

Abstract
Gene conversion between paralogs can alter their patterns of sequence identity, thus obscuring their evolutionary relationships and affecting their propensity to sponsor genomic rearrangements. The details of this important process are poorly understood in the human genome because allelic diversity complicates the interpretation of interparalog sequence differences. Here we exploit the haploid nature of the Y chromosome, which obviates complicating interallelic processes, together with its known phylogeny, to understand the dynamics of conversion between two directly repeated HERVs flanking the 780-kb AZFa region on Yq. Sequence analysis of a 787-bp segment of each of the HERVs in 36 Y chromosomes revealed one of the highest nucleotide diversities in the human genome, as well as evidence of a complex patchwork of highly directional gene conversion events. The rate of proximal-to-distal conversion events was estimated as 2.4 × 10-4 to 1.2 × 10-3 per generation (3.9 × 10-7 to 1.9 × 10-6 per base per generation), and the distal-to-proximal rate as about one-twentieth of this. Minimum observed conversion tract lengths ranged from 1 to 158 bp and maximum lengths from 19 to 1365 bp, with an estimated mean of 31 bp. Analysis of great ape homologs shows that conversion in this hotspot has a deep evolutionary history.
Recent analysis of the human genome sequence has revealed the extraordinary prevalence of segmental duplications, or paralogs. These highly similar nonallelic sequences make up 5.2% of the genome (Bailey et al. 2002), providing plentiful substrates for nonallelic homologous recombination (NAHR). NAHR leads to rearrangements, including a pathogenic subset involving gene disruption through inversion, or gene copy number alteration through deletion or duplication (for review, see Lupski 1998).
Homologous recombination is initiated via strand invasion and can be resolved not only through crossovers but also through gene conversion, the nonreciprocal transfer of genetic information between homologous sequences. Gene conversion is a fundamental process of molecular evolution and has been most intensively studied in yeast, in which all four haploid products of meiosis can be readily examined within tetrads. It is less well understood in humans, because meiotic segregation means that only one product is ever observed, and therefore, nonreciprocal transfer (conversion) cannot be formally distinguished from reciprocal transfer (double crossover).
Gene conversion between paralogs located on the same chromosome can have two effects. First, within a chromosome it can act as a homogenizing force, increasing lengths of identity between duplicated sequences (concerted evolution) and thus providing a more readily used substrate for NAHR. Second, because different conversion events occur in different chromosomes, it can generate excess sequence diversity among them—a phenomenon recognized in organisms as diverse as humans, flies, and protozoans (Giordano et al. 1997; King 1998; Nielsen et al. 2003). In principle, the dynamics of these processes can be inferred by comparative sequencing of paralogs in different individuals, but in most of the genome, such studies are difficult to interpret because of sequence diversity between alleles of paralogs and because the underlying evolutionary relationships between different paralog copies are unknown. Neither of these problems applies to the Y chromosome. Because it is haploid, we do not need to consider interallelic diversity; because there is no crossing over, paralogs cannot have independent evolutionary histories; and because there is an established phylogeny based on binary markers, we know the precise evolutionary relationships of chromosomes belonging to different lineages. The Y chromosome therefore provides a useful model to study gene conversion between paralogs.
Two human endogenous retroviral sequences (HERVs) flank a 780-kb region on Yq containing at least one gene required in spermatogenesis (Fig. 1A). NAHR between these direct paralogous repeats causes AZFa (azoospermia factor a) deletions (Blanco et al. 2000; Kamp et al. 2000; Sun et al. 2000) and, consequently, male infertility; the reciprocal duplications have also been observed (Bosch and Jobling 2003). Although the overall sequence similarity between the HERVs is 94%, deletion and duplication breakpoints are restricted to segments of complete sequence identity, known as blocks A, B, and C (Fig. 1A).
Figure 1.
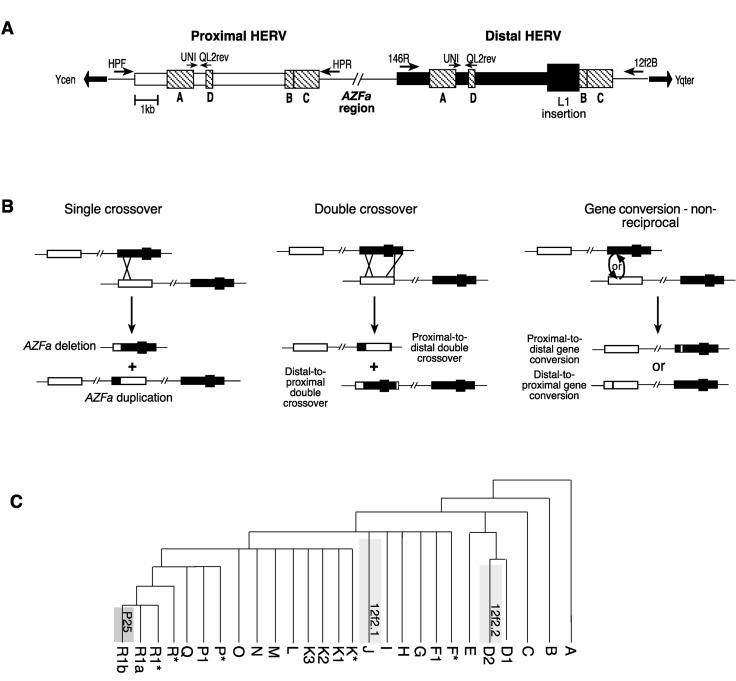
HERV structures, HERV-sponsored rearrangements, and the phylogenetic positions of known double-crossover and gene conversion events. (A) Structures of the proximal and distal AZFa HERVs, showing blocks of identity >200 bp (hatched boxes) and the L1 fragment insertion in the distal HERV. Identity blocks A through D are named according to Bosch and Jobling (2003). Flanking specific primers (large arrows) and primers used for the amplification of the region between identity blocks A and D (small arrows) are shown. (B) Possible sequence rearrangements sponsored by the HERVs: Single meiotic crossover between misaligned sister chromatids (in the same orientation) causes deletion or duplication of the AZFa region, both of which have been observed; double crossover causes the transfer of a large segment of one HERV to the other, observed in the proximal-to-distal case only; and gene conversion causes the nonreciprocal transfer of small segments, observed in both polarities. (C) Phylogeny of the Y chromosome showing known double-crossover and gene conversion events. Two examples of proximal-to-distal transfer through double-crossover are known: The concomitant loss of the L1 segment from the distal HERV is typed as a binary marker (Casanova et al. 1985) and defines haplogroup J (12f2.1). The second occurrence (12f2.2) is phylogenetically equivalent to binary markers defining haplogroup D2 (Blanco et al. 2000). A distal-to-proximal conversion of between 36 and 72 bp is phylogenetically equivalent to the marker P25, defining haplogroup R1b; one example of a chromosome with a proximal-to-distal conversion within this haplogroup is also known (Bosch and Jobling 2003).
