

Summary
By impairing histone demethylation and locking cells into a reprogramming-prone state, oncometabolites can partially mimic the process of induced pluripotent stem cell generation. Using a systems biology approach, combining mathematical modeling, computation, and proof-of-concept studies with live cells, we found that an oncometabolite-driven pathological version of nuclear reprogramming increases the speed and efficiency of dedifferentiating committed epithelial cells into stem-like states with only a minimal core of stemness transcription factors. Our biomathematical model, which introduces nucleosome modification and epigenetic regulation of cell differentiation genes to account for the direct effects of oncometabolites on nuclear reprogramming, demonstrates that oncometabolites markedly lower the “energy barriers” separating non-stem and stem cell attractors, diminishes the average time of nuclear reprogramming, and increases the size of the basin of attraction of the macrostate occupied by stem cells. These findings establish the concept of oncometabolic nuclear reprogramming of stemness as a bona fide metabolo-epigenetic mechanism for generation of cancer stem-like cells.
Keywords: Oncometabolites, reprogramming, stemness, cancer, stem cells, cancer stem cells
Graphical Abstract
Highlights
-
•
Oncometabolites facilitate the reprogramming process evoked by stemness factors
-
•
Oncometabolites lower the epigenetic barriers to nuclear reprogramming
-
•
Cancer stem-like states arise through oncometabolic nuclear reprogramming phenomena
-
•
Oncometabolic regulation of epigenetics can drive stemness in cancer tissues
Using a systems biology approach combining mathematical and computational modeling with proof-of-concept experiments in cultured cells, Menendez and colleagues provide evidence that oncometabolic versions of nuclear reprogramming phenomena are capable of generating cancer stem-like states from more differentiated counterparts. These findings offer an innovative stochastic modeling tool as well as a conceptual framework for investigating the underexplored link between cellular metabolism and cancer-initiating alterations of the epigenome at the stem cell level.
Introduction
The correct functioning of the epigenome ensures fidelity in the establishment of gene-expression programs that are compatible with specific cell identities. The need for tightly controlled epigenetic landscapes is of critical importance for stem cells, which are able to both self-renew and generate differentiated progeny (Barrero et al., 2010, Chen and Dent, 2014, Papp and Plath, 2013, Spivakov and Fisher, 2007). The inability to stabilize stem cell states and functions by maintaining epigenome integrity, a process in which DNA methylation plays a major role, can trigger pathological self-renewal processes that ultimately lead to cancer (Ohnishi et al., 2014, Suva et al., 2013, Tung and Knoepfler, 2015). Interestingly, remodeling of DNA methylation is a cancer-initiating event manifesting in the presence of particular types of cancer-driving metabolites, termed oncometabolites, and in the nuclear reprogramming process of transcription factor-generated induced pluripotent stem cell (iPSC) derivation.
The shared mechanism by which abnormal accumulation of the oncometabolites 2-hydroxyglutarate (2HG), succinate, and fumarate causes potential transformation to malignancy is the ability to promote DNA hypermethylation through suppression of histone demethylation, which, in turn, results in the repression of genes involved in the epigenetic rewiring of lineage-specific differentiation and in the promotion of stem cell-like transcriptional signatures (Chowdhury et al., 2011, Killian et al., 2013, Letouzé et al., 2013, Lu et al., 2012, Terunuma et al., 2014, Saha et al., 2014, Xiao et al., 2012, Xu et al., 2011, Yang et al., 2013). The transient expression of stemness-associated transcription factors, i.e., OCT4, SOX2, KLF4, and c-MYC, in vivo generates tumors consisting of undifferentiated dysplastic cells exhibiting global changes in DNA methylation (Ohnishi et al., 2014), suggesting that the epigenetic regulatory machinery associated with iPSC derivation might initiate cancer development in a manner that does not require mutational changes in the genomic sequence (Ben-David and Benvenisty, 2011, Ohnishi et al., 2014, Knoepfler, 2009, Tung and Knoepfler, 2015).
Because oncometabolites partially mimic the process of iPSC generation, a metabolically driven pathological version of nuclear reprogramming might represent an underappreciated epigenetic mechanism of enrichment for cellular states with increased tumor-initiating capacities and aberrant self-renewal potential (Goding et al., 2014, Menendez and Alarcón, 2014, Menendez et al., 2014b), often termed cancer stem cells. However, although a role for oncometabolite-driven changes in the epigenetic landscape is mechanistically attractive (Lu and Thompson, 2012, Yun et al., 2012, Johnson et al., 2015), the existence of bona fide oncometabolic reprogramming of differentiated cells into cancer stem-like states has never been demonstrated. In an attempt to resolve this issue, we have used a systems biology approach that combined mathematical modeling, computation, and proof-of-concept experimental validation of stochastic predictions in vitro.
Results
We initially developed methods and procedures for the mathematical modeling of oncometabolo-epigenetic regulatory networks involved in the acquisition of stemness (see Supplemental Information). Our stochastic model considers the interactions between a minimal core of stemness-associated transcription factors (OCT4 and SOX2) and two generic lineage-specific genes (LSG1 and LSG2) (Shu et al., 2013) (Figure 1A). The basic effector mechanism of the coupling between metabolism and the epi-transcriptional reprogramming system relies on histone- and nucleosome-modifying enzymes (Dodd et al., 2007). In particular, we considered a metabolo-epigenetic link in which the oncometabolite 2HG drastically inhibits the activity of DNA histone demethylases (HDMs), thus restricting the methylation plasticity that is required for the transition between stem cells and differentiated cells (Lu and Thompson, 2012, Lu et al., 2012).
Figure 1.

