

Abstract
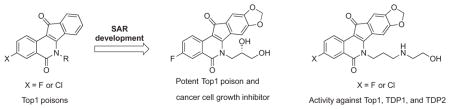
Fluorine and chlorine are metabolically stable, but generally less active replacements for a nitro group at the 3-position of indenoisoquinoline topoisomerase IB (Top1) poisons. A number of strategies were employed in the present investigation to enhance the Top1 inhibitory potencies and cancer cell growth inhibitory activities of halogenated indenoisoquinolines. In several cases, the new compounds’ activities were found to rival or surpass those of similarly substituted 3-nitroindenoisoquinolines, and several unusually potent analogues were discovered through testing in human cancer cell cultures. A hydroxyethylaminopropyl side chain on the lactam nitrogen of two halogenated indenoisoquinoline Top1 inhibitors was found to also impart inhibitory activity against tyrosyl DNA phosphodiesterases 1 and 2 (TDP1 and TDP2), which are enzymes that participate in the repair of DNA damage induced by Top1 poisons.
Graphical Abstract
1. Introduction
Topoisomerase IB (Top1) is a nuclear enzyme that catalyzes the relaxation of supercoiled DNA. To do so, it cleaves one strand of the DNA duplex and forms a transient, covalent complex with the cut strand.1, 2 Top1 poisons intercalate between the base pairs at the DNA cleavage site and thereby trap the covalent Top1-DNA intermediate by preventing its reversal.3 DNA replication forks that encounter a trapped cleavage complex are unable to proceed past it and leave behind a DNA double-strand break and a Top1-DNA adduct. Cells that are unable to efficiently repair this DNA damage ultimately undergo cell death.4 Cancer cells are especially vulnerable to the cytotoxic effects of Top1 poisons because they express higher levels of Top1 to support rapid cell division,5 while having compromised DNA repair and checkpoint capabilities.4
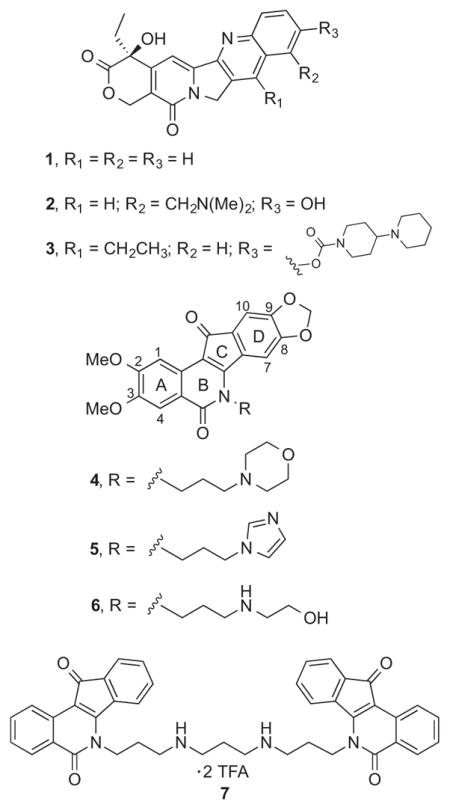
Safer and more effective Top1 poisons outside of the camptothecin (1) structural class are needed for cancer chemotherapy. The approved drugs topotecan (2) and irinotecan (3) suffer from a number of problems related to the development of resistance, chemical instability, drug-DNA-Top1 ternary cleavage complex instability, and dose-limiting side effects, and these might be mitigated by the indenoisoquinoline class of Top1 poisons.4, 6, 7 Some of the advantages of the indenoisoquinolines include chemical stability, longer drug residence times in the cleavage complex, and greater activity vs. camptothecin-resistant enzyme mutants. The limitations observed for the approved drugs have stimulated the design and synthesis of a variety of camptothecin analogues, including gimatecan, belotecan, lurtotecan, exatecan, and diflomotecan, as well as non-camptothecin analogues such as the indolocarbazole edotecarin and the azabenzophenanthridine topovale, but none have obtained FDA approval.6, 8 The present report details the development of highly active analogues of the indenoisoquinoline Phase 1 clinical trial drugs indotecan (LMP400, 4) and indimitecan (LMP776, 5), as well as the clinical trial candidate MJ-III-65 (LMP744, 6).7, 9–14 The new compounds display potent cancer cell growth and Top1 enzyme inhibitory activities and their halogenated A-rings are likely to produce less genotoxic metabolites than the corresponding nitro compounds.15 In addition, the halogenated Top1 inhibitors were screened for inhibition of the DNA repair enzymes tyrosyl DNA phosphodiesterase 1 and 2 (TDP1 and 2). Because many cancer cell types have an already compromised capacity for DNA repair, inhibition of TDP1 and/or TDP2 may selectively reduce the ability of cancer cells to overcome the cytotoxic effects of Top1 poisons, and triple Top1/TDP1/TDP2 inhibitors would therefore be especially interesting.16 A limited number of indenoisoquinolines, such as dimer 7,17 are already known to inhibit Top1 as well as one or more of these DNA repair enzymes.18
2. Chemistry
Structural modifications of fluorinated and chlorinated indenoisoquinolines19 were implemented to enhance their Top1 poisoning activities. The first was the fusion of a dioxolane ring to the 8- and 9-positions of the scaffold. Previous S.A.R. studies documented a modest improvement in Top1 poisoning activity with this substitution.20 A second modification was made to the 2-position. Molecular modeling indicated that a second fluorine or chlorine atom at this position would be tolerated by the surrounding environment. It was hypothesized that the addition of a second electronegative halogen atom could improve the “π-π-stacking” interactions between the ligand and the flanking base pairs in the ternary drug-DNA-Top1 cleavage complex by facilitating charge transfer complex formation.
A series of pentacyclic lactone intermediates (e.g. 13 and 14, Scheme 1) were therefore prepared that could be used to probe the effects of having different A-ring substitution patterns and different side chains on the lactam nitrogen. 3-Hydroxyphthalides 819 and 919 were each condensed with phthalide 10 under basic conditions.21 The 1,3-indanedione intermediates 11 and 12 were each cyclized in situ in refluxing Ac2O to yield lactones 13 and 14,22 which were condensed with primary amines 15–20 to yield the new indenoisoquinolines 21–30. The carbohydrate-derived primary amines 16 and 17 were prepared in two steps from D-xylose or D-ribose, respectively, according to published procedures.23, 24 The amines 15–17 were selected because they contributed to the synthesis of very active carbohydrate-substituted indenoisoquinolines,25 while 18, 19, and 20 were chosen because they would lead to compounds with side chains present in the indotecan (LMP400), indimitecan (LMP776), and MJ-III-65 (LMP744).7, 11, 13
Scheme 1.

a. aReagents and conditions: (a) i. NaOMe, MeOH, EtOAc, reflux; ii. Ac2O, reflux; (b) CHCl3, MeOH, reflux.
2,3-Difluoroindenoisoquinolines 37 and 38 were synthesized starting from the benzoic acid derivative 31 (Scheme 2). Reduction of 31 with BH3 provided the benzylic alcohol 32, which was subjected to a Rosenmund-von Braun reaction followed by hydrolysis and lactonization to yield phthalide 33. Oxidation of the methylene carbon of phthalide 33 was carried out using NBS, and the resulting 3-bromophthalide 34 was hydrolyzed following column chromatography purification to deliver the 3-hydroxyphthalide 35. Condensation of precursors 35 and 10 under basic conditions, and treatment of the unisolated 1,3-indanedione intermediate (not shown, similar to 11 and 12) in heated Ac2O yielded lactone 36. Lactone 36 was condensed with primary amines 15 or 18 to yield 37 and 38, respectively.
Scheme 2.

a. aReagents and conditions: (a) BH3, THF, reflux; (b) i. CuCN, DMF, reflux; ii. H2O, reflux; (c) i. NBS, AIBN, CCl4, reflux; (d) H2O, reflux; (e) i. NaOMe, MeOH, EtOAc, reflux; ii. Ac2O, 90 °C; (f) CHCl3, MeOH, reflux.
3,4-Dichloroindenoisoquinolines 45 and 46 were synthesized starting from phthalic acid 39 (Scheme 3), which was converted to its anhydride 40 with AcCl. Anhydride 40 was reduced with excess NaBH4 and the unisolated intermediate was treated with catalytic pTSA in refluxing PhMe to yield phthalide 41. Oxidation of phthalide 41 with NBS, and hydrolysis of 3-bromophthalide 42 gave them 3-hydroxypthalide 43. 3-Hydroxyphthalide 43 and phthalide 10 were used to make lactone 44 by the two-step sequence described before. Derivatization of lactone 44 with primary amines 15 or 18 provided 45 and 46, respectively.
Scheme 3.

a. aReagents and conditions: (a) AcCl, reflux; (b) NaBH4, THF, 0 °C to room temp.; (c) NBS, AIBN, CCl4, reflux; (d) i. KOH in H2O, reflux; ii. NaHSO4 (e) i. NaOMe, MeOH, EtOAc, reflux; ii. Ac2O, 90 °C; (f) CHCl3, MeOH, reflux.
