

Abstract
Fixed differences of chromosomal rearrangements between isolated populations may promote speciation by preventing between-population gene flow upon secondary contact, either because hybrids suffer from lowered fitness or, more likely, because recombination is reduced in rearranged chromosomal regions. This chromosomal speciation hypothesis thus predicts more rapid genetic divergence on rearranged than on colinear chromosomes because the former are less porous to gene flow. A number of studies of fungi, plants, and animals, including limited genetic data of humans and chimpanzees, support the hypothesis. Here we reexamine the hypothesis for humans and chimpanzees with substantially more genomic data than were used previously. No difference is observed between rearranged and colinear chromosomes in the level of genomic DNA sequence divergence between species. The same is also true for protein sequences. When the gorilla is used as an outgroup, no acceleration in protein sequence evolution associated with chromosomal rearrangements is found. Furthermore, divergence in expression pattern between orthologous genes is not significantly different for rearranged and colinear chromosomes. These results, showing that chromosomal rearrangements did not affect the rate of genetic divergence between humans and chimpanzees, are expected if incipient species on the evolutionary lineages separating humans and chimpanzees did not hybridize.
The chromosomal speciation hypothesis asserts that chromosomal rearrangements cause reproductive isolation between populations and lead to speciation (White 1978). The traditional model assumes that recombination between rearranged chromosomes generates unbalanced gametes that have lowered fitness, which creates a reproductive barrier (White 1978). This model has been criticized because if a rearrangement causes a substantial reduction in fitness of heterozygotes, it cannot be fixed in a population unless the population is very small (Walsh 1982; Lande 1985; Rieseberg 2001, and references therein). The newly developed model (Noor et al. 2001; Rieseberg 2001; Navarro and Barton 2003a) differs from the traditional one in assuming that recombination is suppressed in rearranged chromosomal regions and the fitness effect of rearrangements is minimal. As gene flow requires recombination in the presence of incompatibility genes, the reduction in recombination associated with chromosomal rearrangements results in lowered gene flow in rearranged chromosomal regions compared with colinear chromosomes and generates a partial reproductive barrier. Several studies in fungi, plants, and animals provided substantial evidence for the new model of chromosomal speciation (Rieseberg et al. 1999; Noor et al. 2001; Rieseberg 2001; Machado et al. 2002; Delneri et al. 2003). For instance, genetic loci responsible for hybrid male sterility and female species preferences in Drosophila pseudoobscura and D. persimilis, two occasionally hybridizing species, are located in rearranged chromosomal regions (Noor et al. 2001). A multilocus analysis of the same species pair also indicated reduced gene flow in rearranged chromosome regions (Marchado et al. 2002). The rate of introgression in colinear chromosomes between sunflowers Helianthus petiolaris and H. annuus are twice that in rearranged chromosomes (Rieseberg et al. 1999). Yeasts Saccharomyces cerevisiae and S. mikatae have reciprocal translocations involving three chromosomes, and their hybridization produces sterile progenies. Delneri et al. (2003) engineered the chromosomes of S. cerevisiae to make them colinear with those of S. mikatae. Interestingly, the interspecific hybrids are now fertile.
Nine chromosomes (1, 4, 5, 9, 12, 15, 16, 17, 18) contain pericentric inversions between humans and chimpanzees, and human chromosome 2 resulted from a fusion of two acrocentric chromosomes common to other great apes (Yunis and Prakash 1982). An examination of the influence of these chromosomal rearrangements on the genetic divergence between humans and chimpanzees may reveal the history of the speciation events that eventually led to modern humans and chimpanzees. Navarro and Barton (2003b) recently addressed this question and found some evidence for the effect of chromosomal rearrangements. Most strikingly, they found in an analysis of 115 genes of humans and chimpanzees that the rate of protein sequence evolution is 120% higher for genes located on rearranged chromosomes than for those on colinear chromosomes. However, their data were few, and some of the results were inconclusive (Navarro and Barton 2003b). Concerns have also been raised with regard to their data analysis and interpretation (Bowers 2003; Hey 2003; Lu et al. 2003). By using substantially larger genomic data sets, we here examine four predictions of the chromosomal speciation hypothesis for humans and chimpanzees. We show that there is no difference between rearranged and colinear chromosomes in the level of genomic DNA sequence divergence or protein-coding sequence divergence between species. When the gorilla is used as an outgroup, no acceleration in protein sequence evolution associated with chromosomal rearrangements is found. Divergence in expression pattern between orthologous genes is also similar for rearranged and colinear chromosomes. We discuss these results in the context of human and chimpanzee evolution.