Computation Simulation of Oncometabolic Nuclear Reprogramming Phenomena
(A–C) A stochastic model of oncometabolic nuclear reprogramming. (A) Top: Schematic representation of the minimal gene regulatory network considered in our stochastic model, consisting of a coupled pluripotency module (self-activation of Oct4 and Sox2) and a differentiation module (mutual antagonism between LSGs). Arrows denote activation and blunt-ended lines denote inhibitory interactions. Bottom: Schematic representation of the competitive binding model for activation/repression in the minimal gene regulatory network. (B) A realization path in which our stochastic model was run under baseline conditions (baseline HDM activity and lack of induction of stemness-related transcription factors, i.e., hi-values as per values given in Table S7 [Supplemental Appendix E] and ρ1 = ρ2 = 0). Since the system is symmetric with respect to LSG1 and LSG2, a state where O = 0, S = 0, and L2 = 0, whereas L1 > 0, is also an absorbing state. (C) A realization path in which our stochastic model was run under induction of stemness-related transcription factors (parameter values ρ1 = ρ2 = 1.85 × 107). At the onset of stemness factor induction, i.e., we let ρ1 > 0 and ρ2 > 0, the absorbing states observed in the simulations shown in (B) are not absorbing any longer and, therefore, there is a positive probability for the system to go from the differentiated cell state to the stem cell state. Left: Normal-like metabolism, baseline HDM activity; right: 2HG-induced reduction of HDM activity by 5% with respect to the baseline scenario.
(D–F) Epigenetic landscapes and reprogramming performance in response to 2HG. (D) 2HG-induced inhibition of HDM activity affects the depth of the stem cell attractors by lowering the barriers of the epigenetic landscape. Figures show the joint probability of the random variables O + S (stemness factors) and L1 + L2 (LSGs) for different values of the relative oncometabolic-induced reduction of HDM activity with respect to the baseline scenario. To obtain the epigenetic landscapes for different degrees of 2HG-induced reduction of HDM activity in shorter computational time, we considered the following parameter values: ρ1 = ρ2 = 5.55 × 10−7 and ϑo = ϑs = 0.2. We have also considered that the expression of the LSGs is induced at certain rates given by the following parameter values ρL1 = ρL2 = 2.78 × 10−7. The landscapes with 4%, 5%, and 6% reduced HDM activity correspond to h2 = 0.96, h2 = 0.95, and h2 = 0.94, respectively. The remaining parameter values are given in Table S7 (Supplemental Appendix E). (E) 2HG-induced inhibition of HDM activity affects the kinetic efficiency of the reprogramming process. The panel shows statistics of the average reprogramming time, TE, as well as its probability density, P(TE), as a function of the 2HG-induced reduction of HDM activity. The top panels illustrate that the predicted probability distribution of TE, P(TE) is approximately exponential. The lower panel shows the average and SD (error bars) of the predicted reprogramming time, illustrating that the reprogramming rate increases exponentially with the 2HG-induced reduction of HDM activity. HDM activity reductions of 0%, 2.5%, 5%, and 7.5% correspond to h2 = 1.00, h2 = 0.975, h2 = 0.95, and h2 = 0.925, respectively. (F) 2HG-induced inhibition of HDM activity affects the size of the basin of attraction of the induced stem cell state. The graphic shows a solution of the semiclassical QSSA approximation (see Equations 29–32 and 40–44 in Supplemental Information) for the baseline scenario with no HDM inhibition (h2 = 1.00, red line), and for the case with a 2HG-induced 5% reduction in HDM activity (h2 = 0.95, blue line). The uninhibited scenario converges to the differentiated cell state (red line), whereas the inhibited scenario converges to the stem cell state (blue line). Parameter values ρ1 = ρ2 = 1.85 × 107, cE1 = cE2 = 2, Co = CS = C1 = C2 = 1, and ϑo = ϑs = 0.2. The remaining parameter values are given in Table S7 (Supplemental Appendix E).
The first consistency check we performed was that the “normal metabolism” scenario, defined as baseline HDM activity and lack of induction of stemness transcription factors, should lead to cell differentiation. Figure 1B shows that, after an initial transient regime, the system settles down to a steady state whereby the protein levels of OCT4, SOX2, and LSG1 decay to zero, whereas LSG2 protein climbs to its stationary positive value. In the absence of induction, this cellular state is an absorbing state, i.e., once reached by the system it is not possible to exit. Figure 1C (left panel) shows a particular sample path where reprogramming of stemness is accomplished by merely adding OCT4 and SOX2 to the baseline scenario. Importantly, under baseline conditions both LSG1 and LSG2 are predominantly acetylated and, therefore, their promoters remain accessible to transcription factors, with short-lived journeys into the methylated state (Supplemental Appendix D). Despite the fact that during episodes of transient methylation the expression levels of the LSGs become downregulated, this does not necessarily lead to reprogramming since the stochastic dynamics of the gene regulatory network has to pass through an unstable saddle point (Supplemental Appendix E). Therefore, several episodes of transient methylation should occur before a period of transient methylation of sufficient duration allows the gene-regulation system to successfully pass through the bottleneck.
We then introduced the ability of 2HG to reduce HDM activity. Figure 1C (right panel) shows a realization of the stochastic model whereby reprogramming is achieved with a 5% reduction of HDM activity with respect to the baseline scenario (Figure 1C, left panel). Oncometabolic reduction of HDM activity increases the characteristic duration of the transient episodes of methylation (Supplemental Appendix D) which, in turn, increases the likelihood of one such period of sufficient duration for the gene regulatory system to overcome the bottleneck. In this scenario, the system must transit from the differentiated state to the stem cell state. Figure 1D illustrates how the relative height of the peak corresponding to the stem cell state increases in relation to the peak corresponding to the differentiated cell state, thus implying that the epigenetic barriers are significantly lowered in response to 2HG-induced reduction of HDM activity. There is a “third peak” that arises from the fact that, while attempting reprogramming, the gene regulatory system spends a long time in the vicinity of the saddle point, trying to overcome the bottleneck. The height of this third peak appears to be rather insensitive to the 2HG-regulated activity of HDM, as it depends on the kinetic parameters of the gene regulatory system alone. Figure 1E shows the immediate and significant consequences for the kinetic efficiency of nuclear reprogramming; specifically, the reduction in average reprogramming time varies exponentially with the 2HG-induced reduction of HDM activity. Therefore, even modest 2HG-driven reductions of HDM activity are predicted to drive a considerable increase in the reprogramming efficiency.