3. Results and discussion
A Top1-mediated DNA cleavage assay26 was used to evaluate the Top1 poisoning activities of the compounds, which were tested at 0.1, 1, 10, and 100 μM concentrations in the presence of a 3′-[32P]-labeled, double-stranded DNA fragment and recombinant Top1 enzyme. When a compound poisons Top1-DNA cleavage complexes, the labeled cleaved strands are detected as shorter strands in the electrophoretic assay.26 To measure the activity of a new compound in the Top1-mediated DNA cleavage assay, the number and intensity of the bands observed in test agent lanes is compared to that seen with the positive control, camptothecin (1), at 1 μM. The relative activity is estimated and used to assign a semiquantitative score, which ranges from 0 (no activity) to ++++ (activity equal to that of 1 μM 1; see Table 1 footnotes for the complete rubric). A representative gel is shown in Figure 1. Aside from the determination of relative Top1 poisoning activities, competing Top1 suppression activity can be observed. For example, compound 25 appears to suppress Top1-mediated DNA cleavage at high drug concentration (100 μM), because the band density at several cleavage sites decreases. Suppression may be caused by the compound binding to free DNA, which makes the DNA a poorer substrate for the Top1 cleavage reaction.13
Table 1.
Antiproliferative and Top1 Poisoning Activities of Indenoisoquinolines
| Cytotoxicity (GI50, μM)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | Top1 Cleavagea | Lung HOP-62 | Colon HCT-116 | CNS SF-539 | Melanoma UACC-62 | Ovarian OVCAR-3 | Renal SN12C | Prostate DU-145 | Breast MCF7 | MGMb | |
| 411 | ++++ | 1.78 | 1.15 | 0.040 | 0.030 | 74.1 | 0.813 | 0.155 | N.D.c | 4.64 ± 1.25 | |
| 511 | ++++ | <0.010 | <0.010 | 0.037 | <0.010 | 0.085 | <0.010 | <0.010 | N.D. | 0.079 ± 0.023 | |
| 613 | ++++ | 0.01 | 0.15 | 0.09 | 0.03 | 1.50 | 0.01 | 0.02 | N.D | 0.21 ± 0.19 | |
|
| |||||||||||
| 3-NO2 | 4728 | ++++ | < 0.010 | < 0.010 | < 0.010 | 0.017 | 0.302 | < 0.010 | < 0.010 | 0.025 | 0.090 ± 0.04 |
|
| |||||||||||
| 3-F | 21 | ++++ | 0.049 | 0.079 | 0.051 | 0.045 | 0.182 | 0.076 | 0.102 | 0.032 | 0.105 |
| 23 | ++ | 0.032 | 0.035 | 0.029 | 0.025 | 0.100 | 0.079 | 0.051 | 0.012 | 0.078 | |
| 25 | +++ | < 0.010 | < 0.010 | < 0.010 | < 0.010 | 0.019 | < 0.010 | < 0.010 | < 0.010 | 0.011 | |
| 27 | + | N.S.d | |||||||||
| 29 | ++ | 0.036 | 0.019 | 0.041 | 0.028 | 0.076 | 0.107 | 0.033 | < 0.010 | 0.055 | |
|
| |||||||||||
| 3-Cl | 22 | ++ | 0.022 | 0.020 | 0.025 | 0.012 | 0.062 | 0.027 | 0.032 | < 0.010 | 0.042 |
| 24 | + | N.T.e | |||||||||
| 26 | ++ | < 0.010 | < 0.010 | < 0.010 | < 0.010 | 0.040 | < 0.010 | < 0.010 | < 0.010 | 0.024 | |
| 28 | + | N.S. | |||||||||
| 30 | +++ | 0.071 | 0.105 | 0.240 | 0.043 | 1.32 | 1.07 | 0.059 | 0.018 | 0.229 | |
|
| |||||||||||
| 2,3-F | 37 | ++ | 0.022 | 0.037 | 0.029 | 0.015 | 0.224 | 0.033 | 0.041 | < 0.010 | 0.068 |
| 38 | + | < 0.010 | < 0.010 | < 0.010 | < 0.010 | 0.115 | < 0.010 | < 0.010 | < 0.010 | 0.030 | |
|
| |||||||||||
| 2,3-Cl | 45 | ++ | < 0.010 | < 0.010 | < 0.010 | < 0.010 | 0.016 | < 0.010 | < 0.010 | < 0.010 | 0.013 |
| 46 | + | < 0.010 | < 0.010 | < 0.010 | < 0.010 | 0.043 | < 0.010 | < 0.010 | < 0.010 | 0.017 | |
|
| |||||||||||
| 2.75 | 48d, 19 | ++ | |||||||||
| 4919 | +++ | 0.490 | 0.309 | 0.417 | 0.245 | 1.29 | 0.437 | 0.575 | 0.135 | 0.692 | |
| 5019 | +++ | 2.09 | 1.29 | 2.95 | 1.38 | 4.27 | 2.34 | 2.95 | 0.347 | 2.75 | |
| 5119 | ++ | 0.692 | 0.437 | 0.417 | 0.398 | 2.24 | 0.977 | 2.09 | 0.257 | 1.12 | |
| 5219 | +++ | 0.186 | 0.107 | 0.204 | 0.074 | 0.407 | 0.110 | 0.309 | 0.031 | 0.229 | |
| 5319 | ++ | 2.14 | 1.62 | 2.40 | 3.31 | 6.03 | 5.01 | 4.07 | 0.372 | 4.17 | |
Compound-induced DNA cleavage due to Top1 poisoning, with scores given according to the following system based on the activity of 1 μM 1: 0, no activity; +, between 20 and 50% activity; ++, between 50 and 75% activity; +++, between 75 and 95% activity; ++++, equal activity; ++++(+), greater activity.
Mean graph midpoint of growth inhibition from the 5 concentration assay, ranging from 10−8–10−4 M, for all 60 cell lines tested.
N.D. = not determined.
N.S. = not selected for 5 concentration assay.
N.T. = not tested in the NCI-60.
Figure 1.

Top1-mediated DNA cleavage assay gel. From left: lane 1, DNA alone; lane 2, DNA and Top1; lane 3, DNA and Top1 and 1 μM 1; lane 4, DNA and Top1 and 1 μM 6; remaining lanes, DNA and Top1 and indicated concentration (μM) of test compound. The numbers and arrows at the left indicate cleavage site positions on the DNA substrate (see Experimental Section). Gel-based assays are performed twice for each active compound, and they are always run with positive controls (i.e., 1 and 6).
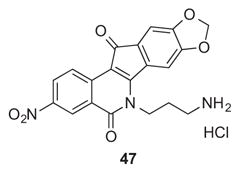
The NCI-60 human tumor cell line screen (“NCI-60”)27 was used to evaluate the cytotoxicities of the new compounds. The NCI-60 triages new compounds after first testing them at 10 μM. If the averaged growth percent across the cell lines is sufficiently low, the compound is promoted to a five-concentration assay. The five testing concentrations range from 10−8 to 10−4 μM, and the resulting concentration-response curves are used to calculate the concentration required for 50% growth inhibition relative to control, or GI50, for each cell line. In cases where a GI50 falls below or above the testing range, it is recorded as either the minimum (10−8 μM) or maximum (10−4 μM) testing concentration. The average GI50 for all 60 cell lines tested under these conditions is called the GI50 mean graph midpoint (MGM). Cytotoxicity data including MGM values and GI50 values for a limited number of specific cell lines, as well as Top1-mediated DNA cleavage assay scores for the tested compounds are displayed in Table 1. Compounds 4–6 and 4728 are included in the table for easy referencing.
Contrary to expectation, the second generation of bioisosteric indenoisoquinolines was not enriched in potent Top1 poisons (i.e., Top1 scores at or above ++++). Of the fourteen new compounds, one achieved a ++++ score, and two achieved a +++ score. Although all of the compounds displayed some Top1 poisoning activity, most were clustered at + or ++ scores. In contrast, the six fluorinated and chlorinated compounds in the first generation (48–53) either achieved a +++ score (49, 50, and 52) or a ++ score (48, 51, and 53). The major structural difference between these two generations was at the D-ring, to which an 8,9-methylenedioxy substitution was added in the second generation. The effect of installation of this group on indenoisoquinoline Top1 poisoning activity was not consistent with observations from previous studies, which have shown that the group can increase activity moderately.20, 29
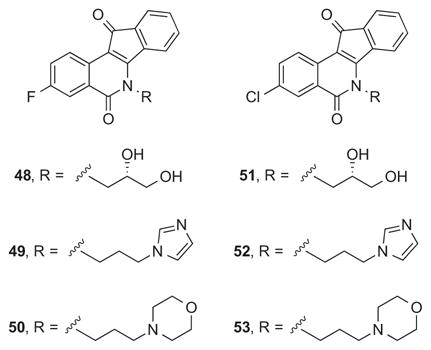
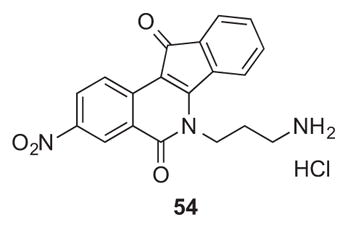
On the other hand, the second generation compounds reported here generally have improved (10-fold or better) GI50 MGM values versus the first generation. Twelve of the fourteen compounds have GI50 MGM values below 100 nM. The remaining two compounds also have low GI50 MGM values (105 nM and 229 nM). It was previously established that particular alkoxy substituents at the 8- and/or 9-positions could enhance antiproliferative potency of indenoisoquinolines. For example, compound 47 has an GI50 MGM value of 90 nM, while its analogue 54, which lacks the 8,9-methylenedioxy group, has an GI50 MGM value of 245 nM.28 The most cytotoxic compound of the present series was the 3-fluoro derivative 25 having an imidazole-containing side chain, which displayed a GI50 MGM of 11 nM. It should be recognized that the actual average GI50 is significantly lower than MGM value of 11 nM, since cases in which the GI50 values were lower than 10 nM were recorded as 10 nM, the lowest concentration tested in the standard NCI-60 assay, for calculation of the GI50 MGM value. The indenoisoquinoline 25 is an attractive analogue of LMP776 (5), which has been shown to undergo oxidative demethylation of the 2-methoxy group in the presence of human liver microsomes.30
The indenoisoquinolines substituted with a 2,3-difluoro (37 and 38) or 2,3-dichloro pattern (45 and 46) were expected to have improved Top1 poisoning activity relative to 3-fluoro- or 3-chloro-substituted analogues. Instead, these four compounds displayed relatively weak Top1 scores (+ or ++). It is not clear from molecular modeling why 2-position substitution with a fluorine or chlorine should be detrimental to Top1 poisoning activity. On the other hand, the 13–69 nM GI50 MGM values for these four compounds indicate very potent cytotoxic activity in human cancer cells.
