

Abstract
Automated planar patch clamp systems are widely used in drug evaluation studies because of their ability to provide accurate, reliable, and reproducible data in a high-throughput manner. Typically, CHO and HEK tumorigenic cell lines overexpressing single ion channels are used since they can be harvested as high-density, homogenous, single-cell suspensions. While human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs) are physiologically more relevant, these cells are fragile, have complex culture requirements, are inherently heterogeneous, and are expensive to produce, which has restricted their use on automated patch clamp (APC) devices. Here, we used high efficiency differentiation protocols to produce cardiomyocytes from six different hPSC lines for analysis on the Patchliner (Nanion Technologies GmbH) APC platform. We developed a two-step cell preparation protocol that yielded cell catch rates and whole-cell breakthroughs of ∼80%, with ∼40% of these cells allowing electrical activity to be recorded. The protocol permitted formation of long-lasting (>15 min), high quality seals (>2 GΩ) in both voltage- and current-clamp modes. This enabled density of sodium, calcium, and potassium currents to be evaluated, along with dose–response curves to their respective channel inhibitors, tetrodotoxin, nifedipine, and E-4031. Thus, we show the feasibility of using the Patchliner platform for automated evaluation of the electrophysiology and pharmacology of hPSC-CMs, which will enable considerable increase in throughput for reliable and efficient drug evaluation.
Introduction
Pharmaceutical drug development is costly and time-consuming, with an average drug development duration of 10–15 years [1] and costs upward of a billion dollars [2]. Furthermore, between 1980 and 2009, approximately one in seven licensed drugs that had demonstrated sufficient efficacies in Phase III trials had to be withdrawn from the market for reasons including unanticipated side effects like cardiotoxicity, hepatotoxicity, and gastrointestinal issues [3]. Unexpected cardiotoxic side effects have been implicated in 28% of drug withdrawals in the United States [4]. It has been calculated that reducing drug attrition by 5% in Phase I clinical development can reduce drug development costs by 5.5%–7.1% [5], equating to savings of about $100 million for drug developers [6]. This has necessitated the development of various in vitro, ex vivo, and/or preclinical models to predict toxicity in humans at earlier stages of the drug development pipeline.
Phase I drug trials are commonly carried out in aneuploid tumor cell lines (eg, CHO or HEK cells) that have been genetically engineered to overexpress an ion channel of choice. However, they cannot replicate the complexity of the working cardiomyocyte, and consequently, multi-channel blocking drugs that are considered safe and “QT-neutral,” such as verapamil (dual blocking of potassium IKr and calcium ICa,L channels) are flagged as potentially harmful in the single ion channel assays [7]. Though ex vivo systems, such as ventricular wedge preparations [8] and Purkinje fibers [9], have been extensively used in physiological and pharmacological studies, their low-throughput nature and inter-species differences limit their suitability as drug screening assays. Use of animals is also not in line with the growing expectation in many countries to address the 3Rs of animal-based research (refinement, reduction, and replacement of animals) [10].
As an example of these issues, the mouse heart beats ∼10 times faster than the human heart and does not utilize the IKr (HERG) potassium channel, which is important in repolarization in human cardiomyocytes [11]. The downstream effects of mutations or drugs implicated in cardiac arrhythmias would ideally be studied in a primary human cardiomyocyte. However, use of such cells as large-scale pharmaceutical screens is hindered by their limited availability, poor consistency, limited proliferation, and almost immediate de-differentiation in culture [6].
Cardiomyocytes derived from human pluripotent stem cells (hPSC-CMs), comprising human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), are now emerging as a viable alternative for drug safety testing (for reviews on the subject refer to [12,13]). Technology improvements over the last decade now allow rapid, cost-effective production of hPSC-CMs at purities that exceed 70% [14].
Even though hPSC-CMs have a relatively immature phenotype, they have shown faithful pharmacological responses to over 60 different compounds that include modulators of ion channels, β (1-, and 2-) receptors, and muscarinic receptors [6]. A blinded study that measured the effects of 12 cardiac and non-cardiac drugs over a 6-log dose–response range, covering estimated unbound therapeutic plasma concentrations, showed a close association between data obtained from hPSC-CMs and the clinic [15]. Work from GlaxoSmithKline that cross-compared the pharmacological responses of hPSC-CMs and animal models concluded that the human cells offered a reliable and cost-effective surrogate to preclinical in vitro testing [16]. hPSC-CMs were also used to show that the anticancer drug doxorubicin, delivered via an HER2-targeted liposomal pathway, reduced the cardiotoxicty observed with anthracyclines, which assisted the decision to advance the drug to Phase I testing [17].
To date, patient-specific hiPSC-CMs have been used to evaluate altered phenotype and drug rescue of various channelopathies affecting the heart, including Long QT syndrome (LQTS) 1 [18], 2 [19–22], 3 [23], 8, [24], and catecholaminergic polymorphic ventricular tachycardia (CPVT) [25,26]. Notably, Terrenoire et al. performed multi-parameter in vitro drug testing on the hPSC-CMs of a patient with complex LQTS to arrive upon a combinatorial regime that proved effective for the patient in the clinic, thus demonstrating the use of hPSC-CMs in personalized medicine [27]. Collectively, such studies have led the CIPA initiative (Comprehensive In Vitro Proarrhythmia Assay) to propose the integration of hPSC-CMs into the ICH (International Conference on Harmonisation) S7a/b and E14 guidelines by the end of 2015. These have been the mainstay over the last decade of preclinical assessment of cardiac electrophysiology for new drugs [28].
Despite these successes, virtually all studies to date have used conventional patch clamp to evaluate the electrophysiology of hPSC-CMs. Given that this approach can only produce 10–15 data points a day [29], there is now a pressing need for automated platforms that can produce high quality data at a throughput relevant to industry and academia. Automated patch clamping (APC), by allowing multiple recordings in parallel, can increase data throughput 10- to 100-fold depending on the ion channel under investigation and the platform used [30]. By reducing the complexity of the process, these platforms also make patch clamping accessible to more users, regardless of previous experience in electrophysiology. General drawbacks of parallel patch clamping include a requirement for high quality, high density, homogenous cell suspensions, which becomes restrictive when using expensive, low-yielding cell types [29].
These challenges are reflected by the paucity of data available for hPSC-CM evaluation using APCs. The PatchXpress planar patch clamp platform has been used for the APC analysis of INa, ICa, and IKr currents using hiPSC-CMs. However, that study used a genetically engineered hiPSC line, which allowed drug selection of the cardiomyocytes. Despite using a genetically enriched near-pure population of hiPSC-CMs, the study reported low seal rates (∼50% across 58 wells) and poor seal qualities (∼200 MΩ) [31], highlighting the need to develop better protocols for the preparation and analysis of cells by APC.
An alternative device is the Patchliner System (Nanion Technologies), which is a medium-throughput patch clamp device, capable of executing up to 48 unattended recordings and generating ∼500 data points a day [32]. However, Patchliner recordings have typically been performed on recombinant HEK and CHO cell lines, which are easy to obtain in large numbers, and form high-quality, high-density, homogenous single-cell suspensions ideal for APC (Table 1). In contrast, hPSC-CMs provide more physiologically relevant model systems for drug efficacy and safety testing (Table 1), but these are expensive to produce in large numbers and very sensitive to enzymatic processing into single-cell suspensions [6].
Table 1.
A Comparison of the Relevance of Recombinant Cell Lines and Human Stem Cell-Derived Cardiomyocytes for Drug Screening and Development
| Attributes | Recombinant cell lines | Stem cell-derived cardiomyocytes |
|---|---|---|
| Cell line generation | ||
| Ease of production | Easy to grow; undergo prolonged, high-density proliferation in culture; high capacity for stable recombinant protein expression | Require specialist know-how and reagents; limited proliferation in culture; low capacity for stable recombinant protein expression |
| Cost of production | ∼£5/L of culture media | ∼£400/L of culture media |
| Cell preparation for planar patch clamp | ||
| Homogeneity of prep | Homogeneous | Heterogeneous |
| Clumpiness of prep | Single cell | A mixture of single cells, cell clusters, and debris |
| Quality of seals | >1 GΩ | 200 MΩ–1 GΩ |
| Quality/relevance of data | ||
| Cell shape | Round | Varied |
| Membrane capacitance | <10 pF for HEK293 | ∼25 pF [29] |
| Multi-ion channel phenotype | No | Yes |
| Contractile machinery | No | Yes |
| Upstroke velocity | ∼400 dV/dt [47] | ∼17 dV/dt [45] |
| RMP | −40 mV [47] | ∼60 mV [45] |
| Maximal peak INa | −264 ± 34 pA/pF [47] | −216.7 ± 18.7 pA/pF [29] |
| Maximal peak ICa,L | 600 pA/pF [47] | −17.1 ± 1.7 pA/pF [29] |
| Peak IKr tail current | 12.8 ± 1.6 pA/pF [48] | 0.95 ± 0.02 pA/pF [29] |
RMP, resting membrane potential.