In a previous study of AZFa duplications (Bosch and Jobling 2003), we identified two examples of apparent gene conversion within a segment lying between the identity block A and a shorter block of identity, D (here referred to as inter-AD). A segment of the proximal HERV, of between 36 and 72 bp, was converted by the distal HERV, and chromosomes carrying this conversion were monophyletic in the Y phylogeny, being found only in haplogroup R1b (Fig. 1C). A second apparent conversion in a single chromosome was also identified; in this example, the transfer was proximal to distal and involved two paralogous sequence variants (PSVs) adjacent to a large tract of inter-HERV identity. Its length is therefore very uncertain (between 5 and 1289 bp), because one end could lie anywhere within the large tract of identity.
Two other apparently nonreciprocal exchanges between the HERVs have been described (Blanco et al. 2000), and each represents a monophyletic group of chromosomes (haplogroups D2 and J; Fig. 1C). These two independent events involve the transfer of ∼5 kb of sequence, lying between identity blocks A and B, from the proximal to the distal HERV, in the process removing the distal-specific 1.5-kb insertion of L1 material (Fig. 1B). These events are regarded as double crossovers rather than as gene conversions, because the transferred segment is so large and because the stretch of complete heterology represented by the L1 fragment is likely to disrupt branch migration.
Here, prompted by the evidence for gene conversion within the inter-AD segment, we investigate further the sequence diversity of the AZFa HERVs in this region. We identify a hotspot for gene conversion of biased directionality, estimate the rate of conversion and the mean conversion tract length by using the framework of the Y phylogeny, and, by analyzing the sequences of great ape homologs, show that conversion in this hotspot has a deep evolutionary history.
RESULTS
Sequence Diversity of Proximal and Distal Inter-AD Regions
We determined the sequence of the 787-bp inter-AD region in proximal and distal HERVs from 33 human Y chromosomes, chosen for their wide coverage of the Y phylogeny. These sequences, together with three previously published examples (Bosch and Jobling 2003), were compared to the database reference sequence, which contains 24 differences between proximal and distal HERVs (PSVs) comprising 22 base substitutions and two single-base indels within the inter-AD segment (Fig. 2). The new sequences reveal no new PSVs; the reference sequence contains all the 24 PSVs observed between the proximal and distal inter-AD sequences in our sample.
Figure 2.

Sequence states of the 24 PSVs in proximal and distal inter-AD segments within 36 Y chromosomes, organized according to the Y phylogeny. Bases shown in white on black are distal specific, and those in black on white are proximal specific, as defined by the reference sequence (Ref). Names of DNA samples are given on the right; on the left, a simplified version of the Y phylogeny (Y Chromosome Consortium 2002) shows the phylogenetic relationships and haplogroup names of the chromosomes analyzed. Sequences within haplogroup J contain a deletion of 13 bp between PSVs 18 and 19, in both proximal and distal sequences. Case 1, case 2, and YCC26 proximal and distal sequences have been reported previously (respectively, AJ511660-1, AJ511663-4, AJ511666-7; Bosch and Jobling 2003). Sequence positions from 103 to 724 (bottom) give coordinates within the inter-AD segments: The position labeled 103 is equivalent to nucleotide no. 11739 on the plus strand of AC002992 for the proximal HERV, and to nucleotide no. 144340 on the minus strand of AC005820 for the distal HERV.
We first consider the diversity of the proximal inter-AD sequences. All six sequences within haplogroup R1b show the distal-to-proximal conversion of four adjacent PSVs (PSV 3–6) described previously (Bosch and Jobling 2003). Two chromosomes within haplogroup J have a deletion of 13 bp between PSVs 18 and 19. All other proximal inter-AD sequences are identical to the reference sequence.
The distal inter-AD sequences present a very different picture. The 36 different chromosomes possess 14 distinct sequences. The two distal sequences within haplogroup D2 are identical to the proximal sequences, reflecting the known double-crossover event transferring ∼5 kb from the proximal to the distal HERV (Blanco et al. 2000). Likewise, the independent double-crossover event within haplogroup J is reflected by the identity of the proximal and distal sequences in the two chromosomes belonging to this haplogroup, including the proximal-to-distal transfer of the 13-bp deletion. We do not consider these four sequences further here, because the absence of PSVs makes them uninformative.