Each of the new compounds was tested in biochemical assays for tyrosyl DNA phosphodiesterase 131 and 232 inhibitory activity, with the rationale being that combining Top1 poisoning with TDP1 and TDP2 inhibition could potentially enhance the therapeutic usefulness of these drugs. TDP1 hydrolyzes 3′-phosphotyrosyl linkages between peptides and DNA that result from degradation of topoisomerase I (Top1)-DNA adducts produced by the action of Top1 inhibitors. TDP2 hydrolyzes 5′-phosphotyrosyl linkages between peptides and DNA that result from degradation of topoisomerase II (Top2)-DNA adducts produced by the action of Top2 inhibitors and to a lesser extent in Top1-DNA adducts produced by Top1 inhibitors.16, 33 TDP2 can also hydrolyze 3′-phosphotyrosyl linkages, like TDP1, and so it can also participate in Top1 poison-induced DNA damage repair. Recent evidence shows that TDP1 and TDP2 may serve as mutual backups in DNA damage repair.16, 33
Two Top1 inhibitors prepared in the current study were found to be active in both recombinant TDP1 and TDP2 inhibition assays (Figures 2 and 3). Indenoisoquinolines 29 and 30 both inhibited TDP1 enzyme with IC50 values of 8.7 and 6.3 μM, respectively (Figure 2 and Table 2). Compounds 29 and 30 also inhibited recombinant TDP2 enzyme with IC50 values of 10.2 and 9.1 μM, respectively (Figure 3 and Table 2). Compounds 29 and 30 are structurally similar, with each possessing a hydroxyethylaminopropyl side chain. This side chain was not previously known to confer TDP1 or TDP2 inhibitory activity to the indenoisoquinolines. This observation is significant, because the same side chain is known to confer Top1 inhibitory activity.7, 12 Compound 29 achieved a ++ score, while compound 30 achieved a +++ score in the Top1 assay, and both compounds are therefore novel triple inhibitors of all three enzymes, Top1, TDP1, and TDP2.
Figure 2.

TDP1 inhibition assay gel. The concentrations of positive control 7 and test compounds were 0.46, 1.4, 4.1, 12.3, 37, 111 μM (left to right). N14Y is 5′-end labeled DNA oligonucleotide with 3′ phosphotyrosyl, and N14P is 5′-end labeled DNA oligonucleotide with a 3′-phosphate group (see Experimental Section).
Figure 3.

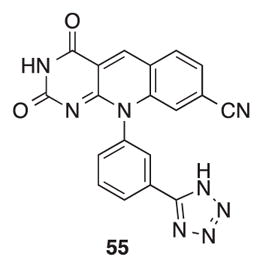
TDP2 inhibition assay gel. The concentrations of positive control 5534 were 0.005, 0.017, 0.05, 0.15, 0.46, 1.4 μM, and the concentrations for the test compounds were 0.46, 1.4, 4.1, 12.3, 37, 111 μM (left to right). The TDP2 substrate Y19 corresponds to a 3′-end labeled DNA oligonucleotide with a 5′ phosphotyrosyl, and the TDP2 product p19 corresponds to a 3′-end labeled DNA oligonucleotide with a 5′ phosphate group (see Experimental Section).
Table 2.
IC50 Determinations for Indenoisoquinolines 29 and 30 versus TDP1 and TDP2
| Compound | TDP1 IC50, μM | TDP2 IC50, μM |
|---|---|---|
| 29 | 6.2, 11.2 (n = 2) | 6.3, 14.1 (n = 2) |
| 30 | 6.3 ± 1.4 (n = 3) | 8.9, 9.3 (n = 2) |
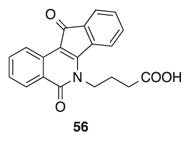
A molecular modeling study was carried out to examine the potential interactions between indenoisoquinoline 25 and its binding site in the Top1-DNA cleavage complex (Figure 4). The fluorine atom is calculated to engage Asn722 in a hydrogen-bonding interaction, and its 11-position ketone oxygen engages Arg364 in a second direct hydrogen-bonding interaction. The binding mode is nearly identical to that observed for indenoisoquinoline 56 in its co-crystal structure with the Top1-DNA cleavage complex.35
Figure 4.

Energy-minimized hypothetical binding pose of 25 (purple) within the X-ray crystal structure of a stalled Top1-DNA cleavage complex co-crystallized with 56 (PDB ID 1SC7).35 Potential hydrogen-bonding interactions are indicated by dashed lines. Heavy atom distances (in Å) appear next to the dashed lines. The stereoview is programmed for wall-eyed (relaxed) viewing.
4. Conclusion
Installation of 8,9-methylenedioxy substitution on the D-ring of 3-position fluorinated and chlorinated indenoisoquinolines and exploration of varied side chains at the lactam nitrogen resulted in compounds with extremely potent growth inhibitory activities, although Top1 poisoning activities were not generally enhanced. Attachment of a fluorine or chlorine atom in the 2-position of 3-halogenated indenoisoquinolines was attempted because of observations made from molecular modeling. It was suspected that addition of a small electronegative halogen atom in this position would be tolerated and allow modulation of the “π-π-stacking” interaction by facilitating charge transfer complex formation. The resulting 2,3-dihalogenated compounds displayed robust cytotoxic activities in cancer cell cultures, although they had relatively weak Top1 poisoning activities. The second generation includes two 3-fluoroindenoisoquinolines 21 and 25 that showed significant growth inhibitory and Top1 poisoning activities. Both compounds compare favorably to the nitrated analogue 47, and are expected to be less genotoxic than 47 because they lack the nitro group.15 It was also found that hydroxyethylaminopropyl side chains can impart inhibitory activity against tyrosyl DNA phosphodiesterase 1 and 2 (TDP1 and 2) without eliminating Top1 poisoning activity. At present, there are few reported dual inhibitors of both TDP1 and TDP2,19, 34 and none that are potent triple inhibitors of both of these enzymes as well as poisons of Top1. Generally speaking, triple inhibitors of Top1, TDP1, and TDP2 are attractive because they not only damage DNA, they also inhibit the repair of the damage.
5. Experimental section
5.1 Chemistry
Reactions were monitored by silica gel analytical thin-layer chromatography, and 254 nm UV light was used for visualization. All yields refer to isolated compounds. Unless otherwise stated, chemicals and solvents were of reagent grade and used as obtained from commercial sources without further purification. Melting points were determined using capillary tubes and are uncorrected. 1H Nuclear magnetic resonance spectroscopy was performed using a 300 MHz or 500 MHz spectrometer. Infrared spectra were obtained using an FTIR spectrometer. Mass spectral analyses were performed at the Purdue University Campus-Wide Mass Spectrometry Center. HPLC analyses were performed on a Waters 1525 binary HPLC pump/Waters 2487 dual λ absorbance detector system, using a 5 μm C18 reversed phase column and UV detection at 254 nm. HPLC purities of all tested compounds were estimated from the major peak area and were ≥ 95% of the combined total peak area.
5.1.1 3-Fluoro-8,9-methylenedioxyindeno[1,2-c]isochromene-5,12-dione (13)
3-Hydroxyphthalide 819 (506 mg, 3.01 mmol) and phthalide 10 (538 mg, 3.02 mmol) were suspended in EtOAc (10 mL) and a freshly prepared solution of NaOMe (0.27 g, 12 mmol of Na) in MeOH (20 mL) was added. The solution was heated at reflux with stirring under an argon atmosphere for 23 h. The mixture was cooled to room temperature, acidified to pH 1 with 37% HCl (1.5 mL), concentrated in vacuo, and azeotroped with PhMe (2 × 10 mL). Ac2O (20 mL) was added to the residue and the mixture was heated at reflux with stirring under an argon atmosphere for 1 h 20 min. The suspension was azeotroped with PhMe (4 × 10 mL) to provide a purple residue, which was sequentially washed with hexanes (50 mL) and H2O (50 mL) and hexanes (20 mL). The impure solid residue was purified by silica gel column chromatography, eluting with CHCl3, to give lactone 13 (140 mg, 15%) as a dull purple solid: mp 324–330 °C. IR (film) 1738, 1715, 1504, 1375, 1160 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.17 (dd, J = 9.0 Hz, J = 5.6 Hz, 1 H), 7.91 (dd, J = 9.0 Hz, J = 2.9 Hz, 1 H), 7.87 – 7.75 (m, 1 H), 7.29 – 7.22 (m, 2 H), 6.20 (s, 2 H); EIMS m/z 310 (M+, 100); HRCIMS calcd for C17H8FO5 311.0350 (MH+), found 311.0353.