In this report we used high efficiency differentiation protocols that do not require genetic selection to produce hPSC-CMs of purities in excess of 80% to enable optimization of the Nanion Patchliner automated planar patch clamp system across cardiomyocytes derived from six hPSC lines that include three healthy lines (one hESC; two hiPSC) and three hiPSC lines harboring disease-causing mutations (one CPVT; two Duchenne muscular dystrophy [DMD]). We developed a two-step cell preparation protocol that proved effective in enabling catch rates of 81.25% ± 6.68%, seal rates of 79.68% ± 7.06%, and seal qualities of up to >2 GΩ. Analysis in voltage clamp mode allowed evaluation of INa, ICa,L, and IK current densities, and the impact on these currents of their respective blockers tetrodotoxin (TTX), nifedipine, and E-4031. Thus, this work paves the way to use APC for high-throughput analysis of the electrophysiology and pharmacology of hPSC-CMs.
Materials and Methods
Isolation of fibroblasts from patient skin biopsies
Skin punch biopsies of 4 mm were transported from the clinic into the laboratory in Transport medium (Table 2) and washed thrice in the same, after which they were transferred into bacterial-grade Petri dishes. Using sterile forceps and scalpels, the samples were cleaned off any adipose and epidermal tissue, minced into 1 mm pieces and then digested—first with 2.5% trypsin (Invitrogen) for 20 min, and then with 1 mg/mL collagenase IV (Invitrogen) for 90 min, both at 37°C. The digested cells were spun down at 200 g for 5 min, plated in Chang's D medium (Table 2), and grown for 2–3 weeks until confluent, with medium changes every 3–4 days.
Table 2.
Media Formulations for Stem Cell Derivation, Culture, and Differentiation
| Acronym | Component | Conc. | Supplier |
|---|---|---|---|
| Transport media | HBSS (10×) | 10% | Invitrogen |
| H2O | 88% | Sigma-Aldrich | |
| Pen/strep | 1% | Invitrogen | |
| Fungizone | 1% | Invitrogen | |
| Chang's D media | Chang's D | 98% | Invitrogen |
| L-glutamine | 2 mM | Invitrogen | |
| HEPES | 100 mM | Invitrogen | |
| Pen/strep | 1% | Invitrogen | |
| Fungizone | 1% | Invitrogen | |
| Fibroblast media | 1× DMEM | 78% | Invitrogen |
| FBS | 20% | Perbio | |
| NEAA | 1% | Invitrogen | |
| L-glutamine | 1% | Invitrogen | |
| β-mercaptoethanol | 100 μM | Sigma-Aldrich | |
| hPSC media | DMEM F12 | 83% | Invitrogen |
| KSR | 15% | Invitrogen | |
| NEAA | 1% | Invitrogen | |
| GlutaMAX | 1% | Invitrogen | |
| β-mercaptoethanol | 100 μM | Sigma-Aldrich | |
| bFGF | 8 ng/mL | R&D | |
| RIP media | 1× RPMI | 98% | Invitrogen |
| ITS | 1% | Gibco | |
| Chemically defined lipid | 1% | Gibco | |
| 1-Thioglycerol | 400 μM | Sigma-Aldrich | |
| PVA | 4 mg/mL | Sigma-Aldrich | |
| bFGF | 6 ng/mL | Peprotech | |
| BMP4 | 20 ng/mL | R&D | |
| RD media | 1× RPMI | 80% | Invitrogen |
| FBS | 20% | Perbio | |
| 1-Thioglycerol | 400 μM | Sigma-Aldrich | |
| RB media | 1× RPMI | 98% | Invitrogen |
| 50× B-27 supplement | 2% | Invitrogen |
Generation of patient-specific hiPSCs
The protocol followed for the generation of hiPSCs was based on a previously published method [33], and media formulations are as detailed in Table 2. In brief, on the day of transduction, fibroblasts were seeded into tissue culture-treated six-well plates in fibroblast medium, at a density of 50,000 cells/well. Once the cells had adhered to the plastic (after 5–6 h), they were infected with streptavidin-conjugated viral particles expressing OCT4, NANOG, SOX2, and LIN28, in the presence of 8 μg/mL of Polybrene (Sigma-Aldrich) at a multiplicity-of-infection of 10. Twenty-four hours post-transduction (p.td), the cells were washed twice with PBS and fed with fresh fibroblast medium. Forty-eight hours p.td., the cells were harvested using 0.05% trypsin (Invitrogen), resuspended in hPSC media supplemented with 10 ng/mL FGF (Peprotech), and transferred to tissue culture treated 90 mm Petri dishes containing mitomycin-C inactivated mouse embryonic fibroblasts (MEFs) at a density of 1.8 × 106 cells/well.
For the first week, cells were maintained in hPSC medium containing 10 ng/mL FGF, after which the medium was switched to MEF-conditioned hPSC medium supplemented with 10 ng/mL FGF. In either case, medium changes were performed daily until around day 25-day 30 p.td.
Cardiac differentiation of hiPSCs
Embryoid body-mediated differentiation
The protocol followed was based on a previously published protocol [34], and all media formulations are detailed in Table 2. In brief, on day 0 of differentiation, embryoid body (EB) formation was initiated by the seeding of hPSCs into 96-well V-bottomed plates (Thermo Scientific), at a density of 6,000 cells/well, in RIP medium. On day 2, the RIP medium was replaced with RD medium. On day 4, the EBs were transferred into 96-well U-bottomed tissue culture-treated plates (Fisher Scientific) and maintained in RB medium from thereon, with medium changes every 3–4 days. Spontaneously beating clusters were observed from day 8 onward.
Monolayer-mediated differentiation
Undifferentiated hPSCs were seeded onto Matrigel™-coated dishes at a density of 1 × 105 cells/cm2, and allowed to expand for 72 h until confluent. At this stage, (day 1 of differentiation) cells were treated with medium A consisting of StemPro-34 (Life Technologies) medium, recombinant human Activin A (Life Technologies) and recombinant human BMP4 (R&D Systems). Media exchange was performed on day 3 with medium B- consisting of RPMI base medium (Life Technologies) supplemented with 1× B-27 (Life Technologies) and a small molecule inhibitor, KY02111 (R&D Systems). From day 5 onward, cells were maintained in RPMI base medium supplemented with B-27 only, and media changes were performed every 2–3 days.
Electrophysiology
Manual patch clamp was performed using an ECP-10 HEKA amplifier. Cells were maintained in NT Buffer and at 37°C during recordings. Patch pipettes were pulled on a Sutter P-97 programmable micropipette puller and had resistances of between 2 and 5 MΩ when filled with ionic solution. Data were recorded using the Pulse software (HEKA) and analyzed using Clampfit v9.0 (Molecular Devices).
Automated electrophysiological analysis of the hPSC-CMs was performed on an NPC-16 Patchliner Octo platform (Nanion Technologies) fitted with two EPC-10 quadro amplifiers (HEKA Elektronik GmBH) using disposable borosilicate glass chips (Nanion Technologies). When filled with intracellular solution the chip resistance was between 1.8–3 MΩ. Data were acquired using the Patchmaster software (HEKA Elektronik GmBH) analyzed offline using IGOR-Pro (WaveMetrics, Inc.).
Patch clamp solutions
Internal solutions for (1) voltage-gated Na and Ca channel recordings: 50 mM CsCl, 10 mM NaCl, 60 mM CsF, 20 mM EGTA, 10 mM HEPES, CsOH pH 7.2, osmolarity 285 mOsmol; (2) voltage-gated K channel recordings: 50 mM KCl, 10 mM NaCl, 60 mM KF, 20 mM EGTA, 10 mM HEPES, KOH pH 7.2, osmolarity 285 mOsmol; (3) action potential (AP) recordings: 145 mM KCl, 5 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, 4 mM ethylene glycol tetra-acetic acid, 10 mM HEPES, KOH pH 7.3. External solutions for (1) voltage-gated K and Na channel recordings: 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM D-glucose monohydrate, 10 mM HEPES, NaOH pH 7.4, osmolarity 298 mOsmol; (2) voltage-gated Ca channel recordings: 125 mM TEA-Cl, 20 mM BaCl2, 10 mM D-glucose monohydrate, 10 mM HEPES/TEA OH pH 7.35, osmolarity: 300–305 mOsmol; (3) AP recordings: 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM D-glucose monohydrate, 10 mM HEPES; Seal enhancer: 80 mM NaCl, 3 mM KCl, 10 mM MgCl2, 35 mM CaCl2, 10 mM HEPES (Na+-salt), HCl pH 7.4, osmolarity: 298 mOsmol
Ethics
This study conformed to the principles outlined in the Declaration of Helsinki. All subjects gave informed consent for blood testing for genetic abnormalities associated with hereditary cardiac channelopathies, and for tissue donation. Isolation and use of patient fibroblasts was approved by the Nottingham Research Ethics Committee (Approval 09/H0408/74), and sample collections are registered with the U.K. Clinical Research Network under IRAS project ID24624. Isolation and use of patient dental pulps were also performed with full patient consent and ethical approval (LREC 180808/HR/12).