RESULTS
Divergence of Genomic Sequences Between Humans and Chimpanzees
The chromosomal speciation hypothesis predicts that the level of neutral sequence divergence between species is higher for rearranged chromosomes than colinear chromosomes because gene flow between populations is hampered for rearranged chromosomes (Rieseberg 2001). To examine this prediction for humans and chimpanzees, we analyzed completely determined chimpanzee bacterial artificial chromosome (BAC) sequences and their human orthologs that are available in GenBank at the time of this study (July 2003). Because >98% of the human genome is noncoding (Venter et al. 2001), the BAC sequences can be effectively regarded as neutrally evolving sequences. After we aligned the orthologous sequences and removed gaps, our data contain ∼1.8 and 2.3 Mb (106 bases), located on seven of the 10 rearranged chromosomes and six of the 14 colinear chromosomes, respectively. We then computed the percentage of sequence divergence between the two species for regions on rearranged and colinear chromosomes. In contrast to the prediction of the chromosomal speciation hypothesis, the divergence is slightly lower on rearranged than colinear chromosomes, although their difference is not significant (P > 0.2, Z test; Table 1). The male-driven evolution hypothesis predicts a higher mutation rate for Y chromosome than autosomes (Li et al. 2002). Indeed, the divergence level is higher for Y than autosomes in our data (P < 0.001, Z test). Nevertheless, even when only autosomes are considered, the sequence divergence for rearranged chromosomes is still slightly lower than that for colinear chromosomes (P > 0.3; Table 1). Because the percentage of G and C nucleotides (GC%) in a chromosomal region may affect the mutation rate (Yi et al. 2002), we examined whether GC% is different between rearranged and colinear chromosomes but found no significant difference (P > 0.5, with or without Y data; Table 1). Furthermore, there is no significant correlation between GC% and sequence divergence in our data (R = -0.1, P > 0.2). Of the 36 BACs analyzed here, two are located within rearranged chromosomal regions (Table 1). The levels of sequence divergence for these two BACs (1.19% and 0.92%, respectively) are not higher than the average of the 36 BACs (1.49%). Our results contrast with those of Navarro and Barton (2003b). In that study, the investigators found significantly higher sequence divergence on rearranged chromosomes than on colinear ones using ∼450 kb (103 bases) of BAC sequences from five chromosomes, of which two (with a total of ∼82 kb) are rearranged chromosomes (Yi et al. 2002). The sequence divergences for the two rearranged chromosomes are the second and third highest, respectively, among the five chromosomes. When the variation in sequence divergence among BACs or chromosomes is considered, the statistical significance that they reported disappears (P > 0.3; Z test). The second data set analyzed by Navarro and Barton (2003b) includes ∼1.9 Mb of sequences (Ebersberger et al. 2002), which showed virtually identical levels of divergence for rearranged (1.25%) and colinear (1.23%) chromosomes (Navarro and Barton 2003b), as in the present case of ∼4.1 Mb sequences. It should be pointed out that although our data are substantially larger than what Navarro and Barton (2003b) had, it still only constitutes ∼0.14% of the genome. It will be interesting to verify our findings when the complete chimpanzee genome sequence becomes available.
Table 1.
Genomic DNA Sequence Divergence Between Humans and Chimpanzees
| Chimpanzee BACs | Corresponding human chromosomes | Length (nt)a | % Divergence for BAC | % Divergence for chromosome | GC% |
|---|---|---|---|---|---|
| Rearranged chromosomes | |||||
| ac092859 | 1 | 132,507 | 0.76 | 0.76 | 44.7 |
| ac097335 | 2 | 98,048 | 1.60 | 1.83 | 37.7 |
| ac120781 | 2 | 165,984 | 1.64 | 43.2 | |
| ac120782 | 2 | 82,520 | 1.99 | 46.7 | |
| ac122175 | 2 | 73,988 | 2.70 | 41.0 | |
| ac122731 | 2 | 83,811 | 2.04 | 42.1 | |
| ac125393 | 2 | 102,840 | 1.43 | 37.4 | |
| ac125392 | 4 | 163,428 | 0.90 | 0.90 | 37.5 |
| ac006582 | 12 | 136,913 | 1.17 | 1.18 | 37.6 |
| ac007214b | 12 | 93,824 | 1.19 | 38.2 | |
| ac123983 | 15 | 141,549 | 1.55 | 1.44 | 41.9 |
| ac120838 | 15 | 112,821 | 1.30 | 42.4 | |
| ac097265 | 16 | 140,954 | 1.23 | 1.35 | 47.9 |
| ac097268 | 16 | 179,913 | 1.28 | 45.5 | |
| ac113435 | 16 | 43,672 | 2.02 | 38.0 | |
| ac097264b | 17 | 78,904 | 0.92 | 0.92 | 46.3 |
| Subtotal | 1,831,676 | 1.20 ± 0.14 | 41.8 | ||
| Colinear chromosomes | |||||
| ac140949 | 7 | 141,018 | 1.16 | 1.19 | 40.5 |
| ac140950 | 7 | 171,457 | 1.34 | 37.8 | |
| ac140951 | 7 | 128,809 | 1.13 | 39.5 | |
| ac140952 | 7 | 169,376 | 0.97 | 38.3 | |
| ac140953 | 7 | 174,528 | 1.36 | 40.8 | |
| ac124148 | 10 | 149,881 | 1.34 | 1.30 | 42.8 |
| ac124219 | 10 | 12,520 | 2.52 | 39.2 | |
| ac125391 | 10 | 91,167 | 1.08 | 42.1 | |
| ac123982 | 19 | 109,950 | 1.28 | 1.28 | 44.6 |
| ac096630 | 20 | 126,851 | 1.35 | 1.35 | 44.1 |
| ac129098 | 22 | 104,792 | 1.47 | 1.56 | 47.3 |
| ac093571 | 22 | 72,564 | 1.68 | 46.3 | |
| ac093573 | 22 | 182,860 | 1.51 | 50.1 | |
| ac119407 | 22 | 101,211 | 1.53 | 46.4 | |
| ac123980 | 22 | 101,606 | 1.70 | 40.1 | |