5.1.2 3-Chloro-8,9-methylenedioxyindeno[1,2-c]isochromene-5,12-dione (14)
3-Hydroxyphthalide 919 (570 mg, 3.09 mmol) and phthalide 10 (561 mg, 3.15 mmol) were suspended in EtOAc (10 mL) and a freshly prepared solution of NaOMe (0.30 g, 13 mmol of Na) in MeOH (20 mL) was added. The solution was heated at reflux with stirring under an argon atmosphere for 18 h. The mixture was cooled to room temperature, acidified to pH 1 with 37% HCl (1.5 mL), concentrated in vacuo, and azeotroped with PhMe (2 mL). Ac2O (20 mL) was added to the residue and the mixture was heated at reflux with stirring under an argon atmosphere for 18 h. The suspension was azeotroped with PhMe (3 × 5 mL) to provide a purple residue that was partitioned between saturated NaHCO3 (150 mL) and CHCl3 (50 mL). The aqueous layer was removed. H2O (25 mL) and hexanes (10 mL) were added to the organic layer, and the mixture was filtered. The solid residue that was collected was washed with 1:1 hexanes-EtOAc (10 mL) and dried under high vacuum to provide lactone 14 (557 mg, 55%) as a purple solid: mp 335–336 °C. IR (film) 1746, 1711, 1490, 1376, 1240, 1025, 838 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.20 – 8.05 (m, 2 H), 8.01 – 7.88 (m, 1 H), 7.26 (d, J = 8.9 Hz, 2 H), 6.21 (s, 2 H); CIMS m/z (rel intensity) 327/329 (MH+, 100/42); HRCIMS calcd for C17H8ClO5 327.0055 (MH+), found 327.0051.
5.1.3 (S)-3-Fluoro-6-(2,3-dihydroxypropyl)-8,9-methylenedioxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (21)
Lactone 13 (46 mg, 0.15 mmol) was suspended in CHCl3 (15 mL) with stirring and a solution of (S)-2,3-dihydroxypropylamine (15, 33 mg, 0.36 mmol) in MeOH (10 mL) was added. The mixture was heated at reflux for 16 h, cooled to room temperature, and concentrated in vacuo. CHCl3 (5 mL) and hexanes (1 mL) were added to the residue, and the suspension was filtered. The product was washed with hexanes (5 mL) and MeOH (5 mL) and dried under high vacuum to provide 21 (38 mg, 66%) as a purple solid: mp 174 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ 8.50 (t, J = 6.8 Hz, 1 H), 7.79 (d, J = 9.7 Hz, 1 H), 7.67 (s, 1 H), 7.08 (s, 1 H), 6.17 (s, 2 H), 5.14 (d, J = 5.0 Hz, 1 H), 5.05 (t, J = 5.3 Hz, 1 H), 4.56 – 4.29 (m, 2 H), 3.95 (s, 1 H), 3.57 (t, J = 5.0 Hz, 2 H); CIMS m/z (rel intensity) 384 (MH+, 100); HRESIMS calcd for C20H14FNO6Na 406.0703 (MNa+), found 406.0705; HPLC purity, 95.14% (MeOH, 100%).
5.1.4 (S)-3-Chloro-6-(2,3-dihydroxypropyl)-8,9-methylenedioxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (22)
Lactone 14 (73 mg, 0.22 mmol) was suspended in MeOH (10 mL) and CHCl3 (15 mL), and a solution of (S)-2,3-dihydroxypropylamine (15, 40 mg, 0.44 mmol) in MeOH (1 mL) was added with stirring. The mixture was heated at reflux with stirring under an argon atmosphere for 14.5 h, cooled to room temperature, and filtered. The solid residue that collected was washed with hexanes (2 mL) and dried under high vacuum to yield 22 (67 mg, 75%) as a brownish-purple solid: mp 303–304 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.44 (d, J = 8.7 Hz, 1 H), 8.04 (d, J = 2.3 Hz, 1 H), 7.78 (dd, J = 8.7, 2.4 Hz, 1 H), 7.70 (s, 1 H), 7.09 (s, 1 H), 6.18 (s, 2 H), 5.13 (d, J = 5.0 Hz, 1 H), 5.11 – 4.97 (m, 1 H), 4.53 – 4.31 (m, 2 H), 4.00 – 3.86 (m, 1 H), 3.57 (t, J = 5.1 Hz, 2 H); MALDI m/z (rel intensity) 400/402 (MH+, 100/38); HRESIMS calcd for C23H17ClN3O4 400.0588 (MH+), found 400.0580; HPLC purity, 100% (MeOH, 100%).
5.1.5 (2′S,3′R,4′R)-3-Fluoro-5,6-dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (23)
D-Xylitylamine (16, 48 mg, 0.32 mmol) was dissolved in H2O (1 mL), and the solution was added to a stirring suspension of lactone 13 (88 mg, 0.28 mmol) in CHCl3 (20 mL) and MeOH (15 mL). The reaction mixture was heated at reflux with stirring for 24 h. Additional D-xylitylamine (16, 20 mg, 0.13 mmol) dissolved in H2O (0.25 mL) was added and heating to reflux was continued for 16 h. The mixture was concentrated in vacuo until ~2 mL of solvent remained, and the suspension was filtered to get a solid, which was dried under high vacuum with heating. Compound 23 (39 mg, 31%) was obtained as a red-violet solid: mp 260–272 °C (dec). 1H NMR (500 MHz, DMSO-d6) δ 8.50 (dd, J = 9.0, 5.4 Hz, 1 H), 7.83 (s, 1 H), 7.78 (dd, J = 9.4, 2.9 Hz, 1 H), 7.65 (td, J = 8.8, 2.9 Hz, 1 H), 7.07 (s, 1 H), 6.14 (s, 2 H), 5.03 (d, J = 5.2 Hz, 1 H), 4.94 (d, J = 5.7 Hz, 1 H), 4.64 (d, J = 5.5 Hz, 1 H), 4.59 – 4.49 (m, 2 H), 4.42 (dd, J = 14.2, 3.6 Hz, 1 H), 4.11 – 4.03 (m, 1 H), 3.69 – 3.62 (m, 1 H), 3.61 – 3.54 (m, 1 H), 3.50 – 3.43 (m, 1 H), 3.42 – 3.35 (m, 1 H); ESIMS m/z (rel intensity) 466 (MNa+, 100); HRESIMS calcd for C22H19FNO8 444.1095 (MH+), found 444.1086; HPLC purity, 100% (MeOH, 100%).
5.1.6 (2′S,3′S,4′R)-3-Chloro-5,6-dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (24)
D-Ribitylamine (17, 78 mg, 0.24 mmol) was dissolved in H2O (0.5 mL), and the solution was added to a stirring suspension of lactone 14 (41 mg, 0.27 mmol) in CHCl3 (20 mL) and MeOH (15 mL). The reaction mixture was heated at reflux with stirring for 24 h. The mixture was concentrated in vacuo to ~2 mL solvent remained and then filtered. The collected solid was sonicated in CHCl3-hexanes (2:1, 2 mL). After sitting for 10 min, additional solid had precipitated from the filtrate. Both of the suspensions were filtered. The combined solid was dried under high vacuum with heating to ~100 °C. Product 24 (15 mg, 12%) was obtained as a purple solid: mp 231 °C (dec). 1H NMR (500 MHz, DMSO-d6) δ 8.41 (d, J = 8.6 Hz, 1 H), 8.00 (d, J = 2.4 Hz, 1 H), 7.86 (s, 1 H), 7.74 (dd, J = 8.6, 2.4 Hz, 1 H), 7.04 (s, 1 H), 6.14 (d, J = 3.3 Hz, 2 H), 5.26 (d, J = 5.5 Hz, 1 H), 4.97 (d, J = 5.2 Hz, 1 H), 4.69 (d, J = 5.2 Hz, 1 H), 4.59 – 4.27 (m, 3 H), 4.13 (s, 1 H), 3.58 (m, 3 H), 3.41 (m, 1 H); ESIMS m/z (rel intensity) 482/484 (MNa+, 100/40); HRESIMS calcd for C22H19ClNO8 460.0800 (MH+), found 460.0801; HPLC purity, 100% (MeOH, 100%).
5.1.7 3-Fluoro-6-(3-(1H-imidazol-1-yl)propyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (25)
Lactone 13 (53 mg, 0.17 mmol) was dissolved in CHCl3 (20 mL) with stirring and 3-imidazolylpropylamine (18, 43 mg, 0.34 mmol) was added. The mixture was heated at reflux for 16 h, cooled, and concentrated to ~5 mL. Hexanes (1 mL) were added and the suspension was filtered. The product was washed with hexanes (5 mL) and MeOH (5 mL) and dried under high vacuum with heating to 125 °C to furnish 25 (54 mg, 76%) as a purple solid: mp 235 °C (dec). IR (film) 1667, 1503, 1377, 1310, 1027 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.50 (dd, J = 8.9, 5.4 Hz, 1 H), 7.83 (dd, J = 9.5, 2.8 Hz, 1 H), 7.78 – 7.64 (m, 2 H), 7.26 (s, 1 H), 7.15 (s, 1 H), 7.10 (s, 1 H), 6.91 (s, 1 H), 6.20 (s, 2 H), 4.41 (t, J = 7.4 Hz, 2 H), 4.18 (t, J = 6.8 Hz, 2 H), 2.32 – 2.09 (m, 2 H); EIMS m/z (rel intensity) 417 (M+, 100); HRESIMS calcd for C23H17FN3O4 418.1203 (MH+), found 418.1208; HPLC purity, 100% (MeOH, 100%).
