

Abstract
Introduction
Primary ciliary dyskinesia (PCD) is a genetically heterogeneous recessive disorder resulting in chronic oto-sino-pulmonary disease. While PCD is estimated to occur in 1 in 20,000 individuals, fewer than 1,000 patients in the US have a well-established diagnosis.
Areas Covered
We provide an overview of the clinical manifestations of PCD, describe the evolution of diagnostic methods, and critique the literature on management of PCD.
Expert Opinion
Although interest in clinical studies in non-CF bronchiectasis has increased in recent years, some of whom enroll patients with PCD, the literature regarding therapy for PCD as a distinct entity is lacking, as the numbers are small, and there have been no sub-analyses published. However, with improved screening and diagnostic methods, the development of clinical and research consortiums, and actively enrolling registries of PCD patients, the environment is conducive to perform longitudinal studies of disease course and therapeutic studies to alter that course.
Keywords: bronchiectasis, genotype, Kartagener syndrome, nasal nitric oxide, phenotype, primary ciliary dyskinesia
1. Introduction
Primary ciliary dyskinesia (PCD) [OMIM #244400] is a genetically heterogeneous recessive disorder of motile cilia resulting in neonatal respiratory distress, chronic oto-sino-pulmonary disease, and male infertility; organ laterality defects are present in approximately 50% of individuals1–5. Initially recognized in 1933, the triad of chronic sinusitis, bronchiectasis, and situs inversus was termed Kartagener syndrome6. It was not until 1976 when Afzelius described the cilia in these patients as being immotile due to defective ciliary ultrastructure7 that some understanding of the etiology of the syndrome was obtained. Subsequent studies have revealed that cilia have a stiff, or even vigorous, but always uncoordinated and ineffective beat consistent with dyskinesia, rather than complete immotility. The term “primary” distinguishes it from secondary or acquired (i.e. non-genetically determined) ciliary defections associated with infection and inflammation.
Limitations imposed by hitherto inadequate diagnostic methods have made it difficult to determine the true prevalence of PCD. Based on population surveys of situs inversus and bronchiectasis in Norway and Japan, the incidence of PCD is estimated at 1 per 10,000–20,000 births8, 9. In the US, there are fewer than 1,000 patients with a well-established diagnosis of PCD5. The development and validation of nasal nitric oxide (nNO) as a screening test for PCD10 and continued discoveries of PCD causing mutations have improved diagnostic certainty5. At this time, there is no therapy specific for PCD; rather therapies are extrapolated from other suppurative lung diseases such as cystic fibrosis (CF) and non-CF bronchiectasis.
1.1 Cilia structure and function in health and disease
Cilia classification in vivo is based on function, creating two categories: motile and sensory. Normal motile cilia contain 9 outer doublets and a central pair (Figure 1). The outer doublets are composed of microtubules of α- and β-tubulin monomers, and possess outer and inner dynein arms along their length, which contain enzymes for ATP hydrolysis, producing ciliary movement. Stability is provided by the nexin links connecting the outer doublets and by the radial spokes that extend from the doublets to the central pair. Mutations of genes that encode any of these components, or those are involved in the cilia assembly mechanism, result in clinical disease. Specialized motile cilia, which lack a central pair, move in a rotatory fashion and are responsible for organ lateralization during embryogenesis, hence organ laterality defects are associated with some (approximately 50%) patients with PCD.
Figure 1. Normal motile cilia (9+2) configuration.

Reprinted with permission of the American Thoracic Society. Copyright © 2014 American Thoracic Society. Knowles MR, Daniels LA, Davis SD, et al. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med 2013 Oct 15;188(8):913–22. Official Journal of the American Thoracic Society.
A single non-motile sensory cilium is present in most cells of the body and contains specialized proteins and receptors, which sense the local environment. This gives the sensory cilia an important role in signaling pathways and planar cell polarity. Mutations in genes involved in the assembly and function of the sensory cilia result in disorders such as Bardet-Biedl syndrome, retinitis pigmentosa, Joubert syndrome, nephronophthisis, and autosomal dominant polycystic kidney disease. For the purposes of this review, we will not further address these clinical entities.
2. Diagnosis (Figure 2)
Figure 2.
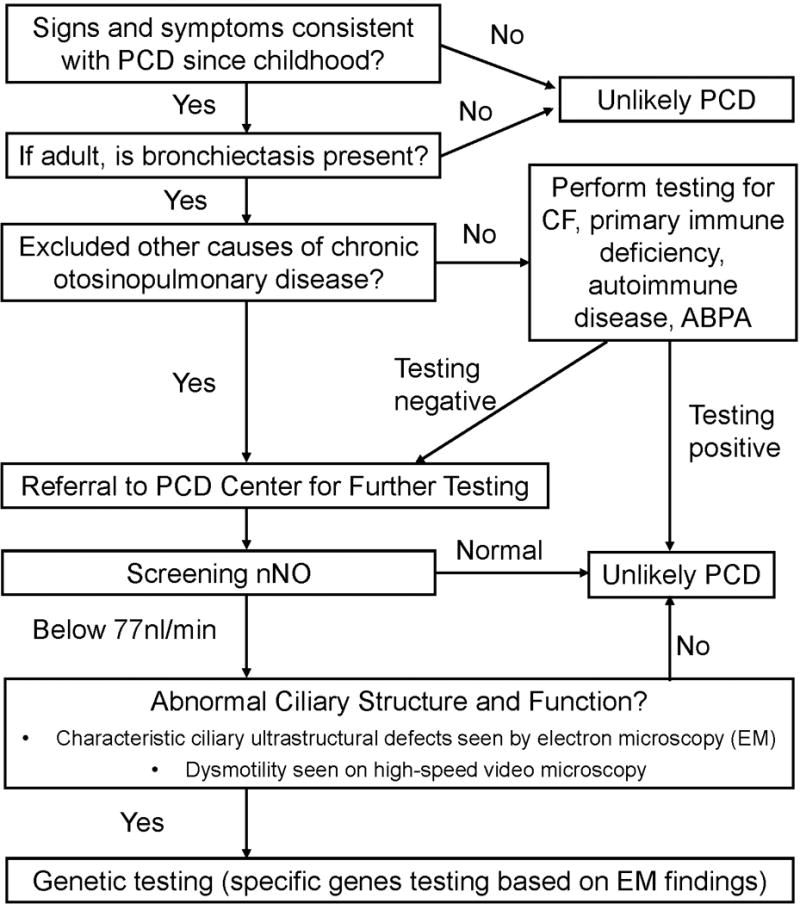
Diagnostic algorithm for PCD diagnosis
A consistent clinical phenotype should prompt a referral to a specialized center for further testing. See next section, but briefly, these include neonatal respiratory distress, persistent cough or recurrent pneumonias from birth, recurrent difficult otitis media, and/or unexplained bronchiectasis in adulthood. The presence of situs inversus or ambiguous, or male infertility and female subfertility in association with the above are particularly suggestive of the likelihood of PCD. Unlike CF, digestive tract abnormalities (pancreatic insufficiency, liver disease, malnutrition) are not usually a feature of PCD. While some features can be seen in isolation in the general population (even situs inversus alone), persistence of hallmark clinical features is highly suggestive of PCD. Because PCD diagnostic testing is expensive and requires expertise, identification of clinical manifestations of PCD is critical to initiate further diagnostic evaluation at specialized centers. Other etiologies of bronchiectasis, including CF, primary immune deficiencies, and alpha1-antitrypsin deficiency, should be ruled out. Since motile cilia are present throughout the respiratory tract (the conductive airways, paranasal sinuses, middle ear), the clinical features of PCD reflect defects in the primary innate defense mechanism of mucociliary clearance, which confers much of the morbidity of PCD on patient’s quality of life (cough with recurrent infections). Laterality defects result from defects in the embryonic nodal motile cilia during embryogenesis. Manifestations of PCD are evident at birth and persist through the individual’s lifetime (Table 1).
Table 1.
Clinical Features of Primary Ciliary Dyskinesia
| Location of Dysfunctional Cilia | Features |
|---|---|
| Embryonic Node | Laterality defects |
| Congenital heart disease | |
| Nose & Sinus | Year-round, daily nasal congestion |
| Pan-sinusitis | |
| Eustachian Tubes | Chronic or recurrent otitis media |
| Transient hearing loss | |
| Trachea & Bronchi | Year-round, daily wet cough |
| Unexplained neonatal respiratory distress | |
| Recurrent lower respiratory infections | |
| Bronchiectasis | |
| Reproductive Tract | Infertility |
| Ectopic pregnancy |
2.1 Clinical presentation
Neonatal respiratory distress, often described as transient tachypnea of the newborn or neonatal pneumonia, is seen in over 80% of term neonates with PCD. Infants have tachypnea, increased work of breathing, and may even need supplemental oxygen or positive pressure ventilation11–13. This phenomenon implies that cilia in the airways are necessary for the clearance of fetal fluid from the lungs.
