity assay (Fig. 4B and C), which is a measure of non-canonical WNT signaling. Taken together, these data suggest that the A219Xfs*49 mutant protein functions in a dominant, non-cell-autonomous manner to repress FZD5 signaling.
Figure 4.

Non-cell-autonomous dominant-negative effect of FZD5 A219Xfs*49 mutant on Wnt signaling. (A) Wnt9b–FZD5 signaling. All experiments were done in triplicates of at least three independent transfections. Left panel: illustration of the experimental scheme. A fixed amount of Wnt9b and FZD5 was co-transfected with pCAG-Renilla luciferase plasmids (RL, used for internal expression control) into STF cells. Different amounts of FZD5 A219Xfs*49 and sCRD plasmids were transfected into HEK293 cells. After 12 h, both STF and HEK293 cells were collected by trypsin-Ethylenediaminetetraacetic acid (EDTA), mixed at 1:1 ratio and seeded into a new plate for another 36 h. Cell extracts were then prepared for Firefly luciferase and Renilla luciferase assay. Middle panel: inhibition of Wnt9b/FZD5 activity by either A219Xfs*49 or sCRD in a dose-dependent manner. Firefly luciferase activities were normalized against Renilla luciferase, and statistics was performed using the Microsoft Excel software. Right panel: the inhibition of FZD5-mediated Wnt signaling by A219Xfs*49 or sCRD was reversed by augmenting FZD5 expression. (B) Wnt5a–FZD5 signaling. RhoA G-lisa assay showed that Wnt5a/FZD5-stimulated accumulation of GTP-RhoA was abolished by A219Xfs*49 mutant or sCRD protein (compare the right three bars). Samples preparation was as described in Figure 3D, G-lisa assay followed instructions of RhoA G-lisa kit (Cytoskeleton, Inc.). Absorbance of horseradish peroxidase colorimetric reaction was measured by SpectraMax M. The data were quantified by the Microsoft Excel. (C) Inhibition of RhoA activation by A219Xfs*49 or sCRD protein was reverted by increased FZD5 expression. Left panel: similar experimental scheme in (A) was used for testing non-cell-autonomous effects of A219Xfs*49 on Wnt5a/FZD5 induced RhoA activation. Right panel: The inhibition of RhoA activation (G-lisa assay) was reverted by increased FZD5 expression. ***P < 0.001, Student's t-test.
Forced expression of FZD5 A219Xfs*49 in mouse retina leads to apical junction defects similar to those observed in Fzd5/8 compound mutants
Previous studies in mice demonstrated apical junction defects in the retinal pigment epithelium of Fzd5/Fzd8 compound mutant retina, and these were likely to contribute to or cause abnormal neurogenesis and coloboma (32). To examine whether the A219Xfs*49 mutation can mimic a FZD5 dominant loss of function, we overexpressed the FZD5 A219Xfs*49 mutation in mouse retina and evaluated FZD5-related downstream molecular events. Mutant constructs were electroporated into E13.5 mouse retina together with a constitutive Ub-GFP expression vector, and the retina was analyzed after 72 h of culture in vitro. Consistent with apical junction defects in Fzd5-/-;Fzd8+/- compound mutant mouse retina (32), the overexpression of the A219Xfs*49 mutant also caused apical junction defects in cultured retinal explants, as indicated by attenuated expression of atypical protein kinase C (aPKC) (Fig. 5A–F) and RhoA (Fig. 5G–L). Both FZD5 and aPKC are expressed in retinal progenitor cells [see Fig. 5 and (27,32)]. Decreased expression of these proteins likely represents the loss of concentrated apical localization of markers, which would not be demonstrated by immunoblotting. Furthermore, both human and mouse FZD5 showed the same apical retinal localization (Fig. 5M–R), supporting the hypothesis that they may mediate similar molecular events during human and mouse retinal development.
Figure 5.
