

Abstract
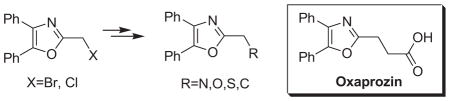
2-(Halomethyl)-4,5-diphenyloxazoles are effective, reactive scaffolds which can be utilized for synthetic elaboration at the 2-position. Through substitution reactions, the chloromethyl analogue is used to prepare a number of 2-alkylamino-, 2-alkylthio- and 2-alkoxy-(methyl) oxazoles. The 2-bromomethyl analogue offers a more reactive alternative to the chloromethyl compounds and is useful in the C-alkylation of a stabilized (malonate) carbanion as exemplified by a concise synthesis of Oxaprozin.
Keywords: heterocycles, oxazoles, click chemistry, ligands, Oxaprozin
Graphical Abstract
1. Introduction
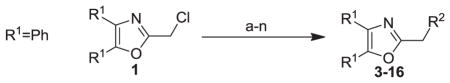
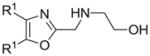
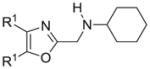
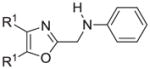
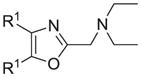
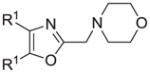
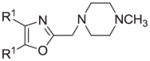
The synthesis and utilization of extended 2-substituted-4, 5-diaryloxazoles has found interesting applications in the synthesis of natural products, medicinal chemistry and photochemistry. In natural products synthesis, the 4,5-diaryloxazole group has functioned as an effective masked carboxyl derivative and functions well when introduced during the early or late stages of a total synthesis.1 Medicinal chemistry groups have investigated the diaryloxazole system in the design and evaluation of prostanoid analogues.2 While the 2-substituted 4,5-diaryloxazole group responds well in photochemical reactions involving singlet oxygen, there is an inherent photochemical response exhibited by these compounds which has potential in scintillation technology.3 Basically three to four general strategies may be followed when preparing extended oxazoles at the 2-position and all these allow for a varied pattern of substituents as well as a varied degree of substituent reactivity or functional group types (Scheme 1). Lithiation of the 2-position of 4,5-diaryloxazoles may be accomplished followed by reactions with a series of electrophiles (Eq 1, Scheme 1), however, the reaction may be complicated by ring-opening to the isonitrile enolate.4 2-methyl-4,5-diaryloxazoles may be deprotonated (LDA) and alkylated to provide extended, fully functionalized oxazoles at the 2-position (Eq 2, Scheme 1). The ring-closure strategy toward 2-extended oxazoles involves the fairly standard benzoin ester formation followed by generation of the heterocycle with ammonium acetate in acetic acid (Eq 3, Scheme 1).5 Typically, the ring-closure strategy is limited by the types of substituted benzoins as well as the carboxylic acid portion of the ester which bears the soon-to-be 2-appendage at the α-position of the carbonyl. While 2-(halomethyl)oxazoles (X=Br, Cl) were first proposed as atom transfer radical polymerization (ATRP) initiators,6 our earlier work showed their synthetic utility in preparing 2-(azidomethyl)oxazole click reactants.5 Considering the facile formation of azides from the title compounds, we now report a diverse manifold of substitution when these halogenated compounds are reacted with appropriate nucleophiles such as amines, alkoxides, thiolates, triphenylphosphine or cyanide ion thereby providing a number of interesting intermediates (Eq 4, Scheme 1). In terms of fundamental nitrogen substitution on the 2-(methylene) position of oxazoles, the simplest, most unambiguous nitrogen nucleophile, i.e. azide ion, was utilized toward the goal of only providing click intermediates. Chain-lengthening of the 2-azidoalkyl group for the purpose of furnishing homologous 2-(aminoalkyl)oxazoles would necessitate oxazole closure of the corresponding homologous 2-(azidoalkyl)esters followed by reduction of the azido group. 