The remaining distal inter-AD sequences are highly diverse: nucleotide diversity (π) for the distal sequences is 5.44 × 10-3, compared with a value for the proximal sequences of 1.60 × 10-3. It is difficult to make a fair comparison of this value with published estimates of π for the Y chromosome, because we ascertained our samples nonrandomly. However, as far as correspondences can be drawn, the published survey of Shen et al. (2000) covers a similar range of diversity to our sample, including chromosomes belonging to all top-level clades in the Y phylogeny with the possible exceptions of F*, H, K*, L, N, and P*. That study found an average nucleotide diversity for noncoding regions of only 9.9 × 10-5. Typical values of nucleotide diversity for autosomal sequences range from 5.3 × 10-4 to 8.8 × 10-4 (Fullerton et al. 2000; Zhao et al. 2000; Yu et al. 2001). The high diversity we observe in distal inter-AD sequences is manifested as a patchy pattern of proximal-like PSVs among the distal PSVs; their total number ranges from a single PSV (e.g., m348) to 13 (m230). Strikingly, 19 of the 24 PSVs are found to be polymorphic among the distal inter-AD sequences, whereas none of the 723 non-PSV sites are polymorphic.
The overall pattern of sequence variation is not concordant with the known Y phylogeny. There are some examples of chromosomes lying within the same haplogroup of the phylogeny that share identical or closely related patterns of proximal-like PSVs (e.g., m230 and YCC23 within haplogroup C). Also, it could be argued that PSV 6 arose as a T-to-C substitution at the root of the clade Y(xA,B). However, there are other cases in which PSVs appear in distinct haplogroups in a way that cannot be explained by common descent. An example is PSV 18, which in the distal HERV reference sequence is an A, but is a G in several independent haplogroups, and therefore displays homoplasy (apparent recurrent mutation).
What mechanism has generated this sequence diversity? In principle there are four possibilities: (1) Recombination among Y chromosomes; this could occur under special circumstances, such as between a segment of Y chromosome from one haplogroup carried as a translocation on a non-Y-chromosome, and a free Y chromosome from another haplogroup (Jobling and Tyler-Smith 2003). However, this has yet to be observed, and it seems unlikely that such an inherently rare event would have occurred multiple times within a small segment of DNA. (2) Another possibility is base mutation: this can be rejected because all changes in the distal HERV affect only the PSVs, resulting in sequence states found in the proximal HERV, and because sequence differences often affect contiguous sets of PSVs (e.g., YCC33, YCC5). (3) A third possibility is redeletion of AZFa duplication chromosomes: these (Bosch and Jobling 2003) contain three HERVs rather than two (see Fig. 1A), and could undergo deletion to generate HERVs containing a patchwork of distal- and proximal-specific PSVs, provided that the breakpoints of the original duplication and subsequent redeletion were different. However, given the restriction of known deletion and duplication breakpoints to the A and B/C blocks of absolute sequence identity, we do not consider that this mechanism provides a viable explanation. (4) The fourth possibility is gene conversion: this remains the most parsimonious explanation for the observed pattern of sequence diversity.
Number and Rate of Gene Conversion Events
Clearly, the gene conversion events are directional, in that there are many more conversions of PSVs within distal inter-AD sequences to proximal-like states than vice versa. We see only one example of distal-to-proximal conversion (in haplogroup R1b). The number of proximal-to-distal events can be counted by considering the phylogeny—when two or more chromosomes within a haplogroup have the same pattern of proximal-to-distal conversion, we regard it as being due to common ancestry rather than to independent events. We also make the simplifying assumptions that each contiguous tract represents a single event, that the reference sequence can be regarded as ancestral (because it contains all 24 PSVs), and that a change at a single PSV represents a gene conversion event, rather than a base mutation, on the basis of arguments given above. In this way we identify a minimum of 22 putative independent conversion events (Fig. 3).
Figure 3.
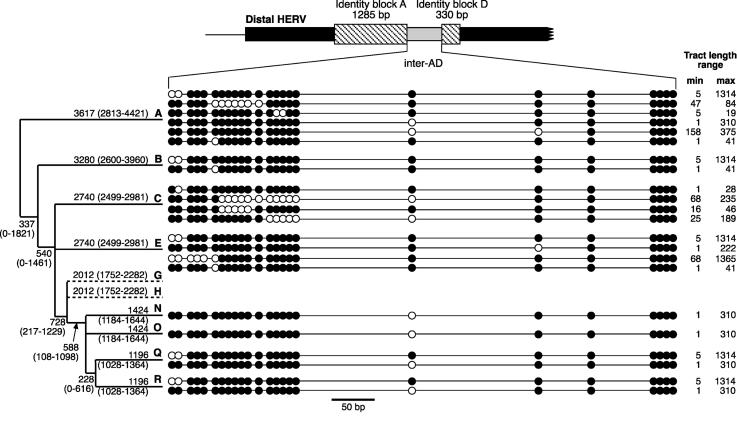
Number of independent proximal-to-distal conversion events within the sample. Sequence states of the PSVs within the distal inter-AD segment are shown schematically as small filled or empty circles and are defined as proximal or distal specific according to the reference sequence; spacing is approximately to scale. Each horizontal line represents one independent event, as deduced from the sequences shown in Fig. 2. The phylogeny to the left shows the haplogroup within which each event occurred and, adjacent to branches, the estimated age in generations (and its range as ±SD) of each branch, taken from published dates (Hammer and Zegura 2002) and assuming a generation time of 25 years. Branches G and H are included, even though they contain no conversions, because their ages are required in the rate calculation. Branches D and J are omitted because their lack of PSVs makes them uninformative.
