

Abstract
Telomerase reverse transcriptase (TERT) is the catalytic subunit of telomerase, an enzyme that elongates telomeres at the ends of chromosomes during DNA replication. Recently it was shown that TERT has additional roles in cell survival, mitochondrial function, DNA repair and Wnt signaling, all of which are unrelated to telomeres. Here we demonstrate that TERT is enriched in Purkinje neurons, but not in the granule cells of the adult mouse cerebellum. TERT immunoreactivity in Purkinje neurons is present in the nucleus, mitochondria and cytoplasm. Furthermore, TERT co-localizes with mitochondrial markers, and immunoblot analysis of protein extracts from isolated mitochondria and synaptosomes confirmed TERT localization in mitochondria. TERT expression in Purkinje neurons increased significantly in response to two stressors: a sub-lethal dose of X-ray radiation and exposure to a high glutamate concentration. While X-ray radiation increased TERT levels in the nucleus, glutamate exposure elevated TERT levels in mitochondria. Our findings suggest that in mature Purkinje neurons TERT is present both in the nucleus and in mitochondria, where it may participate in adaptive responses of the neurons to excitotoxic and radiation stress.
Introduction
Telomerase is a ribonucleoprotein complex that elongates telomeres. The active enzyme consists of telomerase reverse transcriptase (TERT), and an RNA subunit (Terc). The activity of telomerase in vivo requires several additional proteins that are essential for holoenzyme biogenesis and trafficking to telomeres [1]. TERT is expressed in the neonatal brain of mice [2] as well as in distinct regions of the adult mouse brain such as the olfactory bulb, subventricular zone, hippocampus [3], cortex [4] and in Purkinje neurons of the cerebellum [5]. In contrast to its activity in embryos and neonates, telomerase activity in the adult mouse brain is extremely low. Nevertheless, in the last decade TERT has been documented in the cytoplasm of cancerous and normal cells, including neurons [6–8]. Cytoplasmic TERT exhibits additional functions beyond telomere elongation, including inhibition of apoptosis, enhanced mitochondrial functionality, DNA repair and Wnt signaling [9]. However, the roles of TERT in both DNA repair and Wnt signaling are controversial [10]. While defining the function of TERT in mitochondria has been elusive [11], the translocation of TERT from the nucleus to mitochondria was suggested to be crucial for TERT-induced cellular immortalization [12].
Elevated levels of TERT can protect primary neurons in culture from insults including NMDA-induced excitotoxicity and DNA damage induced by etoposide [13, 14]. Additionally, TERT knockdown exacerbates neuronal death caused by exposure to excitotoxins, DNA damaging agents and the β-amyloid protein [14, 15]. Moreover, ectopic TERT expression in mice reduced sensitivity to stroke [16–18], and pharmacological enhancement of TERT expression retarded the onset of symptoms in the SOD1G93A transgenic mouse model of amyotrophic lateral sclerosis [19]. Here we show that in the adult mouse cerebellum TERT is detectable only in Purkinje neurons where it is present in mitochondria, including those in synaptic terminals. Mitochondrial TERT levels increased in response to exposure to a high concentration of glutamate, while nuclear TERT levels increased in response to X-ray radiation.
Results
TERT is present in cerebellar Purkinje neurons
The presence of TERT in Purkinje neurons was previously reported [5]. The specific localization of TERT mRNA in Purkinje neurons can also be seen in the Allen Brain Atlas (http://mouse.brain-map.org/) [20]. Staining the cerebellum with anti-TERT antibodies showed that in Purkinje neurons TERT is found mostly in the cytoplasm and that it can be detected throughout the molecular layer in a pattern that resembles the dendrites of Purkinje neurons (Figure 1A). The specificity of the TERT antibody was demonstrated by the presence of a single band on immunoblots of whole-cell protein extracts derived from the cerebellum of wild type mice, and by the lack of the same band in samples derived from the cerebellum of TERT knockout mice (Figure 1B). Both antibodies did not detect TERT in the cerebellum of the TERT KO mice (supplement figure 1). In addition, human glioblastoma protein extracts showed a slightly higher molecular weight product, corresponding to the well-documented 5 kDa size difference between human and mouse TERT proteins (Figure 1B). Similar staining of TERT in the cerebellum, albeit exhibiting higher background in the granule cell layer, was observed using a different anti-TERT antibody (Figure 1C). Because the higher background observed with the latter antibody could be attributed to the fact that it generated multiple bands in Western blots (data not shown), it was not further used in the present study.
Figure 1. TERT is expressed in cerebellar Purkinje neurons of adult mice.

A. Representative image (n=5 mice) of TERT immunoreactivity in a section of the cerebellum. Note that Purkinje neurons exhibit robust TERT immunoreactivity compared to all other cells. B. Whole-cell protein extracts were prepared from U87 human glioblastoma cells, N2a mouse neuroblastoma cells, the cerebellum of a 16-week old TERT knockout mouse and an age-matched wild-type mouse. Ten micrograms of protein were analyzed by immunoblot assay using anti TERT and anti β-actin antibodies. A representative blot (n=2) illustrates the specificity of the antibody. C. Representative image (n=5) of immune staining with anti-TERT antibody (Rockland). Scale bar=50μm
TERT protein has been reported to be present in the mitochondria of several non-neuronal cell types [7, 21] [22]. Thus, we speculated that the staining pattern we observed in the molecular layer represents TERT in mitochondria located in dendrites of cerebellar Purkinje neurons. This possibility was examined by co-staining for TERT and for the mitochondrial proteins cytochrome c or COX4 (Figures 2A and B, respectively). 3D projection of the colocalization can be seen in Supplemental Figure 2. Line profile analysis of the immunofluorescent signal of TERT and COX4 revealed that the two co-varied (Figure 2C), consistent with co-localization. Indeed, whole-image co-localization analysis revealed significant correlation between TERT and COX4 (R=0.89+/−0.12, Patterson P<0.02; 7 mice, 4–6 images analyzed/mouse). This result indicates that in the molecular layer of the cerebellum TERT is associated with mitochondria. Lack of fluorescence crosstalk is demonstrated by the low magnitude of TERT staining both in the granule cell layer and in the cell bodies of granule cells in the molecular layer, even where mitochondria were present. We further noticed that staining was specific to Purkinje neurons and not to all inhibitory neurons, because parvalbumin-positive inhibitory interneurons located within the molecular layer did not exhibit TERT staining (Figure 2D).