| ac139189 | Y | 141,909 | 1.50 | 1.71 | 37.9 |
| ac139190 | Y | 91,398 | 1.61 | 36.4 | |
| ac139192 | Y | 76,179 | 1.94 | 38.0 | |
| ac139193 | Y | 25,946 | 1.47 | 39.5 | |
| ac139194 | Y | 103,135 | 1.97 | 39.0 | |
| Subtotal | 2,277,157 | 1.40 ± 0.08 | 41.5 | ||
| Subtotal (excluding Y) | 1,838,590 | 1.34 ± 0.06 | 42.6 | ||
| Total | 4,108,833 | 1.29 ± 0.09 | 41.6 |
Alignment length after the removal of gaps
Located within rearranged regions
Divergence of Protein-Coding Sequences Between Humans and Chimpanzees
If rearranged chromosomes are less prone to gene flow, it may be predicted that genes located in these chromosomes can accumulate species-specific adaptive changes more easily and rapidly, resulting in higher rates of protein evolution and more incidences of positive selection compared with those in colinear chromosomes (Navarro and Barton 2003b). Recently, Hey (2003) questioned the validity of this prediction from a theoretical point of view. Here we examined whether there is any empirical evidence for the prediction. Let dS be the number of synonymous nucleotide substitutions (s) divided by the number of potentially synonymous sites (S) between orthologous coding sequences of the human and chimpanzee, and dN be the corresponding number of nonsynonymous nucleotide substitutions (n) divided by the number of potentially nonsynonymous sites (N). Because dS is approximately equivalent to the mutation rate, dN/dS = [(n/N)/(s/S)] = [(n/s)/(N/S)] is a measure of the rate of protein evolution, standardized by the mutation rate (Nei and Kumar 2000). N/S is mainly determined by the ratio of the rate of transitional mutations to that of transversional mutations, which is approximately constant across the nuclear genome (Rosenberg et al. 2003). Thus, N/S is virtually invariant among different genes, and n/s can be used as a proxy of dN/dS when different genes are compared. We analyzed a recently published set of 1126 human–chimpanzee orthologous gene pairs (Hellmann et al. 2003). The chimpanzee sequences in this data set are randomly picked cDNAs from testis and brain cDNA libraries. Because individual cDNA sequences are short (average, ∼360 nucleotides), we concatenated the sequences belonging to the same chromosomes and estimated s and n between the human and chimpanzee for the concatenated sequences of each chromosome. The results (Table 2) show that, opposite to the prediction, the n/s ratio is slightly lower for the rearranged (288/596 = 0.483) than colinear chromosomes (287.5/531.5 = 0.541), with the difference being statistically insignificant (P > 0.1, Fisher's test). Similarly, the average dN/dS ratio for rearranged chromosomes (0.235 ± 0.028) is almost identical to that for colinear chromosomes (0.239 ± 0.019). The same results are obtained when only the autosomes are considered (Table 2). Interestingly, the average dS values for colinear and rearranged chromosomes are close (P > 0.4), indicating similar levels of neutral sequence divergence for the two sets of chromosomes, consistent with the above result from the BAC sequences.
Table 2.
Synonymous and Nonsynonymous Divergences Between Humans and Chimpanzees
| Chromosomes | No. of genes | Total length (nt) | n | s | n/s | dN × 100 | ds × 100 | dN/ds |
|---|---|---|---|---|---|---|---|---|
| Rearranged | ||||||||
| 1 | 104 | 39,495 | 54 | 119 | 0.45 | 0.197 | 0.989 | 0.199 |
| 2 | 83 | 31,017 | 43 | 71 | 0.61 | 0.202 | 0.733 | 0.276 |
| 4 | 50 | 17,919 | 27 | 54 | 0.50 | 0.216 | 0.993 | 0.218 |
| 5 | 56 | 21,402 | 35 | 49 | 0.71 | 0.232 | 0.775 | 0.299 |
| 9 | 33 | 14,163 | 24 | 63 | 0.38 | 0.248 | 1.407 | 0.176 |
| 12 | 67 | 25,017 | 34 | 74 | 0.46 | 0.199 | 0.932 | 0.214 |
| 15 | 70 | 14,052 | 10 | 43 | 0.23 | 0.104 | 0.974 | 0.107 |
| 16 | 35 | 11,307 | 25 | 45 | 0.56 | 0.321 | 1.281 | 0.251 |
| 17 | 56 | 22,179 | 30 | 72 | 0.42 | 0.195 | 1.056 | 0.185 |
| 18 | 14 | 5,904 | 6 | 6 | 1.00 | 0.145 | 0.338 | 0.429 |
| Subtotal | 568 | 202,455 | 288 | 596 | 0.483 | 0.206 ± 0.018 | 0.948 ± 0.093 | 0.235 ± 0.028 |
| Colinear | ||||||||
| 3 | 67 | 26,271 | 38 | 66 | 0.58 | 0.207 | 0.834 | 0.248 |
| 6 | 54 | 21,396 | 35 | 67 | 0.52 | 0.233 | 1.053 | 0.221 |
| 7 | 55 | 20,151 | 34 | 55 | 0.62 | 0.247 | 0.858 | 0.288 |
| 8 | 40 | 14,124 | 14 | 44 | 0.32 | 0.144 | 0.996 | 0.145 |
| 10 | 36 | 12,225 | 18 | 32 | 0.56 | 0.214 | 0.839 | 0.255 |
| 11 | 58 | 20,124 | 27 | 61 | 0.44 | 0.194 | 0.986 | 0.197 |
| 13 | 20 | 7,698 | 15 | 15 | 1.00 | 0.275 | 0.670 | 0.410 |
| 14 | 54 | 20,034 | 39.5 | 56.5 | 0.70 | 0.282 | 0.938 | 0.301 |
| 19 | 38 | 12,192 | 17 | 38 | 0.45 | 0.203 | 0.992 | 0.205 |
| 20 | 24 | 8,181 | 14 | 30 | 0.47 | 0.245 | 1.212 | 0.202 |
| 21 | 9 | 3,081 | 6 | 16 | 0.38 | 0.276 | 1.757 | 0.157 |
| 22 | 21 | 6,234 | 9 | 18 | 0.50 | 0.206 | 0.960 | 0.215 |
| X | 36 | 13,017 | 21 | 33 | 0.64 | 0.228 | 0.867 | 0.263 |
| Subtotal | 512 | 184,728 | 287.5 | 531.5 | 0.541 | 0.227 ± 0.011 | 0.997 ± 0.072 | 0.239 ± 0.019 |
| Subtotal (excluding X) | 476 | 171,711 | 266.5 | 498.5 | 0.535 | 0.227 ± 0.012 | 1.008 ± 0.078 | 0.237 ± 0.021 |
| Total | 1080 | 387,183 | 575.5 | 1127.5 | 0.510 | 0.218 ± 0.010 | 0.976 ± 0.057 | 0.237 ± 0.016 |
There are only two Y-linked genes in the data set, and they are not used here.