To estimate the rate of conversion, we need to know the total amount of time during which the events have occurred (see Supplemental figure). We can place lower and upper bounds on this by estimating the maximum and minimum plausible elapsed time since all the chromosomes analyzed (neglecting those in haplogroups D2 and J) had a common ancestor. First, we consider the lower rate bound. The 32 chromosomes belong to 10 distinct upper-level haplogroups in the Y chromosome phylogeny. Time to most recent common ancestor (TMRCA) for each of these haplogroups has been estimated by coalescent analysis (Hammer and Zegura 2002), so to relate these 10 sets of chromosomes, we take the upper bound on each of these haplogroup ages (Fig. 3). For chromosomes within a haplogroup, we then assume a star-like genealogy; this represents an overestimate of the intra-haplogroup TMRCAs and is conservative in neglecting more recent common ancestry. For a generation time of 25 years, this process yields a maximum total elapsed time of 90,274 generations. To estimate the minimum time in which the conversions could have occurred, we first consider the lower bounds on the published TMRCAs (Hammer and Zegura 2002) for the haplogroups shown in Figure 3 and, to minimize time within-haplogroup, make the reductio ad absurdum assumption that each set of chromosomes within a haplogroup descends from a common ancestor only one generation ago. The minimum summed time is then 18,686 generations. These minimum and maximum times are highly conservative and are therefore highly likely to bracket the actual elapsed time. Twenty-two proximal-to-distal gene conversion events occurring over 18,686 to 90,274 generations represents an average rate of between 2.4 × 10-4 and 1.2 × 10-3 events per generation. By the same argument, the average rate of distal-to-proximal conversion is between 1.1 × 10-5 and 5.3 × 10-5, ∼20-fold lower than conversion in the opposing direction. The overall gene conversion rate between these sequences is the sum of the individual estimates for each direction—between 2.5 × 10-4 and 1.3 × 10-3 events per generation.
The position and directional bias of this hotspot may be due to specific features of the DNA sequence—for example, a recombination signal specific to the proximal segment may make it active as an initiator and donor sequence. We searched the inter-AD segment and adjacent parts of the A and D identity blocks for consensus binding sites for proteins involved in recombination, and for motifs associated with recombination hotspots, with negative results.
Tract Length of Gene Conversion Events
Figure 3 gives the maximum and minimum lengths of the observed conversion tracts: they are highly nonuniform. The observed (minimum) length of a conversion tract is the distance encompassing the outermost converted PSVs, but it will tend to underestimate the true tract length because of the uninformativeness of flanking sites. The relationship between observed and true lengths depends on the frequency of PSVs and also on the true tract length distribution. To derive a maximum-likelihood estimate of the mean true tract length, we use a modified version of the method of Betrán et al. (1997), assuming a geometric distribution of underlying tract lengths. The standard version of this method considers a gene conversion event to have occurred if at least two variant sites have been converted; single-site events are rejected, because they could result from base mutation. Abiding by this restriction would give a maximum-likelihood estimate of the mean true tract length of 62 bp. From the arguments made above, however, we consider single-site events to represent conversion, yielding a mean length of 31 bp. Although we abide by convention in estimating this mean length, it is worth noting that recent work on allelic gene conversion in meiotic recombination hotspots (Jeffreys and May 2004) showed that the dual uncertainties over tract position and length resulting from uneven marker spacing meant that the observed tract lengths were compatible with a range of mean lengths (maximum-likelihood estimates ranging from 63 to 200 bp), under many different models.
A Deeper History of Gene Conversion in the Inter-AD Segment
To ask whether this dynamic conversion process is evolutionarily conserved, we analyzed the sequence of proximal and distal inter-AD segments in a chimpanzee and a gorilla, and compared these to the human reference sequence; note that the latter contains the greatest number of human PSVs, and that the intraspecific diversity of the great ape sequences is unknown. Table 1 shows sequence divergences between the species for proximal and distal segments; all divergences except one (proximal inter-AD between human and chimpanzee) are significantly elevated over a control noncoding sequence from the SMCY gene region (Shen et al. 2000; Chen and Li 2001). This unusually high divergence between all three species suggests that gene conversion in the inter-AD segment has a deep evolutionary history: If conversion elevates sequence diversity within a species, it can also be expected to elevate sequence divergence between species. The observation that the proximal inter-AD human-chimp comparison is not elevated could be explained if conversion in chimpanzees, as in humans, was directional.
Table 1.
Sequence Divergences Among Human, Chimpanzee, and Gorilla Inter-AD Segments, Compared To the Noncoding Region of the SMCY Gene
If gene conversion has been evolutionarily persistent, we expect to see clustering of paralogs, rather than orthologs, in interspecific comparisons—evidence of concerted evolution. In a phylogenetic network produced by split decomposition (Fig. 4A), this is exactly what we observe. Clustering is especially tight for the chimpanzee sequences, suggesting that conversion has been particularly active in chimpanzees. The presence of reticulations within the network (i.e., the absence of a completely tree-like structure) shows that gene conversion has not completely eradicated evidence of the orthologous relationships, although it has resulted in a paucity of PSVs shared between species. The chimpanzee sequences contain 10 PSVs, and the gorilla sequences contain 21; however, the chimpanzee shares no PSVs with either of the other species, and only three PSVs are shared between human and gorilla. The sequence states of these three human/gorilla-shared PSVs indicate that (as suggested above) the directionality of the conversion in the chimpanzee sequences at these sites has been proximal to distal. A further reflection of historical gene conversion is the phylogenetic incompatibility seen among the 21 sites that are not PSVs within any species but differ between species (Fig. 4B). Phylogenetic relationships favoring all three possible sister-clades can be seen at sites scattered along the inter-AD segment: 12 sites favor a human–chimpanzee grouping, six favor human–gorilla, and three favor gorilla–chimpanzee.
Figure 4.