Figure 2. TERT is co-localized with mitochondrial markers in the dendrites of cerebellar Purkinje neurons.

A. Representative image (n=4) of a cerebellar coronal section from an adult mouse immunostained with TERT (red) and Cytochrome C (green) antibodies. Arrows indicate examples of the co-localization of TERT and cytochrome C in the dendrites of Purkinje neurons (notice cell body in lower right corner). B. Representative image (n=7) of a cerebellar coronal section from an adult mouse immunostained with TERT (red) and COX-4 (green) antibodies. C. Example of a line profile analysis that plots the fluorescence intensity of both colors along the light blue line marked in B. Notice the co-variation of TERT (red) with the mitochondrial marker COX-4 (green). D. Representative image (n=3) of a cerebellar coronal section from an adult mouse immunostained with TERT (red) and parvalbumin (green) antibodies. Notice the colocalization of TERT and parvalbumin in the Purkinje neuron layer, but the lack of colocalization in the parvalbumin-positive cell bodies found within the molecular layer. E. Nuclear/cytoplasmic, mitochondrial and synaptosomal protein extracts were prepared from the cerebellum of eight mice (brains were pooled into groups of two in order to provide sufficient protein). Protein extracts (30 μg) were analyzed by immunoblot assay using antibodies against mTERT, synaptophysin and VDAC. The blot is a representative of 3 independent blots. F. The cerebellum was dissected from four 12–16 month old mice and protein extract were isolated from the following compartments: cytoplasm (Cyto), the nucleoplasm (NP, isolated nucleus treated with CHAPS alone) and protein bound to DNA (NDb, CHAPS with 1M NaCl). Protein extracts (30 μg) were analyzed by Western Blot assay using anti-TERT antibody, anti-Top1 antibody, anti-Lamin B and anti β-tubulin. Scale bar=20μm
In order to verify the presence of TERT in the mitochondria, cerebella were isolated from 8 mice and pooled into 4 groups (2 cerebella/group) prior to isolation of mitochondria, synaptosomes (containing synaptic terminal mitochondria) and a mixture of the nucleoplasm and cytoplasm protein extracts. The purity of the fractions was assessed using antibodies against VDAC (mitochondria) and synaptophysin (synaptosomes, Figure 2E). The mitochondrial and synapse markers were present only in the expected fractions (VDAC in the mitochondrial and synaptosomal fraction, and synaptophysin only in the synaptosome fraction), and TERT was present in all three fractions (Figure 2E).
It was previously reported that TERT is bound to DNA by a network of backbone- and solvent-mediated interactions [23]. CHAPS buffer (a commonly used lysis buffer for telomerase extraction) is not expected to extract TERT bound to DNA. Increasing the ionic strength of the solution by adding NaCl disrupts electrostatic DNA/protein interactions [24], thus adding salt to the CHAPS lysis buffer enables the release of DNA-binding proteins. Indeed, we observed that when pellets containing DNA and membrane debris, which had been previously washed three times with CHAPS buffer, were washed once more with CHAPS buffer supplemented with 1M NaCl (final concentration), the quantity of extracted protein increased by three fold (P<0.01), as compared to the previous CHAPS wash (data not shown). The inability of CHAPS alone to extract DNA-binding proteins is also demonstrated by the lack of topoisomerase-1, a known DNA-binding enzyme, in the CHAPS-alone fraction, and its presence in the CHAPS+1M NaCl fraction (Figure 2F). Higher levels of TERT were found in the CHAPS + 1M NaCl fraction than in the one obtained with CHAPS alone (Figure 2F). This result suggests that in the nucleus of Purkinje neurons TERT is mostly found interacting with the DNA (Figure 2F).
TERT expression increases in response to glutamate exposure and X-ray irradiation
It has been shown that TERT expression can protect different cells from oxidative stress in a telomere-independent manner [13–19]. The presence of TERT in the mitochondria and its binding to DNA led us to examine its response to insults such as excitotoxicity and DNA damage.