Statistics
Statistical significance was determined by Student's t-tests, or ANOVA with Tukey post-hoc tests for pairwise comparisons (MS Excel 2010). Data were considered significant where P < 0.05. Results are presented as mean values ± standard error of mean; n denotes the number of cells in which measurements were made.
Results
Generation and characterization of hPSC-CMs
Two healthy hiPSC lines [HUES7-fibroblast-derived FIB-hiPSC and dental pulp-derived BT1-hiPSC], and three diseased hiPSC lines (DMD-afflicted DMD4- and DMD16-hiPSCs and CPVT-afflicted CP1-hiPSCs) were generated in vitro by lentiviral delivery of the OCT4, NANOG, SOX2, and LIN28 reprogramming factors. These putative hiPSC lines were measured against internationally accepted pluripotency criteria to establish their phenotype [35].
Karyotypic analyses revealed that the cells were genetically stable with a normal complement of 46XY or 46XX chromosomes (Fig. 1C), and their population doubling times were similar to those of HUES7 hESCs (Fig. 1A). RT-PCR analysis demonstrated that the hiPSCs had reactivated the reprogramming factors OCT4, NANOG, SOX2, and LIN28 at their endogenous loci, and silenced the lentiviral transgenes (Fig. 1B and Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/scd). Immunostaining confirmed that the hiPSCs had silenced the fibroblast-specific marker FSA; and reactivated the pluripotency markers TRA-1-81, SSEA-4, OCT4, and LIN28 (Supplementary Fig. S2). Flow cytometry further confirmed that the hiPSCs had silenced the differentiation marker SSEA-1; and reactivated the pluripotency marker SSEA-4 (Fig. 1D and Supplementary Fig. S3). These results confirmed the pluripotent nature of the five iPSC lines.
FIG. 1.

Characterization of hiPSC lines. (A) PDs at each passage were calculated using the formula [log10 (total cell counts/cells seeded)/log10 (2)], where the cell number seeded was 48,000 cells/cm2. All five iPSC lines exhibited PDs similar to that of HUES7 hESCs, ∼33.5 h. (B) RT-PCR analysis of OCT4, NANOG, SOX2, and LIN28 expression from endogenous “E” and lentiviral “L” loci in FIB-, BT1-, CP1-, DMD4-, and DMD16-iPS demonstrating the activation of endogenous pluripotency genes and the silencing of lentiviral transgenes, at passage 15. (C) Representative karyograms showing normal 46XY and 46XX karyotypes, at passage 15. (D) Representative flow cytometry data of FIB-iPSCs demonstrating the expression of pluripotency marker SSEA-4, and the absence of differentiation marker SSEA-1. DMD, Duchenne muscular dystrophy; FIB, HUES7-fibroblast-derived; hESCs, human embryonic stem cells; hiPSC, human induced pluripotent stem cells; PD, population doublings. Color images available online at www.liebertpub.com/scd
To generate human cardiomyocytes in vitro, these five hiPSC lines along with a HUES7 hESC control were differentiated via the embryoid-body and monolayer-routes (Supplementary Videos S1 and S2). The presence of cardiac-specific proteins and their spatial organization within the hPSC-CMs were determined by dissociating the beating clusters/sheets and staining the dispersed cells with immunofluorescent cardiac α-actinin and cardiac troponin T (cTnT) antibodies (Fig. 2A, B and Supplementary Figs. S4 and S5). All six lines produced hPSC-CMs with similar morphologies and patterns of myofilament organization. While cTnT appeared as longitudinal, punctuate, fibrous striations, a hallmark of functional cardiomyocytes, α-actinin appeared homogenously throughout the cell as longitudinal fibrous striations. The hPSC-CMs had multi-angular morphologies appearing round, elongated, branched, or triangular, with one or more oval nuclei. Flow cytometry analysis for the cardiac α-actinin marker revealed the cardiac preparations to have purities of 91.2% ± 1.4% (Fig. 2C and Supplementary Fig. S6).
FIG. 2.

Characterization of hiPSC-derived cardiomyocytes (hiPSC-CMs). Representative immunofluorescence images of HUES7-cardiomyocytes showing positive (A) cardiac α-actinin, and (B) cardiac troponin T staining highlighting characteristic cardiac muscle striations. Scale bars represent 32 μm. (C) Representative flow cytometry data of HUES7-CMs demonstrating cardiac purities of 91.2% ± 1.4% (Supplementary Fig. S6). (D) Representative AP traces demonstrating the formation of atrial-like, ventricular-like, and nodal-like cardiomyocytes. The classification of different cardiac cell types was based on the APD90/APD50 ratio. APD90/50 values ≤1.4 were designated as ventricular-like, 1.4–1.7 were designated as nodal-like, and ≥1.7 were designated as atrial-like. AP, action potential. Color images available online at www.liebertpub.com/scd
To assess the electrophysiological functionality of the hPSC-CMs, beating clusters or monolayers of cardiomyocytes were disaggregated into single cells, seeded onto MatTek dishes, and analyzed by whole-cell patch clamp. The APs recorded from these cells demonstrated, on average, AP duration (APD), APD50 (APD at 50% of the repolarisation curve), APD90 (APD at 90% of the repolarization curve), AP amplitude (APA), resting membrane potential (RMP), and upstroke velocity values of 394.9 ± 27.6 ms, 106.7 ± 12.2 ms, 154.9 ± 15.7 ms, 55.8 ± 4.1 mV, −33.2 ± 3.4 mV, and 6.3 ± 1.5 V/s respectively. Table 3 lists the specific values obtained for the AP waveforms of HUES7- (n = 58), FIB-iPS- (n = 44), and BT1-iPS (n = 29)-derived cardiomyocytes.
Table 3.
Action Potential Parameters of Human Pluripotent Stem Cell-Cardiomyocytes as Determined by Manual Patch Clamp
| Cell type | Cell line | Cslow (pF) | RMP (mV) | APA (mV) | Upstroke velocity (V/s) | APD (ms) | APD50 (ms) | APD90 (ms) |
|---|---|---|---|---|---|---|---|---|
| Atrial-like | HUES7 | 34.1 ± 11.1 | −29.2 ± 14 | 44.9 ± 7.1 | 5.6 ± 10.9 | 429.3 ± 2.3 | 84.7 ± 4.5 | 165.2 ± 3.5 |
| FIB-iPS | 26.5 ± 5.1 | −35.23 ± 2.4 | 44.5 ± 4.2 | 5.9 ± 1.6 | 518 ± 129.3 | 105.3 ± 27.9 | 192.3 ± 48.8 | |
| BT1-iPS | 29.2 ± 5.9 | −22.3 ± 1 | 35.1 ± 3.8 | 2.5 ± 0.5 | 379.1 ± 42.9 | 49.4 ± 7.6 | 95 ± 16.1 | |
| Ventricu lar-like | HUES7 | 19.4 ± 4.3 | −36.2 ± 3 | 68.8 ± 5.8 | 1.9 ± 0.5 | 327.2 ± 18.1 | 120.3 ± 10.3 | 149.8 ± 10.5 |
| FIB-iPS | 14.8 ± 3.8 | −53.3 ± 5.1 | 62.7 ± 3.7 | 13.3 ± 3.7 | 336.3 ± 50 | 158.5 ± 27.3 | 127.3 ± 22.3 | |
| BT1-iPS | 21.8 ± 4 | −25.4 ± 2.9 | 69.7 ± 4 | 12.1 ± 2.4 | 252.5 ± 21 | 59.8 ± 4.5 | 77.7 ± 6 | |
| Nodal-like | HUES7 | 29.8 ± 5.5 | −23.1 ± 2.3 | 63.5 ± 3.7 | 9.9 ± 2 | 447.4 ± 24.6 | 115.5 ± 7.7 | 183.6 ± 12.2 |
| FIB-iPS | 10.2 ± 1.6 | −42.5 ± 3.8 | 50.4 ± 5.9 | 3.2 ± 0.9 | 479.3 ± 100.8 | 145.9 ± 30.4 | 220.9 ± 46.1 | |
| BT1-iPS | 14.6 ± 2 | −31.9 ± 3.1 | 62.4 ± 8.3 | 1.9 ± 0.4 | 384.6 ± 86.4 | 121.1 ± 52.6 | 182 ± 78.5 |
The APD90/APD50 ratio, which describes the shape of the repolarization curve, was used to determine the specific subtype of each hPSC-CM analyzed. hPSC-CMs with low APD90/APD50 ratios (≤1.3, indicative of a pronounced plateau phase) were classified as ventricular-like, while those with high APD90/APD50 ratios (≥1.8, indicative of fast phase-I repolarization) were classified as atrial-like. Cardiomyocytes with AP characteristics intermediary to those of the atrial-like and ventricular-like cells, and APD90/APD50 ratios between 1.4 and 1.7 were classified as nodal-like [20]. The HUES7, FIB-iPS, and BT1-iPS lines all produced cardiomyocytes of atrial-like, ventricular-like, and nodal-like morphologies (Table 3), with representative traces shown in Fig. 2D.
