

Temporal transcriptome analysis during drought stress coupled with Bayesian network modeling reveals early drought signaling events and identifies AGL22 as a regulator of primary metabolism.
Abstract
In Arabidopsis thaliana, changes in metabolism and gene expression drive increased drought tolerance and initiate diverse drought avoidance and escape responses. To address regulatory processes that link these responses, we set out to identify genes that govern early responses to drought. To do this, a high-resolution time series transcriptomics data set was produced, coupled with detailed physiological and metabolic analyses of plants subjected to a slow transition from well-watered to drought conditions. A total of 1815 drought-responsive differentially expressed genes were identified. The early changes in gene expression coincided with a drop in carbon assimilation, and only in the late stages with an increase in foliar abscisic acid content. To identify gene regulatory networks (GRNs) mediating the transition between the early and late stages of drought, we used Bayesian network modeling of differentially expressed transcription factor (TF) genes. This approach identified AGAMOUS-LIKE22 (AGL22), as key hub gene in a TF GRN. It has previously been shown that AGL22 is involved in the transition from vegetative state to flowering but here we show that AGL22 expression influences steady state photosynthetic rates and lifetime water use. This suggests that AGL22 uniquely regulates a transcriptional network during drought stress, linking changes in primary metabolism and the initiation of stress responses.
INTRODUCTION
Water limitation in agriculture is poised to intensify in the coming decades due to urbanization, industrialization, depletion of aquifers, and increasingly erratic rainfall patterns exacerbated by climate change (Easterling et al., 2000; Christensen et al., 2007; Seager et al., 2007; Famiglietti and Rodell, 2013). Reduced water availability leads to drought stress, which is a major constraint on the physiology, growth, development, and productivity of plants (Boyer, 1970, 1982; Lobell and Field, 2007; Roberts and Schlenker, 2009; Skirycz et al., 2010; Lobell et al., 2011; Verelst et al., 2013). Therefore, understanding the mechanisms of drought response in plants is essential for the improvement of plant performance under water-limiting conditions and has been the subject of many investigations over the years (Shinozaki and Yamaguchi-Shinozaki, 1997, 2007; Chaves et al., 2009; Nakashima et al., 2009; Pinheiro and Chaves, 2011). Water deficit responses are complex and require stress sensing and signaling to adjust plant growth, maintain water status through osmoregulation, prevent water loss through decreases in stomatal conductance, and activate detoxification processes (Passioura, 1996; Chaves et al., 2003; Pinheiro and Chaves, 2011). An important consideration is that even a slight reduction in water availability can elicit stomatal closure and a reduction in CO2 assimilation and in combination with the diversion of resources toward drought defense mechanisms will affect plant productivity (Chaves et al., 2003).
Plants have adopted different strategies to respond to water limitation, such as drought escape through early flowering and reducing the size of plants to increase water use efficiency or drought avoidance through enhanced soil moisture capture or reduced transpiration (Ludlow, 1989; Blum, 2005; Aguirrezabal et al., 2006; Franks, 2011). In this context, the influence of drought on plant development and growth through its effects on developmental processes such as germination, seedling growth, and leaf development has been studied extensively in the past decade (van der Weele et al., 2000; Finkelstein et al., 2002; Xiong et al., 2006; Yaish et al., 2011). Optimal timing of flowering and inflorescence development are important traits essential in determining plant yield, and these can vary greatly in response to water limitation (Eckhart et al., 2004; Franke et al., 2006; Su et al., 2013; Ma et al., 2014).
At the cellular level, plants respond to drought with changes in gene expression and protein and metabolite abundances (Charlton et al., 2008; Harb et al., 2010; Wilkins et al., 2010; Baerenfaller et al., 2012), which are part of defense mechanisms and detoxification processes (Shinozaki and Yamaguchi-Shinozaki, 2007; Begcy et al., 2011; Ozfidan et al., 2012). Recent progress in genomics, transcriptomics, and bioinformatics has paved the way for dissecting drought-response mechanisms and has enabled the targeted manipulation of drought-responsive genes in plants. For example, the overexpression of a number of genes that code for transcription factors (TFs) leads to drought resistance (Sakuma et al., 2006; Nelson et al., 2007; Chen et al., 2008; Quan et al., 2010; Tang et al., 2012).
In many studies to identify genes important in the regulation of drought responses, the effects of water limitation at the transcriptional level have been analyzed by exposing plants to severe dehydration. This involves treatments such as cutting and air drying leaves and/or roots or induction of osmotic shock through the application of highly concentrated osmotica such as polyethylene glycol or mannitol (Kreps et al., 2002; Seki et al., 2002; Kawaguchi et al., 2004; Kilian et al., 2007; Weston et al., 2008; Fujita et al., 2009; Abdeen et al., 2010; Deyholos, 2010; Mizoguchi et al., 2010). These experiments have substantially increased our knowledge of molecular responses under severe drought stress, but they do not always reflect physiological conditions experienced by drought-stressed soil-grown plants (Bechtold et al., 2010, 2013; Harb et al., 2010; Wilkins et al., 2010; Lawlor, 2013; Zhang et al., 2014). Physiological responses such as stomatal conductance, photosynthetic performance, and metabolic changes are usually not measured during the progression of the drought stress, and the varied nature of the stress induction treatments makes comparative analysis between experiments problematic. Slow developing soil water deficits have different physiological consequences than those induced by rapid tissue dehydration and therefore possibly utilize different gene networks (Chaves et al., 2003, 2009; Pinheiro and Chaves, 2011).
This inconsistency among experiments was first noted in a meta-analysis of microarray experiments comparing air drying, soil drying, and mannitol treatments (Bray, 2004). This analysis found very few differentially expressed genes (DEGs) common to all treatments (Bray, 2004). Consequently, recent experiments have focused on soil-grown plants (Harb et al., 2010; Wilkins et al., 2010; Zhang et al., 2014). From these studies, an overall integrative picture of the temporal responses to drought is emerging slowly, and it is clear that use of a single or a small number of time points and different types of experimental conditions lead to very different outcomes. One consequence of this is that very little is known about the early events in the perception of drought stress signals (Ueguchi et al., 2001; Wohlbach et al., 2008; Pinheiro and Chaves, 2011).
To address the above issues, we set out to gain detailed information on the processes that occur during the transition from well-watered to drought conditions, in which the intensity of the stress becomes gradually greater. We monitored the physiological and metabolic status of plants through a progressive drought experiment and mapped onto these data the temporal responses of the transcriptome. Our intention was to use the highly resolved transcriptional profiling data to construct gene regulatory networks (GRNs) using dynamic Bayesian network modeling (Beal et al., 2005; Breeze et al., 2011; Penfold and Wild, 2011) with the aim of identifying regulatory genes functional during drought perception and signaling. The goal was to link early physiological and metabolic drought avoidance responses with later drought escape and/or tolerance responses (Claeys and Inzé, 2013). This initially required testing of the network modeling to evaluate the capability of these approaches to identify genes important in the regulation of drought responses. This was achieved by selecting a highly connected candidate gene, AGAMOUS-LIKE22 (AGL22; also known as SHORT VEGETATIVE PROTEIN), from the GRNs. AGL22 has an established function in plant development (Gregis et al., 2013; Méndez-Vigo et al., 2013), but in this study, it was shown to play a thus far undiscovered role in the critical early stages of the plant’s response to drought. These results demonstrated the potential value of experimental strategies that combine time-series transcriptomics data with dynamic modeling as a means of identifying stress-responsive genes.
RESULTS
Time-series experiments were performed analyzing physiological, metabolic, and transcriptional changes in Arabidopsis thaliana to reveal the chronology of plant responses to drought stress. A progressive slow-drying experiment starting at 95% relative gravimetric soil water content (rSWC) and drying down to 17% rSWC was performed on 5-week-old Arabidopsis plants (Figure 1). To determine the severity of the stress, daily measurements of relative leaf water content (RWC; Figure 1A) and leaf water potential were also performed (Figure 1B). During the experiment, the average rSWC loss was ∼10% per day, but RWC was maintained throughout the progressive drying period until the point of wilting at 17% rSWC (Figure 1A).
Figure 1.

Plant Responses during a Progressive Drought Experiment.
(A) RWC (open triangles) and rSWC (closed triangles) during a 13-d drying period. The data represent the mean (n = 6; ±se).
(B) Leaf water potential in well-watered (open circles) and drought-stressed (closed circles) plants during a 13-d drying period. The data represent the mean (n = 5; ±se).
(C) Stomatal conductance of well-watered (open circles) and drought-stressed (closed circles) plants, measured at the prevailing growth conditions (see Methods). The data represent the mean (n = 6; ±se).
(D) Carbon assimilation of well-watered (open circles) and drought-stressed (closed circles) plants, measured at the prevailing growth conditions (see Methods). The data represent the mean (n = 6; ±se).
Maximum Photosynthetic Capacity Responds Similarly in Well-Watered and Drought-Stressed Plants during a Progressive Drought Experiment
Stomatal conductance (gs) and photosynthetic carbon assimilation (A) were measured daily on well-watered and drought-stressed plants through the progressive drought treatment (Figures 1C and 1D). Stomatal conductance declined at ∼60% rSWC (day 5; Figure 1C), which was followed by a decline in carbon assimilation at ∼45% rSWC (day 7), indicating that stomatal diffusional limitations affected carbon assimilation (Figure 1D). Plant growth evaluated as rosette fresh weight and rosette area ceased at ∼40% rSWC (Supplemental Figures 1A to 1C).
The light and CO2-saturated maximum photosynthetic rate (Amax), maximum rate of carboxylation (VCmax), and the rate of ribulose-1,5-bisphosphate (RuBP) regeneration (Jmax) showed no difference between well-watered and drought-stressed plants (Figure 2A). In addition, maximum and operating efficiencies of photosystem II (Fv/Fm, Fv′/Fm′, and Fq′/Fm′; Baker, 2008) showed no change during the drought period (Figure 2B; Supplemental Figure 1D), suggesting that the overall primary metabolic capacity was maintained as drought conditions progressed. The sequential changes in photosynthetic physiology, relative water content, and leaf water potential suggest that these conditions allowed us to capture the transition between early physiological changes and later stress responses.
Figure 2.

Potential Photosynthesis in Response to Drought.
(A) A/Ci curves were performed under saturating light conditions of 1000 µmol m−2 s−1 at six selected time points throughout the drying period, and potential photosynthesis was calculated, including light and [CO2]-saturated net CO2 assimilation (Amax), maximum rate of RuBP regeneration (Jmax), and maximum rate of carboxylation (VCmax).
(B) Maximum and operating quantum efficiencies of photosystem II (Fv/Fm, Fq′/Fm′, and Fv′/Fm′). The data represent the mean (n = 3; ±se).
Metabolite Profiling Indicates the Stable Nature of Primary Metabolism during Drought Stress
The decline in stomatal conductance and carbon assimilation led us to perform metabolite analysis to evaluate changes in primary and secondary metabolism (Figure 3). Untargeted liquid chromatography-mass spectrometry metabolite profiling was performed on samples harvested at early (day 2, ∼80% rSWC), mid (day 7, ∼45% rSWC), and late (day 13, 17% rSWC) stages of the drought stress. This analysis showed that the majority of the metabolome was unchanged throughout most of the drought treatment, and distinct clustering between well-watered and drought-stressed samples emerged only by the final day of drought stress (17% rSWC) (Figure 3A). Leaf development was a major factor for sample separation, with days clustered more closely together than treatments (Figure 3A).
Figure 3.

Metabolite Levels during Progressive Drought.
(A) Liquid chromatography/electrospray ionization quadrupole time-of-flight mass spectrometry metabolite profiling of Arabidopsis leaves under well-watered (W) and progressive soil drought (D) conditions. Leaf extracts were analyzed in negative and positive ionization modes. The heat maps show the normalized abundances of all detected chemical features. Samples and chemical features were clustered using a Pearson distance measure and the Ward clustering algorithm (Supplemental Data Set 18).
(B) Relative concentrations of ABA (black bars) and proline (gray bars).
(C) Relative concentrations of galactinol (black bars) and raffinose (gray bars). The data represent the mean of the ratio (n = 4; ±se; *P < 0.05 or **P < 0.01).
Targeted metabolite analysis was performed to determine the foliar levels of 102 stress-associated compounds (Supplemental Data Sets 1 to 3), which also revealed a mainly late response for many of these stress-associated metabolites (Supplemental Data Sets 1 to 3). Often these changes were limited to the last two to three time points (between ∼30 and 17% rSWC; Supplemental Figures 2 and 3). For example, metabolites indicative of drought stress increased only during the late stages of the dehydration period (Figures 3B and 3C). There was a significant increase in abscisic acid (ABA) levels during the last four time points (Xiong et al., 2002; Figure 3B), while proline, a drought stress-responsive compatible solute in vascular plants (Sperdouli and Moustakas, 2012), accumulated to significant levels only during the last two time points (Figure 3B). Additionally, the accumulation of secondary metabolites commonly associated with stress responses such as anthocyanins and flavonols were altered during the late stages of the drought response (Sperdouli and Moustakas, 2012; Supplemental Data Sets 1 and 3 and Supplemental Figure 3). Oligosaccharides/disaccharides associated with osmotic protection during drought and osmotic stresses (galactinol and raffinose; Taji et al., 2002) significantly accumulated during the last 4 days of the drought response (Figure 3C; Supplemental Data Set 1). In conclusion, leaf metabolism remained largely stable during the first 9 d of the experiment, while changes previously associated with drought stress (Taji et al., 2002; Xiong et al., 2002; Sperdouli and Moustakas, 2012) only became evident during the last three to four time points.