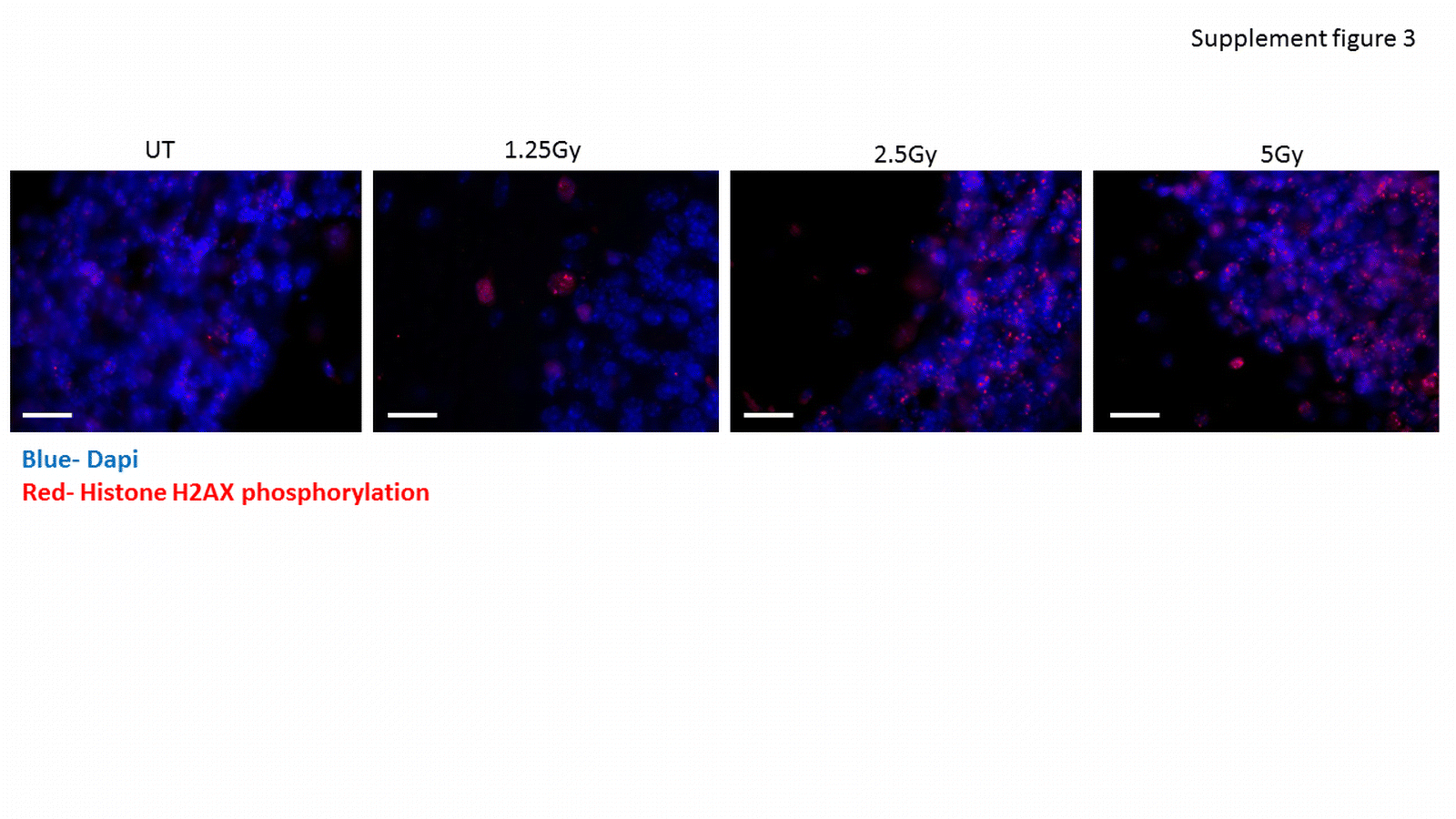
In order to examine the effect of DNA damage, we first aimed to determine a sub-lethal dose of X-rays. For this purpose, we exposed the heads of body-protected anesthetized mice to various X-ray radiation doses (1.25, 2.5 and 5 Gy). The cerebellum was dissected 15 minutes or 4 hours after irradiation, and was stained with a γ-H2AX antibody that detects DNA double-strand breaks caused by radiation. While 4 hours following 1.25 and 2 Gy X-ray irradiation no DNA damage was detected in the cerebellum, clear γ-H2AX foci were detected in mice that received 5 Gy irradiation. These mice also showed signs of unbalanced movement that were not investigated further (data not shown). Fifteen minutes after irradiation DNA damage was detected for all three doses, and quantification of the number of γ-H2AX foci per neuron revealed a dose-dependent increase in foci (representative images are shown in Supplemental Figure 3 and quantification in Figure 3A) [25]. There was an overall effect of irradiation on cerebellar TERT protein levels (Figure 3C), but the effect was not dose-dependent Supplemental Figure 4). Analysis of the pooled results from all three doses of radiation revealed a significant increase of 1.29 ± 0.16 fold, (p < 0.05, n=7 untreated and 9 irradiated mice) (Figure 3C). An increase in the TERT signal in Purkinje neurons of 1.87 ± 0.14 fold (p < 0.05 n=7 untreated and n=9 irradiated mice) was also demonstrated by immunofluorescence staining (a representative image is presented in Figure 3D and quantification in Figure 3E).
Figure 3. TERT levels are elevated in Purkinje neurons in response to radiation and exposure to glutamate.

A. Adult mice were anesthetized and their bodies were shielded before being exposed to different doses of X-ray radiation. 15 minutes after irradiation, the cerebellum was dissected and the levels of DNA damage were analyzed by immunofluorescence with anti-γ-H2AX antibodies. The number of γ-H2AX foci per cell were counted by a blinded observer using image J cell counting software and plotted as a function of radiation dose (n=3 mice per dose, linear regression analysis p<0.05). B. Immunoblot analysis with hTERT and β-actin antibodies. Left panel: protein was extracted from adult mouse cerebellum 15 minutes after X-ray irradiation delivered at the indicated dose. Right panel: protein was extracted from a pool of 4 acute cerebellum slices that were treated with 1mM glutamate or vehicle for 30 min and then with fresh Ringer solution for additional 3 hours. C. Quantification of the immunoblot results (shown in B) by densitometry analysis. The result from the different radiation doses were pooled together and compared to seven mice that were not exposed to radiation (mean±SEM; n=7 untreated, n=9 X-ray radiation). * p<0.05, Students’ t-test analysis. D. Immunofluorescence analysis of TERT (red) expression in cerebellum slices derived from untreated mice (left panel), irradiated mice (middle panel) and glutamate-treated ACS (right panel). The yellow lines mark Region of interest (ROI) that was determined by employing threshold on TERT staining. Note that the larger ROI in images of irradiated and glutamate treated cerebella demonstrate increase TERT staining. E. Quantification of the fluorescence intensity within the thresholded area (as presented in D; mean±SEM; n=7 untreated, n=9 X-ray radiation and n=4 excitotoxicity). At least 50 Purkinje neurons were selected for each mouse. (*p<0.05, t-test). Scale bar=20μm
Excitotoxicity is a neurological insult during which neurons are damaged by excessive stimulation with excitatory neurotransmitters, such as glutamate [26, 27]. We examined whether a high concentration of glutamate affects TERT levels in acute cerebellar slices (ACS). Four ACS, 400 μm each, were prepared from each mouse and were allowed to recover for 1 hour prior to exposure to 1 mM glutamate for 20 minutes. Following glutamate treatment, ACS were re-incubated in fresh medium for an additional three hours. Equivalent amounts of whole protein extracts from glutamate-treated and untreated ACS were analyzed for TERT protein levels by immunoblot. TERT levels increased by 1.7 fold in response to 1 mM glutamate (p < 0.05, n=7 untreated and n=4 glutamate-treated ACS; see Figure 3B, right panel for a representative blot, and Figure 3C for quantification). TERT protein levels were significantly elevated (2.19 ± 0.2 fold, Figure 3E) specifically in Purkinje neurons (a representative image is presented in Figure 3D and quantification in Figure 3E).