2-(Aminoalkyl)-4,5-diphenyloxazoles have been investigated for analgesic and anti-inflammatory activity in rodent models using phenylbutazone and diethamphenazole as standards. Herein, we first show the synthetic variability of the 2-(halomethyl)oxazoles by reaction with suitable amine derivatives under a variety of conditions (Compounds 3–9, Table 1). While nucleophilic substitution of amines on various halogenated centers are well-known reactions,7 we find that the 2-halomethylene unit of the title reactants (1, X=Cl; 2, X=Br) offers reactivity characteristic of a benzylic chloromethyl group. Primary alkyl-/aromatic amines such as ethanolamine, cyclohexylamine and aniline are capable of providing the corresponding N-substituted (2-aminomethyl) oxazoles (3,4 and 5, Table 1), while diethylamine, morpholine, N-methyl piperazine, and imidazole easily form the corresonding N,N-disubstituted products (6–9, Table 1). We further demonstrate the synthetic utility of the 4,5-diphenyl-2-(halomethyl)oxazoles by reaction with various alkoxides or otherwise in situ-generated phenoxide in affording the corresponding alkyl or phenyl ethers (10–12, Table 1). The resulting 2-(alkoxymethyl)- or 2-(phenoxymethyl)-oxazoles have been of interest as anti-inflammatory and analgesic agents whose mechanism of action depends on the modulation of cyclooxygenase activity.8 Sulfur nucleophiles such as thiocyanate and thiophenoxide afford the corresponding 2-(methylthio) cyanate 13 or the 2-(phenylthiomethyl) oxazole 14 in high yield (Table 1). During the formation and purification of 13, no isomerization to the corresponding isothiocyanate was observed.9 With respect to the 2-(phenylthiomethyl) oxazole 14 (thiophenol/NaH), we find that this compound is easily oxidized to the corresponding sulfone,10 a compound which exhibits excellent stabilized anion reactivity for carbon-carbon bond formation. The preparation of triphenylphosphonium salt 15 (PPh3/toluene/heat) was the result of another heterocyclic scaffold modification whereby the potential for carbon-carbon bond formation and oxazole extension exists through Wittig chemistry.11 The 2-(cyanomethyl)oxazole 16 was prepared by cyanide (NaCN/DMF) substitution of 1.12 The nitrile group of 15 should offer excellent potential for carbon-carbon bond formation at the 2-methylene position, through carbanion formation, as well as providing a reactive acceptor for alkyllithiums toward gaining carbonyl products. We demonstrate the usefulness of the 2-halomethyloxazoles 1 and 2 in carbon-carbon bond formation by a synthesis of the non-steroidal anti-inflammatory Oxaprozin (Scheme 2).13 Chloromethyloxazole 1 is reacted with the anion of diethylmalonate (NaH/THF) which affords the diester 17 in 40% isolated yield. Under the same conditions, alkylation with the more reactive bromomethyloxazole 2 provides the diester 17 in 90% isolated yield. Saponification of 17 (aq. NaOH) followed by acidification (dil. HCl/reflux) then gives Oxaprozin in 47% yield.
Scheme 1.

Synthesis of 2-extended oxazoles (X=Cl, Br).
Table 1.
Synthesis of Extended 2-Substituted Oxazoles
| ||
|---|---|---|
| Conditions | Product | Yield (%) |
| a |
|
63 |
| b |
|
40 |
| c |
|
70 |
| d |
|
81 |
| e |
|
90 |
| f |
|
80 |
| g |
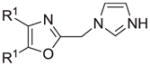
|
85 |
| h |
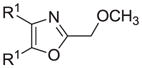
|
96 |
| i |
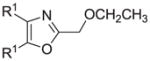
|
80 |
| j |
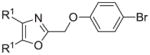
|
72 |
| k |
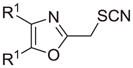
|
93 |
| l |
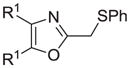
|
90 |
| m |
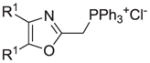
|
30 |
| n |
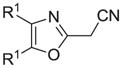
|
41 |
Reagents and conditions: (a) ethanolamine/ethanol/reflux/6h. (b) cyclohexylamine/TEA/THF/60°C/2h. (c) aniline/85°C/12h. (d) diethylamine/benzene/reflux/3h. (e) morpholine/benzene/refux/8h. (f) N-methylpiperazine/TEA/THF/reflux/2h. (g) imidazole/NaH/DMF/5°C/2h. (h) NaOMe/MeOH/5°C to rt/16h. (i) NaOEt/EtOH/5°C to rt/16h. (j) 4-bromophenol/K2CO3/DMF/100°C. (k) KSCN/acetone/reflux/3h. (l) PhSH/NaH/DMF/5°C to rt. (m) PPh3/toluene/reflux/16h. (n) NaCN/DMF/10°C to rt/16h.
Scheme 2.

Synthesis of Oxaprozin:
Reagents and conditions:(a) NaH/diethyl malonate/THF/5°C to rt/16h (40%, X=Cl; 90%, X=Br). (b) 20% aq. NaOH/rt/16h. (c) 10% aq. HCl, pH 3–5/reflux/3h (47% for b,c).