Thus, the structural and electrophysiological properties of hPSC-CMs were consistent with other reports describing these cells, which permitted their use in development of protocols for automated electrophysiology analysis.
Development of a cell harvesting protocol compatible with automated planar patch clamp
As with any planar patch clamp platform, the Patchliner requires freshly dissociated single cells in suspension. To achieve this, intact clusters or monolayers of hPSC-CMs of 91.2% ± 1.4% purity (Fig. 2C and Supplementary Fig. S6) were dissociated into single cells using a trypsin-containing cell dissociation solution at 37°C (Fig. 3A). Trypsin was included in the dissociation solution as it was the only enzyme we found capable of breaking the tightly clustered cardiomyocytes within EBs and monolayers into single cells. Other “gentler” enzymes like accutase, collagenase, and dispase produced cell clumps that were unsuitable for planar patch clamp (Fig. 3B). However, when processed on the Patchliner, trypsin-treated cells demonstrated low catch rates of 54.16% ± 11.89%, and a low incidence of conversion to whole-cell breakthroughs (34.72% ± 8.17%). Furthermore, only 8.33% ± 4.7% of the cells that were successfully patched exhibited functional currents (Table 4). The cardiac differentiation of hPSCs is a costly, time-consuming, and labor-intensive process, and the derived cardiomyocytes are fragile, highly sensitive to the environment, and characterized by low (ie, physiological) levels of endogenous ion channel expression (Table 1). To minimize cell wastage and improve the productivity of the Patchliner, we set about developing a cell preparation protocol that allowed optimal use of our hPSC-CMs on the Patchliner.
FIG. 3.

Development of a cell harvesting method compatible with automated planar patch clamp. (A) The dissociation of intact embryoid bodies or monolayers with trypsin produced single cells with little/no currents (Table 4); Collagenase/accutase/dispase treatment (B) and mechanical dissociation protocols (C) produced cell clumps unsuitable for efficient planar patch clamping. (E) The two-step cell dissociation protocol presented in the study produced (D) single cells with functional currents suitable for efficient analysis of hPSC-CMs on the Patchliner. (F) Shown are the raw current traces on a chip where 100% catch rates and whole-cell breakthroughs were achieved with HUES7 hESC-CMs using the newly developed two-step cell dissociation protocol (E). hPSC-CMs, human pluripotent stem cell-derived cardiomyocytes. Color images available online at www.liebertpub.com/scd
Table 4.
Development of a Cell Harvest Protocol Optimal for Analysis on the Patchliner
| Protocol | % catch rate | % whole-cell | % cells showing APs or currents | Cell quality | Comments |
|---|---|---|---|---|---|
| Trypsin-containing solution | 54 ± 12 | 35 ± 8 | 8 ± 5 | Single cells (Fig. 3A) | Enzyme treatment damaging cells |
| Mechanical dissection | — | — | Cell clusters and debris (Fig. 3C) | No single cells and cell clumps/debris clogging wells | |
| Cold dissociation | 69 ± 11 | 58 ± 10 | 5 ± 2 | Single cells | Better cell viability and membrane integrity, but no currents |
| Two-step protocol | 81 ± 7 | 80 ± 7 | 31 ± 6 | Single cells (Fig. 3D) | Gentler the enzyme treatment on the day of analysis, better the quality of the recordings |
Reasoning that the inclusion of trypsin was damaging the cell membrane [36] and reducing the density of functional ion channels, we evaluated whether hPSC-CMs could be successfully prepared by mechanical dissection using a stem cell knife, a commercially available glass capillary with a 200 μm bore, and a sharpened 45° cutting edge. However, while this approach broke the cardiomyocyte clusters into clumps, it did not yield single cells (Fig. 3C). When these “minced” cell preparations were run on the Patchliner, the clumps blocked the holes of the planar chip preventing any successful recordings (Table 4). As an alternative, we tested a trypsin-including cold dissociation protocol [37] to try and better preserve the integrity of the cell membranes. While this method improved catch rates to 69.44% ± 10.85%, the single cells patched showed little to no currents, suggesting that channel degradation due to enzymatic treatment still remained an issue (Table 4).
To evolve the protocol further, we separated the dissociation protocol into two steps. At least 1 week before use on the Patchliner, hPSC-CM cultures were dispersed with trypsin-containing cell dissociation solution. This relatively harsh treatment produced a single-cell suspension of hPSC-CMs that could be reseeded at a low density onto gelatine-coated tissue culture plastic, with cells recovering their ability to beat spontaneously within 3 days of the procedure. A week after this preseeding step, hPSC-CMs were reharvested using a gentle accutase-assisted cold dissociation protocol to produce a single-cell suspension that was used immediately for Patchliner analysis (Fig. 3D). Relative to the earlier approaches used, this two-step method yielded significant (P < 0.05, n = 5 in each treatment group) improvements in catch rates (81.25% ± 6.68%), whole-cell breakthroughs (79.68% ± 7.06%), and percentage of cells showing APs or ionic currents (31.25% ± 5.79%) (Table 3). In some cases, all eight channels of the Patchliner chips gave productive recordings (Fig. 3F). Therefore, this approach to hPSC-CM preparation (Fig. 3E) was used in all subsequent electrophysiological and pharmacological analyses on the Patchliner.
Study of hPSC-CM APs by automated current-clamp
We next sought to evaluate whether the Patchliner could be used in current clamp mode to record and measure spontaneous and stimulated APs from hPSC-CMs. Following the differentiation and harvest of cardiomyocytes from the hESC HUES7 line, spontaneous APs were recorded from the cells under zero current clamp conditions. However, despite stable seal properties, most cells were electrically quiescent, and any spontaneous activity recorded was erratic and unfit for analysis (Fig. 4A). Based on the data above (Fig. 2, Supplementary Fig. S2–S6 and Table 3), this was not a cell quality issue but rather the requirement for cells in suspension at the time of analysis and storage at 4°C before use.
FIG. 4.