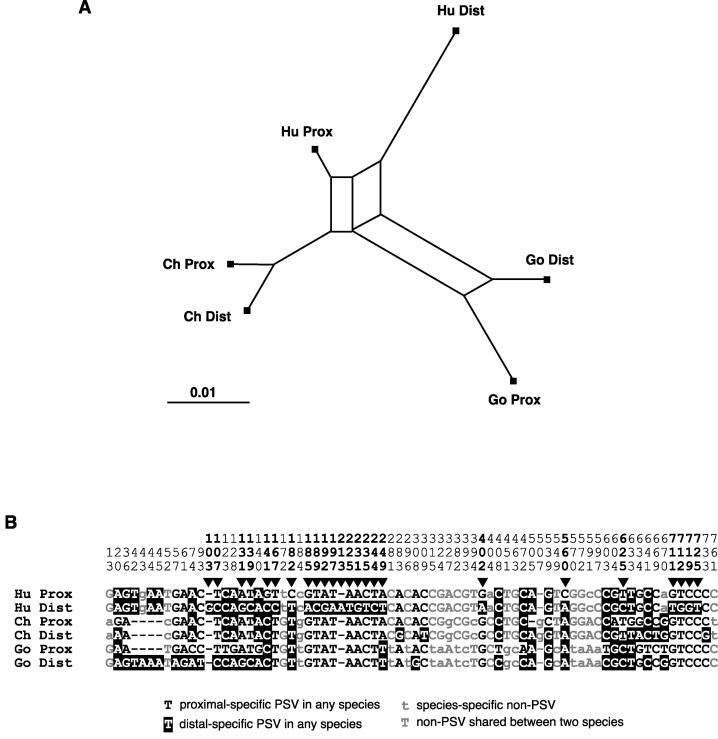
Phylogenetic network analysis and sequence comparison of human, chimp, and gorilla inter-AD segments. (A) Phylogenetic network showing the relationships between inter-AD segments in the three species, produced using SplitsTree. Hu indicates human; Ch, chimpanzee; Go, gorilla; Prox, proximal HERV; and Dist, distal HERV. (B) Sequence states of all variant positions in the three species are shown. Human PSVs are indicated by triangles. Sequence numbering is as in Fig. 2, up to position 507. Beyond this point, one base is added to each number to take account of the gap introduced into the human sequence to facilitate sequence alignments.
DISCUSSION
We have demonstrated the existence of a rapid and directional gene conversion process acting between the paralogous AZFa-HERV repeats, resulting in a hotspot of gene conversion in the distal inter-AD segment. How well delineated is this hotspot? Immediately proximal to inter-AD lies block A, 1285 bp of sequence that is identical between the proximal and distal HERVs. Gene conversion may be going on within this segment but would be undetectable because of the absence of PSVs. Six gene conversion events include PSV 1, adjacent to block A, and so have one endpoint within the identity block; the remaining 16 are contained within inter-AD. We observe a declining frequency of gene conversion events toward the distal end of inter-AD, with the distal-most five PSVs undergoing no conversions at all in our sample, thus marking the distal limit of the hotspot more clearly.
There is no evidence to suggest that high-frequency conversion processes are operating elsewhere within the HERVs. Proximal to identity block A, and distal to the 330-bp block D, the level of sequence similarity between the HERVs declines dramatically, to a degree that is expected to preclude conversion (Lukacsovich and Waldman 1999). Additional sequence data also suggest that conversion cannot be occurring frequently outside the inter-AD segment: In our study of AZFa duplications (Bosch and Jobling 2003), we described the sequence of a 3360-bp segment from distal and proximal HERVs for three chromosomes (case 1, case 2, and YCC26) belonging to haplogroups R1b*, R1b6, and R1b(xR1b6,R1b8). All distal sequences were identical to each other, except for conversions within inter-AD, and all proximal sequences were completely identical to each other. The partial sequence of a 9-kb segment from the distal HERV for two chromosomes belonging to haplogroups J and D2 has also been described (Blanco et al. 2000). Apart from the 13-bp deletion within inter-AD in haplogroup J (Fig. 2), the two sequences were identical. This general absence of variation suggests that detectable gene conversion between the HERVs is indeed confined to the inter-AD segment.
Comparisons of inter-AD sequences between human, chimpanzee, and gorilla indicate that this hotspot is not evolutionarily transient but has been active in all three lineages since their descent from a common ancestor 6 to 9 million years ago. Furthermore, the patterns of interspecific sequence divergence between proximal and distal sequences, together with the sequence states of PSVs, indicate that the proximal-to-distal directionality of the conversion process has also been conserved between the human and chimpanzee lineages.
Previously, indirect evidence for interallelic gene conversion in humans has emerged from observations of lower-than-expected linkage disequilibrium between closely spaced markers (Ardlie et al. 2001; Frisse et al. 2001). The ratio of conversion to crossover was calculated as 7.3 (Frisse et al. 2001), but this was under a model in which tract length was assumed to be 500 bp. Recent direct analysis of interallelic conversion in sperm DNA within three human meiotic recombination hotspots (Jeffreys and May 2004) reveals short (maximum-likelihood mean length, 63 to 200 bp) conversion tracts centered at the peak of crossover activity. At the most intensively studied hotspot, conversion predominates over crossover at a ratio of ∼4 to 15:1; if this were applicable to interparalog conversion in the inter-AD segment (extrapolating from the central value of our mean overall deduced conversion rate, 7.8 × 10-4), we would expect deletion and duplication breakpoints to occur within the inter-AD segment at a frequency of ∼5 × 10-5 to 2 × 10-4 per generation. Note, however, that when mean tract length is small, many gene conversion events are unobserved; our rates do not consider these and therefore represent minimal estimates. Ascertainment of AZFa deletions is good because of the associated infertile phenotype, and a generous estimate of their overall rate is ∼4 × 10-4 per generation (Bosch and Jobling 2003). Although the number of AZFa rearrangements so far described at the molecular level (Blanco et al. 2000; Kamp et al. 2000; Sun et al. 2000; Bosch and Jobling 2003) is only 15, all have their breakpoints within the blocks of identity (A and B/C) and none in the inter-AD segment. Inter-AD breakpoints may be identified in the future, but it seems likely that conversion predominates greatly over crossover in this hotspot.