Transcriptomics Analysis on a Single Leaf Identifies 1815 DEGs during Progressive Drought Stress
Transcriptome profiling was performed on leaf 7 to integrate the complex physiological and metabolic responses with changes at the gene expression level. Leaf 7 was fully expanded at the time of the experiment (Supplemental Figure 1C) and was chosen because a detailed temporal transcriptome analysis of leaf development was available (Breeze et al., 2011). Single leaf 7 samples for transcriptome analysis were taken each day at the midpoint of the light period. RNA from four leaf samples per treatment and time point was hybridized on CATMA v4 arrays (Sclep et al., 2007; see Methods). An adapted MAANOVA (microarray analysis of variance) method was used to analyze the data for each comparison (Wu et al., 2003; Churchill, 2004; Breeze et al., 2011; Windram et al., 2012). This generated a single normalized expression value for each gene. A Gaussian process two-sample test (GP2S; Stegle et al., 2010) was used to identify DEGs. Choosing a Bayes factor value (likelihood of differential expression) of >6 resulted in a total of 1815 DEGs (Supplemental Data Set 4). The upregulated group of genes showed on overrepresentation of Gene Ontology (GO) terms related to carbohydrate biosynthesis, flavonoid, and secondary metabolic processes, while downregulated genes were enriched in protein translation, cell wall-associated processes, pigment biosynthesis, and chloroplast associated processes (Supplemental Data Set 5).
GO terms related to stress, dehydration, and hormonal regulation, including ABA, were not enriched in the complete data set. This result suggested that the overall progressive drought experiment was not a severe dehydration stress response, as indicated by the maintenance of primary metabolic capacity (Figure 2) as well as the late responses of stress-associated metabolites (Figure 3). Leaf water potential has been used as a measure of the progression and effect of drought stress on plants (Zhang et al., 2014). We estimated the cumulative number of DEGs at each time point by determining the time of first differential expression for each gene (Supplemental Data Set 6; Figure 4A). In our experiment, leaf water potential correlated significantly with rSWC (Figure 4B) as well as the number of DEGs (Figure 4C) and showed a weaker correlation with carbon assimilation (Figure 4D). The biggest drop in leaf water potential occurred between 40% (day 8) and ∼30% (day 9) rSWC, which coincided with the biggest increase in DEGs (Figures 4A and 4C), potentially indicating a shift from mild to severe drought stress.
Figure 4.

The Relationship between Stress Severity and Differential Gene Expression.
(A) Number of genes for which the first differential expression was observed at each time point, indicating a late transcriptional response.
(B) Correlation between leaf water potential and rSWC.
(C) Correlation between leaf water potential and number of differentially expressed genes.
(D) Correlation between carbon assimilation and leaf water potential. Line represents the linear regression; r2 and P values are given.
To evaluate the transcriptome data set in the context of other drought experiments, we also compared our data set to two soil-based drought studies in Arabidopsis. The first experiment performed by Harb et al. (2010) comprised a microarray comparison of leaf samples under moderate drought stress (maintaining soil water content at 30% of field capacity) and progressive drought stress at the prewilting (∼15% field capacity) and wilting (∼10% field capacity) stages. In the progressive drought stress treatment, 3005 genes responded >2- and <0.5-fold, in comparison to 441 genes for the moderate drought treatments (Supplemental Data Set 7). The second study analyzed samples at a loss of 25% soil water of field capacity measured at 6-h intervals across a 24-h period (Wilkins et al., 2010) and identified 570 genes that responded across a 24-h interval (hereafter called diel; Supplemental Data Set 7). A general overview of overlapping genes between the different experiments showed that only 30 genes were common to all 4 treatments (Figure 5A). Among the overlapping genes were known stress-responsive, ABA-responsive, and secondary metabolism genes, which predominantly responded during the latter half of the drought experiment (Supplemental Data Set 8). While there were common elements in all four treatments, 63% of the DEGs in our data set were unique to the time series (Figure 5A) and were potentially related to the adjustments to early and moderate drought stress.
Figure 5.

Comparative Meta-Analysis with Publicly Available Data Sets and MapMan Analysis (Thimm et al., 2004) of Primary and Secondary Metabolism Pathways.
(A) Comparative meta-analysis of the 1815 DEGs with publicly available drought data sets. The Venn diagram shows the overlap of time series DEGs with those responsive to moderate (mDr; Harb et al., 2010) or progressive drought (pDr; Harb et al., 2010) and a moderate drought at different times of day (diel; Wilkins et al., 2010).
(B) Comparative meta-analysis of the 1815 DEGs with a publicly available leaf 7 senescence time-series data set (Breeze et al., 2011). The Venn diagram shows the overlap of drought and senescence DEGs.
(C) Overview of antioxidant, photosynthesis. and photorespiration-related gene expression at two different time points (95% rSWC and 17% rSWC).
(D) Overview of oxidation/reduction-related gene expression at two different time points (95% rSWC and 17% rSWC).
(E) Overview of secondary metabolism-related gene expression at two different time points (95% rSWC and 17% rSWC). All MapMan diagrams show gene expression data in leaf 7, where blue indicates increased and yellow indicates decreased gene expression according to the scale. Each square represents a single gene within the pathways.
Slow Soil Drying Induces a Senescence Response in Leaf 7
To assess developmental changes in leaf 7 during drought stress, we compared the drought time series to an Arabidopsis leaf 7 senescence time-series data set (Breeze et al., 2011; Supplemental Data Set 7). In all, 842 genes overlapped with the senescence data set (p-hyper, 4.4E-135; Figure 5B), of which 83% responded between days 8 and 13 (40 to 17% rSWC; Supplemental Data Set 9). The overlap contained genes associated with oxidation/reduction-related processes, pigment biosynthesis, and primary metabolism, which were predominantly downregulated during drought stress (Figures 5C and 5D; Supplemental Data Set 10). The induction of senescence-related processes during drought stress is a known phenomenon (Munné-Bosch and Alegre, 2004) and was further confirmed by a significant overlap of genes between the published drought and senescence data sets (Supplemental Data Set 7 and Supplemental Figure 4A).
In addition, changes in the expression of secondary metabolism genes were observed in the drought time series (Figure 5E), including CHALCONE SYNTHASE, FLAVONOL SYNTHASE1, LEUCOANTHOCYANIDIN DIOXYGENASE, PRODUCTION OF ANTHOCYANIN PIGMENT1, and ANTHOCYANINLESS2 (Supplemental Figure 5 and Supplemental Data Set 9). This coincided with increased accumulation of flavonol and anthocyanin (from day 11; Supplemental Data Set 1 and Supplemental Figure 3) and suggested that the plants had entered a severe stress phase (Vanderauwera et al., 2005). Therefore, it was concluded that slow soil drying induces senescence in leaf 7 but only at the point of severe drought stress. By contrast, early responses to soil drying (days 1 to 7) were mostly unique to the drought time series. Importantly for later considerations, the induction of leaf senescence in response to drought did not affect flowering time (Supplemental Figure 4B).
Temporal Clustering Reveals Coregulated Groups of Genes, but Does Not Reveal Specific Regulatory Mechanisms
The cumulative number of DEGs at each time point (Supplemental Data Set 6) confirmed that major gene expression changes occurred late during the drought experiment as the number of genes that showed first differential expression at each time point was highest between days 8 and 11 (Figure 4A). A total of 336 genes responded during the first half of the experiment, while the majority of genes (1479) showed first differential expression during the latter half of the experiment. A Euclidean distance matrix of the average expression values of the four biological replicates for each data set was generated and used in hierarchical cluster analysis. The resulting dendrogram showed that samples clustered in relation to treatments and within the drought treatment into early and late stage responses (Figure 6A). Dividing the data set into early (days 1 to 7, ∼95 to 45% rSWC) and late responses (days 8 to 13, ∼40 to 17% rSWC) also revealed functional groups of genes responding at different times throughout the progressive drought. Early upregulated genes were associated with carbohydrate and glycoside biosynthetic processes, general carbohydrate metabolic processes, and inorganic cation transporter activities (Table 1). The late upregulated genes encompassed flavonoid and secondary metabolite biosynthesis, while the late downregulated genes were involved in translation, pigment biosynthesis, photosynthesis-related processes, and oxidation/reduction processes (Table 1). To gain insight into the molecular factors underlying this temporal separation of samples, hierarchical cluster analysis of the DEGs was performed using SplineCluster (Heard et al., 2005) on the basis of gene expression patterns in the drought stressed leaf only. Using a prior precision value of 0.01, the 1815 genes were divided into 28 clusters (Figures 6B and 7; Supplemental Data Set 11). The first 14 clusters showed an overall upregulation and contained 1149 genes, while the last 14 downregulated clusters contained 667 genes (Figure 7; Supplemental Data Set 11). The 28 clusters were hypothesized to represent groups of genes that are coregulated during the drought experiment. To explore potential regulatory mechanisms of genes clustered in specific temporal expression profiles, we analyzed each individual cluster for over- and underrepresentation of GO terms in the Biological Process and Molecular Function and for overrepresentation of known TF binding motifs in promoters (Supplemental Data Sets 12 and 13 and Supplemental Figure 6). A few clusters showed enrichment of expected GO terms in response to drought, such as flavonoid biosynthesis, photosynthesis, pigment biosynthesis, and response to stress (Supplemental Data Set 12; Figure 7). However, most clusters did not show any enrichment or underrepresentation of GO terms (Supplemental Data Set 12; Figure 7), and only two clusters (cluster 1 and 9) contained the ABRE binding motif, known to perceive ABA-mediated drought and osmotic stress signals (Supplemental Figure 6 and Supplemental Data Set 13; Kim et al., 2011).
Figure 6.

Temporal Clustering of 1815 Differentially Expressed Genes.
(A) Dendrogram of the hierarchical clustering using a Euclidian distance divides the data set into early and late responses for both the control (white circles) and drought (gray circles) samples.
(B) Heat map of the SplineCluster analysis of the 1815 DEGs on differentially expressed genes (drought samples only) across the time series using normalized and averaged data (Supplemental Data Set 11). The heat map demonstrates expression profiles for genes in each cluster with red representing high expression and green representing low expression.
Table 1. Functional Categorization of Early (95 to 45% rSWC) and Late (40 to 17% rSWC) Responsive Genes.
| Category | GO Term | Biological Process/Molecular Function | Fold | P Value |
|---|---|---|---|---|
| Early upregulated | GO:0034637 | Cellular carbohydrate biosynthetic process | 6.9 | 0.003 |
| Early upregulated | GO:0006812 | Cation transport | 4.3 | 0.014 |
| Early upregulated | GO:0044262 | Cellular carbohydrate metabolic process | 3.7 | 0.018 |
| Early upregulated |
GO:0022890 |
Inorganic cation transmembrane transporter activity |
5.1 |
0.0432 |
| Late upregulated | GO:0009812 | Flavonoid metabolic process | 7.1 | 1.74E-05 |
| Late upregulated | GO:0009813 | Flavonoid biosynthetic process | 7.1 | 5.82E-05 |
| Late upregulated | GO:0019748 | Secondary metabolic process | 2.3 | 0.004 |
| Late upregulated | GO:0009699 | Phenylpropanoid biosynthetic process | 3.8 | 0.006 |
| Late upregulated | GO:0016051 | Carbohydrate biosynthetic process | 2.7 | 0.011 |
| Late upregulated | GO:0006519 | Cellular amino acid and derivative metabolic process | 2.0 | 0.011 |
| Late upregulated | GO:0019438 | Aromatic compound biosynthetic process | 2.9 | 0.021 |
| Late upregulated |
GO:0042398 |
Cellular amino acid derivative biosynthetic process |
2.9 |
0.044 |
| Late downregulated | GO:0006412 | Translation | 2.1 | 4.96E-05 |
| Late downregulated | GO:0015995 | Chlorophyll biosynthetic process | 11.4 | 6.50E-04 |
| Late downregulated | GO:0044085 | Cellular component biogenesis | 2.4 | 0.002 |
| Late downregulated | GO:0042254 | Ribosome biogenesis | 3.6 | 0.003 |
| Late downregulated | GO:0006334 | Nucleosome assembly | 6.9 | 0.003 |
| Late downregulated | GO:0015979 | Photosynthesis | 3.9 | 0.011 |
| Late downregulated | GO:0033014 | Tetrapyrrole biosynthetic process | 7.7 | 0.014 |
| Late downregulated | GO:0055114 | Oxidation reduction | 1.8 | 0.02 |
P value was adjusted using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). “Category” indicates the timing and direction of change in gene expression across the time series. “Fold” indicates the fold enrichment.
Figure 7.