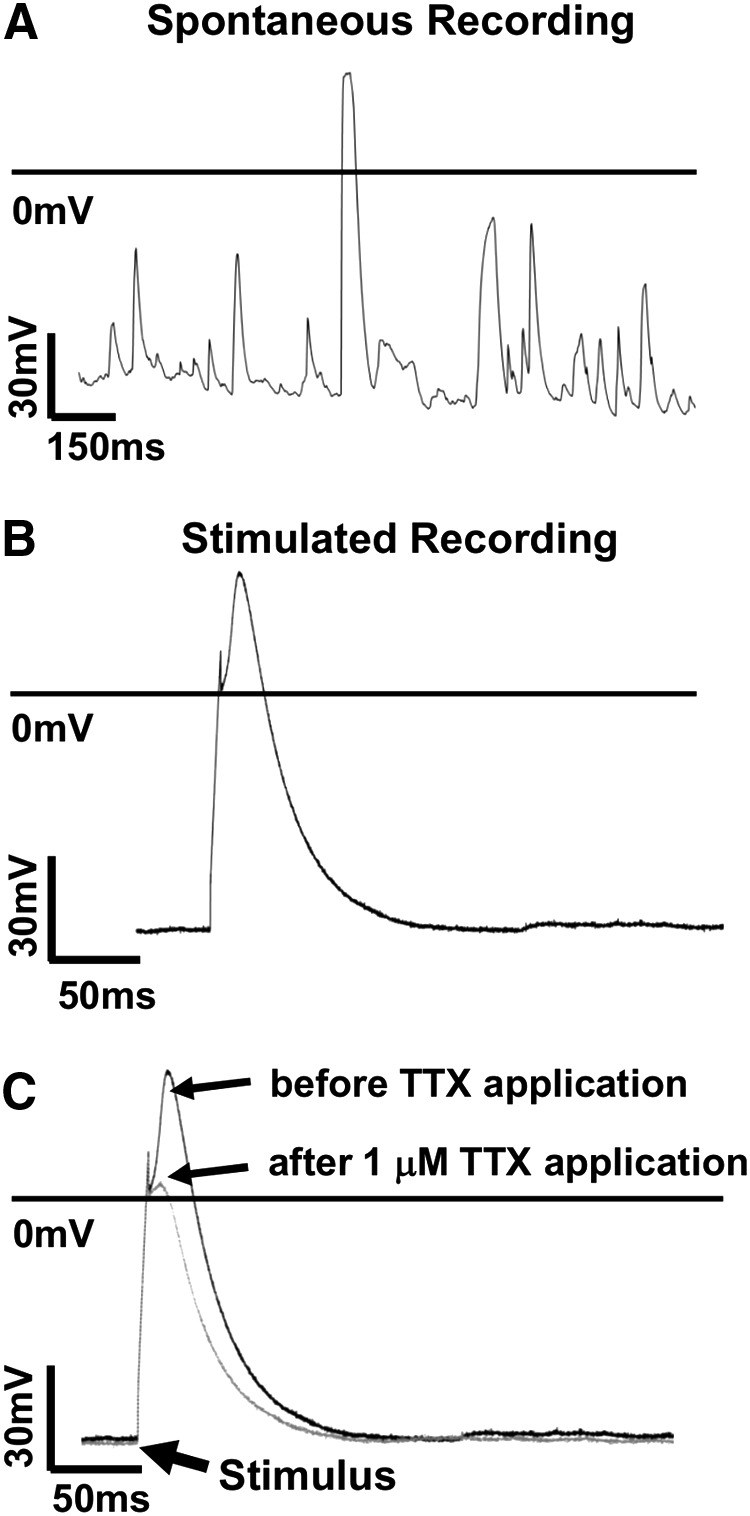
Automated electrophysiological analysis of the hPSC-CMs in current clamp mode. Representative raw data traces of (A) spontaneous APs from HUES7-CMs showing an erratic pattern of activity with individual spikes rarely exceeding 20 ms in duration. (B) In contrast, stimulation protocols produced classical AP traces. Using this approach we estimated that the cells had APD90, upstroke velocity and APA values of 231.4 ± 69 ms, 11.9 ± 1.5 V/s, 96.6 ± 7.5 mV respectively (n = 42). (C) HUES7-CMs when treated with 1 μM TTX (blocker of the fast voltage-gated sodium channel INa), showed a 1.6 ± 0.4-fold reduction in AP amplitude (n = 8), which was restored on washout with external recording solution. TTX, tetrodotoxin.
To determine whether the data quality could be improved by pacing, preparations of HUES7-CMs were subjected to electrical stimulation. An AP can only be initiated when the cell membrane is hyperpolarized beyond a certain value called the “threshold potential,” which varies from cell to cell, but is constant for a given cell at rest [38]. APs were evoked from the HUES7-CMs by subjecting them to a train of 500 ms long depolarizing pulses (Δ20pA) from a holding potential of −70 mV, and analyzing their voltage responses. A depolarizing pulse that caused an amplitude rise >30 mV above RMP was defined as the “threshold current” and was subsequently used to generate APs for analysis (Fig. 4B). Using this approach we estimated that the cells had APD90, upstroke velocity and APA values of 231.4 ± 69 ms, 11.9 ± 1.5 V/s, 96.6 ± 7.5 mV respectively. Table 5 details cell line-specific values for HUES7- (n = 14), FIB-iPS- (n = 15) and BT1-iPS (n = 13)-derived cardiomyocytes.
Table 5.
Action Potential Parameters of Human Pluripotent Stem Cell-Cardiomyocytes as Determined by Automated Planar Patch Clamp
| Cell line | Cslow (pF) | APA (mV) | Upstroke velocity (V/s) | APD (ms) | APD50 (ms) | APD90 (ms) |
|---|---|---|---|---|---|---|
| HUES7 | 16.8 ± 3.6 | 83.3 ± 8.5 | 10.5 ± 2.3 | 369.8 ± 130.6 | 106.2 ± 45.3 | 177.8 ± 68 |
| FIB-iPS | 35.9 ± 21.4 | 93.8 ± 11.5 | 10.4 ± 1.9 | 511.9 ± 256.8 | 202.7 ± 139.1 | 282.1 ± 174 |
| BT1-iPS | 22.5 ± 3.8 | 115.1 ± 18 | 15.1 ± 3.6 | 417.4 ± 146 | 135.8 ± 66 | 238.6 ± 102.4 |
To determine the effects of drug compounds on the AP parameters of HUES7-CMs, the depolarizing Na+ current was inhibited by the selective INa channel blocker TTX. This caused a 1.6 ± 0.4-fold decrease in APA (n = 8), which was restored on washout with external recording solution (Fig. 4C). Thus it was shown that the Patchliner could be used in current clamp mode to investigate the electrophysiology and pharmacology of hPSC-CMs, although only with pacing.
Study of hPSC-CM voltage-dependent currents by automated voltage-clamp
The ability of the Patchliner to implement stepped depolarizing pulses on parallel channels gives it the potential to carry out rapid measurement of ion channel densities. We initially used HUES7-CMs to define the necessary protocols to unveil INa, ICa,L, and IK currents. To record INa, 20 ms long depolarizing pulses between −70 and +40 mV were applied in 10 mV increments from a holding potential of −80 mV. INa was first observed at −60 mV, progressively increased to reach a maximal level at −20 mV, and progressively decreased on further depolarization (Fig. 5A). To record ICa, cells were first held at a potential of −80 mV, after which a 3s long prepulse of −50 mV was applied to voltage inactivate Na+ and any T-type Ca+2 channels that may be present. Then, 200 ms long depolarizing pulses between −70 and +30 mV were applied in 10 mV increments. ICa was first observed at −60 mV, progressively increased to reach a maximal level at −30 mV, and progressively decreased on further depolarization (Fig. 5B). To record IK, 200 ms long depolarizing pulses between −60 and +60 mV were applied in 20 mV increments, from a holding potential of −80 mV. IK was first observed at −40 mV, and progressively increased to reach a maximal level at +60 mV (Fig. 5C). Maximal peak INa, ICa, and IK normalized to cell capacitance (n = 67, mean = 24.38 ± 2.32 pF) were 64.83 ± 9.11 pA/pF (n = 31, −20 mV), 10.2 ± 2.73 pA/pF (n = 12, −30 mV), and 20.95 ± 3.59 pA/pF (n = 24, +40 mV) respectively. V1/2 values for the same were calculated to be −34.76 ± 9.52 mV, −33.4 ± 10.2 mV, and −20.6 ± 12.5 mV respectively.
FIG. 5.

Automated electrophysiological analysis of HUES7 hESC-CMs in the voltage clamp mode. Representative raw data traces of (A) INa, (B) ICa, and (C) IK currents in HUES7-CMs. The maximal current densities for the three currents were determined to be −64.8 ± 9.1 pA/pF (at −20 mV, n = 31), −10.2 ± 3.8 pA/pF (at −30 mV, n = 12), and 20.9 ± 3.6 pA/pF (at +40 mV, n = 24) respectively. (D) Averaged I/V (current-voltage) diagrams of the INa, ICa, and IK currents in HUES7-CMs.
Using the parameters defined for HUES7-CMs, current–voltage curves and current densities were determined for five additional hPSC lines. These included hiPSC lines produced by reprogramming embryonic fibroblasts or patient-derived dental pulp cells from healthy individuals, or from skin fibroblasts derived from patients carrying mutations causing CPVT and DMD. The maximal current densities for cardiomyocytes derived from these cell lines ranged from −19.6 ± 4.5 pA/pF to −37.3 ± 8.9 pA/pF, −12.8 ± 2.4 pA/pF to −22.3 ± 3.3 pA/pF, and 12.2 ± 4.1 pA/pF to 40.3 ± 6.4 pA/pF for INa, ICa, and IK, respectively (Table 6, n = 5 for each cell line, Fig. 6).
Table 6.