5.1.8 3-Chloro-6-(3-(1H-imidazol-1-yl)propyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (26)
Lactone 14 (77 mg, 0.24 mmol) was dissolved in CHCl3 (20 mL) with stirring and a solution of 3-imidazolylpropylamine (18, 59 mg, 0.47 mmol) in CHCl3 (1 mL) was added. The mixture was heated at reflux with stirring under an argon atmosphere for 14.5 h, cooled to room temperature, and filtered. The product was washed with hexanes (2 mL) and dried under high vacuum at 60 °C to provide 26 (81 mg, 79%) as a dark purple solid: mp 286–287 °C. IR (film) 1668, 1537, 1482, 1304, 826 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.46 (d, J = 8.8 Hz, 1 H), 8.31 (s, 1 H), 8.09 (d, J = 2.3 Hz, 1 H), 7.82 (dd, J = 8.6, 2.3 Hz, 1 H), 7.69 (s, 1 H), 7.24 (d, J = 1.1 Hz, 1 H), 7.18 (s, 1 H), 7.13 (s, 1 H), 6.89 (d, J = 1.4 Hz, 1 H), 6.21 (s, 2 H), 4.41 (t, J = 7.3 Hz, 2 H), 4.17 (t, J = 6.9 Hz, 2 H), 2.33 – 2.09 (m, 2 H); MALDI m/z (rel intensity) 434/436 (MH+, 100/83); HRESIMS calcd for C23H17ClN3O4 434.0908 (MH+), found 434.0887; HPLC purity, 100% (MeOH, 100%).
5.1.9 3-Fluoro-5,6-dihydro-6-(3-morpholinopropyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Trifluoroacetate (27)
Lactone 13 (29 mg, 0.093 mmol) was dissolved in CHCl3 (9 mL) with stirring and a solution of 3-morpholinopropylamine (19, 14 mg, 0.096 mmol) in CHCl3 (1 mL) was added. The mixture was heated at reflux with stirring under an argon atmosphere for 20 h and concentrated in vacuo until ~1 mL of solvent remained. Hexanes (1 mL) were added and the suspension was filtered to provide the free base (15 mg, 37%) as a red-violet solid. The trifluoroacetate salt was prepared by treating the free base with neat TFA (5 mL) for 5 min at room temperature with stirring and then evaporating the TFA. Chemical characterization data pertain to the free base: mp 294–296 °C. 1H NMR (300 MHz, CDCl3) δ 8.64 (dd, J = 9.0, 5.3 Hz, 1 H), 7.94 (dd, J = 9.3, 2.9 Hz, 1 H), 7.49 (s, 1 H), 7.43 (td, J = 8.6, 2.8 Hz, 1 H), 7.12 (s, 1 H), 6.11 (s, 2 H), 4.60 – 4.46 (m, 2 H), 3.85 – 3.65 (m, 4 H), 2.64 – 2.40 (m, 6 H), 2.12 – 1.92 (m, 2 H); ESIMS m/z (rel intensity) 437 (MH+, 100); HRESIMS calcd for C24H22FN2O5 437.1513 (MH+), found 437.1493; HPLC purity, 99.35% (MeOH, 100%).
5.1.10 3-Chloro-5,6-dihydro-6-(3-morpholinopropyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Trifluoroacetate (28)
Lactone 14 (34 mg, 0.10 mmol) was dissolved in CHCl3 (9 mL) with stirring and a solution of 3-morpholinopropylamine (19, 15 mg, 0.11 mmol) in CHCl3 (1 mL) was added. The mixture was heated at reflux with stirring under an argon atmosphere for 20 h and concentrated in vacuo until ~1 mL solvent remained. Hexanes (1 mL) was added and the suspension was filtered. The product was dried under high vacuum to provide the free base (15 mg, 33%) as a dark purple solid. The trifluoroacetate salt was prepared by treating the free base with neat TFA (5 mL) for 5 min at room temperature with stirring and then evaporating the TFA. Chemical characterization data pertain to the free base: mp 290–294 °C. 1H NMR (300 MHz, CDCl3) δ 8.56 (d, J = 8.7 Hz, 1 H), 8.26 (d, J = 2.3 Hz, 1 H), 7.62 (dd, J = 8.7, 2.3 Hz, 1 H), 7.50 (s, 1 H), 7.13 (s, 1 H), 6.12 (s, 2 H), 4.59 – 4.46 (m, 2 H), 3.76 (t, J = 4.7 Hz, 4 H), 2.62 – 2.44 (m, 6 H), 2.08 – 1.93 (m, 2 H); ESIMS m/z (rel intensity) 453/455 (MH+, 100/29); HRESIMS calcd for C24H22ClN2O5 (MH+) 453.1217, found 453.1211; HPLC purity, 95.04% (MeOH, 100%).
5.1.11 3-Fluoro-5,6-dihydro-6-(3-((2-hydroxyethyl)amino)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (29)
Lactone 13 (69 mg, 0.22 mmol) was dissolved in CHCl3 (15 mL) with stirring and a solution of 3-(2-hydroxyethyl)propylamine (20, 36 mg, 0.30 mmol) in CHCl3 (1 mL) was added. The mixture was heated at reflux with stirring under an argon atmosphere for 3.5 h and concentrated in vacuo until ~1 mL of solvent remained. Hexanes (1 mL) were added and the suspension was filtered. The product was dried under high vacuum to provide 29 (69 mg, 76%) as a dull purple solid: mp 170 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.45 (dd, J = 9.0, 5.4 Hz, 1 H), 7.77 (dd, J = 9.5, 2.8 Hz, 1 H), 7.71 – 7.53 (m, 2 H), 7.10 (s, 1 H), 6.17 (s, 2 H), 4.56 – 4.27 (m, 3 H), 3.43 (d, J = 6.3 Hz, 2 H), 2.73 – 2.51 (m, 2 H), 1.92 – 1.65 (m, 2 H); ESIMS m/z (rel intensity) 411 (MH+, 100); HRESIMS calcd for C22H20FN2O5 411.1357 (MH+), found 411.1363; HPLC purity, 95.00% (0.1% TFA in MeOH, 100%).
5.1.12 3-Chloro-5,6-dihydro-6-(3-((2-hydroxyethyl)amino)propyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (30)
Lactone 14 (84 mg, 0.26 mmol) was dissolved in CHCl3 (15 mL) with stirring and a solution of 3-(2-hydroxyethyl)propylamine (20, 35 mg, 0.30 mmol) in CHCl3 (1 mL) was added. The mixture was heated at reflux with stirring under an argon atmosphere for 22 h and concentrated in vacuo until ~1 mL of solvent remained. Hexanes (1 mL) were added and the suspension was filtered and dried under high vacuum to provide 30 (93 mg, 84%) as a purple solid: mp 198 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.38 (d, J = 8.7 Hz, 1 H), 8.02 (d, J = 2.4 Hz, 1 H), 7.76 (dd, J = 8.7, 2.4 Hz, 1 H), 7.63 (s, 1 H), 7.10 (s, 1 H), 6.18 (s, 2 H), 4.59 – 4.27 (m, 3 H), 3.44 (t, J = 5.7 Hz, 2 H), 2.67 (t, J = 6.4 Hz, 2 H), 2.58 (t, J = 5.8 Hz, 2 H), 1.92 – 1.71 (m, 2 H); ESIMS m/z (rel intensity) 427/429 (MH+, 100/31); HRESIMS calcd for C22H20ClN2O5 427.1061 (MH+), found 427.1067; HPLC purity, 98.60% (0.1% TFA in MeOH, 100%).
5.1.13 (2-Bromo-4,5-difluorophenyl)methanol (32)
Benzoic acid derivative 31 (5.05 g, 21.3 mmol) was dissolved with stirring in THF (25 mL). A solution of BH3 in THF (1.0 M, 21.3 mL, 21.3 mmol) was added dropwise at room temperature with frequent pausing to allow violent bubbling to subside. The mixture was heated at reflux with stirring for 4 h. A solution of aqueous KOH (2 M, 11 mL) was added and the mixture was cooled to room temperature with stirring for 30 min. The mixture was extracted with Et2O (6 × 25 mL). The combined organic layers were washed with brine (1 × 25 mL) and dried over Na2SO4. The organic solution was concentrated in vacuo to yield a yellow-tinted liquid. Drying under high vacuum with heating to ca. 100 °C afforded 32 (4.01 g, 84%) as an off-white solid: mp 59–66 °C. 1H NMR (300 MHz, CDCl3) δ 7.46 – 7.32 (m, 2 H), 4.68 (d, J = 1.3 Hz, 2 H), 2.12 (br s, 1 H); EIMS m/z (rel intensity) 222/224 (M+, 70/70), 143 [(M − Br)+, 93], 115 [(M − Br − H2O)+, 100].