To explore in more detail the distribution of the enhanced TERT levels, we analyzed separately the effect of either excitotoxicity or DNA damage on TERT levels in nuclei and in mitochondria (Figure 4). Nuclear regions were identified as those stained by DAPI but lacking COX-4 staining (Figure 4A–C), while COX-4 staining served to identify mitochondria (Figure 4C). Figure 4D shows the quantification of TERT fluorescence intensities in the nucleus and in mitochondria. Exposure to glutamate reduced TERT levels in the nucleus, but increased it in mitochondria (Figure 4D), suggesting a possible translocation of TERT from the nucleus to the mitochondria. Similar translocation of TERT was previously reported following oxidative stress [7, 21, 22]. Radiation increased TERT levels in the nucleus as well as in the mitochondria (Figure 4D). The fraction of TERT immunoreactivity associated with the nucleus and mitochondria was determined by dividing the total pixel intensity in the nucleus or mitochondria into the total intensity in the image (without any threshold). The results depicted in figure 4E show that while glutamate induces a significant (P=0.01) reduction in the fraction of TERT in the nucleus, the increase in the mitochondria was not significant (P=0.16).
Figure 4. Intracellular localization of TERT is altered by both DNA damage and excitotoxic glutamate.

A–C. Representative image of a cerebellar section immunostained for TERT (red) and COX4 (green), and counter stained with the nuclear DNA stain DAPI (blue). B. DAPI channel alone. The lines represent the region of interest (ROI) that was used to distinguish the nucleus. C. Binary image of mitochondria that was generated by employing the same threshold on all COX4 images from all treatments. This image was used in order to determine the ROI that represent mitochondrial localization. D. The local expression of TERT was measured by the average red fluorescence intensity (TERT staining) in each selected region (as shown in B and C). The intensity in the nucleus was normalized to the blue fluorescence intensity and the intensity in the mitochondria was normalized to the green fluorescence intensity. E. The fraction (% of total staining) of TERT present in the nucleus and mitochondria was determined by dividing the sum of the red fluorescence intensity (TERT staining) in the ROI represent nucleus or mitochondria (as shown in B and C) to the red fluorescence intensity in the full image. Values are mean±SEM (n=7 untreated, n=9 X-ray radiation and n=4 Glutamate-treated). *p<0.05, t-test. Scale bar=20μm
Discussion
Neurons are post-mitotic, fully differentiated cells with relatively stable telomere length and so may not require TERT for telomere maintenance. Emerging evidence suggests that TERT participates in telomere-independent anti-apoptotic pathways that may be important for neuronal function and survival [9]. TERT is expressed in mouse neonatal brain [2] as well as in distinct regions of the adult mouse brain including the olfactory bulb, subventricular zone, hippocampus [3], cortex [4] and in cerebellar Purkinje neurons [5]. Here we provide evidence that Purkinje neurons express TERT, whereas neurons in the granular and molecular layers do not. Interestingly, immunostaining and subcellular fractionation analysis showed that high levels of TERT are present in the cytoplasm and in dendritic mitochondria. The localization of TERT in the cytoplasm and mitochondria strongly suggests that it possesses a function beyond telomere elongation. We show that X ray irradiation or exposure to a high concentration of the excitatory neurotransmitter glutamate significantly increased TERT levels. The latter findings suggest a role for TERT in maintaining Purkinje neuron survival in response to DNA damage and excitotoxicity, consistent with previous reports from studies of other types of neurons in-vitro [13–14].
We found that TERT is present in mitochondrial of Purkinje neurons. Because mitochondrial TERT has been shown to protect fibroblasts, endothelial cells and cancer cells [28] it is possible that TERT can enhance Purkinje neuron survival. It will therefore be of interest to determine if TERT also regulates mitochondrial function in Purkinje neurons. Recently it was reported that TERT can reduce the sensitivity of hippocampal neurons to tau pathology and oxidative stress [6]. TERT can translocate from the nucleus to the mitochondria, and trapping TERT in the nucleus by deleting its nuclear export sequence increases cellular sensitivity to oxidative stress [7, 12], suggesting that mitochondrial translocation of TERT plays a cytoprotective role. Our findings suggest that when Purkinje neurons are treated with glutamate, TERT levels in the cell nucleus decrease and mitochondria-associated TERT levels may increase. The relatively short time period during which the change in TERT levels in mitochondria was observed (3 hours) is consistent with translocation. TERT levels were quickly (15 min) increased in the mitochondria and nucleus in response to X-ray radiation. Such a short period argues against protein translation and may be a result of the release of TERT from the DNA to the nucleoplasm and its translocation to the mitochondria and/or conformational changes in TERT that expose more epitopes to the antibody [29].
Purkinje neurons have relatively complex, expansive dendritic trees, and each Purkinje neuron receives inputs from about 100,000 synapses. This complicated morphology and connectivity requires high mitochondrial activity, which generates a considerable oxidative load [30]. Accordingly, Purkinje neurons are among the most sensitive neurons in the brain to stress, degenerating early during aging as well as in the context of several neurodegenerative diseases [31]. Whether TERT can protect Purkinje neurons against such pathological conditions is unknown. However, postmortem hippocampal slices from Alzheimer’s disease patients show that TERT localizes to the mitochondria of neurons with low levels of hyperphosphorylated Tau, which suggests that TERT may protect neurons against Tau pathology [6].