Descriptions of the 28 Clusters Derived from SplineCluster.
The blue line indicates the mean expression profile for each of the 28 clusters. Individual genes present in each cluster are available in Supplemental Data Set 11. The red line indicates the switch from early (95 to 45% rSW; days 1 to 7) to late (40 to 17% rSWC; days 8 to 13). Selected enriched GO terms (Supplemental Data Set 12) are indicated on each cluster.
Bayesian State Space Modeling Identifies Genes That Link Drought Responses to Plant Development through Regulating Transcriptional Networks
The data presented so far did not allow us to draw conclusions about a specific regulatory mechanism across the whole time series but suggested that early and late responses to a decline in soil water content are regulated differently. Large-scale transcriptional reprogramming and metabolic adjustment did not play a dominant role during the early phases of the dehydration response. Nevertheless, among the 337 DEGs responding between 95 and 40% rSWC were 33 TF genes (Supplemental Data Set 14), of which 25% had a functional annotation of development (GO:0032502; Supplemental Data Set 14). This suggested that a reprogramming of developmental processes during drought stress may have occurred in response to the observed early physiological changes (closure of stomata and reduction in leaf water potential). We reasoned that the expression of early TF genes must therefore play a role in orchestrating this acclimation to drought stress.
Metropolis Variational Bayesian State Space Modeling (M-VBSSM; Penfold, University of Warwick; Supplemental Methods) was initially performed on 176 differentially expressed TFs in the data set (Supplemental Data Set 14). This approach selects subsets of TFs to generate a network, which is continually updated by probabilistic replacement of TFs to generate a series of networks and provides a consensus model based on the marginal likelihood (Supplemental Methods). From the consensus model, we could calculate the occurrence of each TF (the number of times a particular TF appeared over all models) and a count of the number of downstream connections each TF had across all models at a particular z-score, in this case indicating a 95% confidence threshold (Supplemental Data Set 15). TFs that scored well in both rankings were deemed highly connected hubs with many significant edges. The M-VBSSM consensus model indicated that developmental genes played an important role in the regulation of drought, as four out of the top 10 highly connected TFs were associated with the regulation of plant development (Supplemental Data Set 15). In addition, two of the top 10 TFs were among the group of early responding TFs (Supplemental Data Sets 14 and 15). While the consensus model was useful for the initial ranking of genes, it did not represent a causal model, instead representing a type of averaging of many different network models. For this reason, we opted to model a smaller selection of genes, which was advantageous for two reasons. First, the final model was smaller and sparser, and therefore more interpretable, and second, the resultant interactions could be interpreted causally.
Therefore, the top 10 “hub” TFs with the highest frequency of occurrence and 90 random transcription factors were chosen for analysis with the VBSSM package (Beal et al., 2005; Supplemental Data Set 16). As part of this selection, we also chose early and late responding TFs. In total, 19 early responding TFs (days 1 to 7) were included in the model to establish a potential transcriptional link between early and late responses (Supplemental Data Set 16).
The resultant model placed the early-responding TF gene AGL22 at the center of a 25-TF gene network (Figure 8A). AGL22 was differentially expressed in the drought experiment beginning at day 5 (Supplemental Figure 7A). Fifteen TF genes that were part of the GRN were initially analyzed by qPCR to check for differential expression under drought in wild-type plants (20% rSWC). All genes were differentially expressed in line with the levels observed in the microarray experiment, except for PACLOBUTRAZOL RESISTANCE1, BASIC HELIX-LOOP-HELIX038, and AUXIN RESPONSE FACTOR1 (Figure 8B; Supplemental Figure 7B).
Figure 8.

Constructing and Evaluating a TF Regulatory Network.
(A) Gene regulatory network generated using VBSSM with the drought time-series data (threshold z-score = 1.65). The nodes highlighted in red were upregulated during drought stress including the central hub gene, AGL22. Nodes highlighted in green were genes downregulated during drought stress. Blue nodes signify genes that were not regulated by AGL22 as predicted from the model (see [D]). All red, green, and blue nodes were selected for evaluation after drought stress;
(B) Relative gene expression of selected genes under drought conditions (17% rSWC). Gene expression was analyzed by qPCR. The numbers are expressed as fold changes of drought over control (n = 5 ± se). Significance of the fold changes are indicated by either *P < 0.05 or **P < 0.01. For gene and primer list, see Supplemental Data Set 16 and Supplemental Table 1.
(C) Relative expression levels of AGL22 in two knockout lines, agl22-3 and agl22-4, compared with the wild type determined by qPCR. Significance of the fold changes are indicated **P < 0.01.
(D) Relative gene expression profiles of 16 genes predicted to be regulated by AGL22 under drought stress in agl22-3 (black bars) and agl22-4 (gray bars) compared with the wild type. The data represent the mean (n = 7; ±se), and significance of the fold changes are indicated by either *P < 0.05 or **P < 0.01. Asterisks located centrally indicate both mutants are significantly different to the wild type, while asterisks located over one mutant indicate significance for the specific mutant.
AGL22 is known to affect flowering time and plant development (Supplemental Figure 7C; Gregis et al., 2013; Méndez-Vigo et al., 2013); however, it was not regulated during leaf senescence (Supplemental Data Set 9), suggesting that AGL22 uniquely regulated a transcriptional network during drought stress. This was further explored by performing VBSSM using the control time-series data set of the same transcription factor genes. If AGL22 was a hub gene in the control time series, it would suggest a role in developmental reprogramming over the 13-d experimental period regardless of drought stress. The Bayesian modeling resulted in a number of fragmented connections of a small number of genes (Supplemental Figure 8A), suggesting these genes were not part of a gene regulatory network under well-watered conditions. The highly connected genes in the drought model, AGL22 and RAP2.12, did not feature, not even as peripheral genes (Supplemental Figure 8A).
Therefore, the early responding TF AGL22 was chosen for further analysis to establish how far an unbiased modeling approach can be used to identify genes capable of influencing plant drought phenotypes and downstream network connections. Two independent T-DNA insertion lines were isolated (see Methods), both of which were confirmed knockout mutants for AGL22 (Figure 8C; Supplemental Figure 8B). The mutants were subsequently analyzed for their effect on the AGL22-centered network interactions after drought stress. Eight out of 15 TF genes differentially expressed under drought conditions exhibited altered gene expression in at least one of the agl22 mutants compared with the wild type (Figure 8D). This implied that ∼50% of the network connections were regulated at least partially through AGL22, but also suggested the possibility of redundancy within the network (Figures 8A and 8D). Four late TFs (WRKY20, GIS, DREB1A, and FBH3) were substantially downregulated in both agl22 mutants after drought, suggesting that these TFs were primarily regulated through AGL22 (Figure 8D).
Both agl22 Mutants Had Early-Flowering, Fast-Drying Drought Escape Phenotypes
It is important to note that due to the early-flowering phenotype of the agl22 mutants (Supplemental Figure 7C), drought stress was begun at day 22 after sowing, when there was no visible differences in rosette leaf number (Supplemental Figures 9A to 9C), but with a significant increase in rosette area (Figure 9A).
Figure 9.

Stress and Plant Growth Phenotypes of agl22 Mutants.
Due to the early-flowering phenotype of both agl22 mutant alleles, drought stress was begun at 22 d after sowing.
(A) Rosette area (cm2) of Col-0 (light gray), agl22-3 (black), and agl22-4 (dark gray) plants at different soil water contents (n = 5). The asterisk indicates significant difference compared with the wild type at P < 0.05.
(B) Rate of water loss in agl22-3 and agl22-4 plants compared with the wild type averaged over 13 d of water withdrawal (n = 10). The asterisk indicates significant difference compared with the wild type at P < 0.05.
(C) Stomatal conductance (Gs) at different soil water contents in Co-0 (light gray), agl22-3 (black), and agl22-4 (dark gray) (n = 5). The asterisk indicates significant difference compared with the wild type at P < 0.05.
(D) Light-saturated carbon assimilation (Asat) at different soil water contents in Col-0 (light gray), agl22-3 (black), and agl22-4 (dark gray; n = 5). The asterisk indicates significant difference compared with the wild type at P < 0.05.
(E) Days to flowering in well-watered (light gray) and drought-stressed (black) plants (n = 10). Plants were grown under short-day conditions as described in Methods. At 5 weeks, plants were subjected to progressive drought stress. When 17% rSWC was reached, plants were rewatered and flowering time was recorded as days after sowing. Control plants were maintained well watered. The asterisk indicates significant difference compared with the wild type at P < 0.05.
We observed an increased drying rate in both agl22 mutants, suggesting increased water use (Figure 9B). To determine if this was due to developmental or metabolic changes, we performed light response curve measurements of photosynthesis at specific times throughout the drying period (Supplemental Figure 9D). Due to the small leaf and rosette size, light curves were measured in whole plant chambers at 90, 74, and 25% rSWC (see Methods). The increased water loss was primarily driven by a greater rosette area (Figure 9A), despite a significant reduction in stomatal conductance (Figure 9C). Accordingly, light saturated carbon assimilation (Asat) was significantly reduced throughout the drying period already under well-watered conditions (Figure 9D), leading to a significant reduction in total aboveground biomass (Supplemental Figure 10A). Flowering time remained constant between well-watered and drought treatments in both agl22 mutants, indicating that drought stress conditions did not affect flowering time in the agl22 mutants (Figure 9E).
DISCUSSION
Chronology of the Drought Response Suggests Early Adjustments in Stomatal Conductance and Carbon Assimilation Are Followed by Changes in ABA and Transcriptional Reprogramming
Responses to drought are complex and depend on the type and strength of the drought stress imposed (Harb et al., 2010; Wilkins et al., 2010; Zhang et al., 2014). The slow steady drought experiment performed in this study allowed us to investigate the full range of temporal physiological, transcriptional, and metabolic responses in a single fully expanded Arabidopsis leaf. Additionally, by measuring leaf water potential (Figure 1B) and RWC (Figure 1A), we were able to monitor the progression and degree of drought stress in relation to the physiological, transcriptome, and metabolome changes. The decline in carbon assimilation at ∼45% rSWC was primarily driven by reduced stomatal conductance limiting CO2 diffusion. There were no underlying metabolic constraints, as photosynthetic capacity was unaffected by the drought treatment (Figure 2; Supplemental Figure 1D), and metabolite profiles remained unchanged throughout the majority of the drying period (Figure 3A; Supplemental Figures 2 and 3), suggesting that Arabidopsis Col-0 is a drought-tolerant ecotype.
Stomatal limitation as the primary factor in reducing photosynthesis under mild drought conditions has been observed in other studies; however, severe dehydration stress is believed to lead to metabolic constraints, associated with RuBP availability (Flexas and Medrano, 2002). We did not observe a reduction in the maximum capacity for carbon assimilation (Amax), Rubisco carboxylation (VCmax), and RuBP regeneration (Jmax; Figure 2A). In addition, maximum and operating efficiencies of photosystem II photochemistry (Figure 2B), photochemical and nonphotochemical quenching (Supplemental Figure 1D), were maintained throughout the drying period despite a decline in carbon assimilation (Figures 1D), indicating very little stress on photosystem II. This suggested that an alternative electron sink, most likely photorespiration (reviewed in Chaves et al., 2003; Lawson et al., 2014), must have been operating under drought stress, and increased gene expression in the photorespiratory pathway supports this notion (Figure 5C).
In general, two distinct phases in response to progressive soil drying could be discerned (Figures 6 and 7). Early responses were predominantly adjustments to stomatal conductance leading to restricted CO2 diffusion for photosynthetic carbon assimilation (Boyer, 1970; Passioura, 1996; Figure 1C) with some associated transcriptional changes accounting for 17% of the 1815 DEGs (Supplemental Data Sets 1 to 12; Table 1). By contrast, late responses (from 40% rSWC) encompassed hormonal (ABA), transcriptional, and major metabolic changes associated with senescence (Supplemental Data Sets 1 to 12; Table 1). These later responses corresponded with the many different phenological and physiological changes observed in other studies, including impaired photosynthesis, increased solute accumulation, and growth arrest (Boyer, 1970; Passioura, 1996). At the cellular level, soluble sugars, oligosaccharides, antioxidants, and proline accumulation are known to enhance the tolerance to drought stress by acting as osmolytes or as reactive oxygen species scavengers, especially hydroxyl radicals (Smirnoff and Cumbes, 1989; Cuin and Shabala, 2007). The accumulation of these compounds during the latter stages of the drought period (Figures 3B and 3C), together with the increase in secondary metabolites, such as flavonoids (Supplemental Data Set 1 and Supplemental Figures 3 and 4), suggests a role in the defense against severe drought stress (Tattini et al., 2004; Lei et al., 2006; Xiao et al., 2007; Xu et al., 2008; Harb et al., 2010; Fini et al., 2011; Page et al., 2012). In conclusion, this time series covers all phases during a progressive drought stress and therefore provides the opportunity to study different stages of stress responses in greater detail than has been previously possible (Supplemental Figure 10B).