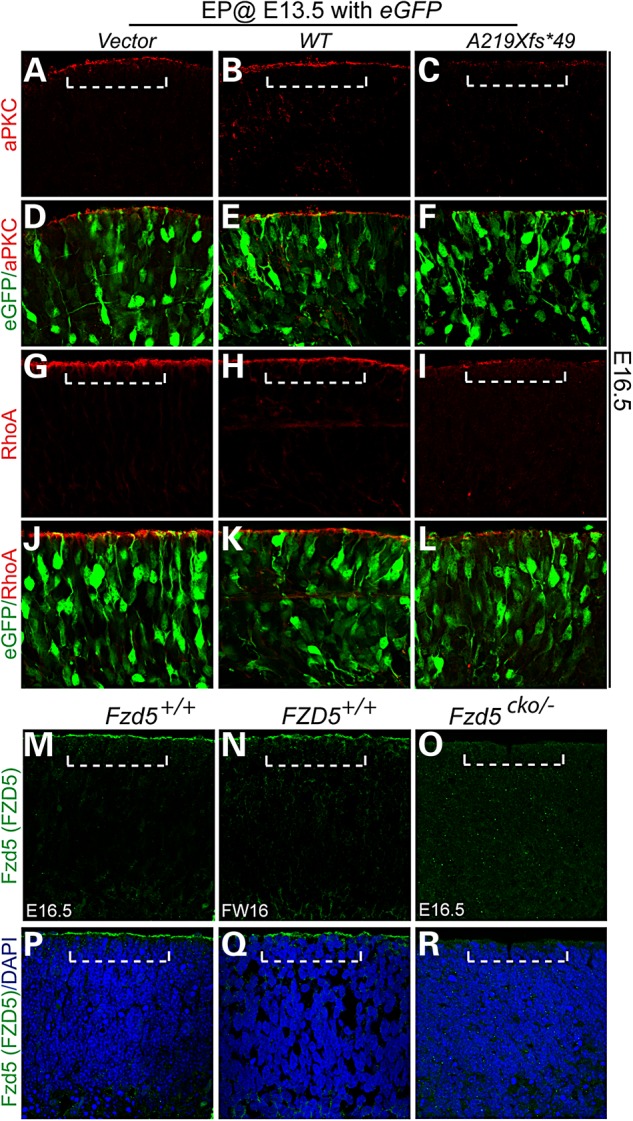
Overexpression of FZD5 A219Xfs*49 led to similar apical junction defects that were reported in mouse Fz5/8 compound mutants. Mouse embryonic (E13.5) retina was dissected and subjected to electroporation supplied with WT FZD5 and FZD5 A219Xfs*49 DNA solution. The retinae were cultured for 72 h and harvested for immunohistochemistry. (A–F), aPKC localization in vector (A), WT FZD5 (WT) (B) and A219Xfs*49 mutant (C) electroporated retinae. Note the loss of apical localization of aPKC in A219Xfs*49-expressing retina (C). (D–F) Images of (A–C) merged with co-elctroporated eGFP, respectively. (G–I) Similar as aPKC, apical RhoA enrichment is also greatly attenuated (compared with G and H). (J–L) Images of (G–I) merged with co-elctroporated eGFP, respectively. (M–R) Localization of FZD5 protein in mouse and human retina. (M) Apical localization of the FZD5 protein in WT mouse retina (above dashed bracket). (N) Same protein localization of FZD5 was detected in human retina. (O) Mouse Fzd5 conditional mutant retina showed the absence of apical FZD5 protein. (P–R) Images from (M–O) merged with 4′,6-diamidino-2-phenylindole, respectively.
Discussion
In the present study, we have identified an ultra-rare frameshift mutation in FZD5 in a large extended family with non-syndromic coloboma segregating as an autosomal dominant disorder. The open reading frame (ORF) of FZD5 is entirely within the second exon, which makes it unlikely that transcript would be subject to nonsense-mediated decay since there is no intron–exon boundary 3′ to the premature termination codon (37). The distinct location of the frameshift in the ORF suggests that the truncated protein would have an antagonistic effect on WNT signaling. This predicted effect was demonstrated in cultured cells, zebrafish retina and mouse retinal explants that establish FZD5 as a strong candidate for human eye malformation(s).