The chromatin of Purkinje neurons is less condensed, which may result in greater sensitivity to DNA damage. TERT may have a role in DNA repair, as demonstrated in human fibroblasts [32]. Sharma et al showed that the binding of TERT to DNA is essential for its role in DNA repair [29, 32]. TERT knockdown also sensitizes cells to radiation and to DNA damage, as observed both in cancer cells and in primary neurons [13]. The significance of TERT in the DNA repair processes is presently controversial, as TERT-knockout mice exhibit an intact DNA repair capacity [10].
The possibility that TERT protects DNA in Purkinje neurons is intriguing, because even though the telomeres of post-mitotic neurons do not shorten due to proliferation they may erode as a result of DNA damage [33–35]. It is therefore conceivable that a TERT-mediated telomere repair mechanism may have evolved in neurons in order to cope with the slow erosion of the telomeres during the long lifespan of the neurons. The ability of TERT to protect neurons from insults such as DNA damage, oxidative stress, NMDA and Aβ42 [14, 15], together with our observations of increased TERT levels in the mitochondria or nucleus of Purkinje neurons in response to insults, support our hypothesis that TERT mediates a protective effect in these cells. The recent finding that neuronal TERT regulates p15INK4B mRNA in RNA granules also suggests roles in neuronal survival and aging [8]. Developmental cerebellar hypoplasia is a phenotype of Hoyeraal Hreidarsson syndrome, which is caused by telomerase dysfunction [36], and short telomeres and degeneration of Purkinje neurons occur in ataxia telangiectasia [37]. Although still controversial, it has been reported that short telomeres in leukocytes are associated with several neurodegenerative diseases [38]. However, more experimental evidence is needed to determine the importance of TERT during normal aging of Purkinje neurons and in diseases in which they degenerate.
Materials and Methods
Animals
CD-1 male mice (– weeks) or C57BL/6 male mice (10–16 weeks) were used for the investigation of TERT localization. Tert null (Tert−/−) mice were a generous gift from Dr. Yie Liu (National Institute on Aging) [39]. Excitotoxicity assays and X-ray radiation were performed with the CD-1 mice. Animal care and experimental procedures followed National Institutes of Health guidelines and were approved either by the National Institute on Aging Animal Care and Use Committee or by the Ben-Gurion University Animal Experimentation Ethics Committee.
Preparation of acute cerebellar slices and exposure to glutamate
Brain slices were prepared as previously described [40]. Briefly, brains were quickly removed from the skull, washed with Ringer solution (150 mM NaCl, 4.6 mM KCl, 26 mM HCO3Cl, 1.3 mM Na2HPO4, 2.5 mM CaCl2, 1 mM MgCl2, 1 mM SO4CH3, and 10 mM glucose (pH 7)., placed in oxygenated Ringer solution at 4°C, and coronal sections (400 μm thick) of the cerebellum were prepared. The sections were incubated in cold oxygenated Ringer solution for 30 min before any further treatments. Slices were treated with Ringer solution supplemented with 1mM Glutamic acid (Sigma, Rehovot, Israel), and then incubated in fresh Ringer solution for an additional 3 hours before being embedded in paraffin or processed for protein extraction.
Mouse irradiation
CD-1 male mice (6–8 weeks) were anesthetized with a ketamine (50 mg/kg)/Xylazine (10 mg/kg) mix and placed in a device that protected their body inside a Faxitron X-ray cabinet. Mice were irradiated for 2 minutes at non-lethal doses: 1.25, 2.5, 5 Gy. The mice were sacrificed 15 min or 4 hours after the irradiation. Protein extracts and paraffin-embedded sections were prepared from the cerebellum.
Preparation of protein extracts
Brains were quickly removed from the skull, washed, and placed in oxygenated Ringer solution at 4°C. The brains were homogenized using a manual homogenizer (pestle B). The homogenates were centrifuged at 500 × g at 4°C for 7 min and the pellets were subjected to nuclear and cytoplasmic extractions, as previously described [40–42]. Briefly, for the cytoplasmic extract, cells were re-suspended in buffer A (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 1 mM EDTA) containing a mixture of protease inhibitors [final concentrations: 2 μg/ml aprotinin, 2 μg/ml antipain, 2 μg/ml leupeptin, 1 μg/ml pepstatin A, 2 μg/ml PMSF (phenylmethylsulfonyl fluoride)], and kept on ice for 15 min. Lysis was performed by passing the solution 10–15 times through a 21 gauge needle. The cytosolic fraction was obtained by centrifugation at 9300 × g at 4°C for 7 min and the supernatant was collected. For preparation of the nuclear extract, the pellet from the cytoplasmic preparation was re-suspended in CHAPS buffer containing 10 mM TRIS HCl, 5 mM 2-β-Mercaptoethanol pH-7.0, 1 mM MgCl2, 1 mM EDTA, 0.1 mM PMSF, 0.5% CHAPS (3[(3 Cholamidopropyl)-dimethylammonio-propanesulfonic acid), and 10% glycerol, followed by incubation at 4°C for 30 minutes. The nuclear extract was centrifuged for 30 minutes at 9300 × g at 4°C, and the supernatant was collected.
Determination of protein concentration
Protein concentration was determined by the Bradford assay using the Bio-Rad protein assay kit (Bio-Rad Laboratories).
Immunoblot analysis
Samples from the various experiments were analyzed by polyacrylamide gel electrophoresis and immunoblotting as previously described [43, 44]. Immunocomplexes were detected by enhanced chemiluminescence (ECL) (Santa Cruz Biotechnology). The following antibodies were used for immunoblots: mice IgG hTERT monoclonal antibody (1531-1, Epitomics, CA), rabit IgG hTERT (600-401-252S Rockland Immunochemicals, PA), Anti-β-actin (Irvine, CA), anti-top1 (goat IgG sc-26167), anti-lamin B (mouse IgG sc-365214) and anti- β-tubulin (mouse IgG sc-58886) (Santa Cruz Biotechnology, CA), anti-VDAC (rabit IgG ab34726) and anti-synaptophysin (mouse IgG ab14692) (Abcam) and anti cytochrome C rabit IgG SAB4502234 Sigma-Aldrich).