We also separated the 1126 cDNA sequences into two groups: those with dN/dS>1 and those with dN/dS<1. We limited our analysis to the sequences that have at least 150 codons, as dN/dS cannot be reliably obtained for short ones. A total of 367 genes were analyzed: 63 of them showed dN = dS = 0 and were excluded from further analysis. Surprisingly, we found that colinear chromosomes harbor disproportionately more genes with dN/dS>1 than rearranged chromosomes do (P < 0.01, Fisher's exact test; Table 3), opposite to the result of Navarro and Barton (2003b). These investigators analyzed 115 genes and found a 120% increase in dN/dS and significantly more genes with dN/dS > 1 for rearranged chromosomes compared with colinear ones. One may argue that the genes analyzed by us do not represent the entire genome equally because they are all expressed in either testis or brain. This appears an unnecessary concern as >40% of human genes are expressed in brain and >40% are expressed in testis (Su et al. 2002). On the other hand, because linked genes tend to have similar rates of protein evolution (Williams and Hurst 2000), it is possible that a small sample such as the one used by Navarro and Barton (2003b) is incidentally biased. One way to examine this possibility is to use another species. If high dN/dS ratios are due to chromosomal rearrangements between humans and chimpanzees, the high ratios should not be observed when the human is compared with another species such as the rhesus monkey. By using this strategy, Lu et al. (2003) found that those genes in Navarro and Barton's (2003b) data with high dN/dS also have high dN/dS between the human and another species, suggesting that the high dN/dS ratio is unrelated to chromosomal rearrangements. Navarro et al. (2003), however, contended that the same chromosomal regions are often rearranged over and over in different primates. They believe that Lu et al.'s observation supports the chromosomal speciation hypothesis rather than rejects it.
Table 3.
Effect of Chromosomal Rearrangements on the Proportion of Genes Under Positive Selection
|
Chromosomes
|
|||
|---|---|---|---|
| dN/ds | Rearranged | Colinear | Total |
| >1 | 23 | 46 | 69 |
| <1 | 125 | 110 | 235 |
| Total | 148 | 156 | 304 |
P = 0.003, Fisher's exact test.
Divergence of Protein-Coding Sequences Among the Human, Chimpanzee, and Gorilla
To resolve the above controversy, we compared dN/dS or n/s ratios for a set of genes before and after chromosomal rearrangements using the gorilla as an outgroup. Because the chromosomal rearrangements among the great apes are known (Yunis and Prakash 1982), there is no controversy as to whether high dN/dS ratios are attributable to chromosomal rearrangements. The 22 autosomes and two sex chromosomes of humans may be classified into four groups. On the unrooted tree of human, chimpanzee, and gorilla, two chromosomes (15 and 18) experienced rearrangements in either the human or chimpanzee branches, but not in the gorilla branch. They are classified as group A chromosomes. Ten chromosomes (3, 6, 11, 13, 19, 20, 21, 22, X, Y) did not experience rearrangements in any of the three branches and are classified as group B. Four chromosomes (7, 8, 10, 14) rearranged only in the gorilla branch, and they are group C. The remaining eight chromosomes (1, 2, 4, 5, 9, 12, 16, 17) are group D. Let nX and sX be the numbers of nonsynonymous and synonymous substitutions in branch X of the tree, where X = H, C, and G for the human, chimpanzee, and gorilla, respectively. Under the hypothesis that chromosomal rearrangements enhance dN/dS or n/s, we expect to see [(nH + nC)/(sH + sC)] > nG/sG for genes on group A chromosomes, because these chromosomes do not have rearrangements on the gorilla branch, but have rearrangements on either human or chimpanzee branches. Similarly, we expect [(nH + nC)/(sH+sC)] = nG/sG for group B genes and [(nH + nC)/(sH + sC)] < nG/sG for group C genes. These predictions can be tested with orthologous gene sequences from the human, chimpanzee, and gorilla. We searched the GenBank and identified 3, 35, and 15 genes, respectively, for group A, B, and C chromosomes. Because we are concerned with chromosomal rearrangements between humans and chimpanzees, the focus is the comparison between groups A and B. To have comparable sets of data for A and B, we sequenced 31 segments of coding sequences in chimpanzee and gorilla from group A chromosomes, bringing the total number of group A genes to 34. The results are shown in Table 4. It is seen that [(nH + nC)/(sH + sC)] is 6.6% higher than is nG/sG for group A chromosomes, but the difference is not significant (P > 0.4, Fisher's test). We then classified the genes on group A chromosomes to those that are located inside the rearranged regions and those outside. The [(nH + nC)/(sH + sC)] value is 3% higher than nG/sG for the former class, but 12% higher for the latter class, opposite to the prediction of the chromosomal speciation hypothesis. Nevertheless, in neither class is the difference between [(nH + nC)/(sH + sC)] and nG/sG statistically significant (P > 0.4). For group B chromosomes, [(nH + nC)/(sH + sC)] is 15% higher than is nG/sG, although the difference is again not significant (P > 0.2). Because group B genes do not have any chromosomal rearrangements among the human, chimpanzee, and gorilla, they serve as a baseline in the comparison between [(nH + nC)/(sH + sC)] and nG/sG. When this baseline is used, group A genes do not show elevated [(nH + nC)/(sH + sC)] over nG/sG. In other words, there is no evidence for an elevation in the rate of protein sequence evolution associated with the chromosomal rearrangements in humans and chimpanzees.