We finally interrogated our stochastic model to examine whether the basins of attractions of each of these cell states, i.e., the set of developmental states which are attracted to each of them, are also altered in response to 2HG-induced reduction of HDM activity. We carried out a semiclassical quasi-steady-state approximation (QSSA) (Supplemental Appendix B) to analyze the existence of initial conditions that, in the absence of HDM inhibition, converge to a differentiated cell state and, upon inhibition, converge to the stem cell state. We found that such initial conditions exist, i.e., a portion of the basin of attraction of the differentiated cell state is transferred to the stem cell state upon HDM inhibition. Figure 1F illustrates how oncometabolic-induced repression of HDM activity actually enlarges the basin of attraction of the stem cell state.
Nishi et al., 2014a, Nishi et al., 2014b have developed a method of inducing cancer stem-like cells (CSCs) through the reprogramming and partial differentiation of the immortalized but otherwise normal MCF10A human mammary epithelial cell line. To experimentally test our computational model of oncometabolic nuclear reprogramming, we similarly employed the MCF10A cell line and an isogenic derivative endogenously heterozygous for the R132H mutation of isocitrate dehydrogenase 1 (IDH1) gene, generating 2HG (Grassian et al., 2012). Quantification of intracellular 2HG showed that the levels of the oncometabolite were more than 30-fold higher in cell lysates from the knockin MCF10A IDH1R132H/WT cells, confirming neomorphic IDH1R132H enzymatic activity (Figure 2A). To corroborate that the sole accumulation of 2HG in an otherwise isogenic background was sufficient to significantly impair histone demethylation, we examined the pattern of histone lysine methylation in IDH1R132H/WT knockin and parental cells. Western blot analysis showed that the global levels of H3K4me3, H3K9me3, and H3K27me3 were increased in 2HG-overproducing MCF10A knockin cells compared with IDH1WT/WT parental cells (Figure 2A). These results were consistent with 2HG-induced broad inhibition of histone demethylation, showing agreement with previous models overexpressing IDH mutants (Duncan et al., 2012, Lu et al., 2012). We then employed a commercially available ELISA-based global DNA methylation able to indirectly provide a global measurement of 5-methtylcytosine (5-mC) levels in genomic DNA obtained from IDH1WT/WT and IDH1R132H/WT knockin cells. Measurement of 5-mC levels of long interspersed nucleotide element 1 (LINE-1) confirmed that LINE-1 methylation significantly increased in 2HG-overproducing IDH1R132H/WT cells when compared with IDH1WT/WT parental cells. Remarkably, a 2-day treatment with the selective R132H-IDH1 inhibitor AGI-5198, which fully suppressed 2HG to background levels (Figure 2A), partially reverted LINE-1 hypermethylation in IDH1R132H/WT cells.
Figure 2.

Effects of 2HG on Histone Demethylation, Activation of Pluripotency Genes, and Genome-wide DNA Methylation
(A) Top: Base-peak chromatograms of extracts from IDH1WT/WT cells (black line), IDH1R132H/WT cells (red line), and IDH1R132H/WT cells treated with 40 μmol/l AGI-5198 for 2 days (blue line). A combined mass spectrum of the region where 2HG was eluted (m/z 349.1317) is shown in the inset (two technical replicates per n; n = 3 biological replicates). Bottom: 2HG promotes broad inhibition of histone demethylation and LINE-1 global methylation. Left panel, western blots for total H3K4me3, H3K9me3, and H3K27me3 histone modifications in parental IDH1WT/WT and IDH1R132H/WT knockin cells. Also shown are total H3 controls (two technical replicates per n; n = 2 biological replicates). Middle and right panels, % of 5-mC relative to either detectable CpG residues or total cytosine content in IDH1WT/WT and IDH1R132H/WT cells, the latter being cultured in the absence or presence of 40 μmol/l AGI-5198 for 2 days. The data are presented as the mean ± SD (error bars); three technical replicates per n; n = 2 biological replicates.
(B and C) 2HG impairs differentiation but does not elevate the baseline expression of core pluripotency factors. (B) Flow cytometry analysis of OCT4/SOX2 expression in IDH1WT/WT and IDH1R132H/WT knockin cells. Representative dot plots showing the distribution of IDH1WT/WT and IDH1R132H/WT cells along the signal obtained with the isotype-specific control antibodies or with the OCT3,4 and SOX2 direct conjugated antibodies (two technical replicates per n; n = 2 biological replicates). (C) Ten top-ranked GO molecular functions and biological processes associated with hypermethylated genes in IDH1R132H/WT cells (accession number GEO: GSE76263). x axis, negative logarithm (-lg) of the p value; y axis, GO category.
Because it could be argued that 2HG-induced chromatin reorganization might promote the pluripotency-associated genes transition from an inactive to an active stage, we assessed whether the overproduction of 2HG promoted the expression of pluripotency regulators in normal breast epithelial cells. Flow cytometry analyses confirmed that the baseline expression of the core transcription factors OCT4 and SOX2 remained essentially unaltered in IDH1R132H/WT knockin cells compared with parental IDH1WT/WT cells (Figure 2B). A preliminary evaluation of the top ten most significant Gene Ontology (GO) “molecular function” and “biological process” term annotations overrepresented in the 290 differentially hypermethylated CpG sites that were identified in IDH1R132H/WT knockin cells (Figure 2C) strongly suggested that 2HG had a significant impact on the transcriptional repression of differentiation programs. Intracellular accumulation of the oncometabolite 2HG due to the heterozygous expression of the IDH1R132H allele is therefore sufficient to notably alter global histone lysine methylation without varying the baseline expression of the most critical reprogramming factors (i.e., OCT4 and SOX2) but altering expression of differentiation genes, thus providing an idoneous experimental model to validate the stochastic predictions of our biomathematical model in vitro.