Immunofluorescence
Paraffin-embedded sections were de-paraffinized in xylene and rehydrated in decreasing concentrations of ethanol and then heated in a microwave for 15 min in 0.01M citric acid buffer, pH 6.0, to reveal antigens. Alternatively, frozen sections were used. The sections were rinsed with PBS and then incubated for 30 min with blocking solution (4% normal horse serum, 0.02% X-triton, 3% BSA diluted in PBS). The sections were then incubated overnight with the suitable primary antibody at the specified dilutions: anti-hTERT 1:400 (Rockland) and 1:800 (Epitomics, CA), Anti-γH2AX 1:1000 (Abcam), anti-cytC 1:500 (BD Bioscience), anti-parvalbumin and anti-COX4 (Santa-cruz) at 4°C. After three rinses in PBS, the sections were again incubated for 1 hour with Alexa Fluor 488 or 568 conjugated anti-rabbit or anti-mouse secondary antibodies (Life Technologies). For nuclear staining, sections were incubated with 0.1 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI; MP Biomedical LLC) for 20 min at room temperature, rinsed with PBS, and mounted with anti-fade mounting medium (Kierkegaard, Gaithersburg, MD, USA). One micrometer Z-stack images of were acquired using a confocal microscopy system FV1000 (Olympus, Tokyo, Japan) or with a Zeiss LSM 510 confocal microscope. Images were acquired using either 20X or 40X magnification objectives. Crossover between the color channels was ruled out by single antibody staining.
Image Analysis
Image analyses were performed using the Fiji software plugins colocalization_finder, colocalization_test, RGB_profile and measure_RGB. The total intensity of TERT staining was divided by the number of Purkinje cells in each image, to account for variability. The summed TERT intensity in nuclei was normalized by the summed intensity of DAPI, while the summed TERT signal in mitochondria was normalized to the summed COX4 intensity in order to reduce image-to-image variability. To measure the fraction of TERT in the nuclei and mitochondria, TERT intensity in these compartments were divided to it entire image intensity. These measurements of TERT intensity were not normalized, because the ratio between the nuclei and mitochondria and total intensity already account for image-to-image variability.
Statistical Analysis
Results are presented as the mean ± standard error of the mean (SEM) of at least three independent experiments. The statistical significance of the results was obtained using the student’s t-test or Two Way ANOVA, using OriginPro7 software. Statistical significance was set at a confidence level of 0.05.
Supplementary Material
Supplemental Figure 1: Expression of TERT in WT and TERT KO mice. Cerebellum slices were taken from adult WT (A) and TERT KO (B) and stained with anti-TERT antibody (green).
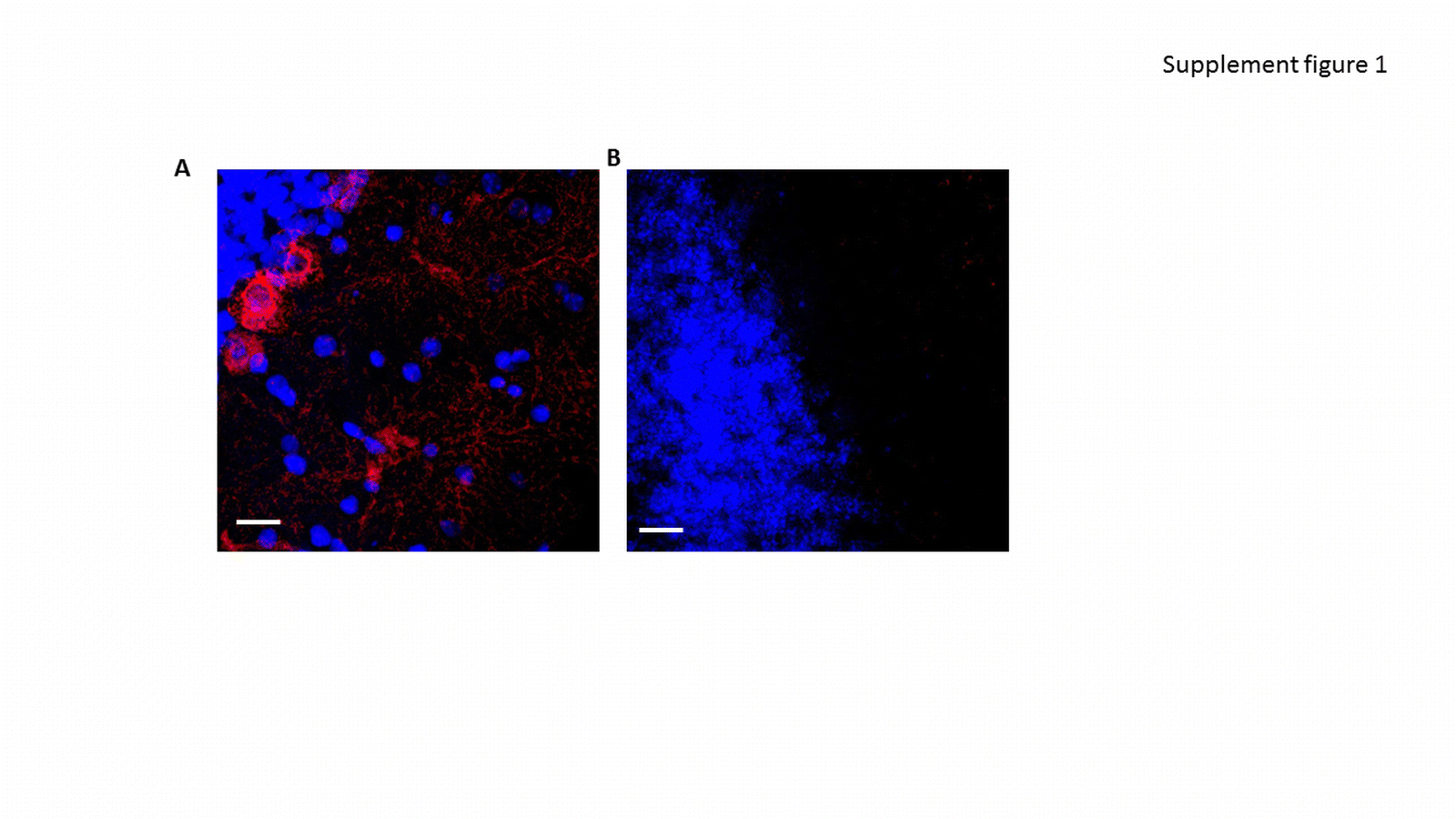{kind=link}
Supplemental Figure 2: TERT co-localizes with Purkinje neuron mitochondria. 3D projection of adult mouse cerebellum slice stained with anti-TERT antibody and anti-cytochrome C antibody.