Table 4.
Synonymous and Nonsynonymous Divergences Among the Human, Chimpanzee, and Gorilla Genes
| Chromosomesa | Number of genes | Total length (nt) | nC | sG | nG/sG | nH + nC | sH + sC | (nH + nC)/(sH + sC) |
|---|---|---|---|---|---|---|---|---|
| Group A | 34b | 19,950 | 36 | 51 | 0.706 | 48 | 64 | 0.750 |
| Within rearranged regions | 14 | 8,199 | 12 | 18 | 0.667 | 22 | 32 | 0.688 |
| Outside rearranged regions | 20 | 11,751 | 24 | 33 | 0.727 | 26 | 32 | 0.813 |
| Group B | 35 | 29,382 | 81.5 | 85 | 0.956 | 121 | 110 | 1.100 |
See text for the classification of chromosomes
Thirty-one of the 34 genes are newly sequenced in this study
In the above analysis, we assumed that gorilla is the outgroup species of human and chimpanzee. Because the time interval between the separation of gorillas from the common ancestor of humans and chimpanzees and the separation of the later two species was relatively short (Chen and Li 2001), ancestral polymorphisms may not have been sorted out at the time of human–chimpanzee speciation. In result, for some genes, the divergence of the human and chimpanzee sequences may pre-date the divergence of either one with the gorilla sequence. But this should not affect our analysis because we considered the unrooted tree of human, chimpanzee, and gorilla, which is unique regardless of the presence or absence of ancestral polymorphisms.
Divergence of Gene Expression Between Humans and Chimpanzees
It may also be predicted by the chromosomal speciation hypothesis that divergence in gene expression patterns between species is greater for genes in rearranged chromosomes than those in colinear chromosomes, regardless of whether the divergence is neutral or adaptive. By using Affymetrix microarrays, Karaman et al. (2003) identified 219 genes that show differential expression levels in cultured fibroblasts among the human, pygmy chimpanzee, and gorilla. They did not find clustering of these genes in rearranged chromosomal regions. In their analysis, however, only those regions with finely mapped breakpoints were considered. We reanalyzed their data, considering all 10 rearranged chromosomes between humans and chimpanzees. The data include 138 genes that exhibit at least twofold difference in expression level between the human and chimpanzee, of which 75 are on rearranged chromosomes and 63 are on colinear chromosomes. Considering that the total length of the rearranged chromosomes is ∼49.3% of the entire genome (Venter et al. 2001), the frequency of gene-expression divergence per Mb is [(75/0.493)/(63/0.507)] = 1.23 times higher in rearranged chromosomes than in colinear chromosomes. But the difference is not significant (P > 0.1, binomial test). Considering that the total number of predicted genes in rearranged chromosomes is ∼48.6% of all genes (Venter et al. 2001), the frequency of expression divergence per gene is ∼[(75/0.486)/(63/0.514)] = 1.26 times higher in rearranged chromosomes than in colinear chromosomes. Again, this difference is not significant (P > 0.1, binomial test). Note that the Y chromosome is not considered in the above analysis because the data do not include any Y-linked genes.
We also analyzed a large set of gene expression data obtained from cortex samples of five humans and four chimpanzees (Caceres et al. 2003). In these data, the expression patterns of ∼10,000 genes were first examined by Affymetrix oligonucleotide arrays, and the genes showing different expressions between species were confirmed by cDNA arrays and real-time RT-PCR. One hundred fifty-two genes were identified and confirmed to have significant expression differences between human and chimpanzee cortexes, among which 80 were located on rearranged chromosomes. The frequency of gene-expression divergence per Mb is about [(80/0.493)/(72/0.507)] = 1.14 times higher in rearranged chromosomes than in colinear chromosomes, their difference being not statistically significant (P > 0.2, binomial test). The frequency of expression divergence per gene is ∼[(80/0.486)/(72/0.514)] = 1.18 times higher in rearranged chromosomes than in colinear chromosomes. Again, this difference is not statistically significant (P > 0.1, binomial test).