MCF10A IDH1R132H/WT and MCF10A IDH1WT/WT cells were then transduced with OCT4 and SOX2 (hereafter called OS) to examine whether endogenously produced 2HG could substitute for combinations of stemness factors (i.e., KLF4 and c-MYC) to reprogram MCF10A mammary cells into iPS-like (iPSL-10A) cells. At day 15 after infection, MCF10A IDH1R132H/WT cells growing on feeder layers showed a >10-fold increase in reprogramming efficiency relative to MCF10A IDH1WT/WT cells, as assessed by counting the number of alkaline phosphatase (AP)-positive (AP+) colonies (Figure 3A). We found that the colonies identified by the highly AP+ criterion were also positive for strong endogenous expression of NANOG (data not shown), which was considered a characteristic of bona fide iPSL-10A cells.
Figure 3.

Effects of 2HG on the Nuclear Reprogramming of Breast Epithelial Cells into CSC-like States
(A) Left panels: Kinetics of reprogramming in the absence or presence of 2HG. MCF10 IDH1WT/WT control cells and 2HG-overproducing MCF10A IDH1R132H/WT isogenic derivatives were reprogrammed by the retroviral delivery of OS (top) or OSKM (bottom) transcription factors. Alternatively, octyl-2HG, a cell-permeable esterified form of 2HG, was added at a final concentration of 1 mmol/l to the culture medium immediately after transduction of IDH1WT/WT parental cells with OS and OSKM, and was maintained for 4 days. The total number of highly AP+ colonies for each condition was counted at different days until day 15 after transduction under feeder conditions. The data are presented as the mean ± SD (error bars); n = 3 biological replicates. Representative microphotographs of AP+ colonies are also shown (scale bar, 5 mm). Middle panels: The reprogramming efficiencies of various conditions were compared with that obtained without octyl-2HG treatment in IDH1WT/WT parental cells, and are presented as relative fold changes (mean [columns] ± SD [error bars]). Insets show microscopy images of the representative cell morphology of IDH1WT/WT, IDH1R132H/WT, and IDH1WT/WT cells growing in the presence of octyl-2HG (scale bar, 10 μm). Right panels: Temporal activation of stemness during reprogramming was analyzed by live-cell staining with an antibody against TRA-1-60 (n = 3 biological replicates).
(B) Western blots for total H3K4me3, H3K9me3, and H3K27me3 histone modifications in parental IDH1WT/WT and IDH1R132H/WT knockin cells at day 6 post-OS or post-OSKM transduction. Also shown are total H3 controls (two technical replicates per n; n = 2 biological replicates). Right panels: Flow cytometry analysis of OCT4/SOX2 expression in CSC-like derivatives obtained from partial differentiation of reprogrammed IDH1WT/WT and IDH1R132H/WT knockin cells. Representative dot plots showing the distribution of IDH1WT/WT and IDH1R132H/WT cells along the signal obtained with the isotype-specific control antibodies or with the OCT3,4 and SOX2 direct conjugated antibodies (two technical replicates per n; n = 2 biological replicates).
For live-cell imaging, we established a 96-well plate-based screening assay to assess expression of the pluripotency-associated surface marker TRA-1-60, a more reliable and specific marker for predicting successful reprogramming and iPS cell derivation than other markers including AP (Mali et al., 2010), during reprogramming. Cell clusters were scored as null, weak, or strong depending on the expression of TRA-1-60. Cell clusters positive for TRA-1-60 were detected in MCF10A IDH1R132H/WT cells 2–3 days earlier relative to MCF10A IDH1WT/WT cells (Figure 3A). When the standard reprogramming protocol was followed with four reprogramming factors, OCT4, SOX2, KLF4, and c-MYC (hereafter OSKM), a significantly greater number of AP+ colonies (∼300) were observed in OSKM-transduced IDH1R132H/WT cells compared with IDH1WT/WT parental cells (∼140) at day 13. TRA-1-60+ clusters were also detected at an earlier stage in OSKM-transduced MCF10A IDH1R132H/WT cells than in MCF10A IDH1WT/WT control cells (Figure 3A).
Because the stimulatory effect of 2HG on the nuclear reprogramming efficiency was more striking in the absence of KLF4 and c-MYC transgenes (>10-fold with OS) than in their presence (2-fold with OSKM), we preliminarily explored the time frame during which 2HG might exert its positive reprogramming effects. Exogenous supplementation of IDH1WT/WT parental cells with 1 mmol/l D-2HG octylester, a concentration of a cell membrane-permeable form of 2HG that has previously been shown to mimic 2HG levels in tumors with aberrant 2HG accumulation by promoting a >100-fold increased intracellular concentration of 2HG (Lu et al., 2012, Xu et al., 2011, Terunuma et al., 2014), beginning soon after OS and OSKM transduction, for 4 days, resulted in a significantly increased number of reprogrammed colonies (Figure 3A). The fact that an early short-term supplementation with exogenous octyl-2HG, which caused prominent epithelial-to-mesenchymal (EMT)-like changes in cell fate (Figure 3A), was sufficient to promote a pro-reprogramming effect similar to that of continued endogenous exposure in 2HG-overproducing EMT-like MCF10A IDH1R132H/WT cells (Grassian et al., 2012), was consistent with the notion that 2HG might contribute to the time-sensitive activation of a mesenchymal stage required during the initiation period of successful reprogramming (Liu et al., 2013, O'Malley et al., 2013).
Furthermore, increased numbers of AP+ colonies and TRA-1-60+ clusters were detected within a shorter period of time in octyl-2HG-treated IDH1WT/WT parental cells (Figure 3A). Interestingly, when monitoring the effect of 2HG on the appearance of tightly packed colonies morphologically resembling human embryonic stem cells (hESCs), 2HG was found to increase the ratio of AP+ iPSL-10A colonies to total hESC-like colonies, i.e., the 2HG-driven increase in the number of AP+ iPSL-10A colonies was not accompanied by changes in the total colony number, thus implying that 2HG enhances the destination of reprogrammed cells to the stem cell fate (data not shown). To confirm that the pre-existing status of histone modifications might differentially regulate the global chromatin environment controlling reprogramming toward a pluripotent state, we reexamined the global histone modification variation that occurred in early stages of reprogramming (i.e., 6 days after OS and OSKM transduction) before genuine AP+ iPS-like patches become apparent in the cultures (Figure 3B). Interestingly, H3K9 methylation, which has been defined as the primary epigenetic determinant for the intermediate pre-iPS state as its removal leads to fully reprogrammed iPS cells (Chen et al., 2013), was notably suppressed by early OS induction in 2HG-overproducing IDH1R132H/WT cells but not in 2HG-negative IDH1WT/WT cells. Moreover, a reduction of H3K27me3 and H3K4me3, a phenomenon that has been associated with the acquisition of a transient open/primed chromatin state during the early transcriptional events of nuclear reprogramming (Hussein et al., 2014), was apparently observed upon early OS induction in 2HG-overproducing IDH1R132H/WT cells but not in 2HG-negative IDH1WT/WT cells. Thus, whereas the baseline levels of functionally opposing histone methylation marks were increased in 2HG-overproducing IDH1R132H/WT cells, consistent with a broad inhibition of histone demethylation, 2HG-induced histone modifications provided a collaborative rewiring of the chromatin organization that responded more rapidly and efficiently to the core stemness factors (OS) at the start of reprogramming.