Daily wet cough, chronic nasal congestion, and recurrent otitis media are nearly universally present in PCD patients11, 14. Recurrent otitis media can result in hearing loss and speech delays. Chronic sinusitis is seen more often in older children and adults as compared to young children secondary to lack of radiographic imaging5. Atelectasis and lobar collapse, particularly of the middle and lower lobes, is seen in infants and young children with PCD11, 14, 15. Bronchiectasis is universally present in adults with PCD; young children with PCD can have airway wall thickening and bronchiectasis visible on high resolution chest CT15–17. Evidence of airflow obstruction may be evident on spirometry18, even in young children19.
Microbial pathogens in PCD tend to parallel those seen in CF. While Haemophilus influenza, Staphylococcus aureus, and Streptococcus pneumoniae dominate the pulmonary bacteriology of children with PCD, Pseudomonas aeruginosa is seen in teens and young adults11, 20. Chronic Pseudomonas tends to present at a later age as compared to CF, and S. pneumoniae is seen more often in PCD than in CF. Similar to CF, some patients culture more than one type of bacteria from the same sample. The prevalence of non-tuberculous mycobacterium (NTM) in adult PCD patients is about 15% and is somewhat lower in PCD children, similar to what is seen in CF20. These observations underscore the need to monitor sputum microbiology (including NTM) on a regular basis (at least twice a year).
Cilia also play a critical role in cardiac development and organ lateralization21–23. Situs inversus totalis occurs in about 50% of PCD patients. Situs ambiguous occurs in about 12% of PCD patients, and manifestations include polysplenia (left isomerism), asplenia (right isomerism), duplication of the inferior vena cava, and complex congenital heart disease24–26. The risk of structural congenital heart disease in PCD patients with situs ambiguous is at least 200-fold higher than the general population with heterotaxy25.
Due to dysmotility of the spermatozoa flagella, nearly all males with PCD are infertile. There are rare reports of men with PCD who have fathered children with27 and without fertility assistance28. PCD females have impaired ciliary function within the fallopian tubes, which delays ovum transit and can result in ectopic pregnancies or infertility29.
2.2 Nitric oxide measurements for screening and diagnosis
The saccharine test was the first screening test for PCD, but due to the lack of specificity and the development of more accurate diagnostic methods, there is no role for this testing in the diagnosis of PCD. The observation in the early 1990s that nitric oxide levels in exhaled nasal air is lower than that of healthy or disease controls was fortuitous, and has led in recent years to the development of measures of nasal NO as a diagnostic test30. Nasal nitric oxide (nNO) is appropriate to screen for PCD in patients with a compatible clinical phenotype and in whom testing has excluded CF20, 31. In PCD, nNO values are typically 10–20% of normal (20.7 nl/min) but individuals with RSPH1 mutations can have higher nNO (98.3 nl/min)32. Using a standardized protocol, a disease specific cutoff of 77nl/min has a sensitivity of 98% and a specificity of >99.9%. The positive predictive value is influenced by age and prevalence, underscoring the importance of obtaining a detailed clinical phenotype3. While the test requires some equipment and technical training/expertise, it is easy to perform down to about age 5 years, and is non-invasive10. Nasal NO has replaced the saccharine test as a reliable screening method.
2.3 Ciliary ultrastructure and ciliary beat
Assessment of ciliary ultrastructure using electron microscopy (EM) has long been considered the PCD diagnostic standard, but this method is limited for several reasons. At least 30% of patients with PCD have normal ultrastructure33, 34. Furthermore, the challenges of obtaining an adequate sample, the technical skill required for sample processing, and the experience necessary to correctly interpret the images impose additional limitations. As many as 15 to 20% of patients referred to the Genetic Disorders of Mucociliary Clearance Consortium (GDMCC) with a diagnosis of PCD based on EM findings did not have PCD5, underscoring the importance of this testing occurring in specialized centers.
Assessment of ciliary motility, including beat frequency and waveform analysis, in experienced hands, can be a useful adjunct to diagnose PCD, particularly if the patient has complete immotility or gross dysmotility of all cilia. Some forms of PCD have subtle or no evidence of abnormal beat or waveform. Secondary ciliary dyskinesia can result from infection and inflammation, increasing the rate of false positive diagnoses when using biologic assessments of ciliary structure and function. Thus, relying on ciliary structural and functional analyses alone, as has traditionally been the practice, does not provide enough specificity nor sensitivity for the diagnosis of PCD.
2.4 Genetics (Table 2)
Table 2.
Mutations in Genes that Cause Human Primary Ciliary Dyskinesia34
| Gene | Chromosomal Location | Axonemal Component | Ciliary Ultrastructure in Subjects with Biallelic Mutations | Laterality Defects Present | Gene MIM# |
|---|---|---|---|---|---|
| DNAH5 | 5p15.2 | ODA dynein HC | ODA defect | yes | 603335 |
| DNAI1 | 9p21-p13 | ODA dynein IC | ODA defect | yes | 604366 |
| DNAI2 | 17q25 | ODA dynein IC | ODA defect | yes | 605483 |
| DNAL1 | 14q24.3 | ODA dynein LC | ODA defect | yes | 610062 |
| CCDC114 | 19q13.32 | ODA DC | ODA defect | yes | 615038 |
| CCDC151 | 19p13.2 | ODA transport component | ODA defect | yes | 615956 |
| ARMC4 | 10p12.1-p11.23 | ODA transport component | ODA defect | yes | 615408 |
| TXNDC3 (NME8) | 7p14-p13 | ODA dynein IC/LC | Partial ODA defect (66% cilia defective) | yes | 607421 |
| DNAAF1 (LRRC50) | 16q24.1 | Cytoplasmic DA preassembly factor | ODA+IDA defect | yes | 613190 |
| DNAAF2 (KTU) | 14q21.3 | Cytoplasmic DA preassembly factor | ODA+IDA defect | yes | 612517 |
| DNAAF3 (C19orf51) | 19q13.42 | Cytoplasmic DA preassembly factor | ODA+IDA defect | yes | 614566 |
| CCDC103 | 17q21.31 | Cytoplasmic DA attachment factor | ODA+IDA defect | yes | 614677 |
| HEATR2 | 7p22.3 | Cytoplasmic DA preassembly or transport | ODA+IDA defect | yes | 614864 |
| LRRC6 | 8q24 | Cytoplasmic DA preassembly or transport | ODA+IDA defect | yes | 614930 |
| C21orf59 | 21q22.1 | Cytoplasmic DA assembly or adaptor for transport | ODA+IDA defect | yes | 615494 |
| SPAG1 | 8q22 | Cytoplasmic DA preassembly or transport | ODA+IDA defect | yes | 603395 |
| DYX1C1 | 15q21.3 | Cytoplasmic DA preassembly factor | ODA+IDA defect | yes | 608706 |
| ZMYND10 | 3p21.3 | Cytoplasmic DA assembly | ODA+IDA defect | yes | 607070 |
| CCDC39 | 3q26.33 | N-DRC | IDA defect + Axonemal disorganization | yes | 613798 |
| CCDC40 | 17q25.3 | N-DRC | IDA defect + Axonemal disorganization | yes | 613799 |
| CCDC164 (DRC1) | 2p23.3 | N-DRC | Nexin link missing; axonemal disorganization in small proportion of cilia | no | 615288 |
| RSPH4A | 6q22.1 | RS component | Mostly normal, CA defects in small proportion of cilia | no | 612647 |
| RSPH9 | 6p21.1 | RS component | Mostly normal, CA defects in small proportion of cilia | no | 612648 |
| RSPH1 | 21q22.3 | RS component | Mostly normal, CA defects in small proportion of cilia | no | 609314 |
| HYDIN | 16q22.2 | CA component | Normal, very occasionally CA defects | no | 610812 |
| DNAH11 | 7p21 | ODA dynein HC | Normal | yes | 603339 |
| CCDC65 (DRC2) | 12q13.12 | N-DRC | Normal | no | 611088 |
| MCIDAS | 5q11.2 | multiciliated cell differentiation | Oligocilia | no | 614086 |
| CCNO | 5q11.2 | multiciliated cell differentiation | Oligocilia | no | 607752 |
| DNAH8 | 6p21.2 | ODA dynein HC | Unknown | Unknown | 60337 |
| RPGR | Xp21.1 | Unknown | Mixed | no | 312610 |
| OFD1 | Xq22 | Unknown | Unknown | no | 300170 |
DA: dynein arm; ODA: outer dynein arm; IDA: inner dynein arm; CA: central apparatus; RS: radial spoke; HC: heavy chain; LC: light chain; IC: intermediate chain; DC: docking complex; N-DRC: nexin-dynein regulatory complex.