Though Jeffreys and May (2004) suggest that similar mechanisms may be operating during both interallelic and interparalog conversion, direct evidence is currently lacking. Data on interparalog conversion come mainly from the occasional detection of variant or pathologically mutated genes in which an altered sequence tract can be shown to have originated in a paralogous gene copy (Collier et al. 1993; Papadakis and Patrinos 1999; Ogasawara et al. 2001). Rates for such processes cannot be easily deduced, but tract lengths are typically estimated as a few hundred base pairs. Studies of interallelic conversion at the HLA-DPB1 locus in sperm DNA (Zangenberg et al. 1995) reveal parameters in similar ranges to those that we observe: a rate of 2.8 to 13.3 × 10-5 per sperm in a 240-bp region, tract lengths between 54 and 132 bp, and a probable conversion to crossover ratio of >20 (Frisse et al. 2001). In principle, direct access to interparalog conversion and recombination events in sperm DNA within the AZFa HERVs is feasible.
Recent studies of the near-complete sequence of the Y euchromatin have shown evidence of high-frequency gene conversion between arms of abundant paralogous sequences arranged as giant palindromes (Rozen et al. 2003). Assuming a steady-state balance between the introduction of new mutations and their homogenization by gene conversion, the observed sequence divergence between palindrome arms was used to calculate a rate of 2.2 × 10-4 conversions per duplicated base per 20-year generation (equivalent to 2.8 × 10-4 for a 25-year generation). The prevalence of these palindromes is a peculiarity of the Y chromosome, and these observations may be of limited relevance to the rest of the genome. However, the rates we calculate here are for direct repeats separated by 780 kb and may be more generally applicable. For proximal-to-distal conversions, they correspond to between 3.9 × 10-7 and 1.9 × 10-6 per base per generation (considering the 622 bp of the inter-AD segment, including the outermost PSVs), which, although much lower than the palindrome rate, is still two to three orders of magnitude higher than the base mutation rate (Thomson et al. 2000). As is the case for the sequences we have studied here, conversions between the palindrome arms are not peculiar to the human species but occur in other tested primates. Comparisons of human–chimpanzee sequence divergence in palindrome arms with that in nonpalindrome regions suggests that gene conversion here is acting as a conservative force (Rozen et al. 2003), reducing sequence divergence rather than increasing it as we observe; in principle, this could occur through natural selection favoring ancestral states or through opposing biases in gene conversion and mutation.
In an early study (Dorit et al. 1995) a 729-bp intron of the ZFY gene was sequenced in 38 Y chromosomes selected for their population diversity and therefore probably covering many of the major haplogroups of the Y phylogeny (Jobling and Tyler-Smith 2003). Famously, this study found no variant sites whatsoever. On a chromosome renowned for its low variability, the presence of 19 variant sites in 36 copies of the similarly sized distal inter-AD segment and one of the highest known values of π in the human genome provide a dramatic illustration of the ability of gene conversion to generate diversity among DNA sequences. Further comparative sequencing of segmental duplications is required to investigate whether the preferential mapping of dbSNP entries to paralogous sequences reflects not only misassignment of sequence reads but also elevated sequence diversity (Hurles 2002).
METHODS
Samples
DNA samples with the prefix “YCC” are from the Y Chromosome Consortium, and haplogroup information is as described (Y Chromosome Consortium 2002). Other samples of known haplogroup were gifts from Chris Tyler-Smith (Wellcome Trust Sanger Institute, UK) and Qasim Mehdi (Dr. A.Q. Khan Research Laboratories, Pakistan), or from collections of the authors.
HERV Amplifications and Sequencing Analysis
Primers and PCR conditions for the amplification of HERV sequences were as described (Bosch and Jobling 2003).
The inter-AD region was reamplified from proximal- and distal-specific human and gorilla HERV amplicons by using the primers UNIV (5′-TTGTGGAGCCTATGGTCTCTAT-3′) and QL2rev (5′-CTCAGCAGGAATCTGTCCTAA-3′). Nested PCR conditions were as described (Bosch and Jobling 2003), except that extension time was reduced to 1 min and the total cycle number reduced to 20. PCR products were purified from agarose gels and used as templates for sequencing by using the same primers.
The UNIV-QL2rev amplification was not possible in chimpanzee HERVs; subsequent sequencing showed several differences in the UNIV primer target sequence. We therefore amplified two overlapping fragments covering the same region from each HERV copy. Primers for the amplification of the first fragment (1873 bp) were SQ3 (5′-ACACAGTATGTAGGGAATGC-3′) and Int2 (5′-CTGTTGCCCTGTGCCTCATA-3′); for the second fragment (1901 bp), SQ4 (5′-TTAACACTCATGCTGCCCTG-3′) and SQ1 (5′-CAAAGGAAGCAGAGGAACCT-3′). Both of these PCR products from each HERV copy were purified from agarose gels and used as template for sequencing with primers SQ3 and SQ4, respectively.
Sequencing reactions were performed by using the ABI-PRISM Big Dye terminator chemistry (version 2), according to the manufacturer's recommendations, and analyzed on a ABI3100 capillary sequencing apparatus (Applied Biosystems).
All sequences were compared with the database reference sequences AC002992 (proximal) and AC005820 (distal). These two sequences derive from different Y chromosomes and therefore do not necessarily lie together on any “real” Y chromosome. However, five chromosomes analyzed here have the same proximal/distal combination of sequences as the database; the database sequences, as a pair, are therefore valid for use as a reference. Nucleotide diversity was calculated by using DnaSP (Rozas and Rozas 1999).
An alternative nomenclature is also used for the blocks of sequence identity within the HERVs: block A in this study is equivalent to block ID1 (Kamp et al. 2000) and blocks B and C together to block ID2, whereas block D has no alternative name. Note that the 13-bp deletion within the inter-AD segment is found here in chromosomes belonging to haplogroup J, not haplogroup D2 as previously reported (Blanco et al. 2000); the haplogroups were switched in error.
