

Abstract
Epithelial-to-mesenchymal transition (EMT) is an essential physiological process that promotes cancer cell migration, invasion, and metastasis. Several lines of evidence from both cellular and genetic studies suggest that AKT1/PKBα, but not AKT2 or AKT3, serves as a negative regulator of EMT and breast cancer metastasis. However, the underlying mechanism by which AKT1 suppresses EMT remains poorly defined. Here, we demonstrate that phosphorylation of Twist1 by AKT1 is required for β-TrCP-mediated Twist1 ubiquitination and degradation. The clinically used AKT inhibitor MK-2206, which possesses higher specificity toward AKT1, stabilized Twist1 and enhanced EMT in breast cancer cells. However, we discovered that resveratrol, a naturally occurring compound, induced β-TrCP-mediated Twist1 degradation to attenuate MK-2206-induced EMT in breast cancer cells. Taken together, our findings demonstrate that resveratrol counteracts the unexpected metastatic potential induced by anti-AKT therapy, and therefore suggest that the addition of resveratrol to an anti-AKT therapeutic regimen may provide extra support for limiting EMT.
Introduction
The highly conserved AKT1/PKBα, AKT2/PKBβ, and AKT3/PKBγ of the AKT/PKB protein kinase family are known to promote tumor initiation and progression and are considered excellent targets for anti-cancer therapy (1-3). However, the oncogenic activities of the AKT isoforms are far more complex as exemplified by several lines of evidence demonstrating that AKT1 but not AKT2 or AKT3 serves as a negative regulator of epithelial-to-mesenchyme transition (EMT) and breast cancer metastasis. For instance, overexpression of AKT1 in breast cancer cells blocks cell motility and invasion (4,5) whereas downregulation of AKT1 in MCF 10A cells decreases miR-200 abundance, thereby promoting transforming growth factor (TGF) β-induced EMT and stem cell-like phenotype (6). In AKT1−/− and ErbB2 transgenic mice, AKT1 activation accelerates mammary tumorigenesis but inhibits tumor invasion and metastasis (7). Moreover, Irie et al. demonstrated that knockdown of AKT1, but not AKT2, promotes insulin-like growth factor-1 (IGF-1)-stimulated EMT and cell migration (4).
EMT is a complex reprogramming process of epithelial cells that plays an indispensable role in tumor invasion and metastasis, and characterized by well-defined features such as the loss of epithelial markers (E-cadherin and α- and γ-catenin), the gain of mesenchymal cell markers (fibronectin, vimentin, and N-cadherin), and the acquisition of migratory and invasive properties (8,9). To date, several transcriptional repressors, e.g., Zeb-1/2, Twist1, and Snail-1/2, have been reported to regulate EMT. Twist1, a highly conserved basic helix-loop-helix (bHLH) transcriptional repressor, induces EMT to promote breast tumor metastasis (10) and is transcriptionally regulated by EGFR/STAT3 (11,12) and NFκB signaling (13). However, the regulatory mechanisms via posttranslational modification of Twist1 remain less understood.
To better understand AKT1-mediated EMT repression, we cross-analyzed gene expression profile of AKT isoforms using several public datasets. We found that low expression of AKT1 was highly associated with aggressive breast cancers and poor disease outcome. Moreover, our results indicated that AKT1 interacts with and phosphorylates Twist1 at S42, T121, and S123 to induce β-TrCP-mediated Twist1 degradation, leading to inhibition of EMT. Importantly, chronic exposure of breast cancer cells to AKT inhibitor, MK-2206, stabilizes Twist1 and induces EMT whereas administration of a β-TrCP inducer, resveratrol, degrades Twist1 and attenuates their metastatic potential. Together, our findings offer new insights into the role of AKT1 in EMT inhibition and also identify a potential for adverse effect of inducing EMT when anti-AKT inhibitor is used in breast cancer treatment.
