

Abstract
Preterm birth (PTB) complicates more than 12% of all deliveries. Despite significant research, the aetiology of most cases of PTB remains elusive. Two major antecedents of PTB, intra-amniotic infection and decidual haemorrhage (abruption), can exhibit dissimilar demographic and genetic predispositions, despite sharing common molecular and cellular pathways. The use of high-throughput, high-dimensional technologies reveals substantial crosstalk between the coagulation and inflammation pathways. Tissue factor, thrombin and cytokines are key mediators of this crosstalk. Abruptions are associated with excess thrombin generated from decidual-cell-expressed tissue factor. Although thrombin is a primary mediator of the coagulation cascade, it can also promote inflammation-associated PTB by enhancing expression of matrix metalloproteinase and neutrophil-chemoattracting and -activating chemokines. Here, we provide novel insights into the molecular mechanisms and pathways leading to PTB in the setting of placental abruption.
The magnitude of the public health problem posed by preterm birth (PTB) is underscored by reports that approximately 540 000 premature infants are born each year and more than $26 billion is spent annually to treat them. This figure is a highly conservative estimate because it does not include rehabilitation or long-term care costs. Early PTB (less than 34 weeks of gestation) accounts for almost 70% of neonatal deaths and 75% of neonatal morbidity (Ref. 1). The US National Institutes of Health made translational medicine a priority (Refs 2, 3). In this context, there is a pressing need to develop novel biomarkers and technologies to predict pregnancies at risk for PTB. The development of targeted primary and secondary prevention strategies and treatments to prevent PTB is vital.
Pregnancy is a critical time during a woman's reproductive life because of the unique adaptation to a new physiological challenge and defiance of the immunological rules of rejection by the placenta and developing fetus. Scientists have investigated ‘the timing of birth’ along with the development, physiology and pathophysiology of the fetus and its environment for many years (Refs 4, 5, 6). Unfortunately the molecular mechanisms set in place to prevent the inappropriate activation of the uterine contractile machinery and PTB continue to be incompletely understood (Ref. 7).
There is compelling evidence to suggest that the PTB syndrome involves several underlying pathogenetic pathways and aetiological factors (Ref. 8). Four distinct pathways probably account for most spontaneous PTBs: (1) maternal or fetal stress, (2) excessive myometrial and cervical stretching, (3) decidual haemorrhage (abruption) and (4) infection or exaggerated inflammatory response to genital tract microbes (Ref. 9). Each has unique genetic and environmental predispositions and most have immunologically mediated or inflammatory components. Understanding of the role of polymorphic maternal–fetal ‘incompatibility’ in triggering PTB and the role of environmental factors in this complex equation is still in its infancy (Ref. 10).
The diverse pathways fuelling PTB-associated uterine contractility, cervical ripening, rupture of membranes and fetal damage have been previously reviewed (Ref. 9). The multiple iterative pathways leading to prematurity and the complex interactions among relevant mechanisms obscure the triggering event for most PTBs. However, irrespective of the initial stimulus, it is believed that inflammation is a unifying signalling pathway leading to most term births and PTBs (Refs 6, 11, 12, 13). Here, we will focus on abruption-induced PTB.
Decidual bleeding (abruption)
Clinical aspects
Placental abruption is defined as complete or partial separation of the placenta from the implantation site (Ref. 14). Its incidence ranges from 0.5% to 2%, depending on the clinical definition and criteria used to characterise abruption intensity (Refs 15, 16), whereas abruption-associated maternal and fetal deaths occur in <1% and 3–10% of cases, respectively (Refs 14, 17). Other major maternal sequelae are renal failure, disseminated intravascular coagulation, transfusion and hysterectomy. The occurrence of abruption peaks at 24–27 weeks of gestation (Ref. 18), with more than 50% of abruption cases associated with PTB (Ref. 17). Consequently, the high rate of abruption-related perinatal mortality of approximately 119 cases per 1000 births is heavily confounded by prematurity (Ref. 19). Neonatal risks of prematurity include intraventricular haemorrhage, bronchopulmonary dysplasia, retinopathy prematurity, deafness and cerebral palsy. Thus, in addition to fetal hypoxic ischaemic injury, abruption increases the likelihood of short- and long-term morbidities for surviving babies.
In cases of abruption, the extent of retroplacental bleeding has a significant impact on the clinical manifestations of this condition (Ref. 19). The classic clinical presentation of acute abruption includes vaginal bleeding, uterine contractions and abnormal fetal heart rate patterns. Concealed abruption, when bleeding is not clinically evident, is of particular concern and presents a challenging diagnostic scenario. In these situations, the diagnosis of placental abruption is generally established intra- or postpartum based on fresh macroscopic or histological examination of the placenta (Refs 15, 20).
Pathophysiology of placental abruption
The haemostatic milieu of human decidua is integral to normal human implantation and placental development (Refs 9, 21). Survival of the blastocyst and embryo, and successful development of the fetus require that extravillous endovascular cytotrophoblasts (EVTs) gain access to the maternal circulation by penetrating the wall of the uterine spiral arteries without inciting haemorrhage (Ref. 22). This process is gradual and well coordinated and provides the developing maternal–fetal unit with a vital source of oxygen and nutrients. Disarray of the highly controlled and synchronised molecular mechanisms of placenta formation at the maternal–fetal interface increases the risk of haemorrhage, leading to both abortion and abruption. The key histological finding in placental abruption is haemorrhage in the decidua basalis (Ref. 14). Such haemorrhage is thought to result from pathological processes damaging the vascular endothelium at the uteroplacental interface (Refs 15, 20, 23).
Placental abruption is traditionally viewed as an acute event. However, histological evaluation of the vasculopathy accompanying decidual haemorrhage provides compelling evidence that the damage is frequently chronic (Refs 15, 20, 24, 25). Indeed, the most frequently encountered histopathological lesions co-associated with chronic decidual bleeding were chronic deciduitis and villitis, hypovascularity, infarct, necrosis or fibrosis of the placenta, blood vessels with absent physiological changes, vascular thrombosis and increased numbers of circulating nucleated erythrocytes (Ref. 20). In addition, a recent study found that acute lesions of chorioamnionitis and funisitis were frequently associated with histological evidence of abruption (haematoma, fibrin deposition, compressed villi and haemosiderin-laden histiocytes) (Ref. 15).
For an adequate understanding of the molecular mechanisms and pathways governing the process of abnormal decidual bleeding, we will first outline the crucial roles of vascular endothelium and haemostatic balance during pregnancy. We will further discuss several mechanisms responsible for the crosstalk between coagulation and inflammation, with special emphasis on abruption. Finally, we will concentrate on genetic risk factors, progesterone and tissue factor (TF)-generated thrombin that modulate the crosstalk between coagulation and inflammation leading to abruption and PTB.
Physiological roles of vascular endothelium and haemostatic balance during pregnancy
Vascular endothelium
Maintenance of pregnancy requires the proper functioning of molecular mechanisms responsible for regulating endothelial permeability and circulatory homeostasis in decidual, placental, umbilical and fetal vessels (Refs 26, 27, 28, 29, 30). In addition to its role as a mechanical barrier, the vascular endothelium maintains normal blood fluidity and vessel wall permeability, regulates leukocyte trafficking (Refs 26, 27, 31) and maintains strict control of the coagulation pathways (Ref. 32).
Tissue factor
TF is a member of a cytokine receptor superfamily comprising a hydrophilic extracellular domain, a transmembrane hydrophobic domain and an intracellular tail (Refs 32, 33, 34). Binding of the extracellular domain of TF to plasma-derived factor VII (FVII) or its active form, FVIIa, markedly enhances the catalytic activity (Fig. 1) (Ref. 35). The TF–FVIIa complex activates FX and FIX, which ultimately generate thrombin that cleaves fibrinogen to fibrin and activates the platelets. Consequently TF acts as the primary initiator of coagulation and is at the centre of the crosstalk between coagulation and inflammation pathways.
Figure 1. Extrinsic and intrinsic pathways of the coagulation cascade.

Tissue factor (TF) is recognised as the main initiator of the coagulation cascade. Once formed, the TF–FVIIa complex initiates clotting by activating both FX and FIX thus, TF carries a unique ability to act through both intrinsic and extrinsic pathways of coagulation. Activated FIX binds to its cofactor VIIIa, to activate FX. Thus, the FVIIIa–FIXa complex of the intrinsic pathway provides an alternative route to generate FXa, which participates in the prothrombinase complex (FVa–FXa). The latter complex activates prothrombin to thrombin, which has a central role in the coagulation protease cascade. Thrombin, in turn, activates FXI, which is an alternative way of generating FIXa. Thrombin also activates FXIII, which stabilised fibrin polymers, but thrombin's most crucial functions are to cleave fibrinogen to fibrin and stimulate platelets by the cleavage of protease-activated receptors. The end result is haemostasis and thrombosis. Abbreviations: FI, factor I; FII, factor II; FIII, factor III; FV, factor V; FVII, factor VII; FIX, factor IX; FX, factor X.