Transcriptional Regulation of Drought Stress Responses
At the gene expression level, a slowly developing soil water deficit is different from rapid tissue dehydration. From this study and in comparison with two other drought experiments (Harb et al., 2010; Wilkins et al., 2010), it is clear that different genes respond depending on the nature of the drought stress applied (Figure 5A). Only few genes overlapped, with the majority of genes, responding in the final 4 d of the experiment (Supplemental Data Set 8).
Previous studies have often focused on the identification of genes coding for TF classes responding to terminal or severe drought stress, including BASIC LEUCINE ZIPPER (bZIPs, e.g., ABA-responsive element binding protein/ABRE binding factor), AP2/EREBP (e.g., DREB/CBF), NAC transcription factors (NAM, ATAF1-2, CUP-SHAPED COTYLEDON2), CCAAT binding (e.g., NUCLEAR FACTOR Y), and ZINC-FINGER (e.g., C2H2 zinc finger protein) families (Umezawa et al., 2004; Bartels and Sukar, 2005; Karaba et al., 2007; Li et al., 2008; Licausi et al., 2010; Jensen et al., 2013). The majority of TF genes in our study also responded relatively late in the drought period (from 40% rSWC), especially those associated with ABA, dehydration, and oxidative stress responses (Supplemental Data Set 14). Similar classes of TF genes have also been shown to respond to drought stress in Medicago truncatula where 8% of the responding genes coding for TFs responded late throughout the drying period (Zhang et al., 2014). By contrast, among the early-responding TF genes (95 to 45% rSWC; Supplemental Data Set 14), ∼25% were linked to plant development, indicating that early physiological changes may influence lifetime traits before the initiation of acute stress defense and senescence responses, highlighting the balancing act between the need to grow and to induce effective stress tolerance mechanisms (Claeys and Inzé, 2013).
Dynamic Bayesian Network Modeling Identifies Genes That Regulate Plant Development
Analysis of promoter binding sites (Supplemental Data Set 13 and Supplemental Figure 6) did not indicate specific regulatory networks or mechanisms during the early events. We therefore assessed the use of a high-throughput gene expression approach coupled with dynamic Bayesian network modeling to identify genes associated with the regulation of early drought responses. To make sense of large high-throughput data sets, network inference algorithms were developed, which are capable of establishing regulatory interactions among genes (Bansal et al., 2007). A number of different inference algorithms were used to successfully reconstruct known gene regulatory networks to validate these approaches (Cantone et al., 2009; Penfold and Wild, 2011). VBSSM is such an algorithm developed specifically for highly resolved temporal gene expression data sets, with the aim of identifying genes that are the key regulators in a given system (Beal et al., 2005). A recent comparison of modeling algorithms used to infer GRNs has shown that VBSSM is competitive with network reconstructions based on experimental data (Penfold and Wild, 2011; Windram et al., 2014; Penfold and Buchanan-Wollaston, 2014). Due to the limited number of experimental observations compared with the much greater number of differentially expressed genes, the system is inherently underdetermined (Penfold and Buchanan-Wollaston, 2014), and previous experience suggested that for these kinds of data sets VBSSM can model around 100 genes (Breeze et al., 2011). However, this type of approach can introduce a bias during the process of gene selection, while the M-VBSSM approach (see Methods) generally avoids this gene selection bias (Supplemental Methods). We therefore opted to use both M-VBSSM and VBSSM to identify key drought-regulatory genes and drought phenotypes associated with those genes.
The initial emergence of several development-associated transcription factors from the M-VBSSM approach (Supplemental Data Set 15) and the subsequent selection of developmental and nondevelopmental TFs for VBSSM (Supplemental Data Set 16) confirmed the flowering time regulator AGL22 as a hub gene in the drought response. Importantly, we did not observe any involvement of AGL22 in leaf senescence (Supplemental Data Set 9 and Supplemental Figure 8A), suggesting that the gene plays a unique role in drought stress responses.
The connection between flowering time and drought in Arabidopsis has been established independently in a number of studies, in which carbon isotope discrimination (Farquhar et al., 1982, 1989) quantitative trait loci colocated with known developmental/flowering time loci (Hausmann et al., 2005; Masle et al., 2005; Juenger et al., 2005; McKay et al., 2008). The identification of a flowering time gene and subsequent verification of its influence on plant water use, photosynthesis, and phenology (discussed below) suggest that this type of dynamic modeling can provide an important means of discovering genes that will produce phenotypes associated with lifetime water use and plant development. However, unlike quantitative trait locus mapping, VBSSM also managed to establish some valid network interactions from time-series transcriptomics data, potentially allowing for a temporal reconstruction of events and biological processes occurring during progressive drought stress.
A Flowering Time Gene Influences Water Use and Photosynthesis under Well-Watered and Drought Stress Conditions
Both agl22 mutants exhibited elevated water loss and rapid development already under nonstress conditions (Figures 9A and 9B). This could imply a trade-off between drought avoidance and escape in environments where drought shortens the growing season (Franks, 2011). Selecting for early flowering may be beneficial for plant survival but not necessarily for achieving high biomass (Supplemental Figure 10A), which suggests that drought survival and the ability to maintain biomass under sustained water-limiting conditions depend on different mechanisms (Skirycz et al., 2011).
The agl22 mutants exhibited 36 and 46% reductions in the steady state light-saturated photosynthetic rate under well-watered conditions (Figure 9D), which appeared to be partly associated with reduced stomatal conductance (Figure 9C). However, during drought stress, the photosynthetic rate in both agl22 mutants was reduced by only 11 and 13%, suggesting that both agl22 mutants were able to maintain substantial photosynthetic rates (Figure 9D). This is supported by the fact that agl22 mutants also maintained rosette growth throughout the drying period in comparison to wild-type plants (Figure 9A), and although total aboveground biomass was significantly reduced in both mutant alleles (Supplemental Figure 10A), biomass distribution shifted from vegetative growth to reproductive growth (Supplemental Figure 10C). The complex links between plant growth, primary metabolism, and flowering time in Arabidopsis are highlighted in a recent article where increased plant growth was positively associated with early-flowering phenotypes (El-Lithy et al., 2010), which may explain the larger rosette area observed prior to flowering in 30-d-old agl22 mutant plants compared with the wild type (Figure 9A; Supplemental Figure 9B). In addition, starch/carbohydrate status and metabolite levels have been linked to rosette growth (Meyer et al., 2007; Sulpice et al., 2009), as well as development and flowering time (Zhou et al., 1998; Moore et al., 2003; Funck et al., 2012). Both photosynthesis and flowering are regulated by the light environment and are clearly linked via the carbohydrate status (Zhou et al., 1998; Moore et al., 2003; Funck et al., 2012), connecting primary metabolism with plant growth and development. However, little is known about the link between photosynthetic performance and flowering time especially in flowering time mutants, and at this point, it is unclear why the agl22 mutants exhibited a substantial reduction in photosynthesis already under well-watered conditions.
Furthermore, the observations regarding AGL22 reinforces that water use in relation to overall plant productivity requires a balance of developmental and physiological processes to successfully complete a lifecycle in the prevailing climatic conditions. AGL22 was identified from moderately drought-stressed plants, which also suggests that not all drought-responsive genes may work in all water deficit scenarios. This may especially be the case for those genes that have been selected under terminal or severe drought conditions (Hu et al., 2006; Nelson et al., 2007; Xiao et al., 2009). It is important to note that none of the TF genes selected as key regulators of the drought response in earlier studies (Hu et al., 2006; Nelson et al., 2007; Xiao et al., 2009) was a hub in the TF GRN (Figure 8A), although many of these TF genes were differentially expressed during drought stress (Supplemental Data Set 14). This is supported by the notion that many genes identified with a role in stress tolerance under severe stress conditions seem to have little effect on plant growth in mild drought conditions (Skirycz et al., 2011).
Interestingly, two targets of AGL22, DREB1A and FBH3 (Figure 8D), have previously been shown to be involved in the regulation of abiotic stress responses. Overexpression of DREB1A leads to drought, salt, and freezing tolerance (Kasuga et al., 1999, 2004), while FBH3 has been shown to regulate stomatal opening (Takahashi et al., 2013) and functions in ABA signaling in response to osmotic stress (Yoshida et al., 2015). Both genes were late-responding targets with few network connections (Figure 8A; Supplemental Data Set 15). The model therefore may allow us to predict the role of AGL22 during drought stress and provide a potential link between mild/moderate and severe drought responses.
This study demonstrates that network inference incorporating highly resolved time-series transcriptomics data is able to predict TF networks and identify genes with regulatory importance during drought stress. Moreover, by focusing on the transition from early physiological changes to drought stress responses, we were able to identify AGL22 as a gene associated with lifetime water use. Consequently, VBSSM as a gene discovery tool promotes the selection of unknown, yet highly connected genes for further phenotypic evaluation.
METHODS
Plant Material, Plant Growth, and Drought Stress
Arabidopsis thaliana plants (Col-0, agl22-3 [SALK_141674], and agl22-4 [SAIL_583_C08]) were obtained from the European Arabidopsis Stock Centre and were grown under a 8:16-h light:dark cycle at 23°C, 60% relative humidity, and light intensity of 150 µmol m−2 s−1, using a mixture of cold and warm white fluorescent tubes. Arabidopsis seed was stratified for 3 d in 0.1% agarose at 4°C before individual seeds were sown onto a soil mix (Scotts Levington’s F2+S compost:fine grade vermiculite in a ratio of 6:1). For the drought time-course experiment, pots (7 × 7 × 9 cm) were filled with the same amount of soil mix. Control pots, to determine 100 and 0% soil water content, were set up at the same time. Plants were transferred into individual pots 2 weeks after the sowing date and were kept well watered until the beginning of the drying episode at 5 weeks after sowing. Half the plants were maintained under well-watered conditions, while for the remaining half, water was withdrawn and pot weight was determined daily. Relative soil water content was calculated for each day and pots were left to dry until 17% rSWC was reached. Five-week-old plants were saturated in water to reach 95% rSWC, and watering was stopped in the treatment plants until ∼17% rSWC was reached. The control plants were maintained under well-watered conditions at ∼95% rSWC. Due to the early-flowering phenotype of both agl22 mutant alleles, drought experiments were performed on 22-d-old plants to ensure the experiments were performed at similar rosette developmental stages and prior to the onset of flowering, as indicated in Supplemental Figures 9B and 9C. The drying rate was determined as the slope of the decline in relative soil water content, measured daily throughout the drying period.
RNA Extractions, Labeling, Microarray Hybridization, and Analysis
Total RNA was extracted, labeled, and hybridized to CATMA v4 arrays (Sclep et al., 2007) as previously described (Breeze et al., 2011; Windram et al., 2012). The experimental design for the drought time-series hybridization is shown in Supplemental Figure 11.
Arrays were hybridized and washed as described (Windram et al., 2012). Arrays were scanned on a 428 Affymetrix scanner at wavelengths of 532 nm for Cy3 and 635 nm for Cy5. Cy3 and Cy5 scans for each slide were combined and processed in ImaGene version 8.0 (BioDiscovery) to extract raw intensity and background corrected data values for each spot on the array. The data have been deposited in the Gene Expression Omnibus under accession number GSE65046.
An adaptation of the MAANOVA package (Wu et al., 2003) was used to analyze the extracted microarray data as described by Breeze et al. (2011) and Windram et al. (2012), using a mixed-model analysis. The MAANOVA fitted model considered dye and array slide as random variables, and time point, treatment, and biological replicate as fixed variables. The model allowed assessment of the main effect of treatment, the main effect of time point, the interaction between these factors, and the nested effect of biological replicate. Predicted means were calculated for each gene in each of the 112 combinations of treatment, time point, and biological replicate and for each of the 28 combinations of treatment and time point, averaged across biological replicates.
Differential Gene Expression Analysis
Genes were ranked based on their Gaussian process two-sample (GP2S) Bayes factor differentially expressed score, a cutoff of ≥6 gave 2496 differentially expressed genes. Genes identified in the F-test as being differentially expressed that were not in the GP2S list of 2496 genes were added manually. The expression profiles of the genes ranked 1800 to 3150 were then plotted and assigned visually as differentially expressed or not. This resulted in a false positive rate of 23.3% for this group, and a final cutoff of 6 for the GP2S was chosen, duplicates were removed, and a list of 1934 differentially expressed genes was produced. Removal of probes and genes with no annotation in TAIR9 left a list of 1815 unique differentially expressed genes. The time at which genes first became differentially expressed (TOFDE) was subsequently determined using the GP2S time-local method (Stegle et al., 2010).
Promoter Analysis
Publically available position-specific scoring matrices (PSSMs) were collected from the PLACE and JASPAR databases (Higo et al., 1999; Sandelin et al., 2004). PSSMs were clustered by similarity, and a representative of each cluster was chosen for screening. Promoter regions corresponding to 200 bp upstream of the transcription start site were retrieved from the Ensembl Plants sequence database (release 50). For any given PSSM and promoter, we scanned the sequence and computed a matrix similarity score (Kel et al., 2003) at each position on both strands. P values for each score were computed from a score distribution obtained by applying the PSSM to randomly generated sequences. We took the top k nonoverlapping hits and performed the binomial test (pbinom function in R Stats package) for the occurrence of k sites with observed P values within a sequence of length 200 bp. The parameter k is optimized within the range 1 to 5 for minimum binomial P value to allow detection of binding sites without a fixed threshold per binding site. To determine the presence or absence of a PSSM in a promoter, in each case the promoters were sorted by binomial P value, and we applied a cutoff to select the top 2000. For each PSSM, its frequency in promoters of each cluster was compared with its occurrence in all promoters in the genome. Motif enrichment was calculated using the hypergeometric distribution (see statistical analysis). For motif enrichment analyses P values ≤ 1e-5 were considered significant, to allow for multiple testing.