Materials and Methods
Western blotting and immunoprecipitation
Western blot analysis using total lysates or immunoprecipitates was performed as previously described (14) using the antibodies listed in Supplementary Table S2.
Gene expression analysis
The following databases were used to compare AKT isoform mRNA expression in breast cancer cells: Cancer Cell Line Encyclopedia (CCLE) (15), Netherlands Cancer Institute (NKI) dataset (16) and the University of North Carolina (UNC) cohort (17,18).
Cell lines and cell culture
All cell lines were obtained from the ATCC (Manassas, VA), independently validated by STR DNA fingerprinting at MD Anderson, and maintained in DMEM/F-12 with 10% fetal bovine serum (FBS). Stable cell lines were grown in the presence of an additional 500 mg/ml G418 (Invitrogen, Carlsbad, CA) or 1 μg/ml puromycin (Calbiochem, San Diego, CA).
In vitro kinase assay
Approximately 5 μg purified GST proteins were incubated with active AKT1 kinase (Upstate Biotechnology, Charlottesville, VA) and analyzed using SDS-PAGE and autoradiography.
Side population analysis
Side population analysis was performed as previously describe (19). Briefly, cells were dissociated with Accutase, washed, and resuspended in ice-cold HBSS/5% FBS at a final concentration of 106 cells/100 μl. The CD44+/CD24− cell population was then determined by fluorescence-activated cell sorting.
Immunohistochemical (IHC) staining
IHC staining was performed as described previously (14). Human breast tumor tissue specimens were incubated with AKT1, β-TrCP, Twist1 or E-cadherin antibodies and a biotin-conjugated secondary antibody and then incubated with an avidin-biotinperoxidase complex. Visualization was performed using amino-ethylcarbazole chromogen. Fisher's exact test and Spearman rank correlation coefficient were used for statistical analysis. A P value < 0.05 is considered statistically significant.
Mouse model of lung metastasis
Tumor metastasis assays were performed using an intravenous breast cancer mouse model as previous described (13). 4T1 cells (1 × 105) were injected into the lateral tail vein of Balb/c mice. Animals were weighed before each experimental end point, and lung nodules were stained with India ink, excised, and counted immediately. All animal procedures were conducted under the approval of the Institutional Animal Care and Use Committee at MD Anderson Cancer Center (protocol number 10-14-07231). Detailed treatment protocol is described in Supplemental Information.
Results
Pathological expression of AKT isoforms in breast cancer cells
AKT activation is correlated with unfavorable clinical prognosis in many cancers including breast cancer. However, the roles of the AKT isoforms in breast cancer pathogenesis and their therapeutic potential have not been established. Thus, we examined the gene expression profile of the AKT isoforms by using a public dataset generated from 917 cancer cell lines (CCLE) (15) and grouped them into epithelial and mesenchymal subtype based on the gene expression profile of CDH1, CDH2, vimentin, and fibronectin. In the mesenchymal subtype, AKT1 was significantly downregulated (Supplementary Fig. S1A) whereas AKT2 remain unchanged in breast and stomach cancer cells (data not shown). AKT3 was upregulated in the mesenchymal subtype in multiple cancers (Supplementary Fig. S1B). To further dissect the expression of AKT isoforms in breast cancer cell lines, unsupervised hierarchical clustering analysis was performed based on the ERBB2, ESR1, PGR, and EMT profiles. Strikingly, we were able to distinguish basal-like from luminal type breast cancer