Because the process of dedifferentiation through the addition of Yamanaka factors is extremely inefficient, the actual contribution of de novo generated “CSC states” via oncometabolic reprogramming to cancer evolution might be a matter of conjecture. To evaluate such a situation, we first confirmed that, upon the introduction of defined reprogramming factors and subsequent partial differentiation, proliferating CSC-like cells stably overexpressing OCT4 and SOX2 with tumor-initiating capacity (Nishi et al., 2014b) likewise arise from non-CSC, OCT4/SOX2-negative MCF10A IDH1R132H/WT cells (Figure 3B). We then designed a mathematical model to investigate the expected dynamics of tumor progression when the presence of oncometabolic signals can favor differentiated cells to revert to a multipotent CSC-like state. When a native, “resident” population sustained by normal stem cells competes with “invader” clones of CSC-like cells generated de novo as the result of nuclear reprogramming, our mathematical model predicts that the chances of prolonged survival increase exponentially with the size of the reprogrammed clones (Supplemental Appendix F). By solely affecting epigenetic events involving histone methylation, oncometabolites such as 2HG can functionally replace stemness transcription factors (e.g., KLF4 and c-MYC) and accelerate the dedifferentiation rates to efficiently drive the de novo generation of reprogrammed CSC-like states, thus confirming that the possibility that metabolically driven nuclear reprogramming-like phenomena contribute to cancer initiation, and that progression cannot be neglected in terms of cancer prognosis and therapeutic planning (Brooks et al., 2015, Leder et al., 2010, Martin-Castillo et al., 2015, Menendez et al., 2014a).
The experimental approach using no-2HG versus 2HG-overproducing cellular models in an identical non-transformed genomic background, functionally confirms the predictions of our stochastic model, demonstrating that an oncometabolite markedly lowers the “energy barriers” separating non-stem and stem cell attractors, diminishes the average time of reprogramming, and increases the size of the basin of attraction of the macrostate occupied by stem cells (Figure 4). For 2HG to improve nuclear reprogramming performance, it is sufficient to be present only during the first few days of reprogramming, when it appears to exert partial functional redundancy with other reprogramming factors that ensure the supply of chromatin-modifying enzymes with metabolic intermediates for the epigenetic activation of stemness-related gene networks (Goding et al., 2014, Gut and Verdin, 2013, Menendez and Alarcón, 2014).
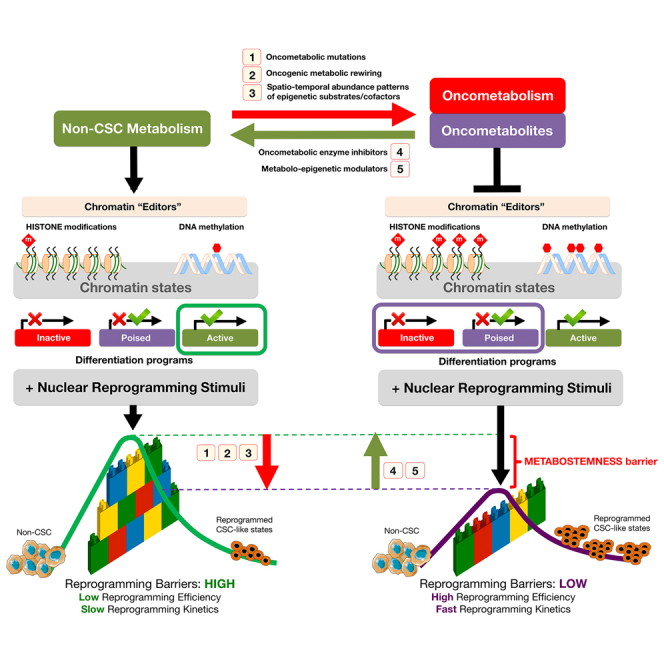
Figure 4.

Oncometabolite-Driven Nuclear Reprogramming of Cancer Stemness: A Framework Proposal
HDMs, such as Jumonji histone demethylases (JHDM) and ten-eleven translocation (TET) family members, remove repressive histone methylation marks and activate the expression of differentiation-related genes by protecting promoters from aberrant DNA methylation. Oncometabolites such as 2HG inhibit the epigenetic “editors” HDMs and TETs, which leads to histone modifications (e.g., increased H3K9me3, H3K27me3, and H3K4me3) and DNA hypermethylation. Oncometabolites reprogram chromatin state to promote the downregulation of genes involved in differentiation as well as bias in developmental gene-expression patterns. This metabolo-epigenetic modification of inactive/poised states of lineage-specific genes is sufficient to significantly alter the efficiency and speed of nuclear reprogramming by lowering the “reprogramming barriers” of the epigenetic landscape and increasing the size of the stem cell state basin of attraction, which results in the acceleration (i.e., higher efficiency and faster kinetics) of the nuclear reprogramming process. Oncometabolites such as 2HG permissively alleviate the unfavorable developmental process of “jumping” from differentiated cell states to CSC-like attractors while concomitantly stabilizing the ground-state self-maintaining character of CSC states. This conceptual figure represents cells stabilized in an initial non-CSC attractor and how nuclear reprogramming can make cells exceed the “reprogramming barrier,” represented as a wall of interlocking bricks, harder or easier in the absence or presence of the oncometabolite 2HG, respectively, and fall down in a final CSC attractor. The cellular reprogramming process is represented as a dashed line from the initial to the final cellular state.