DISCUSSION
Statistical Power
Our analyses of BAC sequences, protein-coding DNA sequences, and gene expression data showed no significant elevation in the rate of genetic divergence between the human and chimpanzee on rearranged chromosomes compared with colinear ones. One question is whether our analyses are of sufficient statistical power to detect the differences if they indeed exist. For the genomic sequence data, we noticed that there is substantial variation in human–chimpanzee sequence divergence among BACs and chromosomes, which is consistent with earlier observations of mutation rate variation among genomic regions (for review, see Ellegren et al. 2003). In our data, the average human–chimpanzee divergence is 1.29%, and the standard error for the difference between the average divergence of rearranged chromosomes and that of colinear chromosomes is 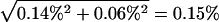 (Table 1). Thus, a 19% increase in the genomic sequence divergence on rearranged chromosomes would result in a Z value of 1.29% × 19%/0.15% = 1.65 in the standard Z test, giving a significance level of 5%. In other words, our BAC data can detect a 19% difference in human–chimpanzee divergence between rearranged and colinear chromosomes. The statistical power, however, is irrelevant here, because the divergence in colinear chromosomes is higher than that in rearranged chromosomes.
(Table 1). Thus, a 19% increase in the genomic sequence divergence on rearranged chromosomes would result in a Z value of 1.29% × 19%/0.15% = 1.65 in the standard Z test, giving a significance level of 5%. In other words, our BAC data can detect a 19% difference in human–chimpanzee divergence between rearranged and colinear chromosomes. The statistical power, however, is irrelevant here, because the divergence in colinear chromosomes is higher than that in rearranged chromosomes.
Similarly, for the cDNA data, the average dN/dS ratio is 0.237, and the standard error for the difference in dN/dS between rearranged and colinear chromosomes is 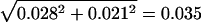 . A 24% increase in dN/dS on rearranged chromosomes would result in a Z value of 0.237 × 24%/0.035 = 1.65 in the standard Z test, giving a significance level of 5%. That is, our cDNA analysis can detect a 24% difference in dN/dS between rearranged and colinear chromosomes. Again, the statistical power is irrelevant here, because dN/dS in colinear chromosomes is higher than that in rearranged chromosomes.
. A 24% increase in dN/dS on rearranged chromosomes would result in a Z value of 0.237 × 24%/0.035 = 1.65 in the standard Z test, giving a significance level of 5%. That is, our cDNA analysis can detect a 24% difference in dN/dS between rearranged and colinear chromosomes. Again, the statistical power is irrelevant here, because dN/dS in colinear chromosomes is higher than that in rearranged chromosomes.
Our further analysis of the cDNA data showed significantly fewer genes with dN/dS > 1 on rearranged chromosomes than expected. This is opposite to what Navarro and Barton (2003b) observed. We think that the classification of genes into groups with dN/dS > 1 and dN/dS < 1 is not biologically meaningful, because for most of the genes analyzed here the numbers of substitutions are so small that the dN/dS ratio is subject to substantial stochastic error. For any individual gene, the dN/dS ratio may not reliably indicate the form and strength of natural selection. For this reason, we believe that the results obtained from the concatenated sequences are more reliable.
Our analyses of the two sets of gene expression data did show relatively higher expression divergences for genes located on rearranged chromosomes than those located on colinear ones, but their differences are not statistically significant. For the data set of Karaman et al. (2003), we estimated that doubling the sample size would result in a statistically significant result (at P = 0.03, binomial test) if the proportion of expression-divergent genes found on rearranged chromosomes is the same as estimated here (i.e., 75/138 = 0.543). For the data set of Caceres et al. (2003), a sample size that is 2.5 times of current size would result in a statistically more or less significant result (at P = 0.06) if the proportion of expression-divergent genes found on rearranged chromosomes is the same as estimated here (i.e., 80/152 = 0.526). Thus, further studies using larger data sets may help resolve the problem.
Previous studies of Drosophila and sunflowers suggested that chromosomal rearrangements reduce gene flow by 40% to 50% (Rieseberg et al. 1999; Machado et al. 2002). This level of difference would have been detected in several of our analyses. We believe that the consistent findings of no significant elevation in the rate of genetic divergence between the human and chimpanzee on rearranged chromosomes are not due to the lack of statistical power.
Implications and Conclusions
The absence of virtually any signature of 10 major chromosomal rearrangements on the rate of genetic divergence suggests that the speciation processes in the evolutionary lineages separating humans and chimpanzees are substantially different from what the chromosomal speciation hypothesis presumes. In particular, the preventive effect of chromosomal rearrangements on gene flow occurs during the secondary contact of populations after an initial period of isolation, but ceases upon the establishment of complete reproductive isolation. Given the number of extinct hominid species described so far (Boyd and Silk 2000; Brunet et al. 2002) and the presence of at least two chimpanzee species, the speciation events on the evolutionary lineages that eventually led to modern humans and chimpanzees may be numerous. Our results thus imply that hybridization has been rare among incipient species on these lineages. It is noteworthy that chimpanzees lack a fossil record (Kelly 1992; Boyd and Silk 2000). This phenomenon suggests that early hominids and chimpanzees probably did not live in proximity (Kelly 1992) because otherwise chimpanzee fossils should be excavated along with hominid fossils. The current distribution of chimpanzees only has a small overlap with the distribution of the early hominid fossils around the Rift Valley in East Africa (Kelly 1992; Gagneux et al. 2001; Brunet et al. 2002). Although it is difficult to infer the historical geographic distribution of chimpanzees without a fossil record, we note that chimpanzees generally favor tropical rain forests whereas hominids lived mostly in open forests and savannas. This difference in their favored ecological environments may have prohibited their secondary contact and hybridization. (Of course, one cannot exclude the possibility that some early hominids also lived in rain forests with chimpanzees, and their fossils are likewise unknown.) Hybridization may also be rare if prezygotic isolation had already evolved when the secondary contact occurred. It is worth noting that the four recognized subspecies of common chimpanzees (P. troglodytes schweinfurthii, P. t. troglodytes, P. t. vellerosus, and P. t. verus) are not known to form hybrid zones, nor do they hybridize with the pygmy chimpanzee (Pan paniscus) in nature. Although this phenomenon is probably due to current geographical isolation, the historical pattern may be assessed by population genetic analysis. Analyses of paternally inherited Y-linked genetic markers and maternally inherited mitochondrial DNA markers show that alleles from each subspecies generally form a monophyletic group (Gagneux et al. 1999; Stone et al. 2002), consistent with lack of hybridization across subspecies. By contrast, autosomal and X-linked markers show intermingling of alleles from different subspecies, a likely result from incomplete lineage sorting and indicative of relatively short separation times (Kaessmann et al. 1999; Deinard and Kidd 2000). A three- to fourfold greater effective population size and longer coalescent time for the bi-sexually transmitted markers than the unisexual markers (Hartl and Clark 1997) can explain the above disparity. Thus, the circumstantial paleontological, ecological, and population genetic evidence is consistent with our genomic data. Together, they suggest that chromosomal rearrangements did not affect the rate of genetic divergence between humans and chimpanzees, and this is likely due to the lack of hybridization among incipient species on the evolutionary lineages separating humans and chimpanzees.