cells with high accuracy (90% properly segregated) from the gene list (Fig. 1A) based on their genetic characteristics. AKT1 was expressed at a lower level in either aggressive basal-like (vs. luminal) or mesenchymal-type breast cancer cells (vs. epithelial-type) (Fig. 1B, middle and right) whereas AKT3 was significantly higher. Similarly, in another breast cancer dataset containing 54 cell lines (20), the basal-like and mesenchymal breast cancer subtypes were positively correlated with higher expression of AKT3 and inversely correlated with AKT1 (Supplementary Figs. S1C and S1D). Next, we asked whether a distinct group of clinical patient samples also share a differential expression pattern of AKT isoforms and found similar correlation using the NKI dataset (16) (Fig. 1C) and the UNC cohort (17,18) (Fig. 1D). While co-expression of AKT1, AKT2, and AKT3 did not correlate with any specific breast cancer subtypes (Figs. 1C and 1D, left panel), a significant negative correlation was observed between AKT1 and aggressive phenotype (Figs. 1C and 1D, middle and right). To consolidate the differential regulation between the AKT isoforms, we first examined the phosphorylation status of GSK3β, which is a well-known substrate of AKT kinases (21), and found that phospho-GSK3β was similar in cells transiently expressing constitutively active form of AKT1 (myr-AKT1) or AKT2 (myr-AKT2) (Supplementary Fig. S1E). Interestingly, only expression of myr-AKT1 activated the E-cadherin promoter in a dose-dependent manner (Fig. 1E), supporting the role of AKT1 in EMT inhibition. Consistently, overexpression myr-AKT1 but not myr-AKT2 or myr-AKT3 in MDA-MB-231 cells demonstrated an association with EMT regulation based on the expression of EMT markers (Fig. 1F). We further examined the protein expression of AKT isoforms in the metastatic breast cancer cohort by immunohistochemical (IHC) staining. The recurrence-free survival of patients with higher expression of p-AKT S473, which represents the phosphorylation level of all three AKT1, 2 and 3 isoforms, was significantly worse than patients with lower p-AKT S473 expression (Fig. 1G). In contrast, total AKT1 expression was correlated with better patient survival (Fig. 1H). These results suggested that each of the three AKT isoforms have a distinct pathological profile that is highly relevant to its functionality.
Figure 1. AKT genes signature co-regulated by EMT discriminates basal-like versus luminal subtype of breast cancers.
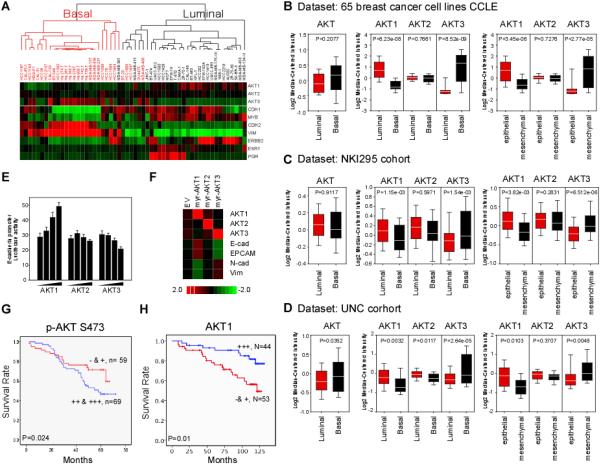
(A) Nonsupervised clustering of 54 breast cancer cell lines based on 10 genes. Gene expression heat map showing distinct expression pattern between basal- and luminal-breast cancer cell lines. TNBC gene signatures ESR1, ERBB2 and PgR are included.
(B) Box plots showing the average expression level of AKT1/2/3 or individual AKT isoforms gene in basal and luminal breast cancer cell lines (left) and (middle) from (A). Gene expression expressed as epithelial/mesenchymal ratio is shown (right).
(C and D) Box plots showing the average expression level of AKT1/2/3 (left), AKT1, AKT2 andAKT3 (middle and right) in basal and luminal cancers of NCI cohort (C) and of UNC cohort (D).
(E) Luciferase reporter assay of HEK-293 cells were transfected with the E-cadherin reporter together with increasing amount of myr-AKT1, myr-AKT2 or myr-AKT3.
(F) Gene expression heat map of E-cadherin, EPCAM, N-cadherin and vimentin in MDA-MB-231 cells expressing the myr-AKT isoforms.
(G) Kaplan-Meier analysis of p-AKT 473 survival of breast cancer patients in the MD Anderson Cancer Center cohort.