5.1.14 5,6-Difluorophthalide (33)
Alcohol 32 (4.01 g, 18.0 mmol) and CuCN (3.33 g, 37.2 mmol) were suspended with stirring in DMF (20 mL) with heating at reflux under Ar atmosphere for 2.5 h. The mixture was cooled to room temperature, H2O (2 mL) was added, and the mixture was heated at 100 °C for 1.5 h. After cooling to room temperature, H2O (80 mL) and EtOAc (100 mL) were added to the mixture, and it was filtered through a Celite plug. The organic layer of the filtrate was removed. The aqueous layer of the filtrate was extracted with EtOAc (2 × 100 mL). The combined organic layers were washed with brine (3 × 50 mL), dried over MgSO4, and concentrated in vacuo. The orange, semisolid residue was purified by silica gel column chromatography, eluting with CHCl3, and the pure product-containing fractions were pooled and concentrated in vacuo. The residue was washed with hexanes (~10 mL) to provide 33 (1.17 g, 38%) as an off-white solid, Rf (SiO2, CHCl3) 0.31: mp 109–112 °C. IR (film) 3062, 1774, 1498, 1455, 1314, 1023 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.72 (dd, J = 8.2, 7.0 Hz, 1 H), 7.32 (dd, J = 8.6, 6.3 Hz, 1 H), 5.29 (s, 2 H); CIMS m/z (rel intensity) 171 (MH+, 100).
5.1.15 5,6-Difluoro-3-hydroxyphthalide (35)
Phthalide 33 (1.00 g, 5.88 mmol), NBS (1.06 g, 5.96 mmol), and AIBN (0.05 g) were suspended with stirring in CCl4 and the mixture was heated at reflux under Ar atmosphere for 1 h 10 min. The suspension was cooled to room temperature and filtered. The filtered solids were washed with CCl4 (10 mL). The combined filtrates were concentrated in vacuo to yield a yellow oil. The oily residue was purified by silica gel column chromatography, eluting with CHCl3. The pure product-containing fractions were pooled and concentrated in vacuo, and the obtained residue was heated at reflux with stirring in H2O (20 mL) for 1 h. The mixture was cooled to room temperature and acidified with NaHSO4 (1.45 g). The product was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine (20 mL) and dried over Na2SO4. The organic solution was concentrated in vacuo and dried under high vacuum with heating to yield 35 (727 mg, 66%) as a white solid, Rf (SiO2, CHCl3) 0: mp 104–107 °C. IR (film) 3401, 1764, 1500, 1353, 1319 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.81 (dd, J = 8.9, 7.0 Hz, 1 H), 7.66 (dd, J = 9.2, 6.8 Hz, 1 H), aldehydic acid peaks not observed; CIMS m/z (rel intensity) 187 (MH+, 94), 169 (MH+ − H2O, 100).
5.1.16 2,3-Difluoro-8,9-methylenedioxyindeno[1,2-c]isochromene-5,12-dione (36)
3-Hydroxyphthalide derivative 35 (408 mg, 2.19 mmol) and phthalide 10 (393 mg, 2.21 mmol) were suspended in EtOAc (10 mL) and a freshly prepared solution of NaOMe (0.21 g, 9.1 mmol of Na) in MeOH (20 mL) was added. The solution was heated at reflux with stirring under Ar atmosphere for 24 h. The mixture was cooled to room temperature, acidified to pH 1 with 37% HCl (2 mL), concentrated in vacuo, and azeotroped with PhMe (10 mL). Ac2O (20 mL) was added to the residue and the mixture was heated at 90 °C with stirring under an argon atmosphere for 2 h. The mixture was cooled to room temperature, H2O (20 mL) was added, and the suspension was filtered. The solid was purified by silica gel column chromatography, eluting with CHCl3, to provide lactone 36 (86 mg, 12%) as a dark purple solid, Rf (SiO2, CHCl3) 0.60: mp 300–301 °C. IR (film) 1744, 1722, 1433, 780 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.12 – 7.99 (m, 2 H), 7.10 (s, 1 H), 6.97 (s, 1 H), 6.12 (s, 2 H); CIMS m/z (rel intensity) 329 (MH+, 100); HRESIMS calcd for C17H7F2O5 329.0256 (MH+), found 329.0244.
5.1.17 (S)-2,3-Difluoro-6-(2,3-dihydroxypropyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (37)
Lactone 36 (28 mg, 0.085 mmol) was suspended in CHCl3 (5 mL), and a solution of (S)-2,3-dihydroxypropylamine (15, 14 mg, 0.15 mmol) in MeOH (2 mL) was added with stirring. The mixture was heated at reflux with stirring under Ar atmosphere for 18.5 h. The suspension was cooled to room temperature, concentrated in vacuo until ~1 mL of solvent remained, and hexanes (0.5 mL) were added. The suspension was filtered and the solid residue that collected was dried under high vacuum to yield 37 (32 mg, 94%) as a red-violet solid: mp 256–261 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.28 (dd, J = 11.6, 7.8 Hz, 1 H), 8.06 (dd, J = 11.1, 8.2 Hz, 1 H), 7.74 (s, 1 H), 7.14 (s, 1 H), 6.19 (dd, J = 2.4, 1.0 Hz, 2 H), 5.16 (d, J = 5.0 Hz, 1 H), 5.06 (t, J = 5.3 Hz, 1 H), 4.50 – 4.37 (m, 2 H), 3.94 (s, 1 H), 3.57 (t, J = 5.0 Hz, 2 H); APCIMS m/z (rel intensity) 402 (MH+, 100); HRESIMS calcd for C20H13F2NO6Na 424.0609 (M + Na+), found 424.0606; HPLC purity, 100% (MeOH, 100%).
5.1.18 2,3-Difluoro-6-(3-(1H-imidazol-1-yl)propyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (38)
Lactone 36 (26 mg, 0.079 mmol) was dissolved in CHCl3 (5 mL) with stirring and a solution of 3-imidazolylpropylamine (18, 13 mg, 0.10 mmol) in CHCl3 (1 mL) was added. The mixture was heated at reflux with stirring under Ar atmosphere for 18.5 h, cooled to room temperature, and filtered. The product was washed with CHCl3 (1 mL) and dried under high vacuum to provide 38 (37 mg, 100%) as a dark purple solid: mp > 400 °C. IR (film) 1673, 1386, 1313 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.25 (dd, J = 11.5, 7.7 Hz, 1 H), 8.07 (dd, J = 11.0, 8.2 Hz, 1 H), 7.71 – 7.67 (m, 1 H), 7.25 (t, J = 1.2 Hz, 1 H), 7.17 (s, 1 H), 7.13 (s, 1 H), 6.95 – 6.85 (m, 1 H), 6.21 (s, 2 H), 4.39 (t, J = 7.2 Hz, 2 H), 4.17 (t, J = 6.8 Hz, 2 H), 2.26 – 2.12 (m, 2 H); ESIMS m/z (rel intensity) 436 (MH+, 100); HRESIMS calcd for C23H16F2N3O4 436.1109 (MH+), found 436.1107; HPLC purity, 98.41% (0.1% TFA in MeOH, 100%).
5.1.19 5,6-Dichlorophthalic Anhydride (40)
Diacid 39 (10.00 g, 42.55 mmol) was suspended in AcCl (50 mL) and the mixture was heated at reflux with stirring for 2 h. The obtained suspension was concentrated in vacuo and azeotroped with hexanes (2 × 10 mL) and PhMe (2 × 10 mL) to provide anhydride 40 (9.23 g, 100%) as a beige powder, Rf (SiO2, CHCl3) 0.64: mp 185–189 °C. 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 2 H); CIMS m/z (rel intensity) 217/219/221 (MH+, 100/69/12).
5.1.20 5,6-Dichlorophthalide (41)
Anhydride 40 (9.53 g, 42.6 mmol) was dissolved with stirring in THF (80 mL) and the solution was cooled to 0 °C under an Ar atmosphere. NaBH4 (1.63 g, 43.1 mmol) was added slowly in a single portion, and the mixture was stirred with cooling to 0 °C for 50 min. The mixture was removed from the ice-water bath and warmed to room temperature for 17 h. MeOH (20 mL) was added slowly and cautiously to the obtained suspension. The mixture was concentrated in vacuo. Dilute, aqueous HCl (0.1 M, 24 mL) was added to the residue. After stirring the mixture for 10 min, it was filtered. The collected solids were vigorously heated at reflux in PhMe (100 mL) with pTsOH·H2O (0.15 g), using a Dean-Stark trap to collect H2O, for 30 h 15 min. The mixture was concentrated in vacuo. The residue was washed with H2O (50 mL) and the solid was azeotroped with PhMe (10 mL) and dried under high vacuum to provide 41 (6.41 g, 74%) as a white solid, Rf (SiO2, CHCl3) 0.55: mp 157–160 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.14 (s, 1 H), 8.04 (s, 1 H), 5.40 (s, 2 H); CIMS m/z (rel intensity) 203/205/207 (MH+, 100/71/14).