The number of genes and mutations identified as causative for PCD has exploded in recent years, allowing a shift away from a biological method of diagnosing the disease and towards a more definitive method of diagnosis (Table 2)34. Over 65% of PCD patients have biallellic mutations identified in one of the 32 published PCD genes5. Continued identification of genetic mutations that result in PCD will provide better understanding of the genotype-phenotype relationship and more clearly define the phenotypic spectrum of disease. For examples, mutations in genes that code for proteins in the ODA complexes result in situs abnormalities; this is not seen with mutations in radial spoke head protein genes due to the unaffected nodal cilium (which lacks the central pair, and thus retains the normal rotatory function important for normal L-R asymmetry). Individuals with mutations in RSPH1 have a milder clinical phenotype and higher nNO measures as compared to other PCD patients32. A recent publication notes the presence of PCD in patients Cri du Chat Syndrome (CdCS) who are hemizygous for a DNAH5 mutation due to the 5p segmental deletion attributed to CdCS on the opposite chromosome35.
3. Disease management
Randomized clinical trials in the PCD population are lacking, and publications of therapies in PCD mainly consist of case reports. Studies of non-CF bronchiectasis often do not indicate if patients with PCD are included, or if they were, how the diagnosis was made. Children with non-CF bronchiectasis are not generally included in non-CF studies. Thus, therapies for PCD are largely extrapolated from other diseases with defective mucociliary clearance, such as cystic fibrosis (CF) and non-CF bronchiectasis. For the purposes of this review, we will address and discuss existing as well as novel treatments that have been studied in the published literature, and review how these treatments might be useful in PCD. The assumption is made that while PCD is a disease associated with defective ciliary function, there are no other complications related to systemic manifestations, and that treatments shown to be useful in CF and non-CF bronchiectasis may therefore be extrapolated to PCD.
The three major focus areas to date are: airway clearance, infection control, and minimizing inflammation. Unlike the situation in CF where approaches have been studied to rectify the basic defect in the disease using gene therapy, and more recently molecular modulators, no such novel strategies have yet been tested in PCD.
3.1 Airway clearance: Devices and pharmacologic approaches (Table 3)
Table 3.
Summary of Therapeutic Trials in non-CF Bronchiectasis
| Author | Study | N | Intervention | Duration | Primary Outcome | Result |
|---|---|---|---|---|---|---|
| O’Donnell et al. 199864 | Randomized | 349 | Aerosolized rhDNase 2.5mg versus placebo | 24 weeks | Incidence of exacerbations and mean % change in FEV1 | rhDNase increased exacerbations and decreased FEV1 |
| Barker et al. 200078 | Randomized | 74 | Tobramycin solution for inhalation (300 mg) twice daily versus placebo | 4 weeks | Change in P aeruginosa density from baseline | Reduction in P aeruginosa bacterial density |
| Drobnic et al. 200579 | Randomized crossover | 30 | Aerosolized tobramycin 300 mg twice daily versus placebo | 6 months | Number of exacerbations and number of hospitalizations | No difference in exacerbations but fewer individuals on tobramycin needed admission and those who were admitted had a shorter duration of hospitalization. |
| Murray et al. 201182 | Randomized | 65 | Gentamicin 80 mg nebulized twice daily versus placebo | 12 months | Sputum bacterial density, sputum purulence, lung function, exercise capacity, LCQ, SGRQ, exacerbation frequency | Reduced sputum bacterial density and purulence; increased exercise capacity; fewer exacerbations and increased time to first exacerbation; improvement in LCQ and SGRQ. No change in sputum volume or lung function. |
| Serisier et al. 201383 | Randomized | 42 | Dual release ciprofloxacin for inhalation versus placebo nebulized once daily for up to 3 treatment cycles of 28 days | 24 weeks | Change in sputum P aeruginosa bacterial density to end of treatment cycle 1 | Reduction in P aeruginosa bacterial density |
| Wilson et al. 201384 | Randomized | 60 | Ciprofloxacin dry powder for inhalation (32.5 mg) twice daily versus placebo | 28 days of therapy and 56 days of follow up | Bacterial density in sputum | Significant reduction in total bacterial load |
| Barker et al. 201480 | Randomized | 540 | Aztreonam for inhalation solution 75 mg three times per day versus placebo (E-flow nebulizer) | 16 weeks | Change in QOL-B-RSS | No difference in QOL-B-RSS |
| Haworth et al. 201481 | Randomized | 144 | Colistin 1 million IU versus placebo (saline 0.45%) via I-neb twice daily | 6 months | Time to exacerbation | No difference. Subanalysis showed benefit in the individuals compliant with colisitin therapy. |
| Wong et al. 201285 | Randomized | 141 | Azithromycin 500 mg three days per week versus placebo | 6 months | event based exacerbations, change in FEV1, and change in SGRQ | Exacerbation rate decreased. No change in FEV1 or SGRQ |
| Altenburg et al. 201387 | Randomized | 83 | Azithromycin 250 mg daily versus placebo | 12 months | Exacerbations over 12 months | Decreased exacerbations |
| Serisier et al. 201386 | Randomized | 117 | Twice daily erythromycin ethylsuccinate 400 mg versus placebo | 12 months | Annualized mean rate of protocol defined pulmonary exacerbations | Decreased exacerbations |
| Mandal et al. 201489 | Randomized | 60 | Atorvastatin 80 mg daily versus placebo | 6 months | Reduction in cough using Leicester Cough Questionnaire | Improved cough on quality of life scale (LCQ). |
LCQ: Leicester Cough Questionnaire; SGRQ: St. George Respiratory Questionnaire; QOL-B-RSS: Quality of Life-Bronchiectasis Respiratory Symptoms Score
Normal mucociliary clearance, a critical component of airway host defense, occurs when the coordinated beating of motile cilia propel the airway surface liquid toward larger airways, expelling infectious and non-infectious particles out of the lungs. Alterations in the airway surface liquid, such as dehydration as seen in CF, and ineffective ciliary beating as seen in primary ciliary dyskinesia, markedly impair this process, leading to clinical disease36–38. This leaves mucus, irritants, and infection in the airways, resulting in increased inflammation and scarring, ultimately causing bronchiectasis. Therefore, it is imperative to augment airway clearance and avoid cough suppressing medications1.
There are a variety of devices and methods available for airway clearance with no clear superiority shown for one over others. Intra-thoracic oscillatory devices generate oscillating positive pressure through a mouthpiece, resulting in vibrations that shake the mucus off the airway walls and intermittent elevation of endobronchial airway pressures that stent the airways open for more effective clearance. Airflow accelerations during exhalation mobilize the freed mucus into the larger airways39, 40. Some of these devices connect to a nebulizer, allowing patients to inhale a medication and generate oscillations on exhalation. High frequency chest compression using vest therapy generate extra-thoracic oscillations at variable frequencies and intensities, which are transmitted to the airways39, 41. Other methods of airway clearance avoid the use of devices and include manual chest physiotherapy, postural drainage, autogenic drainage, active cycle breathing, and exercise41–43. Use of one method does not preclude use of another method, even in combination44. At this time, there is no clear evidence that one device or method is superior to another,45, 46 although one study in children with PCD found that exercise was a more potent bronchodilator than inhaling a β2-agonist47.
Another method of removing airway secretions is through airway hydration via aerosolized therapies, particularly when secretions are inspissated. In an early pilot study including solely PCD patients, aerosolized uridine-5′-triphosphate (UTP), which acts to increase chloride secretion (and thus water) across airway epithelia, significantly increased airway clearance as measured using clearance of radiolabeled particles during cough. Although an increase in ciliary beat would not be expected in PCD (unlike in healthy controls), UTP increased chloride conduction into the airway via an alternative channel, altering the liquid and mucin content within airway secretions, thus facilitating cough clearance48. Along similar lines, several studies have tested whether isotonic saline (0.9%) or hypertonic saline (3 or 7%) improves airway clearance in non-CF bronchiectasis. A small study in non-CF bronchiectasis patients demonstrated improved clearance after nebulizing isotonic saline prior to chest physiotherapy49. Prior to research demonstrating improved airway clearance in CF patients after nebulizing hypertonic saline, evidence emerged that nebulizing hypertonic saline was beneficial in non-CF bronchiectasis. Nebulizing hypertonic saline 7% resulted in significantly reduced sputum viscosity, increased ease of expectoration, and therefore, increased sputum volume expectorated as compared to isotonic saline. Small but significant improvements in lung function were also described50. After three months, individuals with non-CF bronchiectasis using hypertonic saline daily had decreased antibiotic use, lower health care utilization, and improved quality of life measures. There was continued improvement in sputum viscosity, ease of expectoration, and lung function51. In contrast, other studies have found no difference in exacerbation frequency, sputum colonization, lung function or quality of life after nebulizing hypertonic saline 6% or isotonic saline for 12 months52. As an example of why this might be, hypertonic saline is thought to disrupt the interactions between IL-8 and glycosaminoglycans. This makes IL-8 more susceptible to degradation, thereby limiting the concentrations of this pro-inflammatory cytokine within the airway53. The differing impact of hypertonic saline54 may relate to individual variation in airway IL-8 levels as well as the underlying cause of the bronchiectasis. Based on two studies which found significantly higher levels of IL-8 in sputum expectorated from children with PCD as compared to children with CF55, 56, PCD patients may have a greater potential to derive benefit from inhaled hypertonic saline, and deserves further study in this population.