FZD5 mutations with similar predicted dominant-negative effect appear to be extremely rare in human populations. A total of 18 copy number variations (CNVs) encompassing FZD5 locus are listed in the Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources database. Three patients with CNVs have eye abnormalities including cataract (one duplication case) and iris and/or chorioretinal coloboma (two deletion cases). However, a simple phenotype-genotype correlation could not be inferred since the CNV regions are large and include many genes. Only two FZD5 ‘loss-of-function’ alleles, both frameshift, are documented in ExAC. One of these, p.E231Afs*8 is also predicted to generate a secreted WNT-ligand-binding domain with no transmembrane domain. No phenotype information is available for the single individual carrying this mutation in a heterozygous state. Given that non-penetrance has been observed in at least two members of the family presented above, it is possible that this individual is non-penetrant or has microphthalmia, a disorder characterized by reduced ocular size that is closely associated with coloboma. An explanation for the rarity of such mutations may be related to the observation that Fzd5 null mouse embryos die before E11 due to placental angiogenesis defects (38). The non-penetrance of such variants may reflect rescue via genetic background effects and/or compensation by paralogs. The latter effect is prominent in Fzd5/Fzd8 mutant mice (32) although no obvious FZD8 mutations compatible with a digenic effect were identified in WES in the individuals presented here. Notably, similar non-penetrance has been observed in patients with autosomal dominant coloboma due to YAP1 (12) and SHH (8) mutation.
Our results demonstrate a direct role for WNT–FZD signaling in optic fissure closure during human eye development. The A219Xfs*49 mutation converts FZD5 from a membrane-bound WNT receptor to a secreted FZD antagonist that, by competing with WNT ligands or dimerization with WT FZD5 (on the cell surface), might impart dominant-negative characteristics on WNT signaling. As a result of the disrupted WNT signaling, retinal neuroblasts exhibit apical junction defects (Supplementary Material, Fig. S8) (32), which could directly or indirectly impact proliferation, survival and maturation of progenitors (Supplementary Material, Fig. S8), leading to microphthalmia and coloboma. The dominant-negative role of A219Xfs*49 mutant is also consistent with the absence of observable ocular defects in heterozygous Fzd5 null allelic mice.
To date, ocular disorders attributable to mutations in WNT signaling are Norrie disease (16,17), osteoporosis-pseudoglioma syndrome (18) and familial exudative vitreoretinopathy (16,17,19–21). Our study directly implicates perturbed WNT signaling in coloboma and microphthalmia and is consistent with conclusions from mouse models (23,27,32,33). FZD5 mediates both canonical and non-canonical Wnt signaling pathways in different organisms and tissues (22,27,30). However, it is likely that FZD5-mediated non-canonical WNT signaling is the predominant pathway in the developing mammalian retina, as only minimal activity from the canonical pathway has been reported in these cells (39). The retinal apical junction defects observed in Fzd5/Fzd8-knockout mice, and retinal explants expressing the FZD5 mutant protein are likely to be the consequence of interactions between the actin cytoskeleton and components of the apical junctional complexes induced by the loss of non-canonical Wnt activity. The identification of FZD5 as a human coloboma gene extends opportunities to elucidate disease mechanisms and treatment paradigms for ocular malformations.
Materials and Methods
Animal experiments
Animal Care and Use Committee of the National Eye Institute approved all procedures involving the use of mice. Fzd5 and Fzd8 compound mutants were created and maintained as described previously (32).
All zebrafish experiments were consistent with Canadian Council of Animal Care guidelines and approved by the University of Alberta's Animal Care and Use Committee (protocol no. 427). Experiments utilized the WT AB zebrafish strain or Tg(TOP:dGFP)w25 transgenic strain (40). All embryos were grown at 28.5°C and staged according to developmental hallmarks (41).