5.1.21 5,6-Dichloro-3-hydroxyphthalide (43)
Phthalide 41 (1.05 g, 5.17 mmol), NBS (0.93 g, 5.22 mmol), and AIBN (0.10 g) were suspended with stirring in CCl4 (25 mL) and the mixture was heated at reflux under Ar atmosphere for 3 h 15 min. The suspension was cooled to room temperature and filtered. The filtered solids were washed with CCl4 (10 mL). The combined filtrates were concentrated in vacuo to yield a yellow oil. The oily product was purified by silica gel column chromatography, eluting with CHCl3. The pure product-containing fractions were pooled and concentrated in vacuo, and the residue was heated at reflux with stirring in a solution of KOH (0.59 g, 11 mmol) in H2O (15 mL) for 2 h. The mixture was cooled to room temperature and NaHSO4 (1.7 g) was added to acidify. The product was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine (15 mL) and dried over Na2SO4. The organic solution was concentrated in vacuo and dried under high vacuum at room temperature to yield 43 (574 mg, 51%) as a yellow-white solid, Rf (SiO2, CHCl3) 0.03: mp 163 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ 10.41 (br s, 0.2 H), 8.41 (d, J = 7.7 Hz, 1 H), 8.05 (s, 1 H), 7.97 (s, 0.3 H), 6.66 (d, J = 7.8 Hz, 1 H); CIMS m/z (rel intensity) 219/221/223 (MH+, 50/32/6), 201/203/205 (MH+ − H2O, 100/72/14).
5.1.22 2,3-Dichloro-8,9-methylenedioxyindeno[1,2-c]isochromene-5,12-dione (44)
3-Hydroxyphthalide derivative 43 (494 mg, 2.26 mmol) and phthalide 10 (405 mg, 2.27 mmol) were suspended in EtOAc (10 mL) and a freshly prepared solution of NaOMe (0.24 g, 10 mmol of Na) in MeOH (20 mL) was added. The solution was heated at reflux with stirring under Ar atmosphere for 4 d. The mixture was cooled to room temperature, acidified to pH 1 with 37% HCl (1.5 mL), concentrated in vacuo, and azeotroped with PhMe (10 mL). Ac2O (20 mL) was added to the residue and the mixture was heated at 90 °C with stirring under Ar atmosphere for 3 h. The suspension was azeotroped with PhMe (10 mL) and H2O (20 mL) was added to the residue. The mixture was filtered. The residue was washed sequentially with 3:2 CHCl3-hexanes (2 × 30 mL), 1:1 hexanes-EtOAc (25 mL) and 3:2 CHCl3-Et2O (25 mL). The solid was dried under high vacuum to provide lactone 44 (448 mg, 55%) as a dark purple solid, Rf (SiO2, CHCl3) 0.63: mp 301–302 °C. IR (film) 1760, 1714, 1483, 1386, 1317, 780 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.36 (s, 1 H), 8.31 (s, 1 H), 7.11 (s, 1 H), 6.96 (s, 1 H), 6.12 (s, 2 H); CIMS m/z (rel intensity) 361/363/365 (MH+, 100/76/20); HRCIMS calcd for C17H7Cl2O5 360.9665 (MH+), found 360.9670.
5.1.23 (S)-2,3-Dichloro-6-(2,3-dihydroxypropyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (45)
Lactone 44 (77 mg, 0.21 mmol) was suspended in MeOH (10 mL) and CHCl3 (15 mL), and a solution of (S)-2,3-dihydroxypropylamine (15, 44 mg, 0.48 mmol) in MeOH (1 mL) was added with stirring. The mixture was heated at reflux with stirring under an argon atmosphere for 19.5 h, cooled to room temperature, and filtered. The solid residue that collected was washed with hexanes (2 mL) and dried under high vacuum to yield 45 (77 mg, 84%) as a brownish purple solid: mp 314–315 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.46 (s, 1 H), 8.15 (s, 1 H), 7.67 (s, 1 H), 7.06 (s, 1 H), 6.18 (s, 2 H), 5.18 (d, J = 4.9 Hz, 1 H), 5.13 (t, J = 5.2 Hz, 1 H), 4.52 – 4.23 (m, 2 H), 4.01 – 3.85 (m, 1 H); CIMS m/z (rel intensity) 434/436/438 (MH+, 100/82/28); HREIMS calcd for C20H13Cl2NO6 433.0114 (M+), found 433.0112; HPLC purity, 100% (MeOH, 100%).
5.1.24 2,3-Dichloro-6-(3-(1H-imidazol-1-yl)propyl)-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (46)
Lactone 44 (82 mg, 0.23 mmol) was dissolved in CHCl3 (20 mL) with stirring and a solution of 3-imidazolylpropylamine (18, 56 mg, 0.45 mmol) in CHCl3 (1 mL) was added. The mixture was heated at reflux with stirring under an argon atmosphere for 19.5 h, cooled to room temperature, and filtered. The product was washed with hexanes (2 mL) and dried under high vacuum to provide 46 (90 mg, 84%) as a brownish purple solid: mp 309–310 °C. IR (film) 1670, 1603, 1478, 1308, 1293, 1031, 830, 756 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.54 (s, 1 H), 8.32 (s, 1 H), 8.25 (s, 1 H), 7.69 (s, 1 H), 7.25 (s, 1 H), 7.19 (s, 1 H), 7.16 (s, 1 H), 6.91 (s, 1 H), 6.23 (s, 2 H), 4.40 (t, J = 7.0 Hz, 2 H), 4.18 (t, J = 6.9 Hz, 2 H), 2.27 – 2.14 (m, 2 H); CIMS m/z (rel intensity) 468/470/472 (MH+, 100/92/31); HRESIMS calcd for C23H16Cl2N3O4 468.0518 (MH+), found 468.0521; HPLC purity, 99.90% (MeOH, 100%).
5.2 Topoisomerase I-Mediated DNA Cleavage Reactions
A 3′-[32P]-labeled 117-bp DNA oligonucleotide was prepared as previously described.26 The oligonucleotide contains previously identified Top1 cleavage sites in 161-bp pBluescript SK(−) phagemid DNA. Approximately 2 nM of radiolabeled DNA substrate was incubated with recombinant Top1 in 20 μL of reaction buffer [10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL BSA] at 25 °C for 20 min in the presence of various concentrations of test compounds. The reactions were terminated by adding SDS (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of each reaction mixture were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a phosphoimager and ImageQuant software (Molecular Dynamics). The numbers in Figure 1 represent actual cleavage site positions on the 117 bp oligonucleotide substrate.
5.3 Recombinant TDP1 Assay
A 5′-[32P]-labeled single-stranded DNA oligonucleotide containing a 3′-phosphotyrosine (N14Y)31 was incubated at 1 nM with 10 pM recombinant TDP1 in the absence or presence of inhibitor for 15 min at room temperature in the LMP1 assay buffer containing 50 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween-20.17 Reactions were terminated by the addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to a 16% denaturing PAGE with multiple loadings at 12-min intervals. Gels were dried and exposed to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600 (GE Healthcare), and densitometry analyses were performed using ImageQuant software (GE Healthcare).
5.4 Recombinant TDP2 Assay
TDP2 reactions were carried out as described previously32 with the following modifications. The 19-mer single-stranded oligonucleotide DNA substrate containing a 5′ phosphotyrosine (Y19, α32P-cordycepin-3′-labeled) was incubated at 1 nM with 25 pM recombinant human TDP2 in the absence or presence of inhibitor for 15 min at room temperature in the LMP2 assay buffer containing 50 mM Tris-HCl, pH 7.5, 80 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween 20. Reactions were terminated and treated similarly to recombinant TDP1 reactions (see above).
5.5 Cytotoxicity Testing
The cytotoxicity data reported in Table 1 were generated in the National Cancer Institutes’ collection of sixty human cancer cell lines (NCI-60). The assay procedures have been reported in detail.27, 36
5.6 Molecular Mechanics Calculations
The 1SC735 X-ray crystal structure file was prepared for molecular modeling by correcting several atom types, removing all of the water molecules, and adding hydrogens. The hydrogens were energy minimized while the remaining atoms were frozen in aggregate within SYBYL-X.37 The details of the minimizations for Top1 models were set as follows: Powell method; MMFF94s force field38; MMFF94 charges; and 0.05 kcal/mol·Å energy gradient convergence. Top1 ligands were docked into the prepared 1SC7 crystal structure using GOLD 3.2 software.39 The binding site was defined by the crystallized ligand, 56. Ten GOLD algorithm runs were executed per ligand, using default parameters. The top ten docking poses per ligand were inspected visually following the docking runs. The docking poses that had a favorable GOLD score and similar binding mode to the crystallized ligand were selected for further analysis.
Supplementary Material
Acknowledgments
This work was made possible by the National Institutes of Health (NIH) through support with Research Grants U01CA089566 and P30CA023168. In vitro cytotoxicity testing was performed by the Developmental Therapeutics Program at the National Cancer Institute, under contract NO1-CO-56000. Work done in the NCI Intramural Program, Center for Cancer Research was supported by the NIH Intramural Program (Z01 BC006161).