(H) Kaplan-Meier analysis of AKT1 by stratifying patients with quartiles partition.
Twist1 physically associates with AKT1
Given the distinct regulatory nature of AKT isoform in breast cancer cells, we sought to distinguish their functionality by identifying their specific interacting partners. Immunoprecipitation of MDA-MB-231 cells stably expressing HA-myr-AKT1 and HA-myr-AKT2 indicated AKT1 but not AKT2 pulled down a significant amount of a 20-kDa protein (Supplementary Fig. S2A), which was identified by mass spectrometric analysis as Twist1 (with 72% sequence coverage). The complex formation between endogenous AKT1 and Twist1 was further validated by Co-IP analysis (Supplementary Fig. S2B) and DuoLink labeling (Supplementary Fig. S2C). In vitro GST pull-down assay indicated that Twist1 interacted directly with GST-AKT1 but not GST-AKT2 or GST alone (Supplementary Fig. S2D). Since AKT1 inhibited EMT, we asked whether repression of EMT by AKT1 requires its interaction with EMT mediators, such as Twist1, FOXC2, E12, and Snail. AKT1 associated with Twist1 but not with the other EMT mediators (Supplementary Fig. S2E). A reverse IP also showed that AKT1 but not GSK3β, IKKα or IKKβ interacted with Twist1 (Supplementary Fig. S3A). These results raise an interesting possibility that AKT1-mediated EMT repression maybe regulated through the physical and functional interaction with Twist1.
AKT1 phosphorylates Twist1 in vitro
Twist1 contains two AKT phosphorylation RxRxxS/T motifs (22) at S42 and T121/S123, which are highly conserved across species (Fig. 2A and Supplementary Fig. S3B). To determine which of these two motifs might be phosphorylated by AKT1, we expressed GST-Twist1 in two separate fragments: one containing amino acids 1-112 and the other 113-202 (Fig. 2B) and then subjected them to an in vitro kinase assay with AKT1. AKT1 strongly phosphorylated both GST-Twist1 fragments (Fig. 2C) but phosphorylation was abolished when we mutated the consensus motif on Twist1. To pinpoint the phosphorylation potential in context of full-length Twist1, GST-Twist1 S42A (S42A), VA (T121V/S123A) and AVA (S42A/T121V/S123A) mutants were subjected to an in vitro kinase assay. Substitutions in both AKT phosphorylation motifs (Twist1 AVA) abolished AKT1-mediated phosphorylation (Fig. 2D) whereas substitutions in one or the other motif (S42A or T121V/S123A) had little or no effect on Twist1 phosphorylation (Fig. 2D). These results suggested that complete phosphorylation of Twist by AKT1 in vitro requires phosphorylation at S42, T121, and S123.
Figure 2. AKT1 phosphorylates Twist1 in vitro and in vivo.
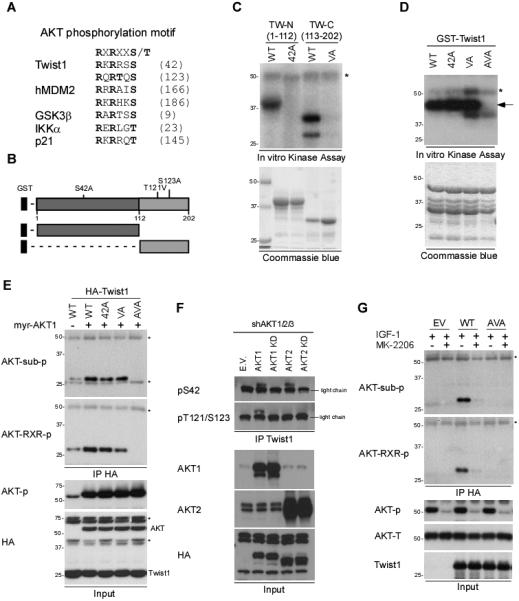
(A) Sequence alignment of AKT1 consensus motif. Twist1 contains two putative AKT1 phosphorylation motifs (RXRXXS/T) at S42 and T121/S123. R, Arginine; S, Serine; T, Threonine; X, any amino acid.