Recombinant human DNase I (rhDNase, dornase alfa) provides significant benefit in CF57, leading some to propose benefit in non-CF bronchiectasis. In the setting of infection, neutrophils accumulate in the airways and undergo necrosis; DNA and actin are released and increase sputum viscosity58–60. Recombinant human DNase I cleaves extracellular DNA, decreasing the DNA concentration, and thereby decreasing sputum viscosity. There are several case reports noting improvement in pulmonary symptoms in PCD patients with both intermittent61 and chronic use62, 63. Although PCD airway secretions contain elevated DNA concentrations55, it is not to the same degree as CF. Importantly, a randomized control trial found that dornase alfa provided no benefit and potentially cause harm when used by idiopathic bronchiectasis patients64. It is unclear how many well-phenotyped PCD patients were enrolled in this study. For the present, inhaled dornase alpha is not recommended for patients with non-CF bronchiectasis, including PCD.
Inhaled mannitol increases mucociliary and cough clearance in patients with asthma, bronchiectasis, and CF by creating an osmotic drive for water to move into the airway and hydrate secretions65. In cell culture, mannitol increases ciliary beat frequency66. This therapy may be beneficial in PCD patients through improved cough clearance as it is unlikely to improve ciliary beat without correcting the underlying structural defect. In a recent publication of non-CF bronchiectasis patients, inhaled dry powder mannitol (400 mg twice daily) over 52 weeks did not decrease annual exacerbation rates (primary outcome) nor improve FEV1 but did increase the time to exacerbation and improved quality of life measures67. Overall, it was well tolerated with similar mild and moderate adverse event frequency to the placebo arm; the treatment arm had less frequent severe adverse events as compared to the control arm.
N-acetylcysteine (NAC) is a sulfhydryl compound that depolymerizes mucus in vitro by breaking the disulfide bonds, decreasing sputum viscosity. Because inhaling NAC can result in bronchospasm in patients with hyperresponsive airways and it possess a distasteful sulfur smell, it is typically taken orally. Unfortunately, there is no evidence that oral NAC reaches therapeutic concentrations in airway secretions68. Additionally, a small study of oral NAC in PCD patients found no improvement in clinical status or lung function69.
Because nNO is low in PCD and NO plays a key role in host defense and up-regulation of ciliary motility, arginine, a precursor to nitric oxide (NO) synthesis, was postulated to have a therapeutic role in PCD. Despite encouraging preliminary data of nasal ciliary motility70, arginine did not normalize airway nitric oxide concentrations nor improve lung function71.
3.2 Infection control
Recommendations from the European Respiratory Society and British Thoracic Society suggest that clinicians monitor airflow mechanics (spirometry) and collect respiratory cultures, including those for NTM, regularly (every 3 to 6 months) in patients with PCD1, 72. Specimens should be processed in the same manner as CF sputum, given the similar microbial pathogens11, 20. Nebulizer equipment and airway clearance devices should be cleaned and disinfected according to CF guidelines.
3.21 Acute anti-microbial therapy
Manifestation of a bronchiectasis exacerbation includes an increase in sputum volume or change in sputum viscosity; an increase in sputum purulence; and an increase in cough, wheeze, and dyspnea72. Sputum should be sent for culture but should not delay initiation of antibiotics, whether oral or intravenous72. Prior sputum culture results can help guide antibiotic choice. Management of the various microbes that usually occur in bronchiectasis including PCD (gram negatives, resistant Staph, fungi, and NTM) is beyond the scope of this article, and the reader is referred to published guidelines72–74. In general, there is little evidence supporting combination antibiotic therapy in the absence of P. aeruginosa. There is some controversy as to whether combination antibiotics are necessary in patients colonized with P. aeruginosa. The addition of inhaled tobramycin to oral ciprofloxacin during exacerbations improved microbial outcomes, but clinical outcomes were no different than treating with oral ciprofloxacin alone75. The optimal length of antibiotic treatment is unknown; the BTS guidelines recommending 14 days of treatment is based on grade D evidence72.
3.22 “Preventive” (chronic) anti-microbials
There is no firm evidence as to when to initiate such treatment, but clinical experience suggests that inhaled antibiotics for prophylaxis should be considered in cases requiring repeated courses of oral antibiotics76, particularly if adequate airway clearance is being performed. It is unlikely that patients who are non-compliant with airway clearance will regularly use inhaled antibiotics. The use of rotational oral antibiotics for long-term outpatient therapy is not recommended routinely72. In the US, tobramycin, aztreonam, and colistin are available in a nebulized form, although are not FDA approved for use in non-CF bronchiectasis as yet. Ciprofloxacin (liposomal and powder preparations), gentamicin, and liposomal amikacin are being tested in clinical trials in international studies. Inhaled tobramycin is both tolerable and beneficial in stable adult non-CF bronchiectasis patient with chronic Pseudomonas infection77–79. The benefit of inhaled aztreonam80, colistin81, and gentamicin82 is less clear (Table 3). Two phase 2 clinical trials of inhaled ciprofloxacin have shown tolerability with significant reductions in total bacterial load and longer time to first exacerbation83, 84.
3.3 Anti-inflammatory approaches
Previously extrapolated from CF literature, the use of macrolides to decrease airway inflammation in idiopathic bronchiectasis has recently been examined in several studies. The EMBRACE trial in New Zealand found that azithromycin 500 mg taken three days per week reduced exacerbation frequency in adults with idiopathic bronchiectasis who experienced at least one exacerbation in the preceding year. The number of individuals who cultured Pseudomonas or who carried a diagnosis of PCD is unknown85. In idiopathic bronchiectasis patients with at least two exacerbations in the previous year, the BLESS trial demonstrated that twice daily erythromycin ethylsuccinate significantly reduced exacerbations in all enrolled patients and in the pre-specified subgroup with baseline Pseudomonas airway infection86. Daily azithromycin 250 mg reduced infectious exacerbation in patients with at least three lower respiratory infections in the preceding year in the BAT trial87. Three and one PCD patient were enrolled in the BLESS and BAT trials respectively. In contrast to CF, most of the non-CF bronchiectasis patients cultured bacteria other than P. aeruginosa, suggesting that the mechanism of azithromycin may be both anti-inflammatory and anti-microbial. Patients should not receive chronic oral macrolides unless they have been tested and proven to not have sputum colonized or infected with NTM5. Overall, it appears that chronic use of a macrolide provides benefits in patients with non-CF bronchiectasis, including patients with PCD. Not all patients with PCD should automatically be prescribed macrolides. In addition to those that may sporadically or chronically expectorate NTM, there are those in whom the potential for side effects (cardiac or gastrointestinal) may exceed the benefits described above, and there are patients with a good quality of life in whom macrolides can be considered in the future.
The use of inhaled corticosteroids, beta-agonists, leukotriene inhibitors, and oral corticosteroids should be considered in individuals with overlapping atopic disease72, 88. There is no clear evidence that use of these agents benefits all patients with PCD5, 72, 74. However, since asthma and allergies are common, they can be present in addition to PCD. Before adding beta-agonists to a treatment regimen, it should be noted that PCD patients can have spirometry measures that meet ATS criteria for repeatability but improve steadily due to cough clearance. If the patient has not reached a plateau during the pre-bronchodilator testing, one may erroneously assume that there is a post-bronchodilator response rather than attributing improvement to continued cough clearance.
Statins have effects beyond reducing cholesterol levels, including reducing inflammation and modulating the innate and adaptive immune systems. A recent publication demonstrated improvement in the Leicester Cough Questionnaire, a quality of life measurement instrument, in non-CF bronchiectasis patients after 6 months of atorvastatin. Further study is needed to determine if long-term statin use can also reduce exacerbations89.
It is critical to avoid exposure to inflammatory triggers. Patients and family members who smoke should receive smoking cessation counseling. Routine immunizations including influenza, pneumococcus, and pertussis should be encouraged1, 5.