RT-PCR and qPCR
Leaves were harvested and frozen in liquid nitrogen. Total RNA was extracted from a pool of three plants using Tri-reagent (Sigma-Aldrich) according to the manufacturer’s instructions. A minimum of five replicates of whole plants from separate experiments was performed for mutant analysis and drought treatments. For cDNA synthesis for real-time qPCR and RT-PCR, 1 µg total RNA was treated with RNase-free DNase (Ambion) according to the manufacturer’s instructions and reverse transcribed as previously described (Ball et al., 2004). qPCR-PCR was performed using a SYBR green fluorescence-based assay as described previously (Bechtold et al., 2010, 2013). Gene-specific cDNA amounts were calculated from threshold cycle (Ct) values and expressed relative to controls and normalized with respect to ACTIN and CYCLOPHILIN cDNA according to Gruber et al. (2001). RT-PCR was performed to amplify the full-length AGL22 gene on both agl22 mutant alleles. The primers used for qPCR and RT-PCR are given in Supplemental Table 1.
Physiological Measurements
Relative Water Content
Whole rosettes of five plants were harvested each day throughout the drying period. The RWC of the leaf was calculated using the formula: rLWC (%) = (FW − DW)/(SW − DW) × 100, where FW is the actual rosette weight at the day of harvest, SW is the fully saturated rosette weight, and DW is the dry weight of the rosette.
Leaf Water Potential
The leaf water potential was measured via the Scholander pressure bomb technique (Scholander et al., 1964) using a SKPM1400 plant moisture system (Skye Instruments). Leaf water potential was measured daily throughout the drying period on both control and drought-stressed plants according to the manufacturer’s instructions.
Plant Development and Biomass Measurements
Arabidopsis development was assessed using the scale developed by Boyes et al. (2001). Once the final flower had opened, watering was ceased and plants were bagged and left to dry out before harvesting. At harvesting, rosettes, stalks, and seeds were separated. The seed weight and dry weight of rosettes and stalks/pods were determined (Bechtold et al., 2010). At least 10 plants per line and watering regime were measured.
Photosynthesis Measurements
Photosynthetic Rate (Snapshot Measurements)
Instantaneous measurements of net CO2 uptake rate (A) and stomatal conductance to water (gs) were made on leaf 7, using an open gas exchange system (PP Systems). Leaves were placed in the cuvette at ambient CO2 concentration (Ca) of 400 µmol mol−1, leaf temperature was maintained at 22 ± 2°C, vapor pressure deficit was ∼1 kPa, and irradiance was set to growth conditions (150 µmol m−2 s−1). A reading was recorded every 3 min when the IRGA conditions had stabilized (∼1.5 min), but before the leaf had a response to the new environment (Parsons et al., 1997).
A/Ci Curves (Maximum Photosynthetic Rates)
Five weeks after emergence, (A) and (gs) were measured on leaf 7, using an infrared gas exchange system (PP Systems). The response of A to changes in the intercellular CO2 concentration (Ci) was measured under a saturating PFD, provided by a combination of red and white LEDs (PP Systems). In addition, the response of A to changes in PFD from saturating to subsaturating levels was measured using the same light source at the current atmospheric CO2 concentration (390 µmol mol−1). All gas analysis was made at a leaf temperature of 20 (±1)°C and a vapor pressure deficit of 1 (±0.2) kPa. Plants were sampled between 1 and 4 h after the beginning of the photoperiod. For each leaf, steady state rates of A and gs at current atmospheric [CO2] were recorded at the beginning of each measurement. The A/Ci parameters, VCmax (maximum RuBP-saturated rate of carboxylation in vivo), Amax (light and CO2 saturated rate of carbon assimilation in vivo), and Jmax (maximum in vivo rate of electron transport contributing to RuBP regeneration) were calculated by fitting equations described by Farquhar et al. (1980) with subsequent modifications described by McMurtrie and Wang (1993).
Chlorophyll Fluorescence Imaging
Plants were analyzed at various stages of the progressive drought stress using the dark (Fv/Fm)- and light (Fv′/Fm′, Fq′/Fm′, and Fq′/Fv′)-adapted chlorophyll a fluorescence parameters, using a chlorophyll fluorescence imaging instrument (Fluorimager; Barbagallo et al., 2003).
Light Response Curves Using Whole-Plant Chambers
A/Q response curves were measured using whole-plant gas exchange system developed at the University of Essex, with a heliospectra LED light source (Heliospectra). Input air was maintained at a relative humidity of 50 to 60%, air temperature of 22°C, and CO2 concentration of 400 mmol mol−1, matching that of the growth conditions. Plants were initially stabilized for 30 min at saturating irradiance 800 µmol m−2 s−1, after which PPFD was reduced in nine steps (Supplemental Figure 9D), with assimilation (A) and stomatal conductance (gs) being recorded at each new PPFD level.
Metabolite and Hormone Analysis
During the experimental period, two leaves per plant were harvested every day, for a total of six plants per treatment. Samples were frozen in liquid nitrogen, freeze-dried overnight, and stored at room temperature in darkness until extraction. Primary metabolites were extracted from frozen tissue with chloroform-methanol as described by Lunn et al. (2006). T6P, other phosphorylated intermediates, and organic acids were measured by high-performance anion-exchange chromatography coupled to tandem mass spectrometry as described by Lunn et al. (2006). Trehalose was measured enzymatically with fluorometric detection as described by Carillo et al. (2013).
For sugars, amino acids, hormones, and secondary metabolites, freeze-dried leaf powder (10 mg) was extracted in 0.8 mL methanol containing 1% acetic acid. After centrifugation (10 min at 16,100g, 4°C), the samples were filtered through a 0.2-µm PVDF syringe filter (Chromacol). For nontargeted Liquid chromatography quadrupole time-of-flight mass spectrometry metabolite profiling, 5 μL extract was injected onto a Zorbax StableBond C18 1.8 mm, 2.1 × 100 mm (QToF) reversed-phase analytical column (Agilent Technologies). Chromatography and mass spectrometry conditions were described by Page et al. (2012). Peaks were extracted and aligned using XCMS (Smith et al., 2006), and statistical analysis and data visualization were performed with MetaboAnalyst 2.0 (Xia et al., 2015). For liquid chromatography-tandem mass spectrometry analysis of hormones, 10 mg freeze-dried leaf powder was extracted in 0.8 mL 10% methanol + 1% acetic acid containing deuterated standards (Forcat et al., 2008). Secondary metabolites and hormones were analyzed with an Agilent 6420B triple quadrupole (QQQ) mass spectrometer (Agilent Technologies) coupled to a 1200 series Rapid Resolution HPLC system. Two microliters of sample extract was loaded onto a Zorbax Eclipse Plus C18 (3.5 mm, 2.1 × 150 mm) for amino acids, sugars, and hormones, respectively. The following gradient was used: 0 min to 0% B; 1 min to 0% B; 5 min to 20% B; 20 min to 100% B; 25 min to 100% B; 27 min to 0% B; 7 min post-time. QQQ source conditions were as follows: gas temperature 350°C, drying gas flow rate 9 liters min−1, nebulizer pressure 35 psig, and capillary voltage 4 kV. The polarity, fragmentor voltage, and collision energies were optimized for each compound. The multiple reaction monitoring modes used for compound identification are shown in Supplemental Data Set 17, and data are reported as peak areas. Flavonoid identification was based on previous tandem mass spectrometry identification of flavonoids in Arabidopsis (Tohge et al., 2005; Stobiecki et al., 2006). For sugar and polyol analysis, 5 μL of sample extract was loaded onto an XBridge amide HILIC column (particle size 3.5 µm, 2.1 mm i.d. × 150 mm; Waters) with a constant flow rate of 0.3 mL min−1 and a column temperature of 35°C for the duration. Mobile phases comprised water:acetonitrile with 0.1% ammonia (mobile phase A was 90% acetonitrile, and B was 10% acetonitrile with 5 mM ammonium formate). Sugars (5 µL) were separated using the following gradient: 0 to 17 min, 0 to 54% B; 17 to 19 min, 54% B; 19 to 20 min, 54 to 0% B, with a 10 min reequilibration time. The QQQ was operated in negative ion mode. Electrospray ionization source conditions were as follows: gas temperature, 350°C; drying gas flow rate, 9 liters min−1; nebulizer pressure, 35 psig; and capillary voltage, 4 kV. Data were acquired in selected ion monitoring mode with a dwell time of 50 ms. The fragmentor voltage was 50 V for all sugars. The sugars were quantified by reference to standards (Supplemental Data Set 17). Amino acids were separated with a ZIC-HILIC column (150 × 2.1 cm, 3.5-µm particle size; Merck SeQuant). Sample (2 µL) was injected into the column with a flow rate of 0.25 mL min−1. Mobile phases comprised of water:acetonitrile with 0.1% formic acid (mobile phase A was 95% acetonitrile and B was 5% acetonitrile with 5 mM ammonium acetate). Compounds were separated using the following gradient: 0 to 10 min, 5 to 50% B, 10 to 15 min, 50-90% B, 15 to 20 min, 90% B, 20 to 25 min, 90 to 5% B, with an 11-min reequilibration time. The QQQ was operated in positive ion mode and electrospray ionization source conditions were as follows: gas temperature, 350°C; drying gas flow rate, 9 liters min−1; nebulizer pressure, 35 psig; and capillary voltage, 4 kV. Data were acquired in multiple reaction monitoring mode with a dwell time of 50 ms. Amino acids were quantified by multiple reaction monitoring (Supplemental Data Set 17) and data are reported as peak areas (Supplemental Data Set 18).
Analysis of Publicly Available Microarray Data Sets
Publicly available microarray data sets from different experiments in which Arabidopsis was subjected to drought and senescence (Harb et al., 2010; Wilkins et al., 2010; Breeze et al., 2011) were located in supplementary files of already published articles and were compared with the drought time-series data sets using VENNY (Oliveros, 2007). Hypergeometric distributions were calculated for different overlaps using the phyper function in R version 3.0.2. A cutoff of -p(log) of 5 was chosen as highly a significant overlap between two or more data sets.
Analysis of GO
GO annotation analysis was performed using DAVID version 6.7 (Huang et al., 2009), BINGO (Maere et al., 2005), and Agrigo (Du et al., 2010) with the GO_Biological_Process category, as described by Ashburner et al. (2000). Overrepresented GO_Biological_Process and GO_Molecular_Function categories were identified using a hypergeometric test with a significance threshold of 0.05 after Benjamini-Hochberg correction (Benjamini and Hochberg, 1995) or Bonferroni correction (Holm, 1979) with the whole annotated genome as the reference set.
VBSSM and M-VBSSM
Significant numbers of genes can be differentially expressed in response to environmental stress, which, given the limited number of experimental measurements, means that network models are often unidentifiable (Penfold and Buchanan-Wollaston, 2014; Windram et al., 2014, and references therein). Furthermore, the interpretation of large, densely connected networks can often be difficult, and any hypothesis we extract from them can therefore be ambiguous. One solution is to select a more limited number of genes to model, either based upon prior knowledge, heuristic approaches, or random selection. The reduced number of genes means network inference approaches can be applied such as the VBSSM of Beal et al. (2005). Within the VBSSM the expression of the genes can be written in the form:
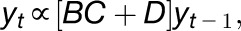 |
where the term 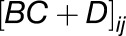 captures all information about how gene j regulates gene i. Rather than infer a point estimate for each interaction
captures all information about how gene j regulates gene i. Rather than infer a point estimate for each interaction 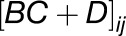 , Beal et al. (2005) infer a posterior distribution and use standard Z-statistics to assess the statistical significance. However, the preselection of genes described above may result in some bias. Here, we chose to additionally build a network model around a particular gene of interest, using random selection via a Metropolis algorithm. At each step in the Metropolis algorithm, a Bayesian state space model is fitted to the time-series gene expression profiles for the selected genes, and the marginal likelihood or “model evidence” used as the selection criteria. In this way, we can infer small network models around each gene that we are interested in (for full details, see Supplemental Methods and Supplemental Figure 12).
, Beal et al. (2005) infer a posterior distribution and use standard Z-statistics to assess the statistical significance. However, the preselection of genes described above may result in some bias. Here, we chose to additionally build a network model around a particular gene of interest, using random selection via a Metropolis algorithm. At each step in the Metropolis algorithm, a Bayesian state space model is fitted to the time-series gene expression profiles for the selected genes, and the marginal likelihood or “model evidence” used as the selection criteria. In this way, we can infer small network models around each gene that we are interested in (for full details, see Supplemental Methods and Supplemental Figure 12).