Current Densities of Human Pluripotent Stem Cell-Cardiomyocytes as Determined by Automated Planar Patch Clamp
| Current density (pA/pF) | ||||||
|---|---|---|---|---|---|---|
| Current (test voltage) | HUES7-hESC | FIB-iPS | BT1-iPS | CP1-iPS | DMD4-iPS | DMD16-iPS |
| Na (−20 mV) | −64.8 ± 9.1 | −32.5 ± 16.1 | −37.3 ± 8.9 | −25.4 ± 9.4 | −32.9 ± 6.4 | −19.6 ± 4.5 |
| Ca (−30 mV) | −10.2 ± 3.8 | n.d. | −22.3 ± 3.3 | n.d. | −12.8 ± 2.4 | n.d. |
| K (40 mV) | 20.9 ± 3.6 | 12.2 ± 4.1 | 22.5 ± 6.1 | 40.3 ± 6.4 | 34.3 ± 5.6 | 29.4 ± 6.2 |
DMD, Duchenne muscular dystrophy; hESCs, human embryonic stem cells; n.d., not determined.
FIG. 6.

A comparison of the electrophysiological properties of hPSC-CMs using the Patchliner. Box plots demonstrating the distribution of (A) INa, (B) ICa, and (C) IK current maxima, at −20,−30, and +40 mV respectively, across six hPSC lines. These cell lines included HUES7-fibroblast-derived “healthy” FIB-hiPSCs, dental pulp-derived BT1-hiPSCs, Duchenne muscular dystrophy (DMD)-afflicted DMD4- and DMD6-hiPSCs, catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted CP1-hiPSCs, and “healthy” HUES7-hESCs. The maximal current densities were shown to range from −19.6 ± 4.5 pA/pF to −37.3 ± 8.9 pA/pF, −12.8 ± 2.4 pA/pF to −22.3 ± 3.3 pA/pF, and 12.2 ± 4.1 pA/pF to 40.3 ± 6.4 pA/pF for INa, ICa, and IK respectively. Error bars represent standard error of mean (n = 5 for each cell line).
Single-factor ANOVA tests were conducted across the different lines, for each channel current. In case of the INa current, there were significant differences in mean current densities across the six lines at P < 0.05 [F(5,65) = 2.73, P = 0.027]. Post hoc comparisons using the Tukey HSD test indicated that the maximal INa current density of HUES7-CMs was significantly different than those of cardiomyocytes derived from FIB-iPS [absolute difference (ABS) = 42.33], BT1-iPS (ABS = 27.53), CP1-iPS (ABS = 39.4), DMD4-iPS (ABS = 31.94), and DMD16-iPS (ABS = 45.21). In case of the ICa maximal current densities, there were no significant differences across the HUES7-, BT1-iPS, and DMD4-iPS-derived cardiomyocytes at P < 0.05 [F(2,10) = 1.92, P = 0.197]. Similarly, no significant differences were found in maximal IK current densities across the six lines at P > 0.05 [F(5,54) = 2.32, P = 0.056]. Taken together, these results suggest that the stem cell-derived cardiomyocytes were a heterogeneous group of cells with variable cardiac phenotypes, as has been reported by several other research groups [39].
Efficient pharmacological screening of hPSC-CMs by automated planar patch clamp
Next, the effects of different ion channel modulators on hPSC-CM currents were determined using the Patchliner. In brief, peak INa, ICa, and IK currents were measured at pulses of −20, −30, and +40 mV respectively. Steady state recordings were conducted for 3 min before the application of escalating doses of their respective blockers. Peak current amplitude was averaged from four voltage pulses for each drug concentration applied and for the baseline recordings. Dose–response data were fitted to the Hill Equations and IC50 values determined for each channel blocker tested.
TTX, a potent blocker of the INa channels, caused a concentration-dependent inhibition of the INa current of the hPSC-CMs, with an IC50 of 289.8 ± 31.9 nM (Fig. 7A, n = 5). Increasing concentrations of nifedipine, an L-type Ca+2 channel blocker, inhibited the current amplitude of the ICa.L channels in a dose-dependent manner, with an IC50 of 1.4 ± 1.5 μM (Fig. 7B, n = 5). Finally, E-4031—a selective IKr (rapidly activating delayed rectifier potassium channel) blocker—was applied to the cells in increasing doses to check for a reduction in total IK. A concentration-dependent inhibition of the IK current was observed and an IC50 of 2 ± 1.4 μM was calculated (Fig. 7C, n = 5). Thus, this feasibility study showed the suitability of the Patchliner APC system for the electrophysiological and pharmacological analyses of hPSC-CMs.
FIG. 7.
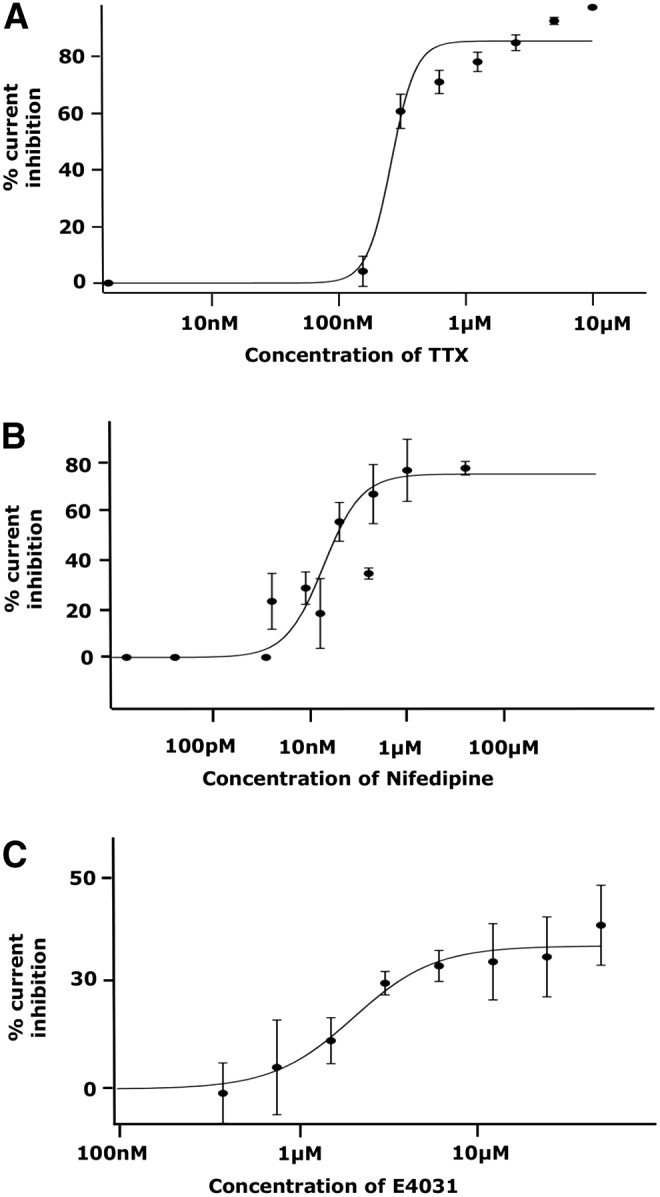
Efficient pharmacological screening of hPSC-CMs by automated planar patch clamp. Patch clamp analysis showing dose-dependent inhibition of the (A) INa, (B) ICa, and (C) IK currents, in hPSC-CMs, on treatment with increasing concentrations of TTX, nifedipine, and terfenadine respectively. IC50 values of 289.8 ± 31.9 nM, 1.4 ± 1.5 μM, and 2 ± 1.4 μM were calculated for TTX, nifedipine, and E-4031 respectively. Error bars represent standard deviation of the mean (n = 5 for each drug).
Discussion
In this study, we demonstrated the potential of the Patchliner planar patch clamp platform for hPSC-CM-based medium-throughput drug safety and efficacy testing. To achieve this, we developed a cell preparation protocol that allowed the dissociation of EB and monolayer cultures of cardiomyocytes into single cells, while maintaining their viability and membrane integrity. When analyzed on the Patchliner, the freshly dissociated cardiomyocytes demonstrated AP generation, depolarizing Na+ and Ca+2 currents, and repolarizing K+ currents, proving their suitability for use in disease modeling and high-throughput drug screening.
In conventional patch clamping the cell is accessed by means of a glass pipette that is manually brought into contact with its membrane [38], while in planar patch clamp the traditional glass pipette is replaced by a hole at the bottom of a well through which an applied negative pressure patches the cell membrane [30]. This allows automation of the patch clamp process that reduces the time and labor costs associated with conventional patch clamp, and parallelization that drastically improves screening efficiency and data output [29].
Today there exist several APC platforms, including the IonWork Quattro (MDS), PatchXpress (Molecular Devices Corporation), Patchliner (Nanion), SynchroPatch (Nanion), QPatch (Sophion), and Flyscreen (Flyion), differing in their technical specifications and level of automation [29]. Initially developed for the analysis of recombinant cell lines used for heterologous channel expression, these are only now starting to be trialled for use with more “complex” cells like stem cell-derived cardiomyocytes [31,37,40] and neurons [37,41].