Zebrafish morpholino and FZD5 mRNA injection experiments
Morpholino oligonucleotides (MOs; GeneTools) were appropriately diluted in Danieau's solution, then heated to 65°C for 10 min and allowed to cool before injection into the 1–2 cell-stage embryos. A previously described translation blocking MO targeting fzd5 (GATGCTCGTCTGCAGGTTTCCTCAT) was injected at a dose of 1.2 pmol (22). Morphological phenotypes were recapitulated by injecting a minimally overlapping MO (TGCAGGTTTCCTCA-TACTGGAAAGC) (data not shown). Human FZD5 WT and A219Xfs*49 cDNA was amplified using pRK5 constructs as template (see below) using the following primer sequences: F—CACAGGATCCACCATGGCTCGGCCTG), R—CACAGAATTCCCTGAACCAAGTGGAA. PCR products were cloned into pCR4-TOPO (Invitrogen) for sequencing confirmation and sub-cloned into pCS2+ for mRNA synthesis. Constructs were linearized with NsiI (New England Biolabs) and mRNA was generated using the SP6 mMessage mMachine kit (Ambion). mRNA was purified using YM-50 Microcon columns (Amicon, Millipore) and the concentration was determined through spectrophotometry. The mRNA was diluted with dimethyl pyrocarbonate-treated water and injected at a dose of 200 pg into one-cell-stage embryos.
Whole mount in situ hybridization, immunofluorescence and live imaging
Live zebrafish embryos were photographed using an Olympus stereoscope with a Qimaging micropublisher camera. Whole mount in situ hybridization was performed as previously described (42). Reverse transcription polymerase chain reaction was used to generate 800–1200 bp templates for probe synthesis or sub-cloned into pCR4-TOPO (Invitrogen). Immunofluorescence was performed as previously described (26) using rabbit polyclonal specific for Laminin (1/100) (Sigma L9393). After either in situ hybridization or immunofluorescence, eyes were dissected off and flat mounted for imaging on Zeiss AxioImager Z1 compound microscope with Axiocam HR digital camera.
Patients and DNA sequencing
Individuals with microphthalmia, anophthalmia and/or coloboma were subjected to exome sequencing and Sanger sequencing. Genomic DNA samples from coloboma probands were analyzed by the National Eye Institute Clinical Eye Center, UK10K consortium, MRC Human Genetics Unit at the Institute of Genetic and Molecular Medicine, University of Edinburgh and the University of Alberta. Informed consent was obtained from each participant, and study approval provided by the relevant ethics boards [National Institutes of Health (NIH) Institutional Review Board (IRB); U of A Health Research Ethics Board (reference no. 01227), UK Multiregional Ethics Committee (reference no. 06/MRE00/76)]. Five affected individuals from family 3483 (UK10K) were subjected to WES, and Sanger sequencing was used to test for the FZD5 mutation in all other available members of this branch of the family. Four affected individuals from Family 111 were Sanger sequenced for the FZD5 gene. Exome sequencing was performed as described (43) with burrows-wheeler alignment 0.5.9 used for alignment, Picard 1.43 for duplicate marking, genome analysis tool kit (GATK) 1.0.5506 for realignment around indels and base quality scores recalibration, and GATK Unified Genotyper for variant calling. The LOD score was calculated using paramlink package in R (44). The oligonucleotides used to PCR amplify FZD5 are listed in Supplementary Material, Table S2.
Immunoblotting, immunofluorescence staining and immunohistochemistry
For examining expression of FZD5 mutant constructs, plasmid DNA of each mutant (D97V or A219Xfs*49) or WT FZD5 construct was transfected into HEK293T cells cultured in six-well dishes. A total of 2 µg DNA was used for transfection for each well, and biological and technical triplicates were made for each transfection. Transfected cells were cultured for 36 h, supplemented with the serum-reduced medium (Opti-minimal essential medium, Life Technologies, Cat. no. 31985), continually cultured for another 24 h. The cell medium was collected and store at −80°C. Total cell extracts were prepared by adding two-time SDS Laemmli buffer (Bio-Rad, Cat. no. 161-0737) onto cells rinsed with phosphate buffered saline (PBS). To prepare extracellular matrix (ECM) proteins, cultured cells were washed with PBS and incubated in PBS containing 10 mm Ethylenediaminetetraacetic acid (EDTA) at 37°C for 30–45 min to remove the cells. ECM proteins are retained on the dish and solubilized in Laemmli buffer.