For the drought data, a total of 176 transcription factors were differentially expressed (Supplemental Data Set 14). We therefore used the Metropolis model selection to systematically build a network of 88 genes around each of the 176 genes in turn. Within the Metropolis selection, each of the 176 network models was run for 2000 iterations in the MCMC chain, by which point the marginal likelihood was seen to be plateau and the algorithm was terminated. The 176 networks at step 2000 were then combined to create a meta-network, which was used to compile summary statistics, such as the number of times a particular gene was found in each of those 176 network models or the number of downstream connections a particular gene had over those 176 models. Ranked lists of genes can be found in Supplemental Data Set 13. Of the top 10 genes with the highest number of downstream connections, four were annotated as being developmental in nature, suggesting a link between drought response and developmental programs. A selection of 99 random differentially expressed transcription factors including the top 10 highly connected genes were selected from the larger pool of 176 TFs that were DE during the drought stress (Supplemental Data Set 16). Both the control and drought time series for these genes were normalized to have zero-mean and unit variance and subsequently modeled using the VBSSM of Beal et al. (2005) using a z-score of 1.65 to select the control and drought-specific networks, and a final VBSSM model (Beal et al., 2005) was fitted to the gene expression for AGL22.
Statistical Analysis
Statistical analyses were performed using SPSS version 19.0. Parameter differences between the wild type and agl22 mutants were determined using one-way ANOVA with appropriate post-hoc analysis. Tukey’s HSD test was used if variances of means were homogenous, and Games Howell test, if variances were not homogenous. The se of the calculated ratios of fold differences for metabolite and gene expression data and errors of individual means were combined “in quadrature” as the final ratio was a combination of the error of the two different means of the control and drought stress samples. Correlations were estimated among drying rate and flowering time as the standard Pearson product-moment correlation between the genotype means. Hypergeometric distributions were analyzed using the phyper function in the R stats package. Generally, P values ≤ 1e-5 were considered significant to allow for multiple testing. The metabolite data were analyzed by between subjects two-way ANOVA for time series data and probability values with false discovery rate multiple testing correction are tabulated. For secondary compounds, amino acids, and sugars, compounds not detected in >50% of samples were discarded and remaining missing data imputed using KNN (Supplemental Data Set 3). Abundance data were normalized to total signal in each sample, log2 transformed, mean-centered, and divided by sd of each variable using Metabolonalyst 3.0 (Xia et al., 2015).
Accession Numbers
Sequence data from this article can be found in the Gene Expression Omnibus data library under accession number GSE65046.
Supplemental Data
Supplemental Figure 1. Plant growth and chlorophyll fluorescence during progressive drought stress.
Supplemental Figure 2. Targeted metabolite analyses of secondary metabolites (except flavonoids), sugars, and amino acids.
Supplemental Figure 3. Targeted metabolite analyses of flavonoids and anthocyanins.
Supplemental Figure 4. The effect of drought stress on plant development.
Supplemental Figure 5. Temporal expression patterns of five selected flavonol biosynthesis genes.
Supplemental Figure 6. Analysis of TF binding sites.
Supplemental Figure 7. Gene expression of selected drought-responsive genes and growth analysis of agl22 mutants.
Supplemental Figure 8. Validation of the knockout phenotype in agl22 insertion mutants and the specific role of AGL22 during drought stress.
Supplemental Figure 9. Growth and photosynthetic phenotype of Col-0 and agl22 mutants during drought stress.
Supplemental Figure 10. Biomass production in agl22 mutants compared with the wild type and timeline of events.
Supplemental Figure 11. Schematic overview of array hybridizations across the 13 time points and two different treatments.
Supplemental Figure 12. Validation of the M-VBSSM approach.
Supplemental Table 1. Primers for qPCR, mutant screen, and RT-PCR analyses.
Supplemental Methods. Model comparison via a Metropolis search.
Supplemental Data Set 1. LC-MS of sugars and LC-MS/MS analysis of secondary metabolites and amino acids.
Supplemental Data Set 2. LC-MS/MS analysis of sugar phosphates, other phosphorylated compounds, and organic acids.
Supplemental Data Set 3. ANOVA of metabolites.
Supplemental Data Set 4. List of differentially expressed genes.
Supplemental Data Set 5. Functional categorization of 1815 DEGs.
Supplemental Data Set 6. Time of first differential expression.
Supplemental Data Set 7. Differentially expressed genes from publicly available drought and senescence data sets.
Supplemental Data Set 8. Comparison with published drought data sets.
Supplemental Data Set 9. Comparison with a published senescence data set.
Supplemental Data Set 10. Gene Ontology analysis of genes overlapping in the drought and senescence time series.
Supplemental Data Set 11. Hierarchical clustering divides the 1815 genes into 28 clusters.
Supplemental Data Set 12. Gene Ontology analysis: over- and underrepresentation of biological process and molecular function of individual clusters.
Supplemental Data Set 13. Analysis of TF binding sites.
Supplemental Data Set 14. Differentially expressed transcription regulators divided into transcription factor families.
Supplemental Data Set 15. Output of the TF Metropolis VBSSM (M-VBSSM).
Supplemental Data Set 16. Selected TFs for VBSSM modeling.
Supplemental Data Set 17. MRMs used for compound identification.
Supplemental Data Set 18. Output from XCMS alignment of peaks from LC-QToF metabolite profiling.
Supplementary Material
Acknowledgments
The authors acknowledge the support of the UK Biotechnology and Biological Science Research Council (BBSRC; Grant BB/F005806/1). S.S. was supported by a University of Essex PhD studentship. J.S.A.M. is supported by an NERC-CASE award (ENV-EATR-DTP: NE/L002582/1), and S.R.M.V-C. is supported by the BBSRC (BB/1001187_1). R.F. and J.E.L. are supported by the Max Planck Society. N.S. and H.F. are supported by Exeter Science Strategy funding, and C.S. is supported by the BBSRC and the Department for Environment, Food, and Rural Affairs through the NORNEX project. D.L.S was supported by a Wellcome Trust Institutional Strategic Support Fund. We thank Susan Corbett and Phillip A. Davey for help with the drought experiments and gas exchange measurements.
AUTHOR CONTRIBUTIONS
U.B. designed, carried out, and analyzed the drought experiments. T.L. designed the gas exchange and fluorescence measurements. J.S.A.M. and S.R.M.V.-C. performed whole-chamber gas exchange experiments on gene mutants. S.S. isolated hub gene mutants. R.F. and J.E.L. measured primary metabolites. H.F., C.S., D.L.S., and N.S. measured and analyzed targeted and untargeted metabolites. C.A.P., D.J.J., L.B., L.B., R.H., S.O., R.L., C.H., and J.D.M. were responsible for data analysis. K.J.D., N.R.B., P.M.M., N.S., A.M., D.L.W., B.F., D.R., J.B., S.O., V.B.-W., and U.B. all had input into the design of the experiments and analysis. U.B. wrote the article with contributions from all authors.
Glossary
- TF
transcription factor
- DEG
differentially expressed gene
- GRN
gene regulatory network
- rSWC
relative gravimetric soil water content
- RWC
relative leaf water content
- RuBP
ribulose-1,5-bisphosphate
- ABA
abscisic acid
- MAANOVA
microarray analysis of variance
- GO
Gene Ontology
- M-VBSSM
Metropolis Variational Bayesian State Space Modeling
- PSSM
position-specific scoring matrix
Footnotes
Articles can be viewed online without a subscription.
References
- Abdeen A., Schnell J., Miki B. (2010). Transcriptome analysis reveals absence of unintended effects in drought-tolerant transgenic plants overexpressing the transcription factor ABF3. BMC Genomics 11: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirrezabal L., Bouchier-Combaud S., Radziejwoski A., Dauzat M., Cookson S.J., Granier C. (2006). Plasticity to soil water deficit in Arabidopsis thaliana: dissection of leaf development into underlying growth dynamic and cellular variables reveals invisible phenotypes. Plant Cell Environ. 29: 2216–2227. [DOI] [PubMed] [Google Scholar]
- Ashburner M., et al. ; The Gene Ontology Consortium (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaller K., et al. (2012). Systems-based analysis of Arabidopsis leaf growth reveals adaptation to water deficit. Mol. Syst. Biol. 8: 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N.R. (2008). Chlorophyll fluorescence: a probe of photosynthesis in vivo. Annu. Rev. Plant Biol. 59: 89–113. [DOI] [PubMed] [Google Scholar]
- Ball L., Accotto G.P., Bechtold U., Creissen G., Funck D., Jimenez A., Kular B., Leyland N., Mejia-Carranza J., Reynolds H., Karpinski S., Mullineaux P.M. (2004). Evidence for a direct link between glutathione biosynthesis and stress defense gene expression in Arabidopsis. Plant Cell 16: 2448–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal M., Belcastro V., Ambesi-Impiombato A., di Bernardo D. (2007). How to infer gene networks from expression profiles. Mol. Syst. Biol. 3: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo R.P., Oxborough K., Pallett K.E., Baker N.R. (2003). Rapid, noninvasive screening for perturbations of metabolism and plant growth using chlorophyll fluorescence imaging. Plant Physiol. 132: 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels D., Sukar R. (2005). Drought and salt tolerance in plants. Crit. Rev. Plant Sci. 24: 23–58. [Google Scholar]
- Beal M.J., Falciani F., Ghahramani Z., Rangel C., Wild D.L. (2005). A Bayesian approach to reconstructing genetic regulatory networks with hidden factors. Bioinformatics 21: 349–356. [DOI] [PubMed] [Google Scholar]
- Bechtold U., Lawson T., Mejia-Carranza J., Meyer R.C., Brown I.R., Altmann T., Ton J., Mullineaux P.M. (2010). Constitutive salicylic acid defences do not compromise seed yield, drought tolerance and water productivity in the Arabidopsis accession C24. Plant Cell Environ. 33: 1959–1973. [DOI] [PubMed] [Google Scholar]
- Bechtold U., et al. (2013). Arabidopsis HEAT SHOCK TRANSCRIPTION FACTORA1b overexpression enhances water productivity, resistance to drought, and infection. J. Exp. Bot. 64: 3467–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begcy K., Mariano E.D., Mattiello L., Nunes A.V., Mazzafera P., Maia I.G., Menossi M. (2011). An Arabidopsis mitochondrial uncoupling protein confers tolerance to drought and salt stress in transgenic tobacco plants. PLoS One 6: e23776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289–300. [Google Scholar]
- Blum A. (2005). Drought resistance, water-use efficiency, and yield potential - are they compatible, dissonant, or mutually exclusive? Aust. J. Agric. Res. 56: 1159–1168. [Google Scholar]
- Boyer J.S. (1970). Leaf enlargement and metabolic rates in corn, soybean, and sunflower at various leaf water potentials. Plant Physiol. 46: 233–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J.S. (1982). Plant productivity and environment. Science 218: 443–448. [DOI] [PubMed] [Google Scholar]
- Boyes D.C., Zayed A.M., Ascenzi R., McCaskill A.J., Hoffman N.E., Davis K.R., Görlach J. (2001). Growth stage-based phenotypic analysis of Arabidopsis: a model for high throughput functional genomics in plants. Plant Cell 13: 1499–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray E.A. (2004). Genes commonly regulated by water-deficit stress in Arabidopsis thaliana. J. Exp. Bot. 55: 2331–2341. [DOI] [PubMed] [Google Scholar]
- Breeze E., et al. (2011). High-resolution temporal profiling of transcripts during Arabidopsis leaf senescence reveals a distinct chronology of processes and regulation. Plant Cell 23: 873–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantone I., Marucci L., Iorio F., Ricci M.A., Belcastro V., Bansal M., Santini S., di Bernardo M., di Bernardo D., Cosma M.P. (2009). A yeast synthetic network for in vivo assessment of reverse-engineering and modeling approaches. Cell 137: 172–181. [DOI] [PubMed] [Google Scholar]
- Carillo P., Feil R., Gibon Y., Satoh-Nagasawa N., Jackson D., Bläsing O.E., Stitt M., Lunn J.E. (2013). A fluorometric assay for trehalose in the picomole range. Plant Methods 9: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton A.J., Donarski J.A., Harrison M., Jones S.A., Godward J., Oehlschlager S., Arques J.L., Ambrose M., Chinoy C., Mullineaux P.M., Domoney C. (2008). Responses of the pea (Pisum sativum L.) leaf metabolome to drought stress assessed by nuclear magnetic resonance spectroscopy. Metabolomics 4: 312–327. [Google Scholar]
- Chaves M.M., Flexas J., Pinheiro C. (2009). Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann. Bot. (Lond.) 103: 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves M.M., Maroco J.P., Pereira J.S. (2003). Understanding plant response to drought – from genes to the whole plant. Funct. Plant Biol. 30: 239–264. [DOI] [PubMed] [Google Scholar]
- Chen J.Q., Meng X.P., Zhang Y., Xia M., Wang X.-P. (2008). Over-expression of OsDREB genes lead to enhanced drought tolerance in rice. Biotechnol. Lett. 30: 2191–2198. [DOI] [PubMed] [Google Scholar]
- Christensen J.H., Carter T.R., Rummukainen M., Amanatidis G. (2007). Evaluating the performance and utility of regional climate models: the PRUDENCE project. Clim. Change 81: 1–6. [Google Scholar]