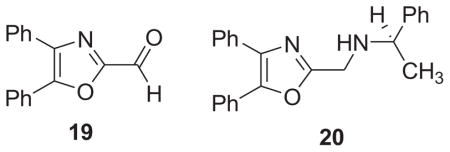
Within the realm of amine substitution at the 2-methylene position of the 4,5-diaryloxazoles, we note that in preliminary experiments, our previously-reported 4,5-diphenyloxazole aldehyde 195a,11 reacts as a convenient partner in a Schiff base formation/reduction sequence to give secondary amines. Therefore the employment of the oxazole aldehyde will provide a useful alternative to the halomethyl intermediates in providing 2-aminomethyl-substituted oxazole scaffolds.14 For example, the reaction of 19 with (+)-R-α-methylbenzylamine (methanol/reflux/16 h) gave the expected intermediate Schiff base (73%) which was directly reduced with sodium borohydride (methanol/rt/1h) to provide the chiral amine 20 (76%).
In summary we have shown that 2-(chloromethyl)-4,5-diphenyloxazoles, which are readily available from the corresponding chloroacetyl esters of benzoin or substituted benzoins, are excellent reactive scaffolds for synthetic elaboration at the 2-(methylene) position. The 2-(bromomethyl)oxazole analogue is best suited for a concise synthesis of Oxaprozin using malonate alkylation as the key step. A number of diverse amine nucleophiles may be used to prepare 2-methyloxazole-derived primary or secondary amines. Similarly, the halomethyloxazoles react well with alkoxides or phenoxides to give the corresponding ethers which have anti-inflammatory or analgesic activity. Sulfur nucleophiles such as thiocyanates and thiophenoxides react in high yield to give the corresponding carbon-sulfur bond motif whereby the 2-phenylthiomethyl analogue will show promise in further reaction scenarios.
Supplementary Material
Acknowledgments
The measurement of high resolution mass spectra by the Texas A&M University Laboratory for Biological Mass Spectrometry is acknowledged. Financial support from the NIH/NIDCR through grant 1RO1DE023206 is gratefully acknowledged.
Footnotes
Supplementary data (1H NMR, FTIR) for compounds 2–18, 20; and additional 13C NMR data for new compounds 5, 9, 12–15, 17, 18, 20. HRMS data are included for compounds 9, 10–16, 20; along with experimental procedures associated with this article can be found, in the online version at http://dx.doi.org/j.tetlet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Wasserman HH, McCarthy KE, Prowse KS. Chem Rev. 1986;86:845–856. [Google Scholar]; (b) Evans DA, Nagorny P, Reynolds DJ, McRae KJ. Angew Chem Int Ed. 2007;46:541–544. doi: 10.1002/anie.200603654. [DOI] [PubMed] [Google Scholar]; (c) Tius MA, Astrab DP, Fauq AH, Ousset JB, Trehan S. J Am Chem Soc. 1986;108:3438–3442. [Google Scholar]
- 2.(a) Ullapu PR, Ku SJ, Choi YH, Park J, Han SY, Baek DJ, Lee J, Pae AN, Min SJ, Cho YS. Bull Korean Chem Soc. 2011;32:3063–3068. [Google Scholar]; (b) Hattori K, Okitsu O, Tabuchi S, Taniguchi K, Nishio M, Koyama S, Seki J, Sakane K. Biorg Med Chem Lett. 2005;15:3279–3283. doi: 10.1016/j.bmcl.2005.04.042. [DOI] [PubMed] [Google Scholar]; (c) Meanwell NA, Rosenfeld MJ, Wright JJ, Kim Brassard CL, Buchanan JO, Federici ME, Fleming JS, Gamberdella M, Hartl KS, Zavoico GB, Seiler SM. J Med Chem. 1993;36:3871–3883. doi: 10.1021/jm00076a017. [DOI] [PubMed] [Google Scholar]; (d) Marchetti E. Ger Offen. 1971:2108437. [Google Scholar]; Chem Abs. 1972;76:46188. [Google Scholar]; (e) Mattaglia G, Marchetti E. Il Farmaco. 1971;26:512–519. [PubMed] [Google Scholar]
- 3.Mahuteau-Betzer F, Piguel S. Tetrahedron Lett. 2013;54:3188–3193. [Google Scholar]
- 4.For recent reviews, See: Haldon E, Nicasio MC, Perez PJ. Org Biomol Chem. 2015;13:9528–9550. doi: 10.1039/c5ob01457c.Thirumurugan P, Matosiuk D, Jozwiak K. Chem Rev. 2013;113:4905–4979. doi: 10.1021/cr200409f.Pedersen DS, Abell A. Eur J Org Chem. 2011:2399–2411.