Immunoblotting was performed as described previously (32) using custom-made rabbit antibody against the N-terminus 143 residues (27–169 amino acids) of the mouse FZD5 protein. The signal intensities were quantified by NIH ImageJ from three representative western blots, and analyzed by the Microsoft Excel. To detect FZD5 and FZD5 A219Xfs*49 cellular or extracellular localization, 2 µg DNA was used for transfection in each well (six-well plate) carrying coated coverslips. Transfected cells were cultured in Dulbecco's modified Eagle's medium (DMEM)-F12 for 48 h. Immunofluorescence staining was conducted using the same antibody for detection of mutated/variant FZD5 proteins. To avoid cytoplasmic staining, live cells were first incubated with anti-Fzd5 antibody in the cultured medium at 4°C for 2 h, washed with PBS and then post-fixed with paraformaldehyde (PFA). After rinse with PBS, secondary antibody was added to further proceed with immunohistochemistry. Standard immunohistochemistry was performed on PFA-fixed frozen retinal sections with anti-Fzd5 antibody (1:500), anti-aPKC (1:500, cell signaling, Cat. no. 9378) and anti-RhoA (Cytoskeleton, Cat. no. ARH03).
FZD5 gene mutagenesis and Wnt/β-catenin pathway activation assay
FZD5 cDNA was cloned into pRK5 expression vector with a Cytomegalovirus promoter and site-directed mutagenesis was performed to generate the FZD5 A219Xfs*49 frameshift mutation. For testing canonical Wnt signaling activity, DNA constructs were transfected into STF HEK293 cells with a seven-time TCF promoter-driven firefly luciferase reporter stably integrated in the genome (27). As shown in Figure 4A, fixed amount of Wnt9b and FZD5 were cotransfected together with pCAG-Renilla luciferase plasmids (RL, used for internal expression control) into STF cells. Different amount of FZD5 A219Xfs*49 and sCRD plasmids were transfected into regular HEK293 cells, respectively. Twelve hours after transfection, both STF and HEK293 cells were lifted off by trypsin-EDTA, and mixed at 1:1 ratio and seeded into new plate for another 36 h culture. Biological and technical triplicates were prepared for each transfection. Cell extracts were then prepared for Firefly luciferase and Renilla luciferase assay using the Dual-luciferase assay system (Promega, E1960). Luminicence was measured sequentially by Turner Biosystem Modulus microplate reader. Firefly luciferase activity was normalized against Renilla luciferase, and P-value calculation was performed using the Microsoft Excel software Student's t-test function.
Active RhoA assay for Wnt5a stimulation
HEK293 cells were cultured to 80% confluence in DMEM:F12 in six-well dishes, transfected with FZD5 WT, A219Xfs*49 and sCRD plasmids, cultured for 24 h, then serum starved for 24 h. Wnt5a recombinant protein conditioned medium (Wnt5a CM, Roche, Cat: 645-WN-010/CF) was applied for 30 min at 250 ng/ml. Cells were lysed and then subjected to active GTP-RhoA assays according to the manufacturer instructions (pulldown assay: RhoA/Rac1/Cdc42 assay kit, Cytoskeleton, Inc., Cat. no. Bk-030; G-lisa assay: RhoA G-lisa kit, Cytoskeleton, Inc., Cat. no. Bk-124). Signal intensity was acquired by NIH ImageJ from three representative immunoblots. Light absorbance/optic density of horseradish peroxidase colorimetric reaction was measured (SpectraMax M), and the data were analyzed in the Microsoft Excel.
Retinal electroporation and explants culture
Mouse embryonic retinas were dissected in the DMEM medium at E13.5 excluding lens and RPE. Retinae were subjected to electroporation with BTX ECM830 electroporator in embryo global positioning system (GPS) chamber (SunIVF, EGPS-010) supplied with 250 ng/µl DNA solution in one-time PBS. The following parameters were set for electroporation: 21 volts for electric field strength; five-time current pulses (50 ms duration); 900 ms intervals between pulses. Retinas were then cultured in DMEM:F12 (Invitrogen, Cat. no. 12660-012) for 72 h, harvested in PBS and fix in 4% PFA, and subjected to sectioning and immunohistochemistry (32).