- Churchill G.A. (2004). Using ANOVA to analyze microarray data. Biotechniques 37: 173–175, 177. [DOI] [PubMed] [Google Scholar]
- Claeys H., Inzé D. (2013). The agony of choice: how plants balance growth and survival under water-limiting conditions. Plant Physiol. 162: 1768–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuin T.A., Shabala S. (2007). Compatible solutes reduce ROS-induced potassium efflux in Arabidopsis roots. Plant Cell Environ. 30: 875–885. [DOI] [PubMed] [Google Scholar]
- Deyholos M.K. (2010). Making the most of drought and salinity transcriptomics. Plant Cell Environ. 33: 648–654. [DOI] [PubMed] [Google Scholar]
- Du Z., Zhou X., Ling Y., Zhang Z., Su Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38: W64–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easterling D.R., Meehl G.A., Parmesan C., Changnon S.A., Karl T.R., Mearns L.O. (2000). Climate extremes: observations, modeling, and impacts. Science 289: 2068–2074. [DOI] [PubMed] [Google Scholar]
- Eckhart V.M., Geber M.A., McGuire C. (2004). Experimental studies of selection and adaptation in Clarkia xantiana (Onagraceae). I. Sources of phenotypic variation across a subspecies border. Evolution 58: 59–70. [DOI] [PubMed] [Google Scholar]
- El-Lithy M.E., Reymond M., Stich B., Koornneef M., Vreugdenhil D. (2010). Relation among plant growth, carbohydrates and flowering time in the Arabidopsis Landsberg erecta x Kondara recombinant inbred line population. Plant Cell Environ. 33: 1369–1382. [DOI] [PubMed] [Google Scholar]
- Famiglietti J.S., Rodell M. (2013). Environmental science. Water in the balance. Science 340: 1300–1301. [DOI] [PubMed] [Google Scholar]
- Farquhar G.D., Ehleringer J.R., Hubick K.T. (1989). Carbon isotope discrimination and photosynthesis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 40: 503–537. [Google Scholar]
- Farquhar G.D., Oleary M.H., Berry J.A. (1982). On the relationship between carbon isotope discrimination and the inter-cellular carbon-dioxide concentration in leaves. Aust. J. Plant Physiol. 9: 121–137. [Google Scholar]
- Farquhar G.D., von Caemmerer S., Berry J.A. (1980). A biochemical model of photosynthesis CO2 fixation in leaves of C-3 species. Planta 149: 78–90. [DOI] [PubMed] [Google Scholar]
- Fini A., Brunetti C., Di Ferdinando M., Ferrini F., Tattini M. (2011). Stress-induced flavonoid biosynthesis and the antioxidant machinery of plants. Plant Signal. Behav. 6: 709–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein R.R., Gampala S.S.L., Rock C.D. (2002). Abscisic acid signaling in seeds and seedlings. Plant Cell 14 (suppl.): S15–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexas J., Medrano H. (2002). Drought-inhibition of photosynthesis in C3 plants: stomatal and non-stomatal limitations revisited. Ann. Bot. (Lond.) 89: 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcat S., Bennett M.H., Mansfield J.W., Grant M.R. (2008). A rapid and robust method for simultaneously measuring changes in the phytohormones ABA, JA and SA in plants following biotic and abiotic stress. Plant Methods 4: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D.M., Ellis A.G., Dharjwa M., Freshwater M., Padron A., Weis A.E. (2006). A steep cline in flowering time for Brassica rapa in Southern California: population-level variation in the field and the greenhouse. Int. J. Plant Sci. 167: 83–92. [Google Scholar]
- Franks S.J. (2011). Plasticity and evolution in drought avoidance and escape in the annual plant Brassica rapa. New Phytol. 190: 249–257. [DOI] [PubMed] [Google Scholar]
- Fujita Y., et al. (2009). Three SnRK2 protein kinases are the main positive regulators of abscisic acid signaling in response to water stress in Arabidopsis. Plant Cell Physiol. 50: 2123–2132. [DOI] [PubMed] [Google Scholar]
- Funck D., Clauß K., Frommer W.B., Hellmann H.A. (2012). The Arabidopsis CstF64-like RSR1/ESP1 protein participates in glucose signaling and flowering time control. Front. Plant Sci. 3: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregis V., et al. (2013). Identification of pathways directly regulated by SHORT VEGETATIVE PHASE during vegetative and reproductive development in Arabidopsis. Genome Biol. 14: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber F., Falkner F.G., Dorner F., Hämmerle T. (2001). Quantitation of viral DNA by real-time PCR applying duplex amplification, internal standardization, and two-color fluorescence detection. Appl. Environ. Microbiol. 67: 2837–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb A., Krishnan A., Ambavaram M.M.R., Pereira A. (2010). Molecular and physiological analysis of drought stress in Arabidopsis reveals early responses leading to acclimation in plant growth. Plant Physiol. 154: 1254–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann N.J., Juenger T.E., Sen S., Stowe K.A., Dawson T.E., Simms E.L. (2005). Quantitative trait loci affecting delta13C and response to differential water availibility in Arabidopsis thaliana. Evolution 59: 81–96. [PubMed] [Google Scholar]
- Heard N.A., Holmes C.C., Stephens D.A., Hand D.J., Dimopoulos G. (2005). Bayesian coclustering of Anopheles gene expression time series: study of immune defense response to multiple experimental challenges. Proc. Natl. Acad. Sci. USA 102: 16939–16944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo K., Ugawa Y., Iwamoto M., Korenaga T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 27: 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S. (1979). A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6: 65–70. [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Hu H., Dai M., Yao J., Xiao B., Li X., Zhang Q., Xiong L. (2006). Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 103: 12987–12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M.K., Lindemose S., de Masi F., Reimer J.J., Nielsen M., Perera V., Workman C.T., Turck F., Grant M.R., Mundy J., Petersen M., Skriver K. (2013). ATAF1 transcription factor directly regulates abscisic acid biosynthetic gene NCED3 in Arabidopsis thaliana. FEBS Open Bio 3: 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juenger T.E., McKay J.K., Hausmann N., Keurentjes J.J.B., Sen S., Stowe K.A., Dawson T.E., Simms E.L., Richards J.H. (2005). Identification and characterization of QTL underlying whole-plant physiology in Arabidopsis thaliana: delta C-13, stomatal conductance and transpiration efficiency. Plant Cell Environ. 28: 697–708. [Google Scholar]
- Karaba A., Dixit S., Greco R., Aharoni A., Trijatmiko K.R., Marsch-Martinez N., Krishnan A., Nataraja K.N., Udayakumar M., Pereira A. (2007). Improvement of water use efficiency in rice by expression of HARDY, an Arabidopsis drought and salt tolerance gene. Proc. Natl. Acad. Sci. USA 104: 15270–15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga M., Liu Q., Miura S., Yamaguchi-Shinozaki K., Shinozaki K. (1999). Improving plant drought, salt, and freezing tolerance by gene transfer of a single stress-inducible transcription factor. Nat. Biotechnol. 17: 287–291. [DOI] [PubMed] [Google Scholar]
- Kasuga M., Miura S., Shinozaki K., Yamaguchi-Shinozaki K. (2004). A combination of the Arabidopsis DREB1A gene and stress-inducible rd29A promoter improved drought- and low-temperature stress tolerance in tobacco by gene transfer. Plant Cell Physiol. 45: 346–350. [DOI] [PubMed] [Google Scholar]
- Kawaguchi R., Girke T., Bray E.A., Bailey-Serres J. (2004). Differential mRNA translation contributes to gene regulation under non-stress and dehydration stress conditions in Arabidopsis thaliana. Plant J. 38: 823–839. [DOI] [PubMed] [Google Scholar]
- Kel A.E., Gössling E., Reuter I., Cheremushkin E., Kel-Margoulis O.V., Wingender E. (2003). MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 31: 3576–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian J., Whitehead D., Horak J., Wanke D., Weinl S., Batistic O., D’Angelo C., Bornberg-Bauer E., Kudla J., Harter K. (2007). The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 50: 347–363. [DOI] [PubMed] [Google Scholar]
- Kim J.S., Mizoi J., Yoshida T., Fujita Y., Nakajima J., Ohori T., Todaka D., Nakashima K., Hirayama T., Shinozaki K., Yamaguchi-Shinozaki K. (2011). An ABRE promoter sequence is involved in osmotic stress-responsive expression of the DREB2A gene, which encodes a transcription factor regulating drought-inducible genes in Arabidopsis. Plant Cell Physiol. 52: 2136–2146. [DOI] [PubMed] [Google Scholar]
- Kreps J.A., Wu Y., Chang H.-S., Zhu T., Wang X., Harper J.F. (2002). Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 130: 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor D.W. (2013). Genetic engineering to improve plant performance under drought: physiological evaluation of achievements, limitations, and possibilities. J. Exp. Bot. 64: 83–108. [DOI] [PubMed] [Google Scholar]
- Lawson T., Davey P.A., Yates S.A., Bechtold U., Baeshen M., Baeshen N., Mutwakil M.Z., Sabir J., Baker N.R., Mullineaux P.M. (2014). C3 photosynthesis in the desert plant Rhazya stricta is fully functional at high temperatures and light intensities. New Phytol. 201: 862–873. [DOI] [PubMed] [Google Scholar]
- Lei Y., Yin C., Li C. (2006). Differences in some morphological, physiological, and biochemical responses to drought stress in two contrasting populations of Populus przewalskii. Physiol. Plant. 127: 182–191. [Google Scholar]
- Li W.X., Oono Y., Zhu J., He X.J., Wu J.M., Iida K., Lu X.Y., Cui X., Jin H., Zhu J.K. (2008). The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20: 2238–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licausi F., van Dongen J.T., Giuntoli B., Novi G., Santaniello A., Geigenberger P., Perata P. (2010). HRE1 and HRE2, two hypoxia-inducible ethylene response factors, affect anaerobic responses in Arabidopsis thaliana. Plant J. 62: 302–315. [DOI] [PubMed] [Google Scholar]
- Lobell D.B., Bänziger M., Magorokosho C., Vivek B. (2011). Nonlinear heat effects on African maize as evidenced by historical yield trials. Nat. Clim. Chang. 1: 42–45. [Google Scholar]
- Lobell D.B., Field C.B. (2007). Global scale climate–crop yield relationships and the impacts of recent warming. Environ. Res. Lett. 2: 014002. [Google Scholar]
- Ludlow M.M. (1989). Strategies of response to water stress. In Structural and Functional Responses to Environmental Stress, Kreeb K.H., Richter H., Hinckley T.M., eds (Amsterdam: SPB Academic; ), pp. 269–281. [Google Scholar]
- Lunn J.E., Feil R., Hendriks J.H.M., Gibon Y., Morcuende R., Osuna D., Scheible W.-R., Carillo P., Hajirezaei M.-R., Stitt M. (2006). Sugar-induced increases in trehalose 6-phosphate are correlated with redox activation of ADPglucose pyrophosphorylase and higher rates of starch synthesis in Arabidopsis thaliana. Biochem. J. 397: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Sukiran N.L., Ma H., Su Z. (2014). Moderate drought causes dramatic floral transcriptomic reprogramming to ensure successful reproductive development in Arabidopsis. BMC Plant Biol. 14: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S., Heymans K., Kuiper M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21: 3448–3449. [DOI] [PubMed] [Google Scholar]
- Masle J., Gilmore S.R., Farquhar G.D. (2005). The ERECTA gene regulates plant transpiration efficiency in Arabidopsis. Nature 436: 866–870. [DOI] [PubMed] [Google Scholar]
- McKay J.K., Richards J.H., Nemali K.S., Sen S., Mitchell-Olds T., Boles S., Stahl E.A., Wayne T., Juenger T.E. (2008). Genetics of drought adaptation in Arabidopsis thaliana II. QTL analysis of a new mapping population, KAS-1 x TSU-1. Evolution 62: 3014–3026. [DOI] [PubMed] [Google Scholar]
- McMurtrie R.E., Wang Y.P. (1993). Mathematical models of the photosynthetic response of tree stands to rising CO2 concentrations and temperatures. Plant Cell Environ. 16: 1–13. [Google Scholar]
- Méndez-Vigo B., Martínez-Zapater J.M., Alonso-Blanco C. (2013). The flowering repressor SVP underlies a novel Arabidopsis thaliana QTL interacting with the genetic background. PLoS Genet. 9: e1003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer R.C., Steinfath M., Lisec J., Becher M., Witucka-Wall H., Törjék O., Fiehn O., Eckardt A., Willmitzer L., Selbig J., Altmann T. (2007). The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 104: 4759–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi M., Umezawa T., Nakashima K., Kidokoro S., Takasaki H., Fujita Y., Yamaguchi-Shinozaki K., Shinozaki K. (2010). Two closely related subclass II SnRK2 protein kinases cooperatively regulate drought-inducible gene expression. Plant Cell Physiol. 51: 842–847. [DOI] [PubMed] [Google Scholar]
- Moore B., Zhou L., Rolland F., Hall Q., Cheng W.-H., Liu Y.-X., Hwang I., Jones T., Sheen J. (2003). Role of the Arabidopsis glucose sensor HXK1 in nutrient, light, and hormonal signaling. Science 300: 332–336. [DOI] [PubMed] [Google Scholar]
- Munné-Bosch S., Alegre L. (2004). Die and let live: leaf senescence contributes to plant survival under drought stress. Funct. Plant Biol. 31: 203–216. [DOI] [PubMed] [Google Scholar]
- Nakashima K., Ito Y., Yamaguchi-Shinozaki K. (2009). Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiol. 149: 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D.E., et al. (2007). Plant nuclear factor Y (NF-Y) B subunits confer drought tolerance and lead to improved corn yields on water-limited acres. Proc. Natl. Acad. Sci. USA 104: 16450–16455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros J.C. (2007). VENNY. An interactive tool for comparing lists with Venn diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html.