3.4 Other management methods
Since PCD is a disease that affects cilia throughout the entire lung, lobectomy should be carefully considered and is rarely indicated. Patients in whom lobectomy should be considered include those with severe localized bronchiectasis with recurrent febrile relapses despite aggressive therapy and those with uncontrolled severe hemoptysis90. PCD patients with end stage lung disease are candidates for lung transplantation. A small number have been successfully performed in PCD patients, both with and without situs inversus91.
3.5 Upper airway disease
The management of ear and nose disease is similar to the management of lung disease. Sinus rinses augment clearance of mucus from the nasal passages and sinuses1. Antibiotic rinses have been used to treat persistent bacterial colonization in PCD. Nasal steroids can be helpful in patients with significant allergic rhinitis and sinusitis. In patients with sinus disease refractory to medical management, functional endoscopic sinus surgery is helpful, particularly if there is aggressive postsurgical treatment to maintain adequate drainage92. Tympanostomy tube placement is controversial. Although tubes may improve hearing93, otitis media can improve during the teen years, and tubes may result in persistent otorrhea94. Because conductive hearing loss due to chronic otitis media can result in speech and language development delay, audiology assessments, hearing aids, and adjunctive therapies should be encouraged1.
4. Conclusion
In conclusion, primary ciliary dyskinesia is a genetically heterogeneous recessive disorder of motile cilia which results in chronic oto-sino-pulmonary disease. Diagnostic methods have advanced to now include nNO as a screening test in individuals with a compatible clinical phenotype and other known causes of bronchiectasis and chronic oto-sino-pulmonary disease have been excluded. Ciliary ultrastructural abnormalities as seen on electron microscopy and videomicroscopy are useful adjunctive diagnostic modalities as the genetic mutations involved in PCD are further identified. Management of PCD is primarily extrapolated from studies in cystic fibrosis and non-CF bronchiectasis. The foundation of managing disease in PCD includes airway clearance, infection control, and reduction of inflammation. With the development of clinical and research consortiums and actively enrolling registries of PCD patients, the environment is conducive to initiate longitudinal studies of disease course as well as therapeutic trials to alter disease course.
5. Expert Opinion
The method of diagnosing PCD has dramatically changed over the past decade. Through large cross-sectional studies, the clinical phenotype of PCD has been refined with better definitions and new knowledge, for example, the high prevalence of neonatal respiratory distress in PCD, and the association with complex congenital heart disease. It is now known that mutations that preserve the ciliary central apparatus do not result in situs abnormalities, and that PCD individuals with situs ambiguous are more likely to have complex congenital heart disease. Nasal NO has proven to be an excellent screening test for PCD. However, like sweat chloride in CF, nNO levels in PCD can include false positives as well as false negatives, underscoring the importance of determining the genetic mutations. The diagnosis of PCD had hinged on the visualization of ciliary ultrastructural defects on EM, an imperfect diagnostic method. At least 30% of individuals with PCD have normal ciliary ultrastructure (DNAH11 and CCDC65 mutations). Other mutations result in ultrastructural defects in only a subset of cilia; this may not be appreciated without the skill and expertise in sample processing and image interpretation. Elucidating genetic mutations for all patients with PCD is rapidly becoming a reality. The number of known PCD causing genes has more than doubled in the past three years, and a PCD research genetic panel is under development through the GDMCC.
Treatment of PCD patients lacks evidence and standardization. Many patients receive sub-optimal management including lack of sputum cultures and pathogen directed antibiotic therapy. The Bronchiectasis Severity Index (BSI) can help clinicians identify patients, including those with PCD bronchiectasis, who are at high risk of developing bronchiectasis complications and therefore, may benefit from intensification of therapies such as adding inhaled antibiotics or oral macrolides95.
Working together, the GDMCC and the PCD Foundation have developed the PCD Foundation Clinical and Research Center Network (CRCN), which includes the eight original GDMCC sites, four new sites, and nine affiliate sites. The goal of the CRCN is to provide a reliable diagnosis; consistent high-quality, appropriate care; and comprehensive data collection through the PCD registry96. BESTCILIA is a research consortium comprised of ten European clinical centers and two US clinical centers with the goal of improving the clinical practice in PCD care through standardized care protocols and diagnostic testing as well as observational and randomized control trials97. The development and validation of a PCD quality of life instrument98, 99 leaves researchers well-poised to capitalize on patient participation in the CRCN and PCD registry with longitudinal studies to more accurately describe disease course and randomized control studies to alter this course.
Therapeutic studies directly assessing the benefit of hypertonic saline, azithromycin, or even dornase alfa are potential initial research directions. Looking to the future, given the success with modulatory therapeutics for CF, it is conceivable that genetic modulators could improve or fully correct ciliary defects in PCD. Extrapolating from the chronic bronchitic COPD phenotype, roflumilast may decrease the airway inflammation seen in PCD. A new orphan drug candidate, NM001 (Lynovex®; NovaBiotics) is a mucolytic agent being tested in CF, and potentially other chronic lung diseases. When considering these types of therapeutic trials, it is critical to choose the correct primary outcome. Exacerbation rate was a sound outcome in the macrolide studies, given that exacerbations manifest with increased inflammation and macrolides are presumably acting as an anti-inflammatory agent. A quality of life measurement was used in the atorvastatin trial and reached significance. However, quality of life measures and exacerbation frequency have not yielded the same outcomes in the inhaled antibiotic studies.
PCD is a genetically heterogenous disease with a variety of mutations in numerous genes resulting in a shared phenotype, sparing no race or ethnic group. Despite general perceptions, PCD lung disease may not necessarily be mild. A delay in recognizing PCD features and formal diagnosis can result in the development and progression of irreversible lung disease. In CF, early identification and diagnosis leads to early treatment and frequent monitoring to decrease morbidity and mortality. Although CF and PCD are both recessive disorders that result in bronchiectasis and sinusitis, the underlying pathogenesis differs. While the CF model is appropriate for disease follow up and development of a clinical network of disease expertise, PCD is not the same as CF, and management is not identical100. This is evidence in prior studies of UTP, which demonstrated improved airway clearance in PCD patients but no benefit in CF patients. Without further PCD patients to test the therapy, there was little incentive for pharmaceutical companies to pursue drug development. Improved identification and more accurate diagnosis of PCD provides a large cohort of PCD patients who are eager to participate in research with the goal of improving clinical care.
Article Highlights.
Primary ciliary dyskinesia is a genetically heterogeneous recessive disorder of motile cilia that results in chronic oto-sino-pulmonary disease as well as organ laterality defects and congenital heart disease.
Nasal nitric oxide is a useful screening test in individuals with a compatible clinical phenotype and in whom other known cause of chronic oto-sino-pulmonary disease have been excluded.
Although useful, biologic diagnostic methods, such as assessment of ciliary ultrastructure using electron microscopy and visualization of ciliary motility using videomicroscopy, are limited by technical skill and availability of equipment.
Continued identification of genetic mutations causing PCD provides a more definitive diagnostic method. Over 65% of PCD patients have biallelic mutations identified in one of the 30 published PCD genes.
The foundation of managing disease in PCD includes airway clearance, infection control, and reduction of inflammation. Use of the Bronchiectasis Severity Index can help identify individuals at high risk of complications from their bronchiectasis who may benefit from intensification of therapies.
With the development of clinical and research consortiums and actively enrolling registries of PCD patients, the environment is conducive to initiate longitudinal studies of disease course as well as therapeutic trials to alter disease course.
Acknowledgments
Thanks to Dr. Michael R. Knowles and Dr. Maimoona Zariwala for their guidance and support, to all of the GDMCC investigators and study coordinators for their hard work, and to the PCD patients for their participation.
MLD is supported by US National Institute of Health (NIH) – National Heart, Lung, and Blood Institute K12 (1K12HL119998-01) and ORDR (U54HL096458). The Genetic Disorders of Mucociliary Clearance Consortium (U54HL096458) is part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS), and the National Heart, Lung & Blood Institute (NHLBI). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Financial disclosure
P Noone has participated in industry sponsored studies for Gilead, Insmed and Aragigm Pharmaceuticals.