Tight control of TF gene (F3) expression is crucial to maintain the balance between blood fluidity and haemostasis (Ref. 36). The mechanisms in control of TF gene expression and its biology have recently been evaluated (Refs 37, 38, 39). However, several aspects should be emphasised. First, inhibition of the TF-FVIIa proteolytic activity by specific plasma and vascular endothelium TF pathway inhibitors (TFPIs) is a crucial protective mechanism against aberrant coagulation (i.e. thrombosis) (Refs 37, 40, 41). The haemostatic balance between TF and TFPI seems to be operational in the human placenta and endothelial cells of the umbilical vein (Ref. 42). Second, TF is constitutively expressed on the cell surface of mesenchymal and epithelial cells localised in the brain, heart, kidney, lung, uterus and placenta (Ref. 35). However, as demonstrated by immunohistochemical studies, endothelial cells that are in contact with the circulation do not normally express TF as a surface antigen (Refs 26, 43). Given that TF is primarily localised and sequestered within the vascular adventitia and tissue parenchymal cells, clotting factors cannot penetrate the intact endothelial wall to initiate the coagulation pathway. Thus, the vascular endothelium serves as a haemostatic ‘envelope’ (Ref. 44).
The expression and functional role of TF in human pregnancy have been previously studied (Ref. 45). In humans, decidual-cell-expressed TF prevents haemorrhage, initially as blastocyst-derived EVTs breach decidual capillaries to establish the fetal–maternal circulation (Ref. 9). Subsequently, TF continues to have a crucial role during placenta formation because EVTs mediate decidual vessel remodelling to increase blood flow to the developing fetal–placental unit (Ref. 10). At the maternal–fetal interface at term, immunoreactive TF levels are highest in decidual cells, with the trophoblast virtually devoid of TF immunostaining (Fig. 2) (Ref. 46). These observations were also made in idiopathic PTB specimens (Ref. 11). Thus, we can conclude that decidual-cell-expressed TF is strategically positioned at the maternal–fetal interface to meet the haemostatic demands of parturition (Ref. 46).
Figure 2. Immunohistochemical analysis of tissue factor expression at the decidual–placental interface.
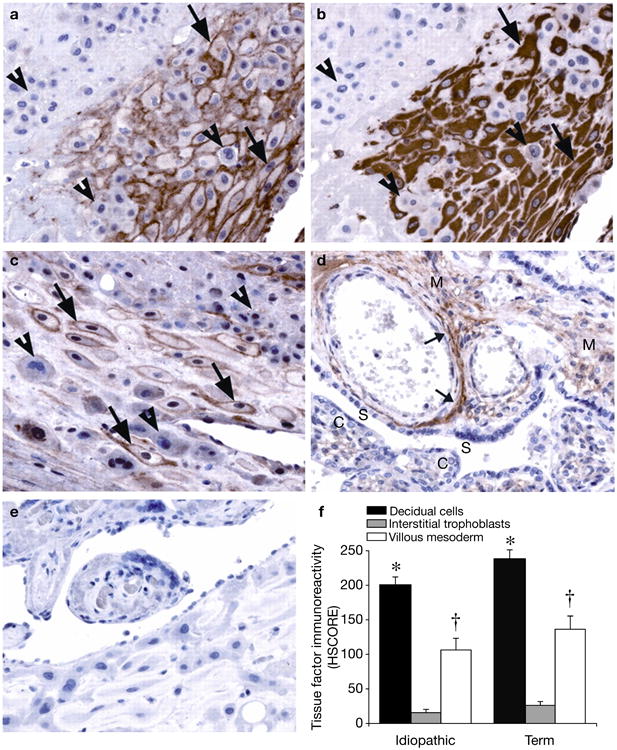
Serial sections of decidual basalis specimens immunostained for tissue factor (TF) and vimentin, as shown in an idiopathic preterm specimen: (a) TF and (b) vimentin. Decidual cells (arrows), identified by positive vimentin staining, exhibit strong perimembranous TF staining. TF staining is absent in the interstitial trophoblast (arrowheads). Similar results are seen in term specimens (c). Placental villi show moderate TF staining in the mesoderm (M), in which the staining is primarily localised to perivascular adventitia (small arrows), as shown in a preterm specimen (d). The syncytiotropoblast (S) and cytotrophoblast (C) show no TF staining. Similar results are seen in term specimens (not shown). Negative control immunostaining using a nonspecific isotype-matched antibody revealed no positive signals (e). TF staining intensity (f) [bars indicate histological score (HSCORE) mean ± s.e.m.] is highest in decidual cells (*P<0.05, versus interstitial trophoblast and the villous mesoderm of the same specimen group), with moderate staining in the villous mesoderm (†P<0.05, versus interstitial trophoblast of the same specimen group). There are no significant differences in HSCORE values between specimen groups. Published with permission from Ref. 46 (© 2009, The Endocrine Society).
A relevant question relates to the stimulus driving the decidual expression of TF in pregnancy. There are data to suggest that both progesterone and oestrogen have key modulatory roles (Refs 47, 48). The concerted actions of oestradiol and progesterone induce decidualisation of stromal cells derived from the human endometrium (Refs 47, 48). During decidualisation, the stromal cells begin to express TF. However, the absence of oestrogen or progesterone receptor (PR) recognition motifs on the TF gene promoter region prompted research aimed at exploring the involvement of other paracrine factors (Ref. 49). Indeed, it was shown that progestin-enhanced TF expression ultimately requires activation of the epidermal growth factor receptor pathway (Ref. 49). Moreover, in term human decidual cell monolayers, TF mRNA and protein expression were enhanced by incubation with exogenous progestin.
Observations on transgenic mice indicate that TF is crucial in pregnancy
No TF-deficient condition has been described in humans (Ref. 39). This emphasises the importance of TF expression to species survival while obviating the possibility of direct assessment of the effects of human TF deficiency. Adult mice that express low levels of TF suffer from haemostatic defects in the heart, lung, uterus and placenta, which are tissues that constitutively express high levels of TF (Ref. 50). Pregnant mice with reduced TF expression experience blood pools in the placental labyrinth and are highly susceptible to postpartum haemorrhage and death (Ref. 50). The importance of TF expression to survival is underscored by the observation that fetuses of murine TF gene knockouts develop fragile aberrant vessels and die of haemorrhage in utero (Ref. 50). However, incorporation of a human minigene expressing TF at only 1% of the wild-type level was sufficient to rescue these TF-knockout mice and enable them to produce live-born pups.
Antithrombotic mechanisms and TF
In addition to TFPIs, several other physiological mechanisms block the aberrant activation of coagulation by TF. Among these are the following: (1) a blood flow profile consistent with high laminar shear stress (Ref. 51); (2) cell-surface expression of heparin sulfate proteoglycans, which bind to antithrombin to greatly enhance thrombin inactivation (Ref. 52); (3) expression of thrombomodulin, a membrane protein that shifts the specificity of thrombin from a procoagulant converter of fibrinogen to fibrin to an anticoagulant activator of protein C (Ref. 53); (4) activated protein C and its cofactor protein S that inactivate FVa and FVIIIa, which are key cofactors of FXa and FIXa, respectively (Ref. 54); and (5) maintenance of platelets in a quiescent state by preventing endothelial cell activation (Refs 26, 55). All these mechanisms are present in uteroplacental circulation and have functionally relevant roles in the human placenta and in normal fetal development (Refs 56, 57).
Adaptive versus maladaptive inflammation
Inflammation is a highly coordinated process designed to guard and ensure the survivability of vertebrate organisms (Refs 58, 59). When properly controlled, the inflammatory response is beneficial to the host, but when occurring in disarray its effects become detrimental (Ref. 60). Traditionally, it was thought that infection, bleeding and trauma that lead to tissue injury are the primary triggers of inflammation. It is thus not surprising to learn that substantial progress has been made in understanding the inflammatory response to the above three factors. For instance, studies of sepsis and haemorrhagic shock in humans and animal models show that both conditions are characterised by the acute release of a variety of inflammatory mediators, which can result in damaging effects on the host (Refs 61, 62). What we have learned from both conditions is that the balance between the expression of pro- and anti-inflammatory molecules determines the magnitude of the damage.