- 5.(a) Patil PC, Luzzio FA, Demuth DR. Tetrahedron Lett. 2015;56:3039–3041. doi: 10.1016/j.tetlet.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Loner CM, Luzzio FA, Demuth DR. Tetrahedron Lett. 2012;53:5641–5644. [Google Scholar]
- 6.Zhang L, Xu Q, Lu J, Xia X, Wang L. Euro Poly J. 2007;43:2718–2724. [Google Scholar]
- 7.Smith MB. March’s Advanced Organic Chemistry. John Wiley and Sons; Hoboken: 2013. pp. 481–487. [Google Scholar]
- 8.Talley JJ. WO 96/36617. World Patent Application. 1995; Chem Abstr. 1997;126:74828. [Google Scholar]
- 9.(a) Bound DJ, Bettadaiah BK, Srinivas P. Synth Commun. 2013;43:1138–1144. [Google Scholar]; (b) Meshram HM, Thakur PB, Babu BM, Bangade VM. Tetrahedron Lett. 2012;53:1780–1785. [Google Scholar]; (c) Gorjizadeh M, Sayyahi S. Chinese Chem Lett. 2011;22:659–662. [Google Scholar]; (d) Li J, Cao JJ, Wei JF, Shi YY, Zhang LH, Feng JJ, Chen ZG. Eur J Org Chem. 2011:229–233. [Google Scholar]; (e) Mohanazadeh F, Aghvami M. Tetrahedron Lett. 2007;48:7240–7242. [Google Scholar]; (f) Ju Y, Kumar D, Varma RS. J Org Chem. 2006;71:6697–6700. doi: 10.1021/jo061114h. [DOI] [PubMed] [Google Scholar]; (g) Kamal A, Chouan G. Tetrahedron Lett. 2005;46:1489–1491. [Google Scholar]; (h) Erian A, Sherif SM. Tetrahedron. 1999;55:7957–8024. [Google Scholar]; (i) Ando T, Clark JH, Cork DG, Fujita M, Kimura T. J Org Chem. 1987;52:681–685. [Google Scholar]
- 10.For a sulfonyl(methylene) oxazole with similar activating potential, See: Sakamoto T, Kondo Y, Suginome T, Ohba S, Yamanaka H. Synthesis. 1992:552–554.
- 11.For heterocyclic-substituted (methyl) triphenylphosphonium salts of heterocycles, See for example: Morais GR, Miranda HV, Santos IC, Santos I, Outeiro TF, Paulo A. Bioorg Med Chem. 2011;19:7698–7710. doi: 10.1016/j.bmc.2011.09.065.Hoffman JM, Smith AM, Rooney CS, Fisher TE, Wai JS, Thomas CS, Bamberger DL, Barnes JL, Williams TM, Jones JH, Olson BD, O’Brien JA, Goldman ME, Nunberg JH, Qintero JC, Schleif WA, Emini EA, Anderson PA. J Med Chem. 1993;36:953–966. doi: 10.1021/jm00060a002.
- 12.(a) Meanwell NA, Rosenfeld MJ, Wright JJK, Brassard CL, Buchanan JO, Federici ME, Fleming JS, Gamberdella M, Hartl KS, Zavoico GB, Seiler SM. J Med Chem. 1993;36:3871–3883. doi: 10.1021/jm00076a017. [DOI] [PubMed] [Google Scholar]; (b) Kotani E, Kobayashi S, Adachi M, Tsujioka T, Nakamura K, Tobinaga S. Chem Pharm Bull. 1989;37:606–609. [Google Scholar]; (c) Kim TY, Shik H, Chung YM, Kim JN. Bull Korean Chem Soc. 2000;21:673–674. [Google Scholar]
- 13.(a) Imai S, Kikui H, Moriyama K, Togo H. Tetrahedron. 2015;71:5267–5274. [Google Scholar]; (b) Saito Akio, Taniguchi A, Kanbara Y, Hanzawa Y. Org Lett. 2013;15:2672–2675. doi: 10.1021/ol4009816. [DOI] [PubMed] [Google Scholar]; (c) Zheng Y, Li X, Ren C, Zhang-Negrerie D, Du Y, Zhao K. J Org Chem. 2012;77:10353–10361. doi: 10.1021/jo302073e. [DOI] [PubMed] [Google Scholar]; (d) Božić BD, Trišović NP, Valentić NV, Ušćumlić GS, Petrović SD. Hem Ind. 2011;65:551–562. [Google Scholar]
- 14.(a) Ref. 5a. Šagud I, Faraguna F, Marinić Z, Šindler-Kulyk M. J Org Chem. 2011;76:2904–2908. doi: 10.1021/jo1025942.Šagud I, Antol I, Marinić Z, Šindler-Kulyk M. J Org Chem. 2015;80:9535–9641. doi: 10.1021/acs.joc.5b01504.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.