METHODS
Completely sequenced and assembled BAC sequences of the chimpanzee (P. troglodytes) were downloaded from GenBank and BLASTed (Altschul et al. 1990) against the human genome sequence to find their human orthologs. We analyzed those cases in which the best hit and query overlap for at least 90% of the query sequence. Segments of BLAST alignments between the query and the best hit with lengths >10 kb were subsequently analyzed.
The 1126 alignments of human and chimpanzee cDNA sequences were from Hellmann et al. (2003), and the gene locations were determined from BLAST and MapViewer searches (http://www.ncbi.nlm.nih.gov/mapview/). Y-linked genes were not analyzed because there are only two of them. We also removed those genes with chromosomal locations that cannot be confidently placed or with sequence lengths <50 nucleotides. The final data include 1080 genes. The modified Nei-Gojobori method (Zhang et al. 1998) was used for the analysis of synonymous and nonsynonymous substitutions.
Thirty-one segments of coding regions each with ∼550 nucleotides were randomly chosen from group A chromosomes (see main text), with the only requirement being that each segment is fully contained within an exon at least 600 nucleotides long. The 31 segments were from 31 different genes. Primers were designed following the human sequences. The chimpanzee (P. troglodytes) and gorilla (Gorilla gorilla) orthologs were PCR amplified and sequenced in both directions. Other genes with orthologous sequences of the human, chimpanzee, and gorilla available in the GenBank were downloaded and analyzed. A list of all the genes is in Supplementary Table 1. The numbers of nucleotide substitutions on branches of the tree of human, chimpanzee, and gorilla were estimated from pairwise nucleotide distances using the least-squares method (see Nei and Kumar 2000).
Acknowledgments
We thank Svante Paabo for sharing the 1126 cDNA sequences and Douglas Futuyma and David Webb for valuable comments. This work was supported by a start-up fund of the University of Michigan and a National Institutes of Health grant (GM67030) to J.Z.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1891104.
Footnotes
[Supplemental material is available online at www.genome.org. The DNA sequences from this study have been submitted to GenBank under accession nos. AY561437–AY561498. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: S. Paabo.]
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- Bowers, E.J. 2003. Chromosomal speciation. Science 301: 764-765. [DOI] [PubMed] [Google Scholar]
- Boyd, R. and Silk, J.B. 2000. How humans evolved. Norton, New York.
- Brunet, M., Guy, F., Pilbeam, D., Mackaye, H.T., Likius, A., Ahounta, D., Beauvilain, A., Blondel, C., Bocherens, H., Boisserie, J.R., et al. 2002. A new hominid from the Upper Miocene of Chad, Central Africa. Nature 418: 145-151. [DOI] [PubMed] [Google Scholar]
- Caceres, M., Lachuer, J., Zapala, M.A., Redmond, J.C., Kudo, L., Geschwind, D.H., Lockhart, D.J., Preuss, T.M., and Barlow, C. 2003. Elevated gene expression levels distinguish human from non-human primate brains. Proc. Natl. Acad. Sci. 100: 13030-13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F.C. and Li, W.H. 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68: 444-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinard, A.S. and Kidd, K. 2000. Identifying conservation units within captive chimpanzee populations. Am. J. Phys. Anthropol. 111: 25-44. [DOI] [PubMed] [Google Scholar]
- Delneri, D., Colson, I., Grammenoudi, S., Roberts, I.N., Louis, E.J., and Oliver, S.G. 2003. Engineering evolution to study speciation in yeasts. Nature 422: 68-72. [DOI] [PubMed] [Google Scholar]
- Ebersberger, I., Metzler, D., Schwarz, C., and Paabo, S. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 70: 1490-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren, H., Smith, N.G., and Webster, M.T. 2003. Mutation rate variation in the mammalian genome. Curr. Opin. Genet. Dev. 13: 562-568. [DOI] [PubMed] [Google Scholar]
- Gagneux, P., Wills, C., Gerloff, U., Tautz, D., Morin, P.A., Boesch, C., Fruth, B., Hohmann, G., Ryder, O.A., and Woodruff, D.S. 1999. Mitochondrial sequences show diverse evolutionary histories of African hominoids. Proc. Natl. Acad. Sci. 96: 5077-5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux, P., Gonder, M.K., Goldberg, T.L., and Morin, P.A. 2001. Gene flow in wild chimpanzee populations: What genetic data tell us about chimpanzee movement over space and time. Philos. Trans. R. Soc. Lond. B. 356: 889-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl, D.L. and Clark, A.G. 1997. Principles of population genetics. Sinauer, Sunderland, MA.