Physiological versus pathological inflammation
As our understanding of the inflammatory process has evolved, we have gained a greater appreciation for not only the pathological but also the physiological aspects of the inflammatory process. Whereas the teleological rationale of an inflammatory response to pathological states is obvious, there is less understanding of the physiological role of inflammation, especially in reproductive tissues. For example, studies have shown that maintenance of normal cellular homeostasis in the human decidua, placenta and amniochorionic membranes involves molecules normally associated with either immune or inflammatory processes, such as interleukin (IL)-1, IL-8, tumour necrosis factor (TNF)-α, nitric oxide (NO), vascular endothelium growth factor (VEGF), angiopoietins, high-mobility group box protein-1 (HMGB-1), matrix metalloproteinases (MMPs), complement factors and soluble receptor for advanced glycation end products (Refs 63, 64, 65, 66, 67, 68, 69, 70, 71). There is evidence to suggest that each of these molecules has critical effects both during normal development and throughout pathologically triggered inflammation.
Human decidua as a distinctive immune tolerance environment
Immune tolerance is necessary at the maternal–fetal interface. The cellular component (i.e. natural killer cells and macrophages) and functional regulation of the cells implicated in controlling the normal silent inflammatory process of the human uterine decidua have been extensively reviewed (Refs 72, 73). The gene expression profiling of human decidual macrophages confirmed that in normal pregnancy these cells display a distinctive transcriptional profile comprising a large number of genes functionally related to immunomodulation and tissue remodeling (Ref. 74). Of the 14 000 genes examined, genes encoding the chemokine CCL-18, CD209 (DC-SIGN, a C-type lectin receptor characteristically present on dendritic cells), insulin-like growth factor (IGF)-1, mannose receptor C-type (MRC)-1, S100 calcium-binding protein (calgranulin A) and fibronectin-1 were conspicuous because of their known immunological function (Ref. 74). The concerted regulation of these genes provided valuable insight into the phenotype of placental and decidual macrophages, which seems to be balanced towards immunosuppression because of the preferential expression of anti-inflammatory cytokines and chemokines (Refs 73, 74). The mechanisms responsible for switching these factors from beneficial to ‘suicide’-assisted molecules are incompletely understood. However, the novel ‘danger’ theory holds that in addition to upregulation of the proinflammatory cytokines, the injured cells release alarm signals (i.e. HMGB-1), which in turn activate and amplify the inflammatory response (Refs 75, 76).
Immunity and coagulation
The innate and the adaptive immune systems display complementary inflammatory networks (Refs 77, 78). During human pregnancy, the innate immune system not only resists infection, but also is poised at the crossroads between inflammation and coagulation. Proteomic studies demonstrated that the process of decidual bleeding could be attended by a robust intra-amniotic inflammatory process in the absence of infectious triggers (Fig. 3) (Ref. 80). Coagulation proteases and free haemoglobin chains control different modules of the innate immunity and a feed-forward mechanism reinforces a sterile inflammatory process (Refs 12, 39, 81, 82, 83). Strategically positioned at the maternal–fetal interface and cervical mucosa, toll-like receptors (TLRs) are major operative agents of the innate immune response against infection (Refs 84, 85, 86). Moreover, recent reports suggest that activation of TLRs also contributes to the inflammatory response mediated by the endogenous release of HMGB-1 and activation of the coagulation cascade during placental abruption (Refs 87, 88).
Figure 3. Representative proteomic profiles characteristic for intra-amniotic inflammation and bleeding.

Three hypervariable areas of representative surface-enhanced laser desorption ionisation time-of-flight tracings of amniotic fluid from women admitted for preterm birth are shown: (a) 3–4 kDa, (b) 10–12.5 kDa and (c) 14–17 kDa. These areas contain proteomic patterns characteristic of intra-amniotic inflammation (MR score composed of four protein biomarkers – P1, P2, P3 and P4) (Ref. 79), intra-amniotic bleeding (Hb chains) or both (bottom tracings). The x-axis of the tracings represents the molecular mass in daltons; the y-axis represents the normalised peak intensity. R denotes a reference protein peak present in all fluid samples, which corresponds to a fragment of beta-2 microglobulin (originally published in Ref. 80; reproduction permitted under Creative Commons Attribution License). Abbreviations: Hb, haemoglobin; MR, mass-restricted; P1, neutrophil defensin-2; P2, neutrophil defensin-1; P3, calgranulin C; and P4, calgranulin A.
Questions about physiological and pathological engagement of the human immune system during pregnancy represent fundamental challenges for reproductive scientists. The literature on the subject is vast. We aim to illustrate the richness of the topic and set the stage for the argument that understanding the mechanisms responsible for the crosstalk between coagulation and inflammation is one critical step towards a better appreciation of the role played by these interacting pathways in abruption-induced PTB.
Significance of the crosstalk between inflammation and coagulation in the mechanism of abruption
Placental abruption can be viewed as a ‘binary’ event in which molecular signals involved in decidual bleeding arise as a consequence of either inflammation or aberrant coagulation (Refs 8, 9, 11). Evidence in support of this theory is derived from studies demonstrating that inflammatory reactions occurring in the setting of infection activate the coagulation system (Ref. 24). Aberrant bleeding and haemostasis are more common in the setting of histological chorioamnionitis (Refs 15, 20, 24, 89), suggesting a model in which proinflammatory cytokines (i.e. IL-1β and IL-6) act on the endothelium of decidual blood vessels to increase the expression of leukocyte interactive proteins [P-selectin, E-selectin, VCAM (vascular cell adhesion protein) and ICAM-1 (intercellular adhesion molecule 1)] (Refs 32, 63). In turn, this phenomenon leads to neutrophil infiltration and decidual vascular damage with perivascular leakage of plasma proteins and coagulation factors. The coagulation pathway is activated when circulating FVII gains access to perivascular adventitial TF and generates thrombin (Fig. 1) (Ref. 90). Therefore, inflammation shifts the decidual haemostatic mechanisms in favour of thrombosis (Ref. 91). In an animal model of endotoxin-induced sepsis, infusion of low thrombin levels protected against lethality (Ref. 92), signifying that this protection was provided by an early anticoagulant and fibrinolytic response to endotoxin. Applying the same process to the decidua, it can be projected that low thrombin levels, generated in the early phase of inflammation-induced abruption, are beneficial. This phenomenon helps to explain the high frequency of histopathological lesions suggestive of chronic decidual bleeding in the absence of clinical manifestations of the disease (Refs 20, 24).
The physiological mechanisms that enable decidual pro- or anticoagulant and fibrinolytic systems to prevent cataclysmic haemorrhage during pregnancy have been reviewed in detail (Ref. 21). Our group previously proposed that when the process of decidual thrombin activation overwhelms the physiological anticoagulant and fibrinolytic response, the abruption process becomes systemic and clinically evident (Ref. 93). We demonstrated for the first time that increased thrombin activation, as assessed by circulating maternal plasma thrombin–antithrombin complexes, predicted adverse pregnancy outcomes related to prematurity (Ref. 93).
Consistent with the ‘binary’ model proposed above during placental abruption, aberrant coagulation is implicated in triggering decidual inflammation (Refs 24, 25, 91). A growing body of research suggests that maternal hypertensive disorders, trauma, idiopathic intrauterine growth restriction and possibly thrombophilias are risk factors for placental abruption (Ref. 40). These conditions are generally associated with reduced uteroplacental blood flow, hypoxia and excess production of reactive oxygen species (ROS). Hypoxia amplifies this vicious feedforward cycle by upregulating the expression of proangiogenic proteins and inducing decidual vasculopathy endothelial cell dysfunction, thrombin generation, thrombosis and inflammation (Fig. 4) (Refs 71, 94). Evidence in support of the ‘binary’ model of abruption was provided by experimental results demonstrating that thrombin promotes decidual vasculogenesis and angiogenesis (Refs 95, 96). Specifically, treatment of primary microvascular endometrial endothelial cells with VEGF or thrombin-induced TF mRNA and protein expression (Ref. 96) and enhanced mRNA levels of several inflammatory cytokines, with the greatest effect seen on macrophage inflammatory protein-3α (MIP3α). Furthermore, in cultured first-trimester decidual cells, thrombin, but not IL-1β or TNF-α, enhanced the mRNA and protein levels of the antiangiogenic factor soluble fms-like tyrosine kinase-1 (sFlt-1) (Ref. 96), with no effects on the expression of the angiogenic VEGF or placental growth factor. This thrombin augmentation of sFlt-1 expression in first-trimester decidual cells did not extend to cultured term decidual cells (Ref. 96). These results indicate that thrombin acts on decidual cells to create an imbalance at the implantation site that interferes with angiogenesis dependent remodelling of decidual vessels and in subsets of abruption cases (Refs 90, 97).
Figure 4. Binary theory of decidual bleeding and inflammation in pathogenesis of placental abruption.