Co-immunoprecipitation
HEK293T cells were transfected with 1.5 µg DNA in each well of six-well dishes. Myc-tagged Wnt3a, HA-tagged WNT7A were coexpressed, respectively, with FZD5 or FZD5 A219Xfs*49. Cell extracts and co-IP procedure were performed essentially as described (34). Antibodies used were mouse anti-HA (TransGene Biotech, HT301), rabbit anti-myc (Sigma, C3956) and rabbit anti-FZD5 (custom-made). Protein A agarose resin was purchased from TansGene Biotech (DP301).
Supplementary Material
Funding
These studies were supported by Intramural Research program of the National Eye Institute (A.S., T.L. and B.B.), a UK Medical Research Council block grant to the University of Edinburgh Medical Research Council Human Genetics Unit (D.R.F., K.A.W. and J.R.) and by grants from Natural Sciences and Engineering Research Council of Canada, Alberta Innovates Technology Futures and Women and Children's Health Research Institute (S.A.W. and A.J.W.), Canadian Institutes of Health Research and WCHRI (O.J.L.) and 100 People Plan of Sun Yat-sen University (C.L.). The Welcome Trust supported UK10K project (no. WT091310).
Supplementary Material
Acknowledgements
We are grateful to Jeremy Nathans for WNT–FZD constructs and reporter cell lines. We thank Arvydas Maminishkis for providing human fetal retina and Suja Hiriyanna for technical assistance.
Conflict of Interest statement. None declared.
References
- 1.Hornby S.J., Gilbert C.E., Rahi J.K., Sil A.K., Xiao Y., Dandona L., Foster A. (2000) Regional variation in blindness in children due to microphthalmos, anophthalmos and coloboma. Ophthalmic. Epidemiol., 7, 127–138. [PubMed] [Google Scholar]
- 2.Gregory-Evans C.Y., Williams M.J., Halford S., Gregory-Evans K. (2004) Ocular coloboma: a reassessment in the age of molecular neuroscience. J. Med. Genet., 41, 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williamson K.A., FitzPatrick D.R. (2014) The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet., 57, 369–380. [DOI] [PubMed] [Google Scholar]
- 4.Bakrania P., Efthymiou M., Klein J.C., Salt A., Bunyan D.J., Wyatt A., Ponting C.P., Martin A., Williams S., Lindley V. et al. (2008) Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am. J. Hum. Genet., 82, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rainger J., van Beusekom E., Ramsay J.K., McKie L., Al-Gazali L., Pallotta R., Saponari A., Branney P., Fisher M., Morrison H. et al. (2011) Loss of the BMP antagonist, SMOC-1, causes ophthalmo-acromelic (Waardenburg anophthalmia) syndrome in humans and mice. PLoS. Genet., 7, e1002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wyatt A.W., Osborne R.J., Stewart H., Ragge N.K. (2010) Bone morphogenetic protein 7 (BMP7) mutations are associated with variable ocular, brain, ear, palate, and skeletal anomalies. Hum. Mutat., 31, 781–787. [DOI] [PubMed] [Google Scholar]
- 7.Asai-Coakwell M., French C.R., Berry K.M., Ye M., Koss R., Somerville M., Mueller R., van Heyningen V., Waskiewicz A.J., Lehmann O.J. (2007) GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am. J. Hum. Genet., 80, 306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schimmenti L.A., de la Cruz J., Lewis R.A., Karkera J.D., Manligas G.S., Roessler E., Muenke M. (2003) Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am. J. Med. Genet. A., 116A, 215–221. [DOI] [PubMed] [Google Scholar]