{kind=link}
Supplemental Figure 3: DNA damage increases in cerebellar Purkinje neurons following irradiation. Cerebellum was dissected from mice 15 minutes after X-ray irradiation in doses of 1.25Gy. 2.5Gy and 5 Gy and stained with anti γ-H2AX antibody.
{kind=link}
Supplemental Figure 4: TERT expression increases in Cerebellar Purkinje neurons following irradiation. Quantification of the TERT expression in response each of the radiation doses.
Footnotes
Conflict of interest: The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
References
- 1.Cohen SB, et al. Protein composition of catalytically active human telomerase from immortal cells. Science. 2007;315(5820):1850–3. doi: 10.1126/science.1138596. [DOI] [PubMed] [Google Scholar]
- 2.Klapper W, Shin T, Mattson MP. Differential regulation of telomerase activity and TERT expression during brain development in mice. J Neurosci Res. 2001;64(3):252–60. doi: 10.1002/jnr.1073. [DOI] [PubMed] [Google Scholar]
- 3.Caporaso GL, et al. Telomerase activity in the subventricular zone of adult mice. Mol Cell Neurosci. 2003;23(4):693–702. doi: 10.1016/s1044-7431(03)00103-9. [DOI] [PubMed] [Google Scholar]
- 4.Flanary BE, Streit WJ. Telomeres shorten with age in rat cerebellum and cortex in vivo. J Anti Aging Med. 2003;6(4):299–308. doi: 10.1089/109454503323028894. [DOI] [PubMed] [Google Scholar]
- 5.Eitan E, et al. Telomerase expression in adult and old mouse Purkinje neurons. Rejuvenation Res. 2012;15(2):206–9. doi: 10.1089/rej.2011.1285. [DOI] [PubMed] [Google Scholar]
- 6.Spilsbury A, et al. The Role of Telomerase Protein TERT in Alzheimer’s Disease and in Tau-Related Pathology In Vitro. J Neurosci. 2015;35(4):1659–74. doi: 10.1523/JNEUROSCI.2925-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos JH, Meyer JN, Van Houten B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum Mol Genet. 2006;15(11):1757–68. doi: 10.1093/hmg/ddl098. [DOI] [PubMed] [Google Scholar]
- 8.Iannilli F, et al. Cytoplasmic TERT Associates to RNA Granules in Fully Mature Neurons: Role in the Translational Control of the Cell Cycle Inhibitor p15INK4B. PLoS One. 2013;8(6):e66602. doi: 10.1371/journal.pone.0066602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cong Y, Shay JW. Actions of human telomerase beyond telomeres. Cell Res. 2008;18(7):725–32. doi: 10.1038/cr.2008.74. [DOI] [PubMed] [Google Scholar]
- 10.Strong MA, et al. Phenotypes in mTERT/ and mTERT/mice are due to short telomeres, not telomere-independent functions of telomerase reverse transcriptase. Mol Cell Biol. 2011;31(12):2369–79. doi: 10.1128/MCB.05312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majerska J, Sykorova E, Fajkus J. Non-telomeric activities of telomerase. Mol Biosyst. 2011;7(4):1013–23. doi: 10.1039/c0mb00268b. [DOI] [PubMed] [Google Scholar]
- 12.Kovalenko OA, et al. A mutant telomerase defective in nuclear-cytoplasmic shuttling fails to immortalize cells and is associated with mitochondrial dysfunction. Aging Cell. 2010 doi: 10.1111/j.1474-9726.2010.00551.x. [DOI] [PubMed] [Google Scholar]
- 13.Fu W, et al. The catalytic subunit of telomerase is expressed in developing brain neurons and serves a cell survival-promoting function. J Mol Neurosci. 2000;14(1–2):3–15. doi: 10.1385/JMN:14:1-2:003. [DOI] [PubMed] [Google Scholar]
- 14.Mattson MP, Klapper W. Emerging roles for telomerase in neuronal development and apoptosis. J Neurosci Res. 2001;63(1):1–9. doi: 10.1002/1097-4547(20010101)63:1<1::AID-JNR1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, et al. TERT promotes cellular and organismal survival independently of telomerase activity. Oncogene. 2008;27(26):3754–60. doi: 10.1038/sj.onc.1211037. [DOI] [PubMed] [Google Scholar]
- 16.Kang HJ, et al. Ectopic expression of the catalytic subunit of telomerase protects against brain injury resulting from ischemia and NMDA-induced neurotoxicity. J Neurosci. 2004;24(6):1280–7. doi: 10.1523/JNEUROSCI.4082-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qu Y, et al. Telomerase reverse transcriptase upregulation attenuates astrocyte proliferation and promotes neuronal survival in the hypoxic-ischemic rat brain. Stroke. 2011;42(12):3542–50. doi: 10.1161/STROKEAHA.111.626325. [DOI] [PubMed] [Google Scholar]
- 18.Zhao F, et al. The neuroprotective role of TERT via an antiapoptotic mechanism in neonatal rats after hypoxia-ischemia brain injury. Neurosci Lett. 2012;515(1):39–43. doi: 10.1016/j.neulet.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 19.Eitan E, et al. Novel telomerase-increasing compound in mouse brain delays the onset of amyotrophic lateral sclerosis. EMBO Mol Med. 2012;4(4):313–29. doi: 10.1002/emmm.201200212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445(7124):168–76. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 21.Li P, et al. Mitochondrial translocation of human telomerase reverse transcriptase in cord blood mononuclear cells of newborns with gestational diabetes mellitus mothers. Diabetes Res Clin Pract. 2014;103(2):310–8. doi: 10.1016/j.diabres.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 22.Haendeler J, et al. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler Thromb Vasc Biol. 2009;29(6):929–35. doi: 10.1161/ATVBAHA.109.185546. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell M, et al. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol. 2010;17(4):513–8. doi: 10.1038/nsmb.1777. [DOI] [PubMed] [Google Scholar]