Discussion
One of the most challenging issues in the field of cancer research is understanding how cellular metabolism influences chromatin structure and the epigenome to drive tumor formation (Johnson et al., 2015, Menendez and Alarcón, 2014, Lu and Thompson, 2012, Yun et al., 2012). To date, however, there have been no attempts to delineate predictive mathematical platforms that operatively integrate the required contribution of certain metabolites for the extensive remodeling of the epigenetic landscape that drives nuclear reprogramming (Morris et al., 2014). From a mathematical standpoint, here we introduce nucleosome modification and epigenetic regulation of lineage-specific genes as an essential element of stochastic modeling that successfully integrate the recognized ability of oncometabolites to competitively inhibit epigenetic regulation of cell differentiation with the process whereby the stemness regulatory circuitry is established during nuclear reprogramming (Ben-David et al., 2013, Shu et al., 2013). By combining mathematical modeling and computation simulation with wet-lab in vitro experiments in an isogenic model, we demonstrate the existence of bona fide oncometabolic nuclear reprogramming phenomena able to efficiently generate CSC-like states (Figure 4). Our model provides a stochastic tool as well as a conceptual framework that should be extremely useful in helping to understand and investigate the underexplored link between cellular metabolism and cancer-driving alterations in the epigenome. Beyond the numerous “common” metabolites that are used as substrates and cofactors for reactions that coordinate epigenetic status (Locasale, 2013, Johnson et al., 2015, Yun et al., 2012), a recent systems approach predicted more than 40 compounds and substructures of potential “oncometabolites” that could result from the loss-of-function and gain-of-function mutations of metabolic enzymes (Nam et al., 2014). In this context, our model can be a starting point for future studies on the processes by which cellular metabolism influences chromatin structure and epi-transcriptional circuits to causally drive stemness in cancer tissues.
Experimental Procedures
Stochastic Model
A detailed mathematical formulation of the stochastic model of oncometabolic nuclear reprogramming can be found in the Supplemental Information.
Reagents
The octyl ester derivative of [2R]-2-hydroxyglutaric acid was purchased from US Biologicals Life Sciences (cat. #01386; Deltaclon). AGI-5198, a highly potent and selective inhibitor of IDH1 R132H/R132C mutants, was purchased from Selleck Chemicals (cat. #S7185).
Cell Lines
MCF10A cells with heterozygous knockin of IDH1 dominant-negative (R132H) point mutation and MCF10A isogenic parental cells were obtained from Horizon Discovery (cat. #HD 101-013 and #HD PAR-058, respectively). IDH1 mutational status was verified by sequencing (Grassian et al., 2012).
Reprogramming of Human Breast Epithelial Cells and Cell Infection
MCF10A IDH1R132H/WT and MCF10A IDH1WT/WT cells were transduced with retroviral vectors encoding nuclear reprogramming factors as previously described (Nishi et al., 2014a, Nishi et al., 2014b (Figure S1). The pMX vectors containing human cDNA for OCT4, SOX2, KLF4, and c-MYC were obtained from Addgene (http://www.addgene.org).
Alkaline Phosphatase Staining
AP staining was performed using the Leukocyte Alkaline Phosphatase kit (cat. #86-R; Sigma-Aldrich) according to the manufacturer's protocol in OS- and OSKM-transduced cells reseeded onto mouse embryo fibroblast feeder layers.
Live Staining by the TRA-1-60 Antibody
A mouse anti-human StainAlive TRA-1-60 antibody (DyLight 488; cat. #09-0068) was employed according to the manufacturer's protocol to identify and track the appearance of iPS-like colonies in OS- and OSKM-transduced cells reseeded onto Matrigel-coated 96-well plates.
Histone Extraction and Western Analysis
Histones were acid-extracted following a modified version of the original protocol published by Sarg et al. (2002). For western blot analyses of H3K4me3, H3K9me3, H3K27me3, and total H3, 12 μg of the histone lysates were electrophoresed on 17% SDS-PAGE gel, transferred to a 0.45-mm polyvinylidene fluoride membrane, and incubated with antibodies against histone H3 (Abcam; cat. #ab1791), H3K4me3 (Abcam; cat. #ab8580), H3K9me3 (Millipore; cat. #CS200604), and H3K27me3 (Upstate-Millipore; cat. #07-449, lot #DAM1421462), followed by horseradish peroxidase-conjugated secondary and chemiluminescence detection.
Targeted Metabolomics
Quantitative measurements of 2HG were performed by employing a method based on gas chromatography coupled to a quadrupole time-of-flight mass spectrometer and an electron impact interface (GC-EI-QTOF-MS). A detailed description of this procedure is given in Cuyàs et al. (2015) and Riera-Borrull et al. (2016).
Global DNA Methylation
DNA was extracted and purified with a DNeasy Blood & Tissue kit (Qiagen; cat. #69504 or #69506) according to the manufacturer's instructions. Global DNA methylation levels were determined using the Global DNA Methylation LINE-1 kit (Active Motif; cat. #55017) according to the manufacturer's instructions.
Genome-wide DNA Methylation: DNA Methylation Microarrays and Data Analysis
Microarray-based DNA methylation profiling was performed using Illumina Infinium HumanMethylation450 BeadChip Array (Bibikova et al., 2011). Methylation levels (beta values) were obtained using Illumina's GenomeStudio Software. The beta value represents a quantitative measure of the DNA methylation level of specific CpG sites and ranges from 0 (completely unmethylated) to 1 (completely methylated). Before analyzing the methylation data (accession number GEO: GSE76263), we excluded possible sources of technical biases that could alter the results. We excluded probes with a detection p value of ≥0.01 and removed the probes containing a SNP at the CpG interrogation site. Because there were only two samples in the experimental design, we used stringent statistical criteria to define differential methylated probes. Thus, we defined a probe to be hypermethylated or hypomethylated if the differences between beta values was larger than 0.5. The HOMER (Hypergeometric Optimization of Motif EnRichment) suite of tools (http://homer.salk.edu/homer/) was used to determine the enrichment of individual ontology terms and create GO maps in the groups of differentially methylated genes.