A reduced or intermittent uteroplacental blood flow causes focal decidual hypoxia and free radicals and ROS (through reperfusion injury). Hypoxia induces the expression of decidual VEGF. This angiogenic factor acts directly on decidual endothelial cells to enhance permeability and degrade the vascular wall through MMP-2 generation. This leads to haemorrhage and aberrant endothelial expression of TF to generate additional thrombin that further induces TF expression and uteroplacental thromboses, which exacerbate reduced blood flow. Free radicals and ROS induce endothelial cell injury, which allows perivascular leakage of coagulation factors, including factor VIIa, which then comes in contact with TF, activating the extrinsic coagulation pathway. The resulting thrombin further induces TF expression as well as expression of inflammatory cytokines, leading to inflammation in the absence of a microbial attack. Abbreviations: MMP, matrix metalloproteinase; ROS, reactive oxygen species; sFlt-1: tyrosine kinase-1; TF, tissue factor; VEGF, vascular endothelium growth factor.
Thrombin altered the expression of the angiopoietin (Ang) proteins in primary cultures of human endometrial cells derived from predecidualised endometrium (Ref. 98). Specifically, thrombin markedly reduced angiogenic Ang-1 mRNA and protein expression in human endometrial stromal cells, while minimally decreasing the production of Ang-2 protein in human endometrial endothelial cells. These results indicate that thrombin generated from aberrant activation of haemostasis can alter decidual vascular angiogenesis, vascular integrity and homeostasis. This creates a feedforward loop, resulting in additional thrombin formation, weakened vasculature and exacerbation of haemorrhage. Thrombin induced by increased expression of TF might act as an autocrine or paracrine enhancer of aberrant vascular remodelling, promoting uteroplacental thromboses and inducing inflammation that causes decidual bleeding in a gestational-age-dependent fashion (Refs 95, 96). On the basis of the above data, we argue that the TF–thrombin unit is central to the crosstalk between inflammation and coagulation in abruption.
Genetic risk factors for placental abruption
A complex genetic predisposition to PTB is generally accepted (Refs 99, 100); however, recent epidemiological studies suggest that genetic risk factors also have an important role in the pathogenesis of abruption (Refs 101, 102, 103). Previous studies show that the recurrence risk of placental abruption in the same woman is higher compared with levels in the general population and that thromboembolic diseases aggregate within female relatives of women with placental abruption (Ref. 104). The observation that thrombophilia has a high prevalence among women with placental abruption supports the idea of a genetic background for this condition (Refs 104, 105, 106). However, there are conflicting genetic studies involving candidate gene polymorphisms (Refs 107, 108). A recent meta-analysis identified the most frequent gene polymorphisms associated with placental abruption (Ref. 109). A positive association was found for coagulation factor FVArg506Gln(OR: 3.4; 95% CI: 1.4–8.3) and for prothrombin G20210A (OR: 6.7; 95% CI: 3.2–13] polymorphisms (Ref. 109), which was confirmed in a large prospective cohort study of nulliparous women (OR: 12.15; 95% CI: 2.45–60.39) (Ref. 110). However, a recent study argues against this association (Ref. 111). The association between fetal thrombophilic mutations (FV Leiden and prothrombin G20210A) and placental abruption suggested a possible fetal genotypic contribution to the occurrence of this obstetrical complication (Ref. 112). Abnormal placental and decidual vasculogenesis (placental infarcts, avascular terminal villi and villi infarcts) is the proposed mechanism (Ref. 113). Larger prospective cohort studies that include gene–gene and gene-environment interactions are needed to increase our understanding of the contribution of genetic factors to complex diseases such as placental abruption.
Progestins and molecular mechanisms leading to placental abruption
Progesterone is essential for pregnancy maintenance from implantation to parturition (Ref. 114). The role of the progesterone ‘block’ (Ref. 115) in promoting uterine quiescence, controlling myometrial gap junctions and cervical ripening, and preventing PTB has been extensively analysed (Refs 116, 117). The decidua is a direct target for progesterone actions during human pregnancy. Several biological effects initiated by progesterone are the consequence of a variety of genomic and nongenomic mechanisms of action (Ref. 118). These effects are facilitated by PR isoforms (PRA and PRB). Several molecular mechanisms, including target gene transcription and cascade signalling pathways, are involved.
Role of progesterone in modulating decidual fibrinolysis and its inhibitors
Decidual haemostasis requires the concerted actions of several proteases, with plasminogen-derived plasmin having an initiating role (Ref. 21). Plasminogen is a circulating zymogen that is converted to the active enzyme plasmin by cleavage at a specific amino acid bond mediated by either urokinase plasminogen activator (uPA) or tissue-type plasminogen activator (tPA). Normally, the actions of uPA and tPA are held in check by plasminogen activator inhibitor-1 (PAI-1), the fast inhibitor of the primary fibrinolytic agent, tPA (Ref. 119), and placental trophoblast-derived PAI-2. Interactions between progesterone and the PA–plasmin system have key roles in the local regulation of decidual haemostasis (Ref. 21). An in vitro model of decidualisation using primary stromal cells from predecidualised human endometrium suggests that first-trimester decidua might express two crucial modulators of haemostasis, TF and PAI-1 (Ref. 120). In these cultured stromal cells, progestins such as medroxyprogesterone acetate (MPA) enhanced mRNA and protein levels of TF (Ref. 120) as well as PAI-1 (Ref. 121) and uPA, but not tPA (Ref. 122). The progestin effects were blocked by the antiprogesterone RU486 (Ref. 123). Consistent with maintenance of decidualisation during gestation, it was observed that progestins also enhanced TF and PAI-1 expression in cultured term decidual cells (Refs 46, 122).
Role of progesterone in the expression of decidual MMP and MMP inhibitor
Decidualisation is associated with stromal- or decidual-cell-mediated turnover of the extracellular matrix (ECM) mediated by interactions between uPA and MMPs (Refs 124, 125). In primary cultures of leukocyte-free term decidual cells, MPA inhibited mRNA and protein levels as well as the enzymatic activity of interstitial collagenase (MMP-1) and stromelysin-1 (MMP-3) (reviewed in Ref. 125). Steroid withdrawal reversed this inhibition, with RU486 eliciting greater reversal. MMP-1 specifically targets fibrillar collagens whereas MMP-3 degrades several ECM proteins and activates the secreted zymogenic form of other MMPs such as pro-MMP-1 Therefore, these in vitro observations suggest that progesterone withdrawal elicits a decidual-cell-expressed ECM-degrading cascade of the decidua and fetal membranes that promotes bleeding and premature separation of the placenta.
Role of progesterone in modulating the expression of decidual cytokines
Coincident with activation of the coagulation cascade, local decidual injury causes the release of cytokines that act as autocrine or paracrine effectors of immunomodulatory, angiogenic and proliferative activities. Specifically, a causal role has been proposed for TNF-α, IL-1β, IL-6, IL-8 and IL-11 in PTB (Ref. 126). A better understanding of the genesis of abruption-induced inflammation was provided by exploring the role of progesterone in modulating the expression of IL-1β and IL-11 in human term decidual cells (Ref. 81). The focus on IL-11 was prompted by its known exertion of complex antiinflammatory effects such as inhibition of endometrial TNF-α production and attenuation of IL-1β, TNF-α, IL-12 and NO production in activated macrophages (Ref. 127). Additionally IL-11 possesses proinflammatory and uterine contraction properties by inducing prostaglandin secretion. Immunohistochemical staining of decidual sections indicated that PTB is associated with significantly enhanced decidual cell IL-11 expression. In term decidual cell cultures, thrombin and IL-1β each augmented IL-11 expression, whereas co-incubation with a progestin significantly blunted the upregulation of IL-11 expression elicited by both agents. Taken together, the results of studies presented above help to account for the role of progesterone in maintaining haemostasis throughout pregnancy by inhibiting the general proteolytic and inflammatory activity at the maternal–fetal interface.
TF and thrombin as regulators of molecular mechanisms leading to placental abruption
A key part of the ‘binary’ model (Fig. 4) of abruption-associated PTB is the evidence that TF-induced thrombin is a critical mediator of the crosstalk between coagulation, angiogenesis and inflammation. This section focuses on other mechanisms by which TF and thrombin might mediate placental abruption and PTB.
TF, thrombin and MMP activity in fetal membranes and decidua
Key to understanding the relationship between decidual bleeding and activation of amniochorion MMPs is the observation that in vivo activation of the thrombin system and histological haemorrhage are frequent complications during preterm premature rupture of the fetal membranes (PPROM) (Refs 20, 93). PPROM is one of the leading risk factors for PTB (Ref. 128). Although anatomically part of the gravid uterus, the amniochorion membrane can be viewed as a separate, complex and heterogeneous organ because it undergoes molecular, enzymatic and biomechanical transformations different from those of the cervix and myometrium (Refs 6, 12). Many risk factors for PPROM have been identified (Refs 9, 129). However, irrespective of the cause, the final unifying pathway is weakness of the amniochorion that allows for rupture.