Cell preparation is a key factor in the performance of an APC device, as it has a direct impact on catch rates, seal qualities and observed current characteristics. Specifically, these systems require high-density, homogenous, single-cell suspensions for optimal performance [29]. Conventional APC systems, like the QPatch [40] and the PatchExpress [31], use between 8,000 and 150,000 PSC-CMs per recording site at a density of 0.5–5 million cells/mL. Such high cell numbers are required to maximize the probability of positioning a cell over the recording aperture, and in many cases increases in catch rates have been shown to positively correlate with increases in cell densities [42].
With recent improvements in cardiac differentiation protocols, hPSC-CMs can be produced in large numbers, but with high reagent/labor costs. To illustrate, the loading of 48 recording wells of an APC, at a density of 150,000 cells/well, would require 7.2 million cells at a cost of USD$7,200 when purchasing them from a commercial source. To reduce costs we used a specially developed “cell stacking” protocol that reduces cell usage to 300–2,000 cells/recording site [37], making it more economical for use in the high-throughput screening of hPSC-CMs.
To obtain single-cell suspensions, cardiomyocytes cultures are usually dissociated with enzymes like trypsin, as gentler nonenzymatic harvest protocols produce cell clusters that block recording sites, lowering the productivity of the system. However, trypsin has been previously shown to induce proteome alterations of mammalian cell membranes, with a subset of these proteins remaining dysregulated even after a 24 h recovery in fresh culture media [36]. Trypsin has also been shown to affect the integrity of membrane phospholipids [43], which could lead to reduced catch rates, low seal resistances, and poor channel activity. In conventional patch clamp, cells are typically seeded onto coverslips following dissociation, and kept in culture for at least 72 h before use [38], which gives them time to recover from enzyme treatment. However, in case of APC, cells are analyzed, in suspension, within a few hours of dissociation and so the dissociation protocol used strongly impacts seal rates and observed current outputs.
In this study, we overcame these problems by developing a two-step cell dissociation protocol. First, intact EB/monolayer cultures of hPSC-CMs were dissociated into single cells using a trypsin-based dissociation method, and then reseeded at low densities onto gelatine-coated tissue-culture plastic. Next, following a week of recovery in culture, the attached single cells were reharvested using a gentle and rapid accutase-assisted cold dissociation protocol for immediate analysis on the Patchliner. This two-step protocol yielded catch rates of 81.25% ± 6.68% and seal resistances of up to 2 GΩ, which are a considerable improvement over previous studies using APC devices like the PatchXpress that reported seal resistances of up to only 200 MΩ using iCell hiPSC-CMs [31].
The cardiomyocytes used in our experiments were derived by both the monolayer and EB-routes. Regular manual patch clamp analysis was used to first validate the suitability of the cardiomyocytes for further use. These experiments revealed atrial-, nodal-, and ventricular-like APs based on generally accepted classification criteria [15,21]. Compared with adult human cardiomyocytes, our cells have hyperpolarized RMPs, low upstroke velocities, smaller APAs, and shorter APs. This is indicative of a fetal/neonatal phenotype and appears to be characteristic of hPSC-CMs [14,16,31,34].
When making automated recordings from our hPSC-CMs in current clamp mode, we found spontaneously beating cells to show erratic beat patterns that were unsuitable for analysis. This could be due to the cells being in suspension and at 4°C immediately before analysis. Though this instability could be controlled by the use of stimulation protocols, we found that different hPSC-CMs displayed different stimulation thresholds. This meant each well required the individual definition of stimulatory conditions, which detracted from the automated nature of the platform. Thus, we would suggest that data collection from hPSC-CMs on the Patchliner platform is most effective in voltage clamp rather than current clamp mode. We also found the cells to express higher APAs and lower RMPs using APC, which could be due to differences in ionic conditions, temperatures, and holding potentials between the two platforms.
On automated voltage clamp analysis, our hPSC-CMs displayed current densities and activation kinetics similar to that of other hPSC-CMs and fetal/neonatal cardiomyocytes. They also predictably responded to the three ion channel blockers used in the study. However, our voltage clamp experiments did not include the recording of cellular APs beforehand to allow for data classification into atrial-, ventricular-, and nodal-like subtypes. This could explain the wide variations in current densities within the same line. Further studies also need to be performed to identify whether the observed inter-line variations in current densities is due to differences in the expression of ion channels, or due to the presence of the channels in different conductance states.
It is important to note that when comparing our data with the existing literature, we found big differences in ionic conditions, experimental protocols, tissue heterogeneity, and disease status across studies [31,44–49], which raises the question whether results are reproducible across research groups [50]. Nevertheless, currently available high-efficiency cardiac differentiation protocols [12,34] in concert with the high-content screening abilities of APCs provide a platform for reliable and efficient drug candidate safety profiling and efficacy testing in industrial and academic settings.
In conclusion, this study compared the electrophysiological properties of cardiomyocytes derived from six different hPSC lines on an APC system. We presented an optimized cell preparation protocol that markedly improved the productivity of the Patchliner when using hPSC-CMs, thus making APC recordings compatible with the study of expensive and heterogeneous cell preparations. This will substantially enhance the feasibility of using APC platforms for drug development using hPSC-CMs.
Supplementary Material
Acknowledgments
We thank Dr. David Darling for providing the BL15 cell line; Dr. Nigel Smith for karyotype analysis; Dr. Mojgan Reza for supplying skin fibroblast cells; Dr. Andrew Staniforth and Dr. Dominic Abrams for providing us with patient skin samples for hiPSC generation; Dr. Nadine Becker and Dr. Sonja Stoeleze for their expertise on the Patchliner platform. This work was supported by EPSRC, MRC, European Union Seventh Framework Programme, British Heart Foundation (Centre for Regenerative Medicine and Programme/Project Grants), Heart Research UK, and National Centre for the Replacement, Refinement, and Reduction of Animals in Research (NC3Rs).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Hutchinson L. and Kirk R. (2011). High drug attrition rates—where are we going wrong? Nat Rev Clin Oncol 8:189–190 [DOI] [PubMed] [Google Scholar]
- 2.Ng R. (2015). An overview of the drug discovery to approval process. In: Drugs: From Discovery to Approval. John Wiley and Sons, Inc., Hoboken, NJ, pp 2–6 [Google Scholar]
- 3.Qureshi ZP, Seoane-Vazquez E, Rodriguez-Monguio R, Stevenson KB. and Szeinbach SL. (2011). Market withdrawal of new molecular entities approved in the United States from 1980 to 2009. Pharmacoepidemiol Drug Saf 20:772–777 [DOI] [PubMed] [Google Scholar]
- 4.Gwathmey JK, Tsaioun K. and Hajjar RJ. (2009). Cardionomics: a new integrative approach for screening cardiotoxicity of drug candidates. Expert Opin Drug Metab Toxicol 5:647–660 [DOI] [PubMed] [Google Scholar]
- 5.Schachter AD. and Ramoni MF. (2007). Clinical forecasting in drug development. Nat Rev Drug Discov 6:107–108 [DOI] [PubMed] [Google Scholar]
- 6.Rajamohan D, Matsa E, Kalra S, Crutchley J, Patel A, George V. and Denning C. (2013). Current status of drug screening and disease modelling in human pluripotent stem cells. Bioessays 35:281–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer T, Leisgen C, Gonser B. and Gunther E. (2004). QT-Screen: high-throughput cardiac safety pharmacology by extracellular electrophysiology on primary cardiac myocytes. Assay Drug Dev Technol 2:507–514 [DOI] [PubMed] [Google Scholar]
- 8.Yan GX, Shimizu W. and Antzelevitch C. (1998). Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation 98:1921–1927 [DOI] [PubMed] [Google Scholar]
- 9.Varro A, Balati B, Iost N, Takacs J, Virag L, Lathrop DA, Csaba L, Talosi L. and Papp JG. (2000). The role of the delayed rectifier component IKs in dog ventricular muscle and Purkinje fibre repolarization. J Physiol 523 Pt 1:67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dick E, Rajamohan D, Ronksley J. and Denning C. (2010). Evaluating the utility of cardiomyocytes from human pluripotent stem cells for drug screening. Biochem Soc Trans 38:1037–1045 [DOI] [PubMed] [Google Scholar]