Multivariate Permeabilized Cell Flow Cytometry
OCT3,4 and SOX2 protein levels were analyzed by intracellular staining using the Fix & Perm Cell Permeabilization kit (Invitrogen, cat. #GAS004), and flow cytometry using the anti-hOct4-PE (Becton Dickinson, cat. #560186) and anti-hSox2-AF488 (BD, cat. #561593) primary antibodies. Corresponding isotype antibodies MsIgG1 PE (BD, cat. #556650) and MsIgG1 AF488 (BD, cat. #551954) were used as controls. Plots show the fluorescence intensity distribution and percentages of cells above or below the thresholds determined by the staining with the isotype antibodies.
Author Contributions
J.A.M. and T.A. conceived the idea for this project and wrote the manuscript. T.A. developed the stochastic mathematical model. J.A.M. designed and analyzed the experiments on living cells. B.C.F. and E.C. conducted reprogramming experiments and data analysis. M.G.G. conducted the western blot experiments and data analysis. A.F.F. and M.F.F. conducted DNA methylation arrays and data analysis.
Acknowledgments
This work was supported by grants from the Ministerio de Ciencia e Innovación (Grant SAF2012-38914), Plan Nacional de I + D + I, Spain and the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) (Grant 2014 SGR229), Departament d’Economia i Coneixement, Catalonia, Spain to J.A.M. T.A. acknowledges financial support from the Ministerio de Ciencia e Innovación (MICINN) under grant MTM2011-29342 and Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) under grant 2014SGR1307. We are thankful to José M.A. Vaquero (Center of Regenerative Medicine, CMR[B] Core Facility-Cytometry Unit, Barcelona, Spain) for excellent assistance with flow cytometry.
Published: February 11, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, Supplemental data, Supplemental appendices, three figures, and eight tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.12.012.
Contributor Information
Javier A. Menendez, Email: jmenendez@iconcologia.net.
Tomás Alarcón, Email: talarcon@crm.cat.
Accession Numbers
The accession number for the DNA methylation data reported in this paper is GEO: GSE76263.
Supplemental Information
References
- Barrero M.J., Boué S., Izpisúa Belmonte J.C. Epigenetic mechanisms that regulate cell identity. Cell Stem Cell. 2010;7:565–570. doi: 10.1016/j.stem.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Ben-David U., Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer. 2011;11:268–277. doi: 10.1038/nrc3034. [DOI] [PubMed] [Google Scholar]
- Ben-David U., Nissenbaum J., Benvenisty N. New balance in pluripotency: reprogramming with lineage specifiers. Cell. 2013;153:939–940. doi: 10.1016/j.cell.2013.04.051. [DOI] [PubMed] [Google Scholar]
- Bibikova M., Barnes B., Tsan C., Ho V., Klotzle B., Le J.M., Delano D., Zhang L., Schroth G.P., Gunderson K.L. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Brooks M.D., Burness M.L., Wicha M.S. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. 2015;17:260–271. doi: 10.1016/j.stem.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Dent S.Y. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet. 2014;15:93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Liu H., Liu J., Qi J., Wei B., Yang J., Liang H., Chen Y., Chen J., Wu Y. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- Chowdhury R., Yeoh K.K., Tian Y.M., Hillringhaus L., Bagg E.A., Rose N.R., Leung I.K., Li X.S., Woon E.C., Yang M. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuyàs E., Fernández-Arroyo S., Corominas-Faja B., Rodríguez-Gallego E., Bosch-Barrera J., Martin-Castillo B., De Llorens R., Joven J., Menendez J.A. Oncometabolic mutation IDH1 R132H confers a metformin-hypersensitive phenotype. Oncotarget. 2015;6:12279–12296. doi: 10.18632/oncotarget.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd I.B., Micheelsen M.A., Sneppen K., Thon G. Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell. 2007;129:813–822. doi: 10.1016/j.cell.2007.02.053. [DOI] [PubMed] [Google Scholar]
- Duncan C.G., Barwick B.G., Jin G., Rago C., Kapoor-Vazirani P., Powell D.R., Chi J.T., Bigner D.D., Vertino P.M., Yan H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012;22:2339–2355. doi: 10.1101/gr.132738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goding C.R., Pei D., Lu X. Cancer: pathological nuclear reprogramming? Nat. Rev. Cancer. 2014;14:568–573. doi: 10.1038/nrc3781. [DOI] [PubMed] [Google Scholar]
- Grassian A.R., Lin F., Barrett R., Liu Y., Jiang W., Korpal M., Astley H., Gitterman D., Henley T., Howes R. Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition (EMT) J. Biol. Chem. 2012;287:42180–42194. doi: 10.1074/jbc.M112.417832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P., Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- Hussein S.M., Puri M.C., Tonge P.D., Benevento M., Corso A.J., Clancy J.L., Mosbergen R., Li M., Lee D.S., Cloonan N. Genome-wide characterization of the routes to pluripotency. Nature. 2014;516:198–206. doi: 10.1038/nature14046. [DOI] [PubMed] [Google Scholar]
- Johnson C., Warmoes M.O., Shen X., Locasale J.W. Epigenetics and cancer metabolism. Cancer Lett. 2015;256:309–314. doi: 10.1016/j.canlet.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian J.K., Kim S.Y., Miettinen M., Smith C., Merino M., Tsokos M., Quezado M., Smith W.I., Jr., Jahromi M.S., Xekouki P. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3:648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler P.S. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009;27:1050–1056. doi: 10.1002/stem.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder K., Holland E.C., Michor F. The therapeutic implications of plasticity of the cancer stem cell phenotype. PLoS One. 2010;5:e14366. doi: 10.1371/journal.pone.0014366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letouzé E., Martinelli C., Loriot C., Burnichon N., Abermil N., Ottolenghi C., Janin M., Menara M., Nguyen A.T., Benit P. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Liu X., Sun H., Qi J., Wang L., He S., Liu J., Feng C., Chen C., Li W., Guo Y. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat. Cell Biol. 2013;15:829–838. doi: 10.1038/ncb2765. [DOI] [PubMed] [Google Scholar]