The fibrillar collagens (types I, III and V)provide tensile strength to the amnion (Ref. 129). The strength of the fetal membranes reflects a balance between synthesis and protease-induced degradation of ECM components. The molecular mechanisms responsible for the latter involve tight regulation by MMP-1, MMP-2, MMP-3 and MMP-9 and the tissue inhibitors of MMPs (TIMP-1 and TIMP-2) (Ref. 129). In vitro, thrombin targets and enhances the expression of MMP-1, MMP-3 and MMP-9 in term fetal membranes (Refs 83, 130, 131). Progesterone partially reverses the process. Interestingly, thrombin augments expression of PAI-1 but not uPA or tPA, as was recently shown (Ref. 122). This effect was abrogated by hirudin. Collectively, these results suggest that thrombin exercises its stimulatory proteolytic effect by targeting MMPs directly and not by stimulating the expression of plasminogen system activators.
A marked decidual infiltrate of neutrophils, which are a rich source of ECM-degrading proteases, is associated with abruption-induced PPROM in the presence or absence of infection (Refs 20, 24). Moreover, as previously shown, thrombin enhances the expression of IL-8 and other neutrophil chemoattractant and -activating chemokines in term decidual cell monolayers (Ref. 83). Thus, it is reasonable to propose that during abruption, thrombin generated from decidual-cell-expressed TF can promote PPROM either directly, by enhanced expression of MMPs, or indirectly, by augmenting decidual expressed neutrophil chemoattractants and activators.
The mechanism of PPROM includes tissue damage secondary to both hypoxia and inflammation, as well as an increased state of oxidative stress (Refs 95, 132). For example, MMP activity in human fetal membranes is reduction–oxidation (redox) regulated (Ref. 132). MMP-9 activity was directly increased by the superoxide anion, a byproduct of macrophages and neutrophils, which are abundantly present in both the amniochorion and decidua in the setting of decidual bleeding. N-acetylcysteine (a glutathione precursor and direct antioxidant) dramatically decreased the amniochorionic MMP-9 activity.
Research in progress and outstanding research questions
The events that initiate activation of the immune and coagulation systems in the setting of abruption-induced PTB are incompletely understood. Unravelling the complexity of the clinical management of acute and chronic placental abruption requires elucidation of the highly controlled and synchronised biochemical circuits that exist in the decidua and amniochorionic membranes. For many years, research in decidual bleeding has concentrated on identifying, localising and evaluating the regulation of individual factors. This approach ignores the reality that abruption involves complex molecular protein-to-protein interactions that function in interconnected cellular networks regulated by specific receptors. A shift in mindset is thus required. The discovery of key biomarkers that can specifically determine whether the inflammatory and coagulation networks are silent or activated in women at risk for decidual bleeding is crucial. This approach is consistent with the idea of taking an individualised medicine approach aimed at identifying patients who might benefit from targeted interventions rather than a ‘one-size-fits-all’ therapy. The challenge is to use our knowledge to develop pharmacological agents that can specifically inhibit the proteolytic activity of thrombin, boost the antiinflammatory activity of progesterone or lower the heightened state of oxidative stress at the maternal–fetal interface.
Acknowledgments
Acknowledgements and funding: We are indebted to the nurses, residents and fellows from the Departments of Obstetrics, Gynecology and Reproductive Sciences of Yale University, Mount Sinai Medical Center – New York, New York University School of Medicine and University of Maryland – Baltimore. This work was supported by National Institutes of Health grants RO1 HD 29540 (C.J.L.), HL 33937 (C.J.L.) and HD 047321 (I.A.B.), March of Dimes grant 21-FY05-1249 (C.J.L.), and March of Dimes Basil O'Connor Award (I.A.B.). We thank the reviewers for their helpful suggestions.
References
- 1.Stoll BJ, et al. National Institute of Child Health and Human Development Neonatal Research Network. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. Journal of the American Medical Association. 2004;292:2357–2365. doi: 10.1001/jama.292.19.2357. [DOI] [PubMed] [Google Scholar]
- 2.Zerhouni EA. US biomedical research: basic, translational, and clinical sciences. Journal of the American Medical Association. 2005;294:1352–1358. doi: 10.1001/jama.294.11.1352. [DOI] [PubMed] [Google Scholar]
- 3.Fagnan LJ. Linking practice-based research networks and clinical and translational science awards: new opportunities for community engagement by academic health centers. Academic Medicine. 2010;85:476–483. doi: 10.1097/ACM.0b013e3181cd2ed3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLean M, et al. A placental clock controlling the length of human pregnancy. Nature Medicine. 1995;1:460–463. doi: 10.1038/nm0595-460. [DOI] [PubMed] [Google Scholar]
- 5.Zenclussen AC, et al. Immunology of pregnancy: cellular mechanisms allowing fetal survival within the maternal uterus. Expert Reviews in Molecular Medicine. 2007;9:1–14. doi: 10.1017/S1462399407000294. [DOI] [PubMed] [Google Scholar]
- 6.Mendelson CR. Minireview: fetal–maternal hormonal signaling in pregnancy and labor. Molecular Endocrinology. 2009;23:947–954. doi: 10.1210/me.2009-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muglia LJ, Katz M. The enigma of spontaneous preterm birth. New England Journal of Medicine. 2010;362:529–535. doi: 10.1056/NEJMra0904308. [DOI] [PubMed] [Google Scholar]
- 8.Buhimschi CS, et al. Multidimensional system biology: genetic markers and proteomic biomarkers of adverse pregnancy outcome in preterm birth. American Journal of Perinatology. 2008;25:175–187. doi: 10.1055/s-2008-1061497. [DOI] [PubMed] [Google Scholar]
- 9.Lockwood CJ, Kuczynski E. Risk stratification and pathological mechanisms in preterm delivery. Paediatric and Perinatal Epidemiology. 2001;2:78–89. doi: 10.1046/j.1365-3016.2001.00010.x. [DOI] [PubMed] [Google Scholar]
- 10.Anum EA, et al. Genetic contributions to disparities in preterm birth. Pediatric Research. 2009;65:1–9. doi: 10.1203/PDR.0b013e31818912e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Challis JR, et al. Inflammation and pregnancy. Reproductive Sciences. 2009;16:206–215. doi: 10.1177/1933719108329095. [DOI] [PubMed] [Google Scholar]
- 12.Norman JE, et al. Inflammatory pathways in the mechanism of parturition. BMC Pregnancy Childbirth. 2007;7(Suppl 1):S7. doi: 10.1186/1471-2393-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiner CP, et al. Human effector/initiator gene sets that regulate myometrial contractility during term and preterm labor. American Journal of Obstetrics and Gynecology. 2010;202:474.e1–474.e20. doi: 10.1016/j.ajog.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Seminars in Perinatology. 2009;33:189–195. doi: 10.1053/j.semperi.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Elsasser DA, et al. Diagnosis of placental abruption: relationship between clinical and histopathological findings. European Journal of Obstetrics, Gynecology, and Reproductive Biology. 2010;148:125–130. doi: 10.1016/j.ejogrb.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ananth CV, et al. Placental abruption in the United States, 1979 through 2001: temporal trends and potential determinants. American Journal of Obstetrics and Gynecology. 2005;192:191–198. doi: 10.1016/j.ajog.2004.05.087. [DOI] [PubMed] [Google Scholar]
- 17.Ananth CV, et al. Placental abruption and adverse perinatal outcomes. Journal of the American Medical Association. 1999;282:1646–1651. doi: 10.1001/jama.282.17.1646. [DOI] [PubMed] [Google Scholar]
- 18.Oyelese Y, Ananth CV. Placental abruption. Obstetrics and Gynecology. 2006;108:1005–1016. doi: 10.1097/01.AOG.0000239439.04364.9a. [DOI] [PubMed] [Google Scholar]
- 19.Ananth CV, Wilcox AJ. Placental abruption and perinatal mortality in the United States. American Journal of Epidemiology. 2001;153:332–337. doi: 10.1093/aje/153.4.332. [DOI] [PubMed] [Google Scholar]
- 20.Salafia CM, et al. Histologic evidence of old intrauterine bleeding is more frequent in prematurity. American Journal of Obstetrics and Gynecology. 1995;173:1065–1070. doi: 10.1016/0002-9378(95)91327-0. [DOI] [PubMed] [Google Scholar]