(B) A schematic diagram of Twist1 fragments.
(C) GST-Twist1-N (WT), GST-Twist1-N (S42A), GST-Twist1-C (WT), or GST-Twist1-C (TS121/123VA) proteins were incubated with recombinant AKT1 kinase. The phosphorylation was then analyzed by SDS-PAGE and autoradiography.
(D) Full-length GST-Twist1 WT, S42A, TS121/123VA, or AVA proteins were incubated with recombinant AKT1 kinase. Arrow, phosphorylated Twist1. *AKT1 auto-kinase activity.
(E) HEK-293T cells coexpressing HA-myr-AKT1 and HA-Twist1 WT, S42A, VA, or AVA were subjected to IP/Western blotting with the indicated antibodies. *Cross-reaction from the IgG heavy chain and light chain.
(F) MDA-MB-231 cells expressing AKT1, AKT1 KD, AKT2, or AKT2 KD by lentivirus infection were subjected to IP/Western blotting with the indicated antibodies.
(G) HEK-293T cells expressing HA-Twist1 WT or AVA were subjected IP/Western blotting with the indicated antibodies. *Cross-reaction from the IgG heavy chain and light chain.
AKT1 phosphorylates Twist1 in vivo
To recapitulate AKT1-mediated Twist1 phosphorylation in vivo, we co-expressed Flag-Twist1 and HA-tagged wild-type AKT1 (WT), dominant negative AKT1 (DN), or myr-AKT1 followed by IP with anti-Flag antibody and immunoblotting with phospho-AKT substrate antibody and phospho-RxRxxS/T antibody (Supplementary Fig. S3C). Twist1, but not other EMT regulators, was phosphorylated in the presence of myr-AKT1 (Supplementary Fig. S3D). Similar to the observations in vitro (Fig. 2D), mutation of either S42 alone or both T121/S123 in Twist1 did not affect its overall level of phosphorylation by AKT1, whereas mutation of all three sites completely abrogated its phosphorylation (Fig. 2E). Myr-AKT1 induced a noticeable mobility shift of wild-type and S42A mutant Twist1 but not Twist1-AVA or Snail (Supplementary Fig. S3E). In addition, AKT1 and AKT2 governed Twist1 functionality through differential phosphorylation in which AKT2 primarily phosphorylated Twist1 at S42 (Supplementary Fig. S3F) whereas AKT1 phosphorylated Twist1 at all three sites both in vitro and in vivo (Figs. 2D and 2E). The observed Twist1 S42 by AKT2 is in agreement with previously reported Twist1 S42 phosphorylation by AKT (23).
To further differentiate AKT1- from AKT2-mediated Twist1 phosphorylation, we re-expressed AKT1 or AKT2 in the AKT1/2/3 knockdown cells. AKT1, AKT2, and AKT3 were specifically knocked down in MDA-MB-231 cells using pGZIP vector (Supplementary Fig. S3G). AKT1 WT, AKT1 KD, AKT2 WT, and AKT2 KD were then re-expressed (Supplementary Fig. S3H). Cells re-expressing AKT1 had Twist1 S42, T121 and S123 phosphorylation, whereas those re-expressing AKT2 only had Twist1 phosphorylation at S42 (Fig. 2F). IGF-1-induced phosphorylation of Twist1 was inhibited in those treated with MK-2206, suggesting that Twist1 phosphorylation is regulated by PI3K signaling cascade (Fig. 2G).