- 9.Pasutto F., Sticht H., Hammersen G., Gillessen-Kaesbach G., Fitzpatrick D.R., Nurnberg G., Brasch F., Schirmer-Zimmermann H., Tolmie J.L., Chitayat D. et al. (2007) Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet., 80, 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srour M., Chitayat D., Caron V., Chassaing N., Bitoun P., Patry L., Cordier M.P., Capo-Chichi J.M., Francannet C., Calvas P. et al. (2013) Recessive and dominant mutations in retinoic acid receptor beta in cases with microphthalmia and diaphragmatic hernia. Am. J. Hum. Genet., 93, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fares-Taie L., Gerber S., Chassaing N., Clayton-Smith J., Hanein S., Silva E., Serey M., Serre V., Gerard X., Baumann C. et al. (2013) ALDH1A3 mutations cause recessive anophthalmia and microphthalmia. Am. J. Hum. Genet., 92, 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williamson K.A., Rainger J., Floyd J.A., Ansari M., Meynert A., Aldridge K.V., Rainger J.K., Anderson C.A., Moore A.T., Hurles M.E. et al. (2014) Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am. J. Hum. Genet., 94, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye M., Berry-Wynne K.M., Asai-Coakwell M., Sundaresan P., Footz T., French C.R., Abitbol M., Fleisch V.C., Corbett N., Allison W.T. et al. (2010) Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet., 19, 287–298. [DOI] [PubMed] [Google Scholar]
- 14.Morcillo J., Martinez-Morales J.R., Trousse F., Fermin Y., Sowden J.C., Bovolenta P. (2006) Proper patterning of the optic fissure requires the sequential activity of BMP7 and SHH. Development, 133, 3179–3190. [DOI] [PubMed] [Google Scholar]
- 15.Reis L.M., Tyler R.C., Schilter K.F., Abdul-Rahman O., Innis J.W., Kozel B.A., Schneider A.S., Bardakjian T.M., Lose E.J., Martin D.M. et al. (2011) BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet., 130, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z.Y., Battinelli E.M., Fielder A., Bundey S., Sims K., Breakefield X.O., Craig I.W. (1993) A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet., 5, 180–183. [DOI] [PubMed] [Google Scholar]
- 17.Nikopoulos K., Venselaar H., Collin R.W., Riveiro-Alvarez R., Boonstra F.N., Hooymans J.M., Mukhopadhyay A., Shears D., van Bers M., de Wijs I.J. et al. (2010) Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum. Mutat., 31, 656–666. [DOI] [PubMed] [Google Scholar]
- 18.Gong Y., Slee R.B., Fukai N., Rawadi G., Roman-Roman S., Reginato A.M., Wang H., Cundy T., Glorieux F.H., Lev D. et al. (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell, 107, 513–523. [DOI] [PubMed] [Google Scholar]
- 19.Robitaille J., MacDonald M.L., Kaykas A., Sheldahl L.C., Zeisler J., Dube M.P., Zhang L.H., Singaraja R.R., Guernsey D.L., Zheng B. et al. (2002) Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet., 32, 326–330. [DOI] [PubMed] [Google Scholar]
- 20.Toomes C., Bottomley H.M., Jackson R.M., Towns K.V., Scott S., Mackey D.A., Craig J.E., Jiang L., Yang Z., Trembath R. et al. (2004) Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am. J. Hum. Genet., 74, 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poulter J.A., Davidson A.E., Ali M., Gilmour D.F., Parry D.A., Mintz-Hittner H.A., Carr I.M., Bottomley H.M., Long V.W., Downey L.M. et al. (2012) Recessive mutations in TSPAN12 cause retinal dysplasia and severe familial exudative vitreoretinopathy (FEVR). Invest. Ophthalmol. Vis. Sci., 53, 2873–2879. [DOI] [PubMed] [Google Scholar]
- 22.Cavodeassi F., Carreira-Barbosa F., Young R.M., Concha M.L., Allende M.L., Houart C., Tada M., Wilson S.W. (2005) Early stages of zebrafish eye formation require the coordinated activity of Wnt11, Fz5, and the Wnt/beta-catenin pathway. Neuron, 47, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagglund A.C., Berghard A., Carlsson L. (2013) Canonical Wnt/beta-catenin signalling is essential for optic cup formation. PLoS One, 8, e81158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lieven O., Ruther U. (2011) The Dkk1 dose is critical for eye development. Dev. Biol., 355, 124–137. [DOI] [PubMed] [Google Scholar]
- 25.Veien E.S., Rosenthal J.S., Kruse-Bend R.C., Chien C.B., Dorsky R.I. (2008) Canonical Wnt signaling is required for the maintenance of dorsal retinal identity. Development, 135, 4101–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holly V.L., Widen S.A., Famulski J.K., Waskiewicz A.J. (2014) Sfrp1a and Sfrp5 function as positive regulators of Wnt and BMP signaling during early retinal development. Dev. Biol., 388, 192–204. [DOI] [PubMed] [Google Scholar]