- 21.Lockwood CJ. Pregnancy-associated changes in the hemostatic system. Clinical Obstetrics and Gynecology. 2006;49:836–843. doi: 10.1097/01.grf.0000211952.82206.16. [DOI] [PubMed] [Google Scholar]
- 22.Benirschke K. Anatomical relationship between fetus and mother. Annals of the New York Academy of Sciences. 1994;731:9–20. doi: 10.1111/j.1749-6632.1994.tb55744.x. [DOI] [PubMed] [Google Scholar]
- 23.Dommisse J, Tiltman AJ. Placental bed biopsies in placental abruption. British Journal of Obstetrics and Gynaecology. 1992;99:651–654. doi: 10.1111/j.1471-0528.1992.tb13848.x. [DOI] [PubMed] [Google Scholar]
- 24.Nath CA, et al. Histologic evidence of inflammation and risk of placental abruption. New Jersey-placental abruption study investigators. American Journal of Obstetrics and Gynecology. 2007;197:319.e1–319.e6. doi: 10.1016/j.ajog.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 25.Arias F, et al. Maternal placental vasculopathy and infection: two distinct subgroups among patients with preterm labor and preterm ruptured membranes. American Journal of Obstetrics and Gynecology. 1993;168:585–591. doi: 10.1016/0002-9378(93)90499-9. [DOI] [PubMed] [Google Scholar]
- 26.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nature Reviews Immunology. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 27.Kumar P, et al. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Reviews in Molecular Medicine. 2009;11:e19. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reznik SE, et al. Immunohistochemical localization of carboxypeptidases E and D in the human placenta and umbilical cord. Journal of Histochemistry and Cytochemistry. 1998;46:1359–1368. doi: 10.1177/002215549804601204. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, et al. Advanced glycation end products and lipopolysaccharide synergistically stimulate proinflammatory cytokine/chemokine production in endothelial cells via activation of both mitogen-activated protein kinases and nuclear factor-kappaB. FEBS Journal. 2009;276:4598–4606. doi: 10.1111/j.1742-4658.2009.07165.x. [DOI] [PubMed] [Google Scholar]
- 30.Pinheiro da Silva F, Soriano FG. Neutrophils recruitment during sepsis: critical points and crossroads. Frontiers in Bioscience. 2009;14:4464–4476. doi: 10.2741/3542. [DOI] [PubMed] [Google Scholar]
- 31.Ley K, Reutershan J. Leucocyte–endothelial interactions in health and disease. Handbook of Experimental Pharmacology. 2006;176:97–133. doi: 10.1007/3-540-36028-x_4. [DOI] [PubMed] [Google Scholar]
- 32.Levi M, van der Poll T. Inflammation and coagulation. Critical Care Medicine. 2010;38:S26–S34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- 33.Bach R, Nemerson Y, Konigsberg W. Purification and characterization of bovine tissue factor. Journal of Biological Chemistry. 1981;256:8324–8331. [PubMed] [Google Scholar]
- 34.Spicer EK, et al. Isolation of cDNA clones coding for human tissue factor: primary structure of the protein and cDNA. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:5148–5152. doi: 10.1073/pnas.84.15.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polgar J, Matuskova J, Wagner DD. The P-selectin, tissue factor, coagulation triad. Journal of Thrombosis and Haemostasis. 2005;3:1590–1596. doi: 10.1111/j.1538-7836.2005.01373.x. [DOI] [PubMed] [Google Scholar]
- 36.Moll T, et al. Regulation of the tissue factor promoter in endothelial cells. Journal of Biological Chemistry. 1995;270:3849–3857. doi: 10.1074/jbc.270.8.3849. [DOI] [PubMed] [Google Scholar]
- 37.Rao LV, Mackman N. Factor VIIa and tissue factor – from cell biology to animal models. Thrombosis Research. 2010;125(Suppl 1):S1–S3. doi: 10.1016/j.thromres.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Popescu NI, Lupu C, Lupu F. Role of PDI in regulating tissue factor: FVIIa activity. Thrombosis Research. 2010;125(Suppl 1):S38–S41. doi: 10.1016/j.thromres.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackman N. The many faces of tissue factor. Journal of Thrombosis and Haemostasis. 2009;1:136–139. doi: 10.1111/j.1538-7836.2009.03368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krikun G, Lockwood CJ, Paidas MJ. Tissue factor and the endometrium: from physiology topathology. Thrombosis Research. 2009;124:393–396. doi: 10.1016/j.thromres.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 41.Rao LV, Pendurthi UR. Regulation of tissue factor–factor VIIa expression on cell surfaces: a role for tissue factor–factor VIIa endocytosis. Molecular and Cellular Biochemistry. 2003;253:131–140. doi: 10.1023/a:1026004208822. [DOI] [PubMed] [Google Scholar]
- 42.Aharon A, et al. Tissue factor and tissue factor pathway inhibitor levels in trophoblast cells: implications for placental hemostasis. Thrombosis and Haemostasis. 2004;92:776–786. doi: 10.1160/TH04-01-0033. [DOI] [PubMed] [Google Scholar]
- 43.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. American Journal of Pathology. 1989;134:1087–1097. [PMC free article] [PubMed] [Google Scholar]
- 44.Stassen JM, Arnout J, Deckmyn H. The hemostatic system. Current Medical Chemistry. 2004;11:2245–2260. doi: 10.2174/0929867043364603. [DOI] [PubMed] [Google Scholar]
- 45.Lockwood CJ, et al. Amniotic fluid contains tissue factor, a potent initiator of coagulation. American Journal of Obstetrics and Gynecology. 1991;165:1335–1341. doi: 10.1016/0002-9378(91)90363-v. [DOI] [PubMed] [Google Scholar]
- 46.Lockwood CJ, et al. Progestin and thrombin regulate tissue factor expression in human term decidual cells. Journal of Clinical Endocrinology and Metabolism. 2009;94:2164–2170. doi: 10.1210/jc.2009-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lockwood CJ, et al. Progestational regulation of human endometrial stromal cell tissue factor expression during decidualization. Journal of Clinical Endocrinology and Metabolism. 1993;76:231–236. doi: 10.1210/jcem.76.1.8421090. [DOI] [PubMed] [Google Scholar]
- 48.Schatz F, et al. Progestin-regulated expression of tissue factor in decidual cells: implications in endometrial hemostasis, menstruation and angiogenesis. Steroids. 2003;68:849–860. doi: 10.1016/s0039-128x(03)00139-9. [DOI] [PubMed] [Google Scholar]
- 49.Lockwood CJ, et al. Progestin-epidermal growth factor regulation of tissue factor expression during decidualization of human endometrial stromal cells. Journal of Clinical Endocrinology and Metabolism. 2000;85:297–301. doi: 10.1210/jcem.85.1.6292. [DOI] [PubMed] [Google Scholar]
- 50.Erlich J, et al. Tissue factor is required for uterine hemostasis and maintenance of the placental labyrinth during gestation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8138–8143. doi: 10.1073/pnas.96.14.8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bergh N, et al. Influence of TNF-alpha and biomechanical stress on endothelial anti- and prothrombotic genes. Biochemical and Biophysical Research Communications. 2009;385:314–318. doi: 10.1016/j.bbrc.2009.05.046. [DOI] [PubMed] [Google Scholar]
- 52.Mertens G, et al. Cell surface heparan sulfate proteoglycans from human vascular endothelial cells. Core protein characterization and antithrombin III binding properties. Journal of Biological Chemistry. 1992;267:20435–20443. [PubMed] [Google Scholar]
- 53.Jackson CJ, Xue M. Activated protein C – an anticoagulant that does more than stop clots. International Journal of Biochemistry and Cell Biology. 2008;40:2692–2697. doi: 10.1016/j.biocel.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 54.Castellino FJ, Ploplis VA. The protein C pathway and pathologic processes. Journal of Thrombosis and Haemostasis. 2009;7(Suppl 1):140–145. doi: 10.1111/j.1538-7836.2009.03410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ignarro LJ. Endothelium-derived nitric oxide: actions and properties. FASEB Journal. 1989;3:31–36. doi: 10.1096/fasebj.3.1.2642868. [DOI] [PubMed] [Google Scholar]
- 56.Harris LK. Trophoblast-derived heparanase is not required for invasion. Placenta. 2008;29:332–337. doi: 10.1016/j.placenta.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 57.Dempsey LA, et al. Heparanase expression in invasive trophoblasts and acute vascular damage. Glycobiology. 2000;10:467–475. doi: 10.1093/glycob/10.5.467. [DOI] [PubMed] [Google Scholar]
- 58.Meeusen EN, Bischof RJ, Lee CS. Comparative T-cell responses during pregnancy in large animals and humans. American Journal of Reproductive Immunology. 2001;46:169–179. doi: 10.1111/j.8755-8920.2001.460208.x. [DOI] [PubMed] [Google Scholar]