- Locasale J.W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Thompson C.B. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Ward P.S., Kapoor G.S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C.R., Khanin R., Figueroa M.E., Melnick A. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Ye Z., Chou B.K., Yen J., Cheng L. An improved method for generating and identifying human induced pluripotent stem cells. Methods Mol. Biol. 2010;636:191–205. doi: 10.1007/978-1-60761-691-7_12. [DOI] [PubMed] [Google Scholar]
- Martin-Castillo B., Lopez-Bonet E., Cuyàs E., Viñas G., Pernas S., Dorca J., Menendez J.A. Cancer stem cell-driven efficacy of trastuzumab (Herceptin): towards a reclassification of clinically HER2-positive breast carcinomas. Oncotarget. 2015;6:32317–32338. doi: 10.18632/oncotarget.6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez J.A., Alarcón T. Metabostemness: a new cancer hallmark. Front. Oncol. 2014;4:262. doi: 10.3389/fonc.2014.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez J.A., Alarcón T., Corominas-Faja B., Cuyàs E., López-Bonet E., Martin A.G., Vellon L. Xenopatients 2.0: reprogramming the epigenetic landscapes of patient-derived cancer genomes. Cell Cycle. 2014;13:358–370. doi: 10.4161/cc.27770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez J.A., Corominas-Faja B., Cuyàs E., Alarcón T. Metabostemness: metaboloepigenetic reprogramming of cancer stem-cell functions. Oncoscience. 2014;1:803–806. doi: 10.18632/oncoscience.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R., Sancho-Martinez I., Sharpee T.O., Izpisua Belmonte J.C. Mathematical approaches to modeling development and reprogramming. Proc. Natl. Acad. Sci. USA. 2014;111:5076–5082. doi: 10.1073/pnas.1317150111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam H., Campodonico M., Bordbar A., Hyduke D.R., Kim S., Zielinski D.C., Palsson B.O. A systems approach to predict oncometabolites via context-specific genome-scale metabolic networks. PLoS Comput. Biol. 2014;10:e1003837. doi: 10.1371/journal.pcbi.1003837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M., Akutsu H., Kudoh A., Kimura H., Yamamoto N., Umezawa A., Lee S.W., Ryo A. Induced cancer stem-like cells as a model for biological screening and discovery of agents targeting phenotypic traits of cancer stem cell. Oncotarget. 2014;5:8665–8680. doi: 10.18632/oncotarget.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M., Sakai Y., Akutsu H., Nagashima Y., Quinn G., Masui S., Kimura H., Perrem K., Umezawa A., Yamamoto N. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene. 2014;33:643–652. doi: 10.1038/onc.2012.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi K., Semi K., Yamamoto T., Shimizu M., Tanaka A., Mitsunaga K., Okita K., Osafune K., Arioka Y., Maeda T. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014;156:663–677. doi: 10.1016/j.cell.2014.01.005. [DOI] [PubMed] [Google Scholar]
- O'Malley J., Skylaki S., Iwabuchi K.A., Chantzoura E., Ruetz T., Johnsson A., Tomlinson S.R., Linnarsson S., Kaji K. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature. 2013;499:88–91. doi: 10.1038/nature12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B., Plath K. Epigenetics of reprogramming to induced pluripotency. Cell. 2013;152:1324–1343. doi: 10.1016/j.cell.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera-Borrull M., Rodríguez-Gallego E., Hernández-Aguilera A., Luciano F., Ras R., Cuyàs E., Camps J., Segura-Carretero A., Menendez J.A., Joven J., Fernández-Arroyo S. Exploring the process of energy generation in pathophysiology by targeted metabolomics: performance of a simple and quantitative method. J. Am. Soc. Mass Spectrom. 2016;27:168–177. doi: 10.1007/s13361-015-1262-3. [DOI] [PubMed] [Google Scholar]
- Saha S.K., Parachoniak C.A., Ghanta K.S., Fitamant J., Ross K.N., Najem M.S., Gurumurthy S., Akbay E.A., Sia D., Cornella H. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–114. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarg B., Koutzamani E., Helliger W., Rundquist I., Lindner H.H. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J. Biol. Chem. 2002;277:39195–39201. doi: 10.1074/jbc.M205166200. [DOI] [PubMed] [Google Scholar]
- Shu J., Wu C., Wu Y., Li Z., Shao S., Zhao W., Tang X., Yang H., Shen L., Zuo X. Induction of pluripotency in mouse somatic cells with lineage specifiers. Cell. 2013;153:963–975. doi: 10.1016/j.cell.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivakov M., Fisher A.G. Epigenetic signatures of stem-cell identity. Nat. Rev. Genet. 2007;8:263–271. doi: 10.1038/nrg2046. [DOI] [PubMed] [Google Scholar]
- Suva M.L., Riggi N., Bernstein B.E. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma A., Putluri N., Mishra P., Mathé E.A., Dorsey T.H., Yi M., Wallace T.A., Issaq H.J., Zhou M., Killian J.K. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Invest. 2014;124:398–412. doi: 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung P.Y., Knoepfler P.S. Epigenetic mechanisms of tumorigenicity manifesting in stem cells. Oncogene. 2015;34:2288–2296. doi: 10.1038/onc.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M., Yang H., Xu W., Ma S., Lin H., Zhu H., Liu L., Liu Y., Yang C., Xu Y. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., Ito S., Yang C., Wang P., Xiao M.T. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Soga T., Pollard P.J. Oncometabolites: linking altered metabolism with cancer. J. Clin. Invest. 2013;123:3652–3658. doi: 10.1172/JCI67228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J., Johnson J.L., Hanigan C.L., Locasale J.W. Interactions between epigenetics and metabolism in cancers. Front. Oncol. 2012;2:163. doi: 10.3389/fonc.2012.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.