Footnotes
Competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1••.Barbato A, Frischer T, Kuehni CE, et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur Respir J. 2009 Dec;34(6):1264–76. doi: 10.1183/09031936.00176608. First consensus statement on the diagnosis and treatment of PCD in the pediatric age group. [DOI] [PubMed] [Google Scholar]
- 2.Bush A, Chodhari R, Collins N, et al. Primary ciliary dyskinesia: current state of the art. Arch Dis Child. 2007 Dec;92(12):1136–40. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leigh MW, Pittman JE, Carson JL, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. 2009 Jul;11(7):473–87. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zariwala MA, Knowles MR, Omran H. Genetic defects in ciliary structure and function. Annu Rev Physiol. 2007;69:423–50. doi: 10.1146/annurev.physiol.69.040705.141301. [DOI] [PubMed] [Google Scholar]
- 5••.Knowles MR, Daniels LA, Davis SD, et al. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013 Oct 15;188(8):913–22. doi: 10.1164/rccm.201301-0059CI. Recent review summarizing advances in disease characterization, diagnostic methods, and genetics of PCD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kartagener M. Zur pathogenese der bronkiectasien. Bronkiectasien bei situs viscerum inversus. Beitr Klin Tuberk Spezif Tuberkuloseforsch. 1933;83:489–501. [Google Scholar]
- 7.Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976 Jul 23;193(4250):317–9. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 8.Torgersen J. Transposition of viscera, bronchiectasis and nasal polyps; a genetical analysis and a contribution to the problem of constitution. Acta radiol. 1947 Feb 28;28(1):17–24. [PubMed] [Google Scholar]
- 9.Katsuhara K, Kawamoto S, Wakabayashi T, Belsky JL. Situs inversus totalis and Kartagener’s syndrome in a Japanese population. Chest. 1972 Jan;61(1):56–61. doi: 10.1378/chest.61.1.56. [DOI] [PubMed] [Google Scholar]
- 10••.Leigh MW, Hazucha MJ, Chawla KK, et al. Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia. Ann Am Thorac Soc. 2013 Dec;10(6):574–81. doi: 10.1513/AnnalsATS.201305-110OC. Standardization of nNO as screening test in PCD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sagel SD, Davis SD, Campisi P, Dell SD. Update of respiratory tract disease in children with primary ciliary dyskinesia. Proc Am Thorac Soc. 2011 Sep;8(5):438–43. doi: 10.1513/pats.201103-024SD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferkol T, Leigh M. Primary ciliary dyskinesia and newborn respiratory distress. Semin Perinatol. 2006 Dec;30(6):335–40. doi: 10.1053/j.semperi.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Leigh MW, Shapiro AJ, Pittman JE, et al. B18. RARE PEDIATRIC LUNG DISEASE: EXPANDING OUR UNDERSTANDING. American Thoracic Society; Definition Of Clinical Criteria For Diagnosis Of Primary Ciliary Dyskinesia; pp. A2483–A83. [Google Scholar]
- 14.Pittman JE, Rosenfeld M, LaFave C, et al. A28. CILIA IN HEALTH AND DISEASE: PRIMARY CILIARY DYSKINESIA AND BRONCHIECTASIS. American Thoracic Society; 2011. Characteristics Of Primary Ciliary Dyskinesia In Children Under 5 Years Of Age; pp. A1213–A13. [Google Scholar]
- 15.Kennedy MP, Noone PG, Leigh MW, et al. High-resolution CT of patients with primary ciliary dyskinesia. AJR Am J Roentgenol. 2007 May;188(5):1232–8. doi: 10.2214/AJR.06.0965. [DOI] [PubMed] [Google Scholar]
- 16.Jain K, Padley SP, Goldstraw EJ, et al. Primary ciliary dyskinesia in the paediatric population: range and severity of radiological findings in a cohort of patients receiving tertiary care. Clin Radiol. 2007 Oct;62(10):986–93. doi: 10.1016/j.crad.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 17.Santamaria F, Montella S, Tiddens HA, et al. Structural and functional lung disease in primary ciliary dyskinesia. Chest. 2008 Aug;134(2):351–7. doi: 10.1378/chest.07-2812. [DOI] [PubMed] [Google Scholar]
- 18.Marthin JK, Petersen N, Skovgaard LT, Nielsen KG. Lung function in patients with primary ciliary dyskinesia: a cross-sectional and 3-decade longitudinal study. Am J Respir Crit Care Med. 2010 Jun 1;181(11):1262–8. doi: 10.1164/rccm.200811-1731OC. [DOI] [PubMed] [Google Scholar]
- 19.Brown DE, Pittman JE, Leigh MW, et al. Early lung disease in young children with primary ciliary dyskinesia. Pediatr Pulmonol. 2008 May;43(5):514–6. doi: 10.1002/ppul.20792. [DOI] [PubMed] [Google Scholar]
- 20••.Noone PG, Leigh MW, Sannuti A, et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004 Feb 15;169(4):459–67. doi: 10.1164/rccm.200303-365OC. First publication describing the clinical and microbiologic phenotype of adults and children with PCD. [DOI] [PubMed] [Google Scholar]
- 21.Francis RJ, Christopher A, Devine WA, et al. Congenital heart disease and the specification of left-right asymmetry. Am J Physiol Heart Circ Physiol. 2012 May 15;302(10):H2102–11. doi: 10.1152/ajpheart.01118.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakhleh N, Francis R, Giese RA, et al. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation. 2012 May 8;125(18):2232–42. doi: 10.1161/CIRCULATIONAHA.111.079780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brueckner M. Impact of genetic diagnosis on clinical management of patients with congenital heart disease: cilia point the way. Circulation. 2012 May 8;125(18):2178–80. doi: 10.1161/CIRCULATIONAHA.112.103861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shapiro AJ, Davis SD, Ferkol T, et al. Laterality Defects other than Situs Inversus Totalis in Primary Ciliary Dyskinesia: Insights into Situs Ambiguus and Heterotaxy. Chest. 2014 Feb 27; doi: 10.1378/chest.13-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Kennedy MP, Omran H, Leigh MW, et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007 Jun 5;115(22):2814–21. doi: 10.1161/CIRCULATIONAHA.106.649038. First publication linking PCD with congenital heart disease and other forms of heterotaxy. [DOI] [PubMed] [Google Scholar]
- 26.Brueckner M. Heterotaxia, congenital heart disease, and primary ciliary dyskinesia. Circulation. 2007 Jun 5;115(22):2793–5. doi: 10.1161/CIRCULATIONAHA.107.699256. [DOI] [PubMed] [Google Scholar]
- 27.Sha YW, Ding L, Li P. Management of primary ciliary dyskinesia/Kartagener’s syndrome in infertile male patients and current progress in defining the underlying genetic mechanism. Asian J Androl. 2014 Jan-Feb;16(1):101–6. doi: 10.4103/1008-682X.122192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munro NC, Currie DC, Lindsay KS, et al. Fertility in men with primary ciliary dyskinesia presenting with respiratory infection. Thorax. 1994 Jul;49(7):684–7. doi: 10.1136/thx.49.7.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halbert SA, Patton DL, Zarutskie PW, Soules MR. Function and structure of cilia in the fallopian tube of an infertile woman with Kartagener’s syndrome. Hum Reprod. 1997 Jan;12(1):55–8. doi: 10.1093/humrep/12.1.55. [DOI] [PubMed] [Google Scholar]
- 30.Lundberg JO, Weitzberg E, Nordvall SL, et al. Primarily nasal origin of exhaled nitric oxide and absence in Kartagener’s syndrome. Eur Respir J. 1994 Aug;7(8):1501–4. doi: 10.1183/09031936.94.07081501. [DOI] [PubMed] [Google Scholar]
- 31.Leigh MW, O’Callaghan C, Knowles MR. The challenges of diagnosing primary ciliary dyskinesia. Proc Am Thorac Soc. 2011 Sep;8(5):434–7. doi: 10.1513/pats.201103-028SD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32•.Knowles MR, Ostrowski LE, Leigh MW, et al. Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am J Respir Crit Care Med. 2014 Mar 15;189(6):707–17. doi: 10.1164/rccm.201311-2047OC. Recent study reporting milder phenotype in individuals with RSPH1 mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knowles MR, Leigh MW, Carson JL, et al. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax. 2012 May;67(5):433–41. doi: 10.1136/thoraxjnl-2011-200301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine. Johns Hopkins University; Baltimore, MD: [cited 09/16/2014]; Available from: http://omim.org. [Google Scholar]