- 59.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annual Review of Immunology. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 60.Reddy RC, et al. Sepsis-induced immunosuppression: from bad to worse. Immunologic Research. 2001;24:273–287. doi: 10.1385/IR:24:3:273. [DOI] [PubMed] [Google Scholar]
- 61.Cinel I, Opal SM. Molecular biology of inflammation and sepsis: a primer. Critical Care Medicine. 2009;37:291–304. doi: 10.1097/CCM.0b013e31819267fb. [DOI] [PubMed] [Google Scholar]
- 62.Benhamou Y, et al. Toll-like receptors 4 contribute to endothelial injury and inflammation in hemorrhagic shock in mice. Critical Care Medicine. 2009;37:1724–1728. doi: 10.1097/CCM.0b013e31819da805. [DOI] [PubMed] [Google Scholar]
- 63.Arcuri F, et al. Mechanisms of leukocyte accumulation and activation in chorioamnionitis: interleukin 1 beta and tumor necrosis factor alpha enhance colony stimulating factor 2 expression in term decidua. Reproductive Sciences. 2009;16:453–461. doi: 10.1177/1933719108328609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lockwood CJ, et al. Matrix metalloproteinase 9 (MMP9) expression in preeclamptic decidua and MMP9 induction by tumor necrosis factor alpha and interleukin 1 beta in human first trimester decidual cells. Biology of Reproduction. 2008;78:1064–1072. doi: 10.1095/biolreprod.107.063743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buhimschi IA, et al. The nitric oxide pathway in pre-eclampsia: pathophysiological implications. Human Reproduction Update. 1998;4:25–42. doi: 10.1093/humupd/4.1.25. [DOI] [PubMed] [Google Scholar]
- 66.Lee S, et al. The interleukin-6 (IL-6) trans-signaling system: evidence for presence and activation in pregnancies complicated by intra-amniotic infection. American Journal of Obstetrics and Gynecology. 2008;199:S141. Presented at the Society for Maternal Fetal Medicine 2009 (26-31 January 2009; San Diego, CA, USA) [Google Scholar]
- 67.Buhimschi IA, et al. The receptor for advanced glycation end products (RAGE) system in women with intra-amniotic infection and inflammation. American Journal of Obstetrics and Gynecology. 2007;196:e1–e13. doi: 10.1016/j.ajog.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 68.Dunk C, et al. Angiopoietin-1 and angiopoietin-2 activate trophoblast Tie-2 to promote growth and migration during placental development. American Journal of Pathology. 2000;156:2185–2199. doi: 10.1016/S0002-9440(10)65089-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ahmed A, et al. Role of VEGF receptor-1 (Flt-1) in mediating calcium-dependent nitric oxide release and limiting DNA synthesis in human trophoblast cells. Laboratory Investigation. 1997;76:779–791. [PubMed] [Google Scholar]
- 70.Holmlund U, et al. The novel inflammatory cytokine high mobility group box protein 1 (HMGB1) is expressed by human term placenta. Immunology. 2007;122:430–437. doi: 10.1111/j.1365-2567.2007.02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buhimschi CS, et al. Amniotic fluid angiopoietin-1, angiopoietin-2, and soluble receptor Tie2 levels and regulation in normal pregnancy and intra-amniotic inflammation induced preterm birth. Journal of Clinical Endocrinology and Metabolism. 2010;95:3428–3436. doi: 10.1210/jc.2009-2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bulmer JN, Williams PJ, Lash GE. Immune cells in the placental bed. International Journal of Developmental Biology. 2010;54:281–294. doi: 10.1387/ijdb.082763jb. [DOI] [PubMed] [Google Scholar]
- 73.Mantovani A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 74.Gustafsson C, et al. Gene expression profiling of human decidual macrophages: evidence for immunosuppressive phenotype. PLoS One. 2008;3:e2078. doi: 10.1371/journal.pone.0002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pétrilli V, et al. The inflammasome: a danger sensing complex triggering innate immunity. Current Opinion in Immunology. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 76.Buhimschi CS, et al. Characterization of RAGE, HMGB1, and S100beta in inflammation-induced preterm birth and fetal tissue injury. American Journal of Pathology. 2009;175:958–975. doi: 10.2353/ajpath.2009.090156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kimbrell DA, Beutler B. The evolution and genetics of innate immunity. Nature Reviews Genetics. 2001;2:256–267. doi: 10.1038/35066006. [DOI] [PubMed] [Google Scholar]
- 78.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 79.Buhimschi IA, Christner R, Buhimschi CS. Proteomic biomarker analysis of amniotic fluid for identification of intra-amniotic inflammation. BJOG. 2005;112:173–181. doi: 10.1111/j.1471-0528.2004.00340.x. [DOI] [PubMed] [Google Scholar]
- 80.Buhimschi IA, et al. Multidimensional proteomics analysis of amniotic fluid to provide insight into the mechanisms of idiopathic preterm birth. PLoS One. 2008;3:e2049. doi: 10.1371/journal.pone.0002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cakmak H, et al. Progestin suppresses thrombin- and interleukin-1beta-induced interleukin-11 production in term decidual cells: implications for preterm delivery. Journal of Clinical Endocrinology and Metabolism. 2005;90:5279–5286. doi: 10.1210/jc.2005-0210. [DOI] [PubMed] [Google Scholar]
- 82.Buhimschi CS, Buhimschi IA. Proteomic biomarkers of adverse pregnancy outcome in preterm birth – a theranostics opportunity. Future drugs – expert review. Obstetrics and Gynecology. 2007;2:743–753. [Google Scholar]
- 83.Lockwood CJ, et al. Mechanisms of abruption-induced premature rupture of the fetal membranes: thrombin-enhanced interleukin-8 expression in term decidua. American Journal of Pathology. 2005;167:1443–1449. doi: 10.1016/S0002-9440(10)61230-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krikun G, et al. Expression of Toll-like receptors in the human decidua. Histology and Histopathology. 2007;22:847–854. doi: 10.14670/HH-22.847. [DOI] [PubMed] [Google Scholar]
- 85.Koga K, Mor G. Expression and function of toll-like receptors at the maternal–fetal interface. Reproductive Sciences. 2008;15:231–242. doi: 10.1177/1933719108316391. [DOI] [PubMed] [Google Scholar]
- 86.Buhimschi CS, et al. Insight into innate immunity of the uterine cervix as a host defense mechanism against infection and preterm birth. Expert Review of Obstetrics and Gynecology. 2009;4:9–15. [Google Scholar]
- 87.van Zoelen MA, et al. Role of toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock. 2009;31:280–284. doi: 10.1097/SHK.0b013e318186262d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen MJ, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Critical Care. 2009;13:R174. doi: 10.1186/cc8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ananth CV, et al. Preterm premature rupture of membranes, intrauterine infection, and oligohydramnios: risk factors for placental abruption. Obstetrics and Gynecology. 2004;104:71–77. doi: 10.1097/01.AOG.0000128172.71408.a0. 2004. [DOI] [PubMed] [Google Scholar]
- 90.Lockwood CJ, et al. Involvement of human decidual cell-expressed tissue factor in uterine hemostasis and abruption. Thrombosis Research. 2009;124:516–520. doi: 10.1016/j.thromres.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Esmon CT. Crosstalk between inflammation and thrombosis. Maturitas. 2008;61:122–131. doi: 10.1016/j.maturitas.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 92.Taylor FB, Jr, et al. A model for thrombin protection against endotoxin. Thrombosis Research. 1984;36:177–185. doi: 10.1016/0049-3848(84)90339-6. [DOI] [PubMed] [Google Scholar]
- 93.Rosen T, et al. Plasma levels of thrombin-antithrombin complexes predict preterm premature rupture of the fetal membranes. Journal of Maternal–Fetal and Neonatal Medicine. 2001;10:297–300. doi: 10.1080/714904361. [DOI] [PubMed] [Google Scholar]
- 94.Fiedler U, Augustin HG. Angiopoietins: a link between angiogenesis and inflammation. Trends in Immunology. 2006;27:552–558. doi: 10.1016/j.it.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 95.Krikun G, et al. Thrombin activation of endometrial endothelial cells: a possible role in intrauterine growth restriction. Thrombosis and Haemostasis. 2007;97:245–253. [PubMed] [Google Scholar]