- 11.Wehrens XHT, Kirchhoff S. and Doevendans PA. (2000). Mouse electrocardiography: an interval of thirty years. Cardiovasc Res 45:231–237 [DOI] [PubMed] [Google Scholar]
- 12.Zhang M, Schulte JS, Heinick A, Piccini I, Rao J, Quaranta R, Zeuschner D, Malan D, Kim K-P, Roepke A, et al. (2015). Universal cardiac induction of human pluripotent stem cells in two and three-dimensional formats: implications for in vitro maturation. Stem Cells 33:1456–1469 [DOI] [PubMed] [Google Scholar]
- 13.Denning C, Borgdorff V, Crutchley J, Firth KSA, George V, Kalra S, Kondrashov A, Hoang MD, Mosqueira D, et al. (2015). Cardiomyocytes from human pluripotent stem cells: from laboratory curiosity to industrial biomedical platform. Biochim Biophys Acta Pt 2: S0167-4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burridge PW, Keller G, Gold JD. and Wu JC. (2012). Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 10:16–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R. and Mummery CL. (2010). Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res 4:107–116 [DOI] [PubMed] [Google Scholar]
- 16.Harris K, Aylott M, Cui Y, Louttit JB, McMahon NC. and Sridhar A. (2013). Comparison of electrophysiological data from human-induced pluripotent stem cell–derived cardiomyocytes to functional preclinical safety assays. Toxicol Sci 134:412–426 [DOI] [PubMed] [Google Scholar]
- 17.Reynolds JG, Geretti E, Hendriks BS, Lee H, Leonard SC, Klinz SG, Noble CO, Lücker PB, Zandstra PW, et al. (2012). HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol Appl Pharmacol 262:1–10 [DOI] [PubMed] [Google Scholar]
- 18.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, Dorn T, Goedel A, Hohnke C. and Hofmann F. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 363:1397–1409 [DOI] [PubMed] [Google Scholar]
- 19.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G. and Hammerman H. (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471:225–229 [DOI] [PubMed] [Google Scholar]
- 20.Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A. and Denning C. (2011). Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J 32:952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsa E, Dixon JE, Medway C, Georgiou O, Patel MJ, Morgan K, Kemp PJ, Staniforth A, Mellor I. and Denning C. (2014). Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur Heart J 35:1078–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lahti AL, Kujala VJ, Chapman H, Koivisto AP, Pekkanen-Mattila M, Kerkela E, Hyttinen J, Kontula K, Swan H, et al. (2011). Human disease model for long QT syndrome type 2 using iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech 5:220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma D, Wei H, Zhao Y, Lu J, Li G, Sahib NBE, Tan TH, Wong KY, Shim W, et al. (2013). Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int J Cardiol 168:5277–5286 [DOI] [PubMed] [Google Scholar]
- 24.Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J. and Dolmetsch RE. (2011). Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471:230–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fatima A, Xu G, Shao K, Papadopoulos S, Lehmann M, Arnaiz-Cot JJ, Rosa AO, Nguemo F, Matzkies M, et al. (2011). In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem 28:579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung CB, Moretti A, Mederos M. Schnitzler Y, Iop L, Storch U, Bellin M, Dorn T, Ruppenthal S, et al. (2012). Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med 4:180–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terrenoire C, Wang K, Chan Tung KW, Chung WK, Pass RH, Lu JT, Jean J-C, Omari A, Sampson KJ, et al. (2013). Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol 141:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sager PT, Gintant G, Turner JR, Pettit S. and Stockbridge N. (2014). Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J 167:292–300 [DOI] [PubMed] [Google Scholar]
- 29.Farre C. and Fertig N. (2012). HTS techniques for patch clamp-based ion channel screening—advances and economy. Expert Opin Drug Discov 7:515–524 [DOI] [PubMed] [Google Scholar]
- 30.Brueggemann A, Farre C, Haarmann C, Haythornthwaite A, Kreir M, Stoelzle S, George M. and Fertig N. (2008). Planar patch clamp: advances in electrophysiology. Methods Mol Biol 491:165–176 [DOI] [PubMed] [Google Scholar]
- 31.Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ. and January CT. (2011). High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol 301:H2006–H2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoelzle S, Obergrussberger A, Brueggemann A, Haarmann C, George M, Kettenhofen R. and Fertig N. (2011). State-of-the-art automated patch clamp devices: heat activation, action potentials, and high throughput in ion channel screening. Front Pharmacol 2:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dick E, Matsa E, Young LE, Darling D. and Denning C. (2011). Faster generation of hiPSCs by coupling high-titer lentivirus and column-based positive selection. Nat Protoc 6:701–714 [DOI] [PubMed] [Google Scholar]
- 34.Burridge PW, Thompson S, Millrod MA, Weinberg S, Yuan X, Peters A, Mahairaki V, Koliatsos VE, Tung L. and Zambidis ET. (2011). A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS One 6:e18293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maherali N. and Hochedlinger K. (2008). Guidelines and techniques for the generation of induced pluripotent stem cells. Cell Stem Cell 3:595–605 [DOI] [PubMed] [Google Scholar]
- 36.Huang H-L, Hsing H-W, Lai T-C, Chen Y-W, Lee T-R, Chan H-T, Lyu P-C, Wu C-L, Lu Y-C, et al. (2010). Trypsin-induced proteome alteration during cell subculture in mammalian cells. J Biomed Sci 17:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Becker N, Stoelzle S, Gopel S, Guinot D, Mumm P, Haarmann C, Malan D, Bohlen H, Kossolov E, et al. (2013). Minimized cell usage for stem cell-derived and primary cells on an automated patch clamp system. J Pharmacol Toxicol Methods 68:82–87 [DOI] [PubMed] [Google Scholar]
- 38.Sakmann B. and Neher E. (1984). Patch clamp technique for studying ionic channels in excitable membranes. Annu Rev Physiol 46:455–472 [DOI] [PubMed] [Google Scholar]
- 39.Hartman ME, Dai D-F. and Laflamme MA. (2016). Human pluripotent stem cells: prospects and challenges as a source of cardiomyocytes for in vitro modeling and cell-based cardiac repair. Adv Drug Deliv Rev 96:3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroder RL, Christensen M, Anson B. and Sunesen M. (2012). Exploring stem cell-derived cardiomyocytes with automated patch clamp techniques. Biophys J 102:544A [Google Scholar]
- 41.Haythornthwaite A, Stoelzle S, Hasler A, Kiss A, Mosbacher J, George M, Brueggemann A. and Fertig N. (2012). Characterizing human ion channels in induced pluripotent stem cell-derived neurons. J Biomol Screen 17:1264–1272 [DOI] [PubMed] [Google Scholar]
- 42.Yajuan X, Xin L. and Zhiyuan L. (2012). A comparison of the performance and application differences between manual and automated patch-clamp techniques. Curr Chem Genomics 6:87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirkpatrick CJ, Melzner I. and Göller T. (1985). Comparative effects of trypsin, collagenase and mechanical harvesting on cell membrane lipids studied in monolayer-cultured endothelial cells and a green monkey kidney cell line. Biochim Biophys Acta 846:120–126 [DOI] [PubMed] [Google Scholar]
- 44.Satin J, Kehat I, Caspi O, Huber I, Arbel G, Itzhaki I, Magyar J, Schroder EA, Perlman I. and Gepstein L. (2004). Mechanism of spontaneous excitability in human embryonic stem cell derived cardiomyocytes. J Physiol 559:479–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakakibara Y, Furukawa T, Singer DH, Jia HJ, Backer CL, Arentzen CE. and Wasserstrom JA. (1993). Sodium current in isolated human ventricular myocytes. Am J Physiol 265:H1301–H1309 [DOI] [PubMed] [Google Scholar]
- 46.Pelzmann B, Schaffer P, Bernhart E, Lang P, Machler H, Rigler B. and Koidl B. (1998). L-type calcium current in human ventricular myocytes at a physiological temperature from children with tetralogy of Fallot. Cardiovasc Res 38:424–432 [DOI] [PubMed] [Google Scholar]
- 47.Tipparaju SM, Kumar R, Wang YG, Joyner RW. and Wagner MB. (2004). Developmental differences in L-type calcium current of human atrial myocytes. Am J Physiol Heart Circ Physiol 286:H1963–H1969 [DOI] [PubMed] [Google Scholar]
- 48.Jost N, Acsai K, Horvath B, Banyasz T, Baczko I, Bitay M, Bogats G. and Nanasi PP. (2009). Contribution of IKr and IK1 to ventricular repolarization in canine and human myocytes: is there any influence of action potential duration? Basic Res Cardiol 104:33–41 [DOI] [PubMed] [Google Scholar]
- 49.Sheng X, Reppel M, Nguemo F, Mohammad FI, Kuzmenkin A, Hescheler J. and Pfannkuche K. (2012). Human pluripotent stem cell-derived cardiomyocytes: response to TTX and Lidocain reveals strong cell to cell variability. PLoS One 7:e45963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Priori SG, Napolitano C, Di Pasquale E. and Condorelli G. (2013). Induced pluripotent stem cell-derived cardiomyocytes in studies of inherited arrhythmias. J Clin Invest 123:84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.