- Hellmann, I., Zollner, S., Enard, W., Ebersberger, I., Nickel, B., and Paabo, S. 2003. Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res. 13: 831-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey, J. 2003. Speciation and inversions: Chimps and humans. Bioessays 25: 825-828. [DOI] [PubMed] [Google Scholar]
- Kaessmann, H., Wiebe, V., and Paabo, S. 1999. Extensive nuclear DNA sequence diversity among chimpanzees. Science 286: 1159-1162. [DOI] [PubMed] [Google Scholar]
- Karaman, M.W., Houck, M.L., Chemnick, L.G., Nagpal, S., Chawannakul, D., Sudano, D., Pike, B.L., Ho, V.V., Ryder, O.A., and Hacia, J.G. 2003. Comparative analysis of gene-expression patterns in human and African great ape cultured fibroblasts. Genome Res. 13: 1619-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, J. 1992. Evolution of apes. In The cambridge encyclopedia of human evolution (eds. S. Jones, et al.), pp. 223-230. Cambridge University Press, Cambridge.
- Lande, R. 1985. The fixation of chromosomal rearrangements in a subdivided population with local extinction and colonization. Heredity 54: 323-332. [DOI] [PubMed] [Google Scholar]
- Li, W.H., Yi, S., and Makova, K. 2002. Male-driven evolution. Curr. Opin. Genet. Dev. 12: 650-656. [DOI] [PubMed] [Google Scholar]
- Lu, J., Li, W.H., and Wu, C.I. 2003. Comment on “Chromosomal speciation and molecular divergence-accelerated evolution in rearranged chromosomes.” Science 302: 988. [DOI] [PubMed] [Google Scholar]
- Machado, C.A., Kliman, R.M., Markert, J.A., and Hey, J. 2002. Inferring the history of speciation from multilocus DNA sequence data: The case of Drosophila pseudoobscura and close relatives. Mol. Biol. Evol. 19: 472-488. [DOI] [PubMed] [Google Scholar]
- Navarro, A. and Barton, N.H. 2003a. Accumulating postzygotic isolation genes in parapatry: A new twist on chromosomal speciation. Evolution 57: 447-459. [DOI] [PubMed] [Google Scholar]
- ———. 2003b. Chromosomal speciation and molecular divergence-accelerated evolution in rearranged chromosomes. Science 300: 321-324. [DOI] [PubMed] [Google Scholar]
- Navarro, A., Marquès-Bonet, T., and Barton, N.H. 2003. Response to comment on “Chromosomal speciation and molecular divergence-accelerated evolution in rearranged chromosomes.” Science 302: 988. [DOI] [PubMed] [Google Scholar]
- Nei, M. and Kumar, S. 2000. Molecular evolution and phylogenetics. Oxford University Press, New York.
- Noor, M.A., Grams, K.L., Bertucci, L.A., and Reiland, J. 2001. Chromosomal inversions and the reproductive isolation of species. Proc. Natl. Acad. Sci. 98: 12084-12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg, L.H. 2001. Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16: 351-358. [DOI] [PubMed] [Google Scholar]
- Rieseberg, L.H., Whitton, J., and Gardner, K. 1999. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152: 713-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, M.S., Subramanian, S., and Kumar, S. 2003. Patterns of transitional mutation biases within and among mammalian genomes. Mol. Biol. Evol. 20: 988-993. [DOI] [PubMed] [Google Scholar]
- Stone, A.C., Griffiths, R.C., Zegura, S.L., and Hammer, M.F. 2002. High levels of Y-chromosome nucleotide diversity in the genus Pan. Proc. Natl. Acad. Sci. 99: 43-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, A.I., Cooke, M.P., Ching, K.A., Hakak, Y., Walker, J.R., Wiltshire, T., Orth, A.P., Vega, R.G., Sapinoso, L.M., Moqrich, A., et al. 2002. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. 99: 4465-4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- Walsh, J.B. 1982. Rate of accumulation of reproductive isolation by chromosomal rearrangement. Am. Nat. 120: 510-532. [Google Scholar]
- White, M.J.D. 1978. Modes of speciation. Freeman, San Francisco.
- Williams, E.J. and Hurst, L.D. 2000. The proteins of linked genes evolve at similar rates. Nature 407: 900-903. [DOI] [PubMed] [Google Scholar]
- Yi, S., Ellsworth, D.L., and Li, W.H. 2002. Slow molecular clocks in Old World monkeys, apes, and humans. Mol. Biol. Evol. 19: 2191-2198. [DOI] [PubMed] [Google Scholar]
- Yunis, J.J. and Prakash, O. 1982. The origin of man: A chromosomal pictorial legacy. Science 215: 1525-1530. [DOI] [PubMed] [Google Scholar]
- Zhang, J., Rosenberg, H.F., and Nei, M. 1998. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc. Natl. Acad. Sci. 95: 3708-3713. [DOI] [PMC free article] [PubMed] [Google Scholar]