- 96.Lockwood CJ, et al. Thrombin regulates soluble fms-like tyrosine kinase-1 (sFlt-1) expression in first trimester decidua: implications for preeclampsia. American Journal of Pathology. 2007;170:1398–1405. doi: 10.2353/ajpath.2007.060465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goldman-Wohl D, Yagel S. Regulation of trophoblast invasion: from normal implantation to pre-eclampsia. Molecular and Cellular Endocrinology. 2002;187:233–238. doi: 10.1016/s0303-7207(01)00687-6. [DOI] [PubMed] [Google Scholar]
- 98.Krikun G, et al. Endometrial angiopoietin expression and modulation by thrombin and steroid hormones: a mechanism for abnormal angiogenesis following long-term progestin-only contraception. American Journal of Pathology. 2004;164:2101–2107. doi: 10.1016/S0002-9440(10)63768-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chaudhari BP, et al. The genetics of birth timing: insights into a fundamental component of human development. Clinical Genetics. 2008;74:493–501. doi: 10.1111/j.1399-0004.2008.01124.x. [DOI] [PubMed] [Google Scholar]
- 100.Nesin M. Genetic basis of preterm birth. Frontiers in Bioscience. 2007;12:115–124. doi: 10.2741/2052. [DOI] [PubMed] [Google Scholar]
- 101.Rasmussen S, Irgens LM, Dalaker K. The effect on the likelihood of further pregnancy of placental abruption and the rate of its recurrence. British Journal of Obstetrics and Gynaecology. 1997;104:1292–1295. doi: 10.1111/j.1471-0528.1997.tb10977.x. [DOI] [PubMed] [Google Scholar]
- 102.Peltier MR. Thromboembolic diseases in families of women with placental abruption. Epidemiology. 2009;20:733–737. doi: 10.1097/EDE.0b013e3181aa2d96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rasmussen S, Irgens LM. Occurrence of placental abruption in relatives. BJOG. 2009;116:693–699. doi: 10.1111/j.1471-0528.2008.02064.x. [DOI] [PubMed] [Google Scholar]
- 104.Kinzler WL, et al. The effect of maternal thrombophilia on placental abruption: histologic correlates. Journal of Maternal-Fetal and Neonatal Medicine. 2009;22:243–248. doi: 10.1080/14767050802551795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van der Molen EF, et al. A common mutation in the 5,10-methylenetetrahydro-folate reductase gene as a new risk factor for placental vasculopathy. American Journal of Obstetrics and Gynecology. 2000;182:1258–1263. doi: 10.1067/mob.2000.105199. [DOI] [PubMed] [Google Scholar]
- 106.Procházka M. Factor V Leiden in pregnancies complicated by placental abruption. BJOG. 2003;110:462–466. [PubMed] [Google Scholar]
- 107.Jaaskelainen E, et al. Polymorphism of the interleukin 1 receptor antagonist (IL1Ra) gene and placental abruption. Journal of Reproductive Immunology. 2008;79:58–62. doi: 10.1016/j.jri.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 108.Ananth CV, et al. Associations between 2 polymorphisms in the methylene-tetrahydrofolate reductase gene and placental abruption. American Journal of Obstetrics and Gynecology. 2007;197:385.e1–385.e7. doi: 10.1016/j.ajog.2007.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zdoukopoulos N, Zintzaras E. Genetic risk factors for placental abruption: a HuGE review and meta-analysis. Epidemiology. 2008;19:309–323. doi: 10.1097/EDE.0b013e3181635694. [DOI] [PubMed] [Google Scholar]
- 110.Said JM, et al. Inherited thrombophilia polymorphisms and pregnancy outcomes in nulliparous women. Obstetrics and Gynecology. 2010;115:5–13. doi: 10.1097/AOG.0b013e3181c68907. [DOI] [PubMed] [Google Scholar]
- 111.Silver RM, et al. Prothrombin gene G20210A mutation and obstetric complications. Obstetrics and Gynecology. 2010;115:14–20. doi: 10.1097/AOG.0b013e3181c88918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anteby EY, et al. Fetal inherited thrombophilias influence the severity of preeclampsia, IUGR and placental abruption. European Journal of Obstetrics, Gynecology, and Reproductive Biology. 2004;113:31–35. doi: 10.1016/j.ejogrb.2003.05.002. [DOI] [PubMed] [Google Scholar]
- 113.Redline RW, Pappin A. Fetal thrombotic vasculopathy: the clinical significance of extensive avascular villi. Human Pathology. 1995;26:80–85. doi: 10.1016/0046-8177(95)90118-3. [DOI] [PubMed] [Google Scholar]
- 114.Smith R. Parturition. New England Journal of Medicine. 2007;356:271–283. doi: 10.1056/NEJMra061360. [DOI] [PubMed] [Google Scholar]
- 115.Csapo A. Progesterone “block. American Journal of Anatomy. 1956;98:273–292. doi: 10.1002/aja.1000980206. [DOI] [PubMed] [Google Scholar]
- 116.Tita AT, Rouse DJ. Progesterone for preterm birth prevention: an evolving intervention. American Journal of Obstetrics and Gynecology. 2009;200:219–224. doi: 10.1016/j.ajog.2008.12.035. [DOI] [PubMed] [Google Scholar]
- 117.Mesiano S, Welsh TN. Steroid hormone control of myometrial contractility and parturition. Seminars in Cell and Developmental Biology. 2007;18:321–331. doi: 10.1016/j.semcdb.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 118.Merlino A, et al. Nuclear progesterone receptor expression in the human fetal membranes and decidua at term before and after labor. Reproductive Science. 2009;16:357–363. doi: 10.1177/1933719108328616. [DOI] [PubMed] [Google Scholar]
- 119.Schatz F, Lockwood CJ. Progestin regulation of plasminogen activator inhibitor type 1 in primary cultures of endometrial stromal and decidual cells. Journal of Clinical Endocrinology and Metabolism. 1993;77:621–625. doi: 10.1210/jcem.77.3.8370684. [DOI] [PubMed] [Google Scholar]
- 120.Krikun G, et al. Transcriptional regulation of the tissue factor gene by progestins in human endometrial stromal cells. Journal of Clinical Endocrinology and Metabolism. 1998;83:926–930. doi: 10.1210/jcem.83.3.4616. [DOI] [PubMed] [Google Scholar]
- 121.Krikun G, et al. Regulation of tissue factor gene expression in human endometrium by transcription factors Sp1 and Sp3. Molecular Endocrinology. 2000;14:393–400. doi: 10.1210/mend.14.3.0430. [DOI] [PubMed] [Google Scholar]
- 122.Norwitz ER, et al. Progestin inhibits and thrombin stimulates the plasminogen activator/inhibitor system in term decidual stromal cells: implications for parturition. American Journal of Obstetrics and Gynecology. 2007;196:382.e1–382.e8. doi: 10.1016/j.ajog.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 123.Lockwood CJ. Biological mechanisms underlying RU 486 clinical effects: inhibition of endometrial stromal cell tissue factor content. Journal of Clinical Endocrinology and Metabolism. 1994;79:786–790. doi: 10.1210/jcem.79.3.8077362. [DOI] [PubMed] [Google Scholar]
- 124.Lockwood CJ, et al. Matrix metalloproteinase and matrix metalloproteinase inhibitor expression in endometrial stromal cells during progestin-initiated decidualization and menstruation-related progestin withdrawal. Endocrinology. 1998;139:4607–4613. doi: 10.1210/endo.139.11.6304. [DOI] [PubMed] [Google Scholar]
- 125.Schatz F, et al. Implications of decidualization-associated protease expression in implantation and menstruation. Seminars in Reproductive Endocrinology. 1999;17:3–12. doi: 10.1055/s-2007-1016206. [DOI] [PubMed] [Google Scholar]
- 126.Christiaens I, et al. Inflammatory processes in preterm and term parturition. Journal of Reproductive Immunology. 2008;79:50–57. doi: 10.1016/j.jri.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 127.Trepicchio WL, et al. Recombinant human IL-11 attenuates the inflammatory response through down-regulation of proinflammatory cytokine release and nitric oxide production. Journal of Immunology. 1996;157:3627–3634. [PubMed] [Google Scholar]
- 128.Anum EA, et al. Connective tissue and related disorders and preterm birth: clues to genes contributing to prematurity. Placenta. 2009;30:207–215. doi: 10.1016/j.placenta.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parry S, Strauss JF., III Premature rupture of the fetal membranes. New England Journal of Medicine. 1998;338:663–670. doi: 10.1056/NEJM199803053381006. [DOI] [PubMed] [Google Scholar]
- 130.Rosen T, et al. Thrombin-enhanced matrix metalloproteinase-1 expression: a mechanism linking placental abruption with premature rupture of the membranes. Journal of Maternal–Fetal and Neonatal Medicine. 2002;11:11–17. doi: 10.1080/jmf.11.1.11.17. [DOI] [PubMed] [Google Scholar]
- 131.Stephenson CD, et al. Thrombin-dependent regulation of matrix metalloproteinase (MMP)-9 levels in human fetal membranes. Journal of Maternal–Fetal and Neonatal Medicine. 2005;18:17–22. doi: 10.1080/14767050500123632. [DOI] [PubMed] [Google Scholar]
- 132.Buhimschi IA, et al. Reduction– oxidation (redox) state regulation of matrix metalloproteinase activity in human fetal membranes. American Journal of Obstetrics and Gynecology. 2000;182:458–464. doi: 10.1016/s0002-9378(00)70239-0. [DOI] [PubMed] [Google Scholar]