- 24.Shiffman ML, et al. Protein dissociation from DNA in model systems and chromatin. Nucleic Acids Res. 1978;5(9):3409–26. doi: 10.1093/nar/5.9.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rube CE, et al. DNA double-strand break repair of blood lymphocytes and normal tissues analysed in a preclinical mouse model: implications for radiosensitivity testing. Clin Cancer Res. 2008;14(20):6546–55. doi: 10.1158/1078-0432.CCR-07-5147. [DOI] [PubMed] [Google Scholar]
- 26.Ward MW, et al. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20(19):7208–19. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Platt SR. The role of glutamate in central nervous system health and disease--a review. Vet J. 2007;173(2):278–86. doi: 10.1016/j.tvjl.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Parkinson EK, Fitchett C, Cereser B. Dissecting the non-canonical functions of telomerase. Cytogenet Genome Res. 2008;122(3–4):273–80. doi: 10.1159/000167813. [DOI] [PubMed] [Google Scholar]
- 29.Sharma GG, et al. hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene. 2003;22(1):131–46. doi: 10.1038/sj.onc.1206063. [DOI] [PubMed] [Google Scholar]
- 30.Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32(2):301–13. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- 31.Zhang B, et al. Deficiency of telomerase activity aggravates the blood-brain barrier disruption and neuroinflammatory responses in a model of experimental stroke. J Neurosci Res. 2010 doi: 10.1002/jnr.22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masutomi K, et al. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci U S A. 2005;102(23):8222–7. doi: 10.1073/pnas.0503095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Dilley C, Mattson MP. DNA damage responses in neural cells: Focus on the telomere. Neuroscience. 2007;145(4):1439–48. doi: 10.1016/j.neuroscience.2006.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rochette PJ, Brash DE. Human telomeres are hypersensitive to UV-induced DNA Damage and refractory to repair. PLoS Genet. 2010;6(4):e1000926. doi: 10.1371/journal.pgen.1000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, et al. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 2010;6(5):e1000951. doi: 10.1371/journal.pgen.1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aalfs CM, et al. The Hoyeraal-Hreidarsson syndrome: the fourth case of a separate entity with prenatal growth retardation, progressive pancytopenia and cerebellar hypoplasia. Eur J Pediatr. 1995;154(4):304–8. doi: 10.1007/BF01957367. [DOI] [PubMed] [Google Scholar]
- 37.Metcalfe JA, et al. Accelerated telomere shortening in ataxia telangiectasia. Nat Genet. 1996;13(3):350–3. doi: 10.1038/ng0796-350. [DOI] [PubMed] [Google Scholar]
- 38.Eitan E, Hutchison ER, Mattson MP. Telomere shortening in neurological disorders: an abundance of unanswered questions. Trends Neurosci. 2014;37(5):256–63. doi: 10.1016/j.tins.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, et al. The telomerase reverse transcriptase is limiting and necessary for telomerase function in vivo. Curr Biol. 2000;10(22):1459–62. doi: 10.1016/s0960-9822(00)00805-8. [DOI] [PubMed] [Google Scholar]
- 40.Zehorai E, et al. Glutamate regulates the activity of topoisomerase I in mouse cerebellum. Mol Neurobiol. 2008;38(3):242–52. doi: 10.1007/s12035-008-8044-x. [DOI] [PubMed] [Google Scholar]
- 41.Auer B, et al. Intracellular distribution of DNA topoisomerase I in fibroblasts from patients with Fanconi’s anaemia. Hum Genet. 1982;61(4):369–71. doi: 10.1007/BF00276603. [DOI] [PubMed] [Google Scholar]
- 42.Bendetz-Nezer S, Gazit A, Priel E. DNA topoisomerase I as one of the cellular targets of certain tyrphostin derivatives. Mol Pharmacol. 2004;66(3):627–34. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 43.Maniatis T, et al. The molecular genetics of human hemoglobins. Annu Rev Genet. 1980;14:145–78. doi: 10.1146/annurev.ge.14.120180.001045. [DOI] [PubMed] [Google Scholar]
- 44.Kaufmann SH, Svingen PA. Immunoblot analysis and band depletion assays. Methods Mol Biol. 1999;94:253–68. doi: 10.1385/1-59259-259-7:253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Expression of TERT in WT and TERT KO mice. Cerebellum slices were taken from adult WT (A) and TERT KO (B) and stained with anti-TERT antibody (green).
Supplemental Figure 2: TERT co-localizes with Purkinje neuron mitochondria. 3D projection of adult mouse cerebellum slice stained with anti-TERT antibody and anti-cytochrome C antibody.
Supplemental Figure 3: DNA damage increases in cerebellar Purkinje neurons following irradiation. Cerebellum was dissected from mice 15 minutes after X-ray irradiation in doses of 1.25Gy. 2.5Gy and 5 Gy and stained with anti γ-H2AX antibody.
Supplemental Figure 4: TERT expression increases in Cerebellar Purkinje neurons following irradiation. Quantification of the TERT expression in response each of the radiation doses.