- 35.Shapiro AJ, Weck KE, Chao KC, et al. Cri du Chat Syndrome and Primary Ciliary Dyskinesia: A Common Genetic Cause on Chromosome 5p. J Pediatr. 2014 Jul 24; doi: 10.1016/j.jpeds.2014.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Randell SH, Boucher RC, University of North Carolina Virtual Lung G Effective mucus clearance is essential for respiratory health. Am J Respir Cell Mol Biol. 2006 Jul;35(1):20–8. doi: 10.1165/rcmb.2006-0082SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wanner A, Salathe M, O’Riordan TG. Mucociliary clearance in the airways. Am J Respir Crit Care Med. 1996 Dec;154(6 Pt 1):1868–902. doi: 10.1164/ajrccm.154.6.8970383. [DOI] [PubMed] [Google Scholar]
- 38.Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest. 2002 Mar;109(5):571–7. doi: 10.1172/JCI15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marks JH. Airway clearance devices in cystic fibrosis. Paediatr Respir Rev. 2007 Mar;8(1):17–23. doi: 10.1016/j.prrv.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Elkins MR, Jones A, van der Schans C. Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database Syst Rev. 2006 doi: 10.1002/1465.1858.CD003147.pub3(2):CD003147. [DOI] [PubMed] [Google Scholar]
- 41.Rogers D, Doull IJM. Physiological principles of airway clearance techniques used in the physiotherapy management of cystic fibrosis. Current Paediatrics. 2005;15(3):233–8. [Google Scholar]
- 42.Bradley J, Moran F, Greenstone M. Physical training for bronchiectasis. Cochrane Database Syst Rev. 2002 doi: 10.1002/1465.1858.CD002166(3):CD002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee AL, Hill CJ, Cecins N, et al. The short and long term effects of exercise training in non-cystic fibrosis bronchiectasis–a randomised controlled trial. Respir Res. 2014;15:44. doi: 10.1186/1465-9921-15-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Webber BA, Hofmeyr JL, Morgan MD, Hodson ME. Effects of postural drainage, incorporating the forced expiration technique, on pulmonary function in cystic fibrosis. Br J Dis Chest. 1986 Oct;80(4):353–9. doi: 10.1016/0007-0971(86)90088-4. [DOI] [PubMed] [Google Scholar]
- 45.Morrison L, Agnew J. Oscillating devices for airway clearance in people with cystic fibrosis. Cochrane Database Syst Rev. 2014;(7):CD006842. doi: 10.1002/14651858.CD006842.pub3. [DOI] [PubMed] [Google Scholar]
- 46.Lee AL, Burge A, Holland AE. Airway clearance techniques for bronchiectasis. Cochrane Database Syst Rev. 2013;(5):CD008351. doi: 10.1002/14651858.CD008351.pub2. [DOI] [PubMed] [Google Scholar]
- 47.Phillips GE, Thomas S, Heather S, Bush A. Airway response of children with primary ciliary dyskinesia to exercise and beta2-agonist challenge. Eur Respir J. 1998 Jun;11(6):1389–91. doi: 10.1183/09031936.98.11061389. [DOI] [PubMed] [Google Scholar]
- 48.Noone PG, Bennett WD, Regnis JA, et al. Effect of aerosolized uridine-5′-triphosphate on airway clearance with cough in patients with primary ciliary dyskinesia. Am J Respir Crit Care Med. 1999 Jul;160(1):144–9. doi: 10.1164/ajrccm.160.1.9806146. [DOI] [PubMed] [Google Scholar]
- 49.Sutton PP, Gemmell HG, Innes N, et al. Use of nebulised saline and nebulised terbutaline as an adjunct to chest physiotherapy. Thorax. 1988 Jan;43(1):57–60. doi: 10.1136/thx.43.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kellett F, Redfern J, Niven RM. Evaluation of nebulised hypertonic saline (7%) as an adjunct to physiotherapy in patients with stable bronchiectasis. Respir Med. 2005 Jan;99(1):27–31. doi: 10.1016/j.rmed.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 51.Kellett F, Robert NM. Nebulised 7% hypertonic saline improves lung function and quality of life in bronchiectasis. Respir Med. 2011 Dec;105(12):1831–5. doi: 10.1016/j.rmed.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 52.Nicolson CH, Stirling RG, Borg BM, et al. The long term effect of inhaled hypertonic saline 6% in non-cystic fibrosis bronchiectasis. Respir Med. 2012 May;106(5):661–7. doi: 10.1016/j.rmed.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 53.Reeves EP, Williamson M, O’Neill SJ, et al. Nebulized hypertonic saline decreases IL-8 in sputum of patients with cystic fibrosis. Am J Respir Crit Care Med. 2011 Jun 1;183(11):1517–23. doi: 10.1164/rccm.201101-0072OC. [DOI] [PubMed] [Google Scholar]
- 54.Hart A, Sugumar K, Milan SJ, et al. Inhaled hyperosmolar agents for bronchiectasis. Cochrane Database Syst Rev. 2014;5:CD002996. doi: 10.1002/14651858.CD002996.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bush A, Payne D, Pike S, et al. Mucus properties in children with primary ciliary dyskinesia: comparison with cystic fibrosis. Chest. 2006 Jan;129(1):118–23. doi: 10.1378/chest.129.1.118. [DOI] [PubMed] [Google Scholar]
- 56.Oren K, Brandie W, Michael RK, et al. A96. A TALE OF TWO RARE LUNG DISEASES IN PEDIATRICS: PRIMARY CILIARY DYSKINESIA AND PULMONARY HYPERTENSION. American Thoracic Society; 2013. Airway Inflammation In A Large Cohort Of Children With Primary Ciliary Dyskinesia; pp. A2086–A86. [Google Scholar]
- 57.Konstan MW, Ratjen F. Effect of dornase alfa on inflammation and lung function: potential role in the early treatment of cystic fibrosis. J Cyst Fibros. 2012 Mar;11(2):78–83. doi: 10.1016/j.jcf.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chernick WS, Barbero GJ. Composition of tracheobronchial secretions in cystic fibrosis of the pancreas and bronchiectasis. Pediatrics. 1959 Nov;24:739–45. [PubMed] [Google Scholar]
- 59.Potter JL, Spector S, Matthews LW, Lemm J. Studies on pulmonary secretions. 3. The nucleic acids in whole pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am Rev Respir Dis. 1969 Jun;99(6):909–16. doi: 10.1164/arrd.1969.99.6.909. [DOI] [PubMed] [Google Scholar]
- 60.Picot R, Das I, Reid L. Pus, deoxyribonucleic acid, and sputum viscosity. Thorax. 1978 Apr;33(2):235–42. doi: 10.1136/thx.33.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.ten Berge M, Brinkhorst G, Kroon AA, de Jongste JC. DNase treatment in primary ciliary dyskinesia–assessment by nocturnal pulse oximetry. Pediatr Pulmonol. 1999 Jan;27(1):59–61. doi: 10.1002/(sici)1099-0496(199901)27:1<59::aid-ppul11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 62.Desai M, Weller PH, Spencer DA. Clinical benefit from nebulized human recombinant DNase in Kartagener’s syndrome. Pediatr Pulmonol. 1995 Nov;20(5):307–8. doi: 10.1002/ppul.1950200509. [DOI] [PubMed] [Google Scholar]
- 63.El-Abiad NM, Clifton S, Nasr SZ. Long-term use of nebulized human recombinant DNase1 in two siblings with primary ciliary dyskinesia. Respir Med. 2007 Oct;101(10):2224–6. doi: 10.1016/j.rmed.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 64.O’Donnell AE, Barker AF, Ilowite JS, Fick RB. Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase Study Group. Chest. 1998 May;113(5):1329–34. doi: 10.1378/chest.113.5.1329. [DOI] [PubMed] [Google Scholar]
- 65.Daviskas E, Rubin BK. Effect of inhaled dry powder mannitol on mucus and its clearance. Expert Rev Respir Med. 2013 Feb;7(1):65–75. doi: 10.1586/ers.12.72. [DOI] [PubMed] [Google Scholar]
- 66.Yaghi A, Zaman A, Dolovich MB. The direct effect of hyperosmolar agents on ciliary beating of human bronchial epithelial cells. J Aerosol Med Pulm Drug Deliv. 2012 Apr;25(2):88–95. doi: 10.1089/jamp.2011.0914. [DOI] [PubMed] [Google Scholar]
- 67.Bilton D, Tino G, Barker AF, et al. Inhaled mannitol for non-cystic fibrosis bronchiectasis: a randomised, controlled trial. Thorax. 2014 Sep 21; doi: 10.1136/thoraxjnl-2014-205587. [DOI] [PubMed] [Google Scholar]
- 68.Boogaard R, de Jongste JC, Merkus PJ. Pharmacotherapy of impaired mucociliary clearance in non-CF pediatric lung disease. A review of the literature. Pediatr Pulmonol. 2007 Nov;42(11):989–1001. doi: 10.1002/ppul.20693. [DOI] [PubMed] [Google Scholar]