Analysis of Gene Conversion Tract Length
After determining the minimum number of gene conversion events by using the phylogeny (see text), observed tract lengths were estimated by using the method of Betrán et al. (1997) as implemented in DnaSP (Rozas and Rozas 1999). The method was modified to allow changes at single PSVs (normally discounted) to be considered as gene conversion events. Average true tract length was estimated from observed tract length by using a modified maximum-likelihood method (Betrán et al. 1997), which models gene conversion according to a geometric distribution with parameter φ. After initiation of gene conversion, the tract either extends (unidirectionally) to an additional base (with probability φ), or terminates (with probability [1 - φ]). The probability that a tract will extend to n bases in length is then (1 - φ)φn-1 and mean tract length is 1/(1 - φ).
Phylogenetic Analysis and Jukes-Cantor Distance
Interspecific phylogenetic networks of proximal- and distal-specific inter-AD HERV sequences were constructed by using SplitsTree (Huson 1998; http://www-ab.informatik.uni-tuebingen.de/software/splits/welcome_en.html). Corrected Jukes-Cantor distances between species were calculated in DnaSP (Rozas and Rozas 1999), and standard deviations were estimated as (p[1 - p]/N) - 1, where p is the proportion of nucleotides with substitutions, and N the length of sequence in base pairs (785 bp).
Recombination Motif Searches
The inter-AD segment (including variant sequences as shown in Fig. 2) and the adjacent A and D blocks were searched for a number of recombination and replication motifs using the FIND-PATTERNS program within GCG. We sought the sequences listed and referenced by Badge et al. (2000): the Escherichia coli χ sequence, the Schizosaccharomyces pombe ade6-M26 heptamer, the long terminal repeat element (LTR-IS) motif, the retrotransposon LTR sequence, the XY32 homopurine-pyrimidine H-palindrome motif, the human minisatellite core sequence, two human hypervariable minisatellite sequences, the pur and translin protein binding sites, the human replication origin consensus, the S. cerevisiae and S. pombe ARS consensus sequences, consensus mammalian scaffold attachment regions, and the topoisomerase II binding site. Also sought was the RBP-2N recognition sequence (Dou et al. 1994).
Acknowledgments
We thank Chris Tyler-Smith for supplying DNA samples, Qasim Mehdi for sample PRS015, Peidong Shen for information about markers around the SMCY gene, Zoë Rosser for unpublished haplotyping information on m176, and Francesc Calafell for help with calculations. E.B. was supported by the Wellcome Trust, M.E.H. by the McDonald Institute; M.A.J., by a Wellcome Trust Senior Fellowship in Basic Biomedical Science (grant no. 057559).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2177404.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession numbers AY500148–AY500151 and AY549481–AY549491. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: C. Tyler-Smith, Q. Mehdi, P. Shen, and Z. Rosser.]
References
- Ardlie, K., Liu-Cordero, S.N., Eberle, M.A., Daly, M., Barrett, J., Winchester, E., Lander, E.S., and Kruglyak, L. 2001. Lower-than-expected linkage disequilibrium between tightly linked markers in humans suggests a role for gene conversion. Am. J. Hum. Genet. 69: 582-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badge, R.M., Yardley, J., Jeffreys, A.J., and Armour, J.A.L. 2000. Crossover breakpoint mapping identifies a subtelomeric hotspot for male meiotic recombination. Hum. Mol. Genet. 9: 1239-1244. [DOI] [PubMed] [Google Scholar]
- Bailey, J.A., Gu, Z., Clark, R.A., Reinert, K., Samonte, R.V., Schwartz, S., Adams, M.D., Myers, E.W., Li, P.W., and Eichler, E.E. 2002. Recent segmental duplications in the human genome. Science 297: 1003-1007. [DOI] [PubMed] [Google Scholar]
- Betrán, E., Rozas, J., Navarro, A., and Barbadilla, A. 1997. The estimation of the number and the length distribution of gene conversion tracts from population DNA sequence data. Genetics 146: 89-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco, P., Shlumukova, M., Sargent, C.A., Jobling, M.A., Affara, N., and Hurles, M.E. 2000. Divergent outcomes of intra-chromosomal recombination on the human Y chromosome: Male infertility and recurrent polymorphism. J. Med. Genet. 37: 752-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch, E. and Jobling, M.A. 2003. Duplications of the AZFa region of the human Y chromosome are mediated by homologous recombination between HERVs and are compatible with male fertility. Hum. Mol. Genet. 12: 341-347. [DOI] [PubMed] [Google Scholar]
- Casanova, M., Leroy, P., Boucekkine, C., Weissenbach, J., Bishop, C., Fellous, M., Purrello, M., Fiori, G., and Siniscalco, M. 1985. A human Y-linked DNA polymorphism and its potential for estimating genetic and evolutionary distance. Science 230: 1403-1406. [DOI] [PubMed] [Google Scholar]
- Chen, F.C. and Li, W.H. 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68: 444-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier, S., Tassabehji, M., and Strachan, T. 1993. A de novo pathological point mutation at the 21-hydroxylase locus: Implications for gene conversion in the human genome. Nat. Genet. 3: 260-264. [DOI] [PubMed] [Google Scholar]
- Dorit, R.L., Akashi, H., and Gilbert, W. 1995. Absence of polymorphism at the ZFY locus on the human Y chromosome. Science 268: 1183-1185. [DOI] [PubMed] [Google Scholar]