- 27.Liu C., Nathans J. (2008) An essential role for frizzled 5 in mammalian ocular development. Development, 135, 3567–3576. [DOI] [PubMed] [Google Scholar]
- 28.Borello U., Buffa V., Sonnino C., Melchionna R., Vivarelli E., Cossu G. (1999) Differential expression of the Wnt putative receptors Frizzled during mouse somitogenesis. Mech. Dev., 89, 173–177. [DOI] [PubMed] [Google Scholar]
- 29.Sumanas S., Ekker S.C. (2001) Xenopus frizzled-5: a frizzled family member expressed exclusively in the neural retina of the developing eye. Mech. Dev., 103, 133–136. [DOI] [PubMed] [Google Scholar]
- 30.Van Raay T.J., Moore K.B., Iordanova I., Steele M., Jamrich M., Harris W.A., Vetter M.L. (2005) Frizzled 5 signaling governs the neural potential of progenitors in the developing Xenopus retina. Neuron, 46, 23–36. [DOI] [PubMed] [Google Scholar]
- 31.Burns C.J., Zhang J., Brown E.C., Van Bibber A.M., Van Es J., Clevers H., Ishikawa T.O., Taketo M.M., Vetter M.L., Fuhrmann S. (2008) Investigation of Frizzled-5 during embryonic neural development in mouse. Dev. Dyn., 237, 1614–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu C., Bakeri H., Li T., Swaroop A. (2012) Regulation of retinal progenitor expansion by Frizzled receptors: implications for microphthalmia and retinal coloboma. Hum. Mol. Genet., 21, 1848–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou C.J., Molotkov A., Song L., Li Y., Pleasure D.E., Pleasure S.J., Wang Y.Z. (2008) Ocular coloboma and dorsoventral neuroretinal patterning defects in Lrp6 mutant eyes. Dev. Dyn., 237, 3681–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carmon K.S., Loose D.S. (2008) Secreted frizzled-related protein 4 regulates two Wnt7a signaling pathways and inhibits proliferation in endometrial cancer cells. Mol. Cancer. Res., 6, 1017–1028. [DOI] [PubMed] [Google Scholar]
- 35.Carmon K.S., Loose D.S. (2010) Development of a bioassay for detection of Wnt-binding affinities for individual frizzled receptors. Anal. Biochem., 401, 288–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bodine P.V., Zhao W., Kharode Y.P., Bex F.J., Lambert A.J., Goad M.B., Gaur T., Stein G.S., Lian J.B., Komm B.S. (2004) The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol. Endocrinol., 18, 1222–1237. [DOI] [PubMed] [Google Scholar]
- 37.Popp M.W., Maquat L.E. (2013) Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet., 47, 139–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishikawa T., Tamai Y., Zorn A.M., Yoshida H., Seldin M.F., Nishikawa S., Taketo M.M. (2001) Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development, 128, 25–33. [DOI] [PubMed] [Google Scholar]
- 39.Liu H., Thurig S., Mohamed O., Dufort D., Wallace V.A. (2006) Mapping canonical Wnt signaling in the developing and adult retina. Invest. Ophthalmol. Vis. Sci., 47, 5088–5097. [DOI] [PubMed] [Google Scholar]
- 40.Dorsky R.I., Sheldahl L.C., Moon R.T. (2002) A transgenic Lef1/beta-catenin-dependent reporter is expressed in spatially restricted domains throughout zebrafish development. Dev. Biol., 241, 229–237. [DOI] [PubMed] [Google Scholar]
- 41.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn., 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 42.Gongal P.A., Waskiewicz A.J. (2008) Zebrafish model of holoprosencephaly demonstrates a key role for TGIF in regulating retinoic acid metabolism. Hum. Mol. Genet., 17, 525–538. [DOI] [PubMed] [Google Scholar]
- 43.Olbrich H., Schmidts M., Werner C., Onoufriadis A., Loges N.T., Raidt J., Banki N.F., Shoemark A., Burgoyne T., Al Turki S. et al. (2012) Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet., 91, 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Egeland T., Pinto N., Vigeland M.D. (2014) A general approach to power calculation for relationship testing. Forensic Sci. Int. Genet., 9, 186–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.