- 69.Stafanger G, Garne S, Howitz P, et al. The clinical effect and the effect on the ciliary motility of oral N-acetylcysteine in patients with cystic fibrosis and primary ciliary dyskinesia. Eur Respir J. 1988 Feb;1(2):161–7. [PubMed] [Google Scholar]
- 70.Loukides S, Kharitonov S, Wodehouse T, et al. Effect of arginine on mucociliary function in primary ciliary dyskinesia. Lancet. 1998 Aug 1;352(9125):371–2. doi: 10.1016/S0140-6736(05)60471-0. [DOI] [PubMed] [Google Scholar]
- 71.Grasemann H, Gartig SS, Wiesemann HG, et al. Effect of L-arginine infusion on airway NO in cystic fibrosis and primary ciliary dyskinesia syndrome. Eur Respir J. 1999 Jan;13(1):114–8. doi: 10.1183/09031936.99.13111499. [DOI] [PubMed] [Google Scholar]
- 72.Pasteur MC, Bilton D, Hill AT, British Thoracic Society Bronchiectasis non CFGG British Thoracic Society guideline for non-CF bronchiectasis. Thorax. 2010 Jul;65(Suppl 1):i1–58. doi: 10.1136/thx.2010.136119. [DOI] [PubMed] [Google Scholar]
- 73.Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007 Feb 15;175(4):367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 74.Sidhu MK, Mandal P, Hill AT. Bronchiectasis: an update on current pharmacotherapy and future perspectives. Expert Opin Pharmacother. 2014 Mar;15(4):505–25. doi: 10.1517/14656566.2014.878330. [DOI] [PubMed] [Google Scholar]
- 75.Bilton D, Henig N, Morrissey B, Gotfried M. Addition of inhaled tobramycin to ciprofloxacin for acute exacerbations of Pseudomonas aeruginosa infection in adult bronchiectasis. Chest. 2006 Nov;130(5):1503–10. doi: 10.1378/chest.130.5.1503. [DOI] [PubMed] [Google Scholar]
- 76.Brodt AM, Stovold E, Zhang L. Inhaled antibiotics for stable non-cystic fibrosis bronchiectasis: a systematic review. Eur Respir J. 2014 Aug;44(2):382–93. doi: 10.1183/09031936.00018414. [DOI] [PubMed] [Google Scholar]
- 77.Orriols R, Roig J, Ferrer J, et al. Inhaled antibiotic therapy in non-cystic fibrosis patients with bronchiectasis and chronic bronchial infection by Pseudomonas aeruginosa. Respir Med. 1999 Jul;93(7):476–80. doi: 10.1016/s0954-6111(99)90090-2. [DOI] [PubMed] [Google Scholar]
- 78.Barker AF, Couch L, Fiel SB, et al. Tobramycin solution for inhalation reduces sputum Pseudomonas aeruginosa density in bronchiectasis. Am J Respir Crit Care Med. 2000 Aug;162(2 Pt 1):481–5. doi: 10.1164/ajrccm.162.2.9910086. [DOI] [PubMed] [Google Scholar]
- 79.Drobnic ME, Sune P, Montoro JB, et al. Inhaled tobramycin in non-cystic fibrosis patients with bronchiectasis and chronic bronchial infection with Pseudomonas aeruginosa. Ann Pharmacother. 2005 Jan;39(1):39–44. doi: 10.1345/aph.1E099. [DOI] [PubMed] [Google Scholar]
- 80.Barker AF, O’Donnell AE, Flume P, et al. Aztreonam for inhalation solution in patients with non-cystic fibrosis bronchiectasis (AIR-BX1 and AIR-BX2): two randomised double-blind, placebo-controlled phase 3 trials. Lancet Respir Med. 2014 Sep;2(9):738–49. doi: 10.1016/S2213-2600(14)70165-1. [DOI] [PubMed] [Google Scholar]
- 81.Haworth CS, Foweraker JE, Wilkinson P, et al. Inhaled colistin in patients with bronchiectasis and chronic Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. 2014 Apr 15;189(8):975–82. doi: 10.1164/rccm.201312-2208OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murray MP, Govan JR, Doherty CJ, et al. A randomized controlled trial of nebulized gentamicin in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2011 Feb 15;183(4):491–9. doi: 10.1164/rccm.201005-0756OC. [DOI] [PubMed] [Google Scholar]
- 83.Serisier DJ, Bilton D, De Soyza A, et al. Inhaled, dual release liposomal ciprofloxacin in non-cystic fibrosis bronchiectasis (ORBIT-2): a randomised, double-blind, placebo-controlled trial. Thorax. 2013 Sep;68(9):812–7. doi: 10.1136/thoraxjnl-2013-203207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wilson R, Welte T, Polverino E, et al. Ciprofloxacin dry powder for inhalation in non-cystic fibrosis bronchiectasis: a phase II randomised study. Eur Respir J. 2013 May;41(5):1107–15. doi: 10.1183/09031936.00071312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85•.Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet. 2012 Aug 18;380(9842):660–7. doi: 10.1016/S0140-6736(12)60953-2. First publication reporting benefit from chronic macrolide use in non-CF bronchiectasis. [DOI] [PubMed] [Google Scholar]
- 86•.Serisier DJ, Martin ML, McGuckin MA, et al. Effect of long-term, low-dose erythromycin on pulmonary exacerbations among patients with non-cystic fibrosis bronchiectasis: the BLESS randomized controlled trial. JAMA. 2013 Mar 27;309(12):1260–7. doi: 10.1001/jama.2013.2290. Further evidence of benefit from chronic macrolide use in non-CF bronchiectasis. [DOI] [PubMed] [Google Scholar]
- 87•.Altenburg J, de Graaff CS, Stienstra Y, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA. 2013 Mar 27;309(12):1251–9. doi: 10.1001/jama.2013.1937. Further evidence of benefit from chronic macrolide use in non-CF bronchiectasis. [DOI] [PubMed] [Google Scholar]
- 88.Goyal V, Chang AB. Combination inhaled corticosteroids and long-acting beta2-agonists for children and adults with bronchiectasis. Cochrane Database Syst Rev. 2014;(6):CD010327. doi: 10.1002/14651858.CD010327.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mandal P, Chalmers JD, Graham C, et al. Atorvastatin as a stable treatment in bronchiectasis: a randomised controlled trial. Lancet Respir Med. 2014 Jun;2(6):455–63. doi: 10.1016/S2213-2600(14)70050-5. [DOI] [PubMed] [Google Scholar]
- 90.Smit HJ, Schreurs AJ, Van den Bosch JM, Westermann CJ. Is resection of bronchiectasis beneficial in patients with primary ciliary dyskinesia? Chest. 1996 Jun;109(6):1541–4. doi: 10.1378/chest.109.6.1541. [DOI] [PubMed] [Google Scholar]
- 91.Macchiarini P, Chapelier A, Vouhe P, et al. Double lung transplantation in situs inversus with Kartagener’s syndrome. Paris-Sud University Lung Transplant Group. J Thorac Cardiovasc Surg. 1994 Jul;108(1):86–91. [PubMed] [Google Scholar]
- 92.Parsons DS, Greene BA. A treatment for primary ciliary dyskinesia: efficacy of functional endoscopic sinus surgery. Laryngoscope. 1993 Nov;103(11 Pt 1):1269–72. doi: 10.1288/00005537-199311000-00010. [DOI] [PubMed] [Google Scholar]
- 93.Campbell R. Managing upper respiratory tract complications of primary ciliary dyskinesia in children. Curr Opin Allergy Clin Immunol. 2012 Feb;12(1):32–8. doi: 10.1097/ACI.0b013e32834eccc6. [DOI] [PubMed] [Google Scholar]
- 94.Pruliere-Escabasse V, Coste A, Chauvin P, et al. Otologic features in children with primary ciliary dyskinesia. Arch Otolaryngol Head Neck Surg. 2010 Nov;136(11):1121–6. doi: 10.1001/archoto.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chalmers JD, Goeminne P, Aliberti S, et al. The bronchiectasis severity index. An international derivation and validation study. Am J Respir Crit Care Med. 2014 Mar 1;189(5):576–85. doi: 10.1164/rccm.201309-1575OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Foundation PCD. PCD Clinical and Research Centers: Overview & Updates. Primary Ciliary Dyskinesia Foundation; 2011. [cited September 22, 2014]; Available from: http://www.pcdfoundation.org/en/finding-a-cure/pcd-clinical-center. [Google Scholar]
- 97.BESTCILIA. BESTCILIA: Better Experimental Screening and Treatment for Primary Ciliary Dyskinesia. 2014 [cited September 22, 2014]; Available from: http://www.bestcilia.eu/
- 98.Behan L, DunnGalvin A, Quittner AL, et al. European Respiratory Society International Congress Munich 2014. Munich, Germany: 2014. Development of the QOL-PCD: A cross-cultural patient-reported outcome measure for adults with primary ciliary dyskinesia. [Google Scholar]
- 99.Dell S, Leigh M, Ferkol T, et al. European Respiratory Society International Congress Munich 2014. Munich, Germany: 2014. Development of pediatric cross-cultural patient-reported outcome measures: QOL-PCD. [Google Scholar]
- 100.Lucas JS, Carroll M. Primary ciliary dyskinesia and cystic fibrosis: different diseases require different treatment. Chest. 2014 Apr;145(4):674–6. doi: 10.1378/chest.13-2590. [DOI] [PubMed] [Google Scholar]