- Dou, S., Zeng, X., Cortes, P., Erdjument-Bromage, H., Tempst, P., Honjo, T., and Vales, L.D. 1994. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol. Cell. Biol. 14: 3310-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse, L., Hudson, R.R., Batoszewicz, A., Wall, J.D., Donfack, J., and Di Rienzo, A. 2001. Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am. J. Hum. Genet. 69: 831-843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton, S.M., Clark, A.G., Weiss, K.M., Nickerson, D.A., Taylor, S.L., Stengard, J.H., Salomaa, V., Vartiainen, E., Perola, M., Boerwinkle, E., et al. 2000. Apolipoprotein E variation at the sequence haplotype level: Implications for the origin and maintenance of a major human polymorphism. Am. J. Hum. Genet. 67: 881-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano, M., Marchetti, C., Chiorboli, E., Bona, G., and Richiardi, P.M. 1997. Evidence for gene conversion in the generation of extensive polymorphism in the promoter of the growth hormone gene. Hum. Genet. 100: 249-255. [DOI] [PubMed] [Google Scholar]
- Hammer, M.F. and Zegura, S.L. 2002. The human Y chromosome haplogroup tree: Nomenclature and phylogeny of its major divisions. Annu. Rev. Anthropol. 31: 303-321. [Google Scholar]
- Hurles, M. 2002. Are 100,000 “SNPs” useless? Science 298: 1509. [DOI] [PubMed] [Google Scholar]
- Huson, D.H. 1998. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 14: 68-73. [DOI] [PubMed] [Google Scholar]
- Jeffreys, A.J. and May, C.A. 2004. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nature Genet. 36: 152-156. [DOI] [PubMed] [Google Scholar]
- Jobling, M.A. and Tyler-Smith, C. 2003. The human Y chromosome: An evolutionary marker comes of age. Nat. Rev. Genet. 4: 598-612. [DOI] [PubMed] [Google Scholar]
- Kamp, C., Hirschmann, P., Voss, H., Huellen, K., and Vogt, P.H. 2000. Two long homologous retroviral sequence blocks in proximal Yq11 cause AZFa microdeletions as a result of intrachromosomal recombination events. Hum. Mol. Genet. 9: 2563-2572. [DOI] [PubMed] [Google Scholar]
- King, L.M. 1998. The role of gene conversion in determining sequence variation and divergence in the Est-5 gene family in Drosophila pseudoobscura. Genetics 148: 305-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacsovich, T. and Waldman, A.S. 1999. Suppression of intrachromosomal gene conversion in mammalian cells by small degrees of sequence divergence. Genetics 151: 1559-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski, J.R. 1998. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 14: 417-422. [DOI] [PubMed] [Google Scholar]
- Nielsen, K.M., Kasper, J., Choi, M., Bedford, T., Kristiansen, K., Wirth, D.F., Volkman, S.K., Lozovsky, E., and Hartl, D.L. 2003. Gene conversion as a source of nucleotide diversity in Plasmodium falciparum. Mol. Biol. Evol. 20: 726-734. [DOI] [PubMed] [Google Scholar]
- Ogasawara, K., Yabe, R., Uchikawa, M., Nakata, K., Watanabe, J., Takahashi, Y., and Tokunaga, K. 2001. Recombination and gene conversion-like events may contribute to ABO gene diversity causing various phenotypes. Immunogenet. 53: 190-199. [DOI] [PubMed] [Google Scholar]
- Papadakis, M.N. and Patrinos, G.P. 1999. Contribution of gene conversion in the evolution of the human β-like globin gene family. Hum. Genet. 104: 117-125. [DOI] [PubMed] [Google Scholar]
- Rozas, J. and Rozas, R. 1999. DnaSP version 3: An integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174-175. [DOI] [PubMed] [Google Scholar]
- Rozen, S., Skaletsky, H., Marszalek, J.D., Minx, P.J., Cordum, H.S., Waterston, R.H., Wilson, R.K., and Page, D.C. 2003. Abundant gene conversion between arms of massive palindromes in human and ape Y chromosomes. Nature 423: 873-876. [DOI] [PubMed] [Google Scholar]
- Shen, P., Wang, F., Underhill, P.A., Franco, C., Yang, W.-H., Roxas, A., Sung, R., Lin, A.A., Hyman, R.W., Vollrath, D., et al. 2000. Population genetic implications from sequence variation in four Y chromosome genes. Proc. Natl. Acad. Sci. 97: 7354-7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C., Skaletsky, H., Rozen, S., Gromoll, J., Nieschlag, E., Oates, R., and Page, D.C. 2000. Deletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses. Hum. Mol. Genet. 9: 2291-2296. [DOI] [PubMed] [Google Scholar]
- Thomson, R., Pritchard, J.K., Shen, P., Oefner, P.J., and Feldman, M.W. 2000. Recent common ancestry of human Y chromosomes: Evidence from DNA sequence data. Proc. Natl. Acad. Sci. 97: 7360-7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Y Chromosome Consortium. 2002. A nomenclature system for the tree of human Y-chromosomal binary haplogroups. Genome Res. 12: 339-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, N., Zhao, Z., Fu, Y.X., Sambuughin, N., Ramsay, M., Jenkins, T., Leskinen, E., Patthy, L., Jorde, L.BL., Kuromori, T., et al. 2001. Global patterns of human DNA sequence variation in a 10-kb region on chromosome 1. Mol. Biol. Evol. 18: 214-222. [DOI] [PubMed] [Google Scholar]
- Zangenberg, G., Huang, M.M., Arnheim, N., and Erlich, H. 1995. New HLA-DPB1 alleles generated by interallelic gene conversion detected by analysis of sperm. Nat. Genet. 10: 407-414. [DOI] [PubMed] [Google Scholar]
- Zhao, Z., Jin, L., Fu, Y.X., Ramsay, M., Jenkins, T., Leskinen, E., Pamilo, P., Trexler, M., Patthy, L., and Jorde, L.B. 2000. Worldwide DNA sequence variation in a 10-kilobase noncoding region on human chromosome 22. Proc. Natl. Acad. Sci. 97: 11354-11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www-ab.informatik.uni-tuebingen.de/software/splits/welcome_en.html; SplitsTree software.