Abbreviations
- CPT
camptothecin
- Top1
topoisomerase IB
- TDP1
tyrosyl-DNA phosphodiesterase 1
- TDP2
tyrosyl-DNA phosphodiesterase 2
- PAGE
polyacrylamide gel electrophoresis
- SDS
sodium dodecyl sulfate
Footnotes
The authors declare the following competing financial interest: Mark Cushman is on the Board of Directors of and is an investor in Linus Oncology, Inc., which has licensed indenoisoquinoline intellectual property owned by Purdue University. Neither Linus Oncology, Inc., nor any other commercial company sponsored or provided other direct financial support to the author or his laboratory for the research reported in this article. The remaining authors have no competing and/or relevant financial interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A Model for the Mechanism of Human Topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 2.Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WGJ. Crystal Structures of Human Topoisomerase I in Covalent and Noncovalent Complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 3.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Stewart L. The Mechanism of Topoisomerase I Poisoning by a Camptothecin Analogue. Proc Natl Acad Sci USA. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier Y. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 5.Thomas CJ, Rahier NJ, Hecht SM. Camptothecin: Current Perspectives. Bioorg Med Chem. 2004;12:1585–1604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y. Drugging Topoisomerases: Lessons and Challenges. ACS Chem Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential Induction of Topoisomerase I-DNA Cleavage Complexes by the Indenoisoquinoline MJ-III-65 (NSC 706744) and Camptothecin: Base Sequence Analysis and Activity against Camptothecin-Resistant Topoisomerase I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- 8.Pommier Y. DNA Topoisomerase I Inhibitors: Chemistry, Biology, and Interfacial Inhibition. Chem Rev. 2009;109:2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. [accessed July 13, 2015];Clinical Study: 10-C-0056, a Phase I Study of Indenoisoquinolines LMP400 and LMP776 in Adults with Relapsed Solid Tumors and Lymphomas. http://clinicalstudies.info.nih.gov/cgi/detail.cgi?A_2010-C-0056.html.
- 10. [accessed July 13, 2015];Indenoisoquinoline LMP400 for Advanced Solid Tumors and Lymphomas. http://clinicaltrials.gov/ct2/show/NCT01794104.
- 11.Nagarajan M, Morrell A, Ioanoviciu A, Antony S, Kohlhagen G, Agama K, Hollingshead M, Pommier Y, Cushman M. Synthesis and Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Substituted with Nitrogen Heterocycles. J Med Chem. 2006;49:6283–6289. doi: 10.1021/jm060564z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, Rustum YM, Cushman M, Pommier Y. Cellular Topoisomerase I Inhibition and Antiproliferative Activity by MJ-III-65 (NSC 706744), an Indenoisoquinoline Topoisomerase I Poison. Mol Pharmacol. 2005;67:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 13.Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y. Synthesis of New Indeno[1,2-c]isoquinolines: Cytotoxic Non-Camptothecin Topoisomerase I Inhibitors. J Med Chem. 2000;43:3688–3698. doi: 10.1021/jm000029d. [DOI] [PubMed] [Google Scholar]
- 14.Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, Pommier Y. Novel Indenoisoquinolines NSC 725776 and NSC 724998 Produce Persistent Topoisomerase I Cleavage Complexes and Overcome Multidrug Resistance. Cancer Res. 2007;67:10397–10405. doi: 10.1158/0008-5472.CAN-07-0938. [DOI] [PubMed] [Google Scholar]
- 15.Smith GF. Designing Drugs to Avoid Toxicity. In: Lawton G, Witty DR, editors. Prog Med Chem. Vol. 50. 2011. pp. 1–47. [DOI] [PubMed] [Google Scholar]
- 16.Pommier Y, Huang S-yN, Gao R, Das BB, Murai J, Marchand C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2) DNA Repair. 2014;19:114–129. doi: 10.1016/j.dnarep.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen TX, Morrell A, Conda-Sheridan M, Marchand C, Agama K, Bermingam A, Stephen AG, Chergui A, Naumova A, Fisher R, O’Keefe BR, Pommier Y, Cushman M. Synthesis and Biological Evaluation of the First Dual Tyrosyl-DNA Phosphodiesterase I (Tdp1)-Topoisomerase I (Top1) Inhibitors. J Med Chem. 2012;55:4457–4478. doi: 10.1021/jm300335n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lv PC, Agama K, Marchand C, Pommier Y, Cushman M. Design, Synthesis, and Biological Evaluation of O-2-Modified Indenoisoquinolines as Dual Topoisomerase I-Tyrosyl-DNA Phosphodiesterase I Inhibitors. J Med Chem. 2014;57:4324–4336. doi: 10.1021/jm500294a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beck DE, Abdelmalak M, Lv W, Reddy PVN, Tender GS, O’Neill E, Agama K, Marchand C, Pommier Y, Cushman M. Discovery of Potent Indenoisoquinoline Topoisomerase I Poisons Lacking the 3-Nitro Toxicophore. J Med Chem. 2015;58:3997–4015. doi: 10.1021/acs.jmedchem.5b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagarajan M, Morrell A, Fort BC, Meckley MR, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis and Anticancer Activity of Simplified Indenoisoquinoline Topoisomerase I Inhibitors Lacking Substituents on the Aromatic Rings. J Med Chem. 2004;47:5651–5661. doi: 10.1021/jm040025z. [DOI] [PubMed] [Google Scholar]
- 21.Shapiro SL, Freedman L, Youlus J, Geiger K. Indandiones 2. Modified Dieckmann Reaction. J Org Chem. 1961;26:3580. [Google Scholar]
- 22.Pailer M, Worthen H, Meller A. Some Reactions of 2-Aryl-1,3-indandiones. Monatsh Chem. 1961;92:1037–1047. [Google Scholar]
- 23.Peterson KE, Cinelli MA, Morrell AE, Mehta A, Dexheimer TS, Agama K, Antony S, Pommier Y, Cushman M. Alcohol-, Diol-, and Carbohydrate-Substituted Indenoisoquinolines as Topoisomerase I Inhibitors: Investigating the Relationships Involving Stereochemistry, Hydrogen Bonding, and Biological Activity. J Med Chem. 2011;54:4937–4953. doi: 10.1021/jm101338z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinstock C, Plaut GWE. Synthesis and Properties of Certain Substituted Lumazines. J Org Chem. 1961;26:4456–4462. [Google Scholar]
- 25.Beck DE, Agama K, Marchand C, Chergui A, Pommier Y, Cushman M. Synthesis and Biological Evaluation of New Carbohydrate-Substituted Indenoisoquinoline Topoisomerase I Inhibitors and Improved Syntheses of the Experimental Anticancer Agents Indotecan (LMP400) and Indimitecan (LMP776) J Med Chem. 2014;57:1495–1512. doi: 10.1021/jm401814y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dexheimer TS, Pommier Y. DNA Cleavage Assay for the Identification of Topoisomerase I Inhibitors. Nat Protoc. 2008;3:1736–1750. doi: 10.1038/nprot.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoemaker RH. The NCI60 Human Tumour Cell Line Anticancer Drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 28.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2004;14:3659–3663. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 29.Morrell A, Placzek M, Parmley S, Antony S, Dexheimer TS, Pommier Y, Cushman M. Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors: A Systematic Study and Optimization. J Med Chem. 2007;50:4419–4430. doi: 10.1021/jm070361q. [DOI] [PubMed] [Google Scholar]
- 30.Cinelli MA, Reddy PVN, Lv PC, Liang JH, Chen L, Agama K, Pommier Y, van Breemen RB, Cushman M. Identification, Synthesis, and Biological Evaluation of Metabolites of the Experimental Cancer Treatment Drugs Indotecan (LMP400) and Indimitecan (LMP776) and Investigation of Isomerically Hydroxylated Indenoisoquinoline Analogues as Topoisomerase I Poisons. J Med Chem. 2012;55:10844–10862. doi: 10.1021/jm300519w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchand C, Lea WA, Jadhav A, Dexheimer TS, Austin CP, Inglese J, Pommier Y, Simeonov A. Identification of Phosphotyrosine Mimetic Inhibitors of Human Tyrosyl-DNA Phosphodiesterase I by a Novel AlphaScreen High-throughput Assay. Mol Cancer Ther. 2009;8:240–248. doi: 10.1158/1535-7163.MCT-08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao R, Huang S-yN, Marchand C, Pommier Y. Biochemical Characterization of Human Tyrosyl-DNA Phosphodiesterase 2 (TDP2/TTRAP) A Mg2//Mn2+-dependent Phosphodiesterase Specific for the Repair of Topoisomerase Cleavage Complexes. J Biol Chem. 2012;287:30842–30852. doi: 10.1074/jbc.M112.393983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng Z, Sharma A, Ju L, Murai J, Umans L, Vermeire L, Pommier Y, Takeda S, Huylebroeck D, Caldecott KW, El-Khamisy SF. TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic Acids Res. 2012;40:8371–8380. doi: 10.1093/nar/gks622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raoof A, Depledge P, Hamilton NM, Hamilton NS, Hitchin JR, Hopkins GV, Jordan AM, Maguire LA, McGonagle AE, Mould DP, Rushbrooke M, Small HF, Smith KM, Thomson GJ, Turlais F, Waddell ID, Waszkowycz B, Watson AJ, Ogilvie DJ. Toxoflavins and Deazaflavins as the First Reported Selective Small Molecule Inhibitors of Tyrosyl-DNA Phosphodiesterase II. J Med Chem. 2013;56:6352–6370. doi: 10.1021/jm400568p. [DOI] [PubMed] [Google Scholar]
- 35.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 36.Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved Anticancer Agents in the NCI60 Panel of Human Tumor Cell Lines. Mol Cancer Ther. 2010;9:1451–1460. doi: 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.SYBYL-X. Tripos, Inc; St. Louis, MO: 2009. [Google Scholar]
- 38.Halgren TA. Merck Molecular Force Field. 1. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J Comput Chem. 1996;17:490–519. [Google Scholar]
- 39.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved Protein-Ligand Docking Using GOLD. Protein Struct Funct Genet. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.