- Ozfidan C., Turkan I., Sekmen A.H., Seckin B. (2012). Abscisic acid-regulated responses of aba2-1 under osmotic stress: the abscisic acid-inducible antioxidant defence system and reactive oxygen species production. Plant Biol (Stuttg) 14: 337–346. [DOI] [PubMed] [Google Scholar]
- Page M., Sultana N., Paszkiewicz K., Florance H., Smirnoff N. (2012). The influence of ascorbate on anthocyanin accumulation during high light acclimation in Arabidopsis thaliana: further evidence for redox control of anthocyanin synthesis. Plant Cell Environ. 35: 388–404. [DOI] [PubMed] [Google Scholar]
- Parsons R., Weyers J.D.B., Lawson T., Godber I.M. (1997). Rapid and straightforward estimates of photosynthetic characteristics using a portable gas exchange system. Photosynthetica 34: 265–279. [Google Scholar]
- Passioura J.B. (1996). Drought and drought tolerance. Plant Growth Regul. 20: 79–83. [Google Scholar]
- Penfold C.A., Buchanan-Wollaston V. (2014). Modelling transcriptional networks in leaf senescence. J. Exp. Bot. 65: 3859–3873. [DOI] [PubMed] [Google Scholar]
- Penfold C.A., Wild D.L. (2011). How to infer gene networks from expression profiles, revisited. Interface Focus 1: 857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro C., Chaves M.M. (2011). Photosynthesis and drought: can we make metabolic connections from available data? J. Exp. Bot. 62: 869–882. [DOI] [PubMed] [Google Scholar]
- Quan R., Hu S., Zhang Z., Zhang H., Zhang Z., Huang R. (2010). Overexpression of an ERF transcription factor TSRF1 improves rice drought tolerance. Plant Biotechnol. J. 8: 476–488. [DOI] [PubMed] [Google Scholar]
- Roberts M.J., Schlenker W. (2009). World supply and demand of food commodity calories. Am. J. Agric. Econ. 91: 1235–1242. [Google Scholar]
- Sakuma Y., Maruyama K., Osakabe Y., Qin F., Seki M., Shinozaki K., Yamaguchi-Shinozaki K. (2006). Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell 18: 1292–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A., Alkema W., Engström P., Wasserman W.W., Lenhard B. (2004). JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32: D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholander P.F., Hammel H.T., Hemmingsen E.A., Bradstreet E.D. (1964). Hydrostatic pressure and osmotic potential in leaves of mangroves and some other plants. Proc. Natl. Acad. Sci. USA 52: 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclep G., Allemeersch J., Liechti R., De Meyer B., Beynon J., Bhalerao R., Moreau Y., Nietfeld W., Renou J.-P., Reymond P., Kuiper M.T.R., Hilson P. (2007). CATMA, a comprehensive genome-scale resource for silencing and transcript profiling of Arabidopsis genes. BMC Bioinformatics 8: 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seager R., et al. (2007). Model projections of an imminent transition to a more arid climate in southwestern North America. Science 316: 1181–1184. [DOI] [PubMed] [Google Scholar]
- Seki M., et al. (2002). Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 31: 279–292. [DOI] [PubMed] [Google Scholar]
- Shinozaki K., Yamaguchi-Shinozaki K. (1997). Gene expression and signal transduction in water-stress response. Plant Physiol. 115: 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki K., Yamaguchi-Shinozaki K. (2007). Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 58: 221–227. [DOI] [PubMed] [Google Scholar]
- Skirycz A., De Bodt S., Obata T., De Clercq I., Claeys H., De Rycke R., Andriankaja M., Van Aken O., Van Breusegem F., Fernie A.R., Inzé D. (2010). Developmental stage specificity and the role of mitochondrial metabolism in the response of Arabidopsis leaves to prolonged mild osmotic stress. Plant Physiol. 152: 226–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skirycz A., et al. (2011). Survival and growth of Arabidopsis plants given limited water are not equal. Nat. Biotechnol. 29: 212–214. [DOI] [PubMed] [Google Scholar]
- Smirnoff N., Cumbes Q.J. (1989). Hydroxyl radical scavenging activity of compatible solutes. Phytochemistry 28: 1057–1060. [Google Scholar]
- Smith C.A., Want E.J., O’Maille G., Abagyan R., Siuzdak G. (2006). XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78: 779–787. [DOI] [PubMed] [Google Scholar]
- Sperdouli I., Moustakas M. (2012). Interaction of proline, sugars, and anthocyanins during photosynthetic acclimation of Arabidopsis thaliana to drought stress. J. Plant Physiol. 169: 577–585. [DOI] [PubMed] [Google Scholar]
- Stegle O., Denby K.J., Cooke E.J., Wild D.L., Ghahramani Z., Borgwardt K.M. (2010). A robust Bayesian two-sample test for detecting intervals of differential gene expression in microarray time series. J. Comput. Biol. 17: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobiecki M., Skirycz A., Kerhoas L., Kachlicki P., Muth D., Einhorn J., Mueller-Roeber B. (2006). Profiling of phenolic glycosidic conjugates in leaves of Arabidopsis thaliana using LC/MS. Metabolomics 2: 197–219. [Google Scholar]
- Su Z., Ma X., Guo H., Sukiran N.L., Guo B., Assmann S.M., Ma H. (2013). Flower development under drought stress: morphological and transcriptomic analyses reveal acute responses and long-term acclimation in Arabidopsis. Plant Cell 25: 3785–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpice R., et al. (2009). Starch as a major integrator in the regulation of plant growth. Proc. Natl. Acad. Sci. USA 106: 10348–10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taji T., Ohsumi C., Iuchi S., Seki M., Kasuga M., Kobayashi M., Yamaguchi-Shinozaki K., Shinozaki K. (2002). Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J. 29: 417–426. [DOI] [PubMed] [Google Scholar]
- Takahashi Y., Ebisu Y., Kinoshita T., Doi M., Okuma E., Murata Y., Shimazaki K. (2013). bHLH transcription factors that facilitate K⁺ uptake during stomatal opening are repressed by abscisic acid through phosphorylation. Sci. Signal. 6: ra48. [DOI] [PubMed] [Google Scholar]
- Tang Y., Liu M., Gao S., Zhang Z., Zhao X., Zhao C., Zhang F., Chen X. (2012). Molecular characterization of novel TaNAC genes in wheat and overexpression of TaNAC2a confers drought tolerance in tobacco. Physiol. Plant. 144: 210–224. [DOI] [PubMed] [Google Scholar]
- Tattini M., Galardi C., Pinelli P., Massai R., Remorini D., Agati G. (2004). Differential accumulation of flavonoids and hydroxycinnamates in leaves of Ligustrum vulgare under excess light and drought stress. New Phytol. 163: 547–561. [DOI] [PubMed] [Google Scholar]
- Thimm O., Bläsing O., Gibon Y., Nagel A., Meyer S., Krüger P., Selbig J., Müller L.A., Rhee S.Y., Stitt M. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37: 914–939. [DOI] [PubMed] [Google Scholar]
- Tohge T., et al. (2005). Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 42: 218–235. [DOI] [PubMed] [Google Scholar]
- Ueguchi C., Koizumi H., Suzuki T., Mizuno T. (2001). Novel family of sensor histidine kinase genes in Arabidopsis thaliana. Plant Cell Physiol. 42: 231–235. [DOI] [PubMed] [Google Scholar]
- Umezawa T., Yoshida R., Maruyama K., Yamaguchi-Shinozaki K., Shinozaki K. (2004). SRK2C, a SNF1-related protein kinase 2, improves drought tolerance by controlling stress-responsive gene expression in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 101: 17306–17311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weele C.M., Spollen W.G., Sharp R.E., Baskin T.I. (2000). Growth of Arabidopsis thaliana seedlings under water deficit studied by control of water potential in nutrient-agar media. J. Exp. Bot. 51: 1555–1562. [DOI] [PubMed] [Google Scholar]
- Vanderauwera S., Zimmermann P., Rombauts S., Vandenabeele S., Langebartels C., Gruissem W., Inzé D., Van Breusegem F. (2005). Genome-wide analysis of hydrogen peroxide-regulated gene expression in Arabidopsis reveals a high light-induced transcriptional cluster involved in anthocyanin biosynthesis. Plant Physiol. 139: 806–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verelst W., Bertolini E., De Bodt S., Vandepoele K., Demeulenaere M., Pè M.E., Inzé D. (2013). Molecular and physiological analysis of growth-limiting drought stress in Brachypodium distachyon leaves. Mol. Plant 6: 311–322. [DOI] [PubMed] [Google Scholar]
- Weston D.J., Gunter L.E., Rogers A., Wullschleger S.D. (2008). Connecting genes, coexpression modules, and molecular signatures to environmental stress phenotypes in plants. BMC Syst. Biol. 2: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins O., Bräutigam K., Campbell M.M. (2010). Time of day shapes Arabidopsis drought transcriptomes. Plant J. 63: 715–727. [DOI] [PubMed] [Google Scholar]
- Windram O., et al. (2012). Arabidopsis defense against Botrytis cinerea: chronology and regulation deciphered by high-resolution temporal transcriptomic analysis. Plant Cell 24: 3530–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windram O., Penfold C.A., Denby K.J. (2014). Network modeling to understand plant immunity. Annu. Rev. Phytopathol. 52: 93–111. [DOI] [PubMed] [Google Scholar]
- Wohlbach D.J., Quirino B.F., Sussman M.R. (2008). Analysis of the Arabidopsis histidine kinase ATHK1 reveals a connection between vegetative osmotic stress sensing and seed maturation. Plant Cell 20: 1101–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Kerr K., Cui X., Churchill G. (2003). MAANOVA: A software package for the analysis of spotted cDNA microarray experiments. In The Analysis of Gene Expression Data: Methods and Software, Parmigiani G., Garrett E., Irizarry R., Zeger S., eds (New York: Springer; ), pp. 313–341. [Google Scholar]
- Xia J., Sinelnikov I.V., Han B., Wishart D.S. (2015). MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 43: W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B., Huang Y., Tang N., Xiong L. (2007). Over-expression of a LEA gene in rice improves drought resistance under the field conditions. Theor. Appl. Genet. 115: 35–46. [DOI] [PubMed] [Google Scholar]
- Xiao B.Z., Chen X., Xiang C.B., Tang N., Zhang Q.F., Xiong L.Z. (2009). Evaluation of seven function-known candidate genes for their effects on improving drought resistance of transgenic rice under field conditions. Mol. Plant 2: 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L., Schumaker K.S., Zhu J.K. (2002). Cell signaling during cold, drought, and salt stress. Plant Cell 14 (Suppl): S165–S183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L., Wang R.G., Mao G., Koczan J.M. (2006). Identification of drought tolerance determinants by genetic analysis of root response to drought stress and abscisic acid. Plant Physiol. 142: 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.Q., Huang J., Guo S.Q., Yang X., Bao Y.M., Tang H.J., Zhang H.S. (2008). Overexpression of a TFIIIA-type zinc finger protein gene ZFP252 enhances drought and salt tolerance in rice (Oryza sativa L.). FEBS Lett. 582: 1037–1043. [DOI] [PubMed] [Google Scholar]
- Yaish M.W., Colasanti J., Rothstein S.J. (2011). The role of epigenetic processes in controlling flowering time in plants exposed to stress. J. Exp. Bot. 62: 3727–3735. [DOI] [PubMed] [Google Scholar]
- Yoshida T., Fujita Y., Maruyama K., Mogami J., Todaka D., Shinozaki K., Yamaguchi-Shinozaki K. (2015). Four Arabidopsis AREB/ABF transcription factors function predominantly in gene expression downstream of SnRK2 kinases in abscisic acid signalling in response to osmotic stress. Plant Cell Environ. 38: 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.Y., Cruz de Carvalho M.H., Torres-Jerez I., Kang Y., Allen S.N., Huhman D.V., Tang Y., Murray J., Sumner L.W., Udvardi M.K. (2014). Global reprogramming of transcription and metabolism in Medicago truncatula during progressive drought and after rewatering. Plant Cell Environ. 37: 2553–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Jang J.-C., Jones T.L., Sheen J. (1998). Glucose and ethylene signal transduction crosstalk revealed by an Arabidopsis glucose-insensitive mutant. Proc. Natl. Acad. Sci. USA 95: 10294–10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.