Phosphorylation by AKT1 is required for Twist1 degradation
Next, we asked whether AKT1-mediated phosphorylation of Twist1 affects its expression levels. In AKT1-deficient mouse embryonic fibroblasts (MEFs), Twist1 expression was higher compared with that in WT MEFs (Fig. 3A). In contrast, Twist1 expression was downregulated in the HeLa cells stably expressing myr-AKT1 but not myr-AKT2, even though GSK3β was appropriately phosphorylated by both AKT1 and AKT2 (Fig. 3B). Downregulation of AKT1 but not AKT2 by shRNA upregulated Twist1 expression (Fig. 3C, right). Consistent with an earlier study (6), silencing AKT1 but not AKT2 expression induced EMT phenotypic change in MCF 10A cells (Fig. 3C, left).
Figure 3. AKT1 enhances Twist1 ubiquitination and degradation.
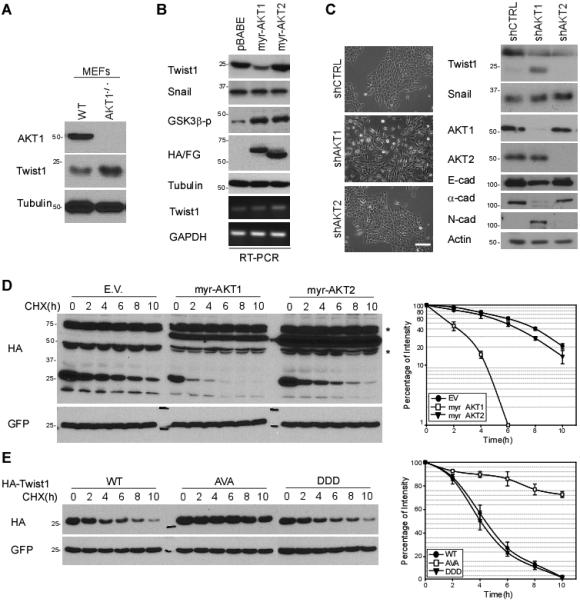
(A) Protein expression of Twist1 in wild-type and AKT1-deficient MEFs.
(B) Overexpression of myr-AKT1 and myr-AKT2 in HeLa cells by retrovirus infection. Cell lysates from these stable clones were subjected to Western blotting with the indicated antibodies.
(C) Phase contrast imaging of EMT morphotypic changes in MCF 10A shCTRL, shAKT1, and shAKT2 stable cells treated with 100 nM TGFβ for 6 h. Protein expression was analyzed by Western blot (right).
(D) Western blot analysis of Twist1 protein stability with indicated transient transfection. Cells were then treated with 20 mM cycloheximide as the indicated intervals.
(E) Protein half-life of Twist1 WT, AVA and DDD was determined as described in Fig. 3D.
Next, we measured the protein turnover rate in the presence of cycloheximide to determine the effect of AKT1-mediated phosphorylation on Twist1 protein stability. The turnover rate of Twist1 in myr-AKT1 expressing cells was much faster compared with those in myr-AKT2-expressing or empty vector control cells (Fig. 3D). We further examined the protein half-life of Twist1 and its three phospho-deficient variants (S42A, VA, and AVA) and found that the half-life of Twist1 AVA was significantly longer compared to that of Twist1 WT and Twist1 DDD phosphorylation-mimic mutant in the presence of myr-AKT1 (Fig. 3E).
AKT1 modulates β-TrCP-mediated Twist1 degradation
We noted that Twist1 contains one β-TrCP destruction box, DSΨXXS, which is also present in β-catenin, IκB, and Snail (Fig. 4A) (24,25). Overexpression of β-TrCP in HEK293 cells led to a rapid degradation of Twist1 (Supplementary Fig. S4A). Mutation of this motif (DSLSNS to DALSNA) (Fig. 4B) or knockdown of β-TrCP (Supplementary Fig. S4B) stabilized Twist1 expression. Similar to many β-TrCP substrates, induction of β-TrCP expression in MDA-MB-468 (Fig. 4C) and HeLa cells (Supplementary Fig. S4C) by ciglitazone (CG), troglitazone (TG), or resveratrol (26,27) reduced Twist1 expression, supporting that β-TrCP upregulation mediates Twist1 degradation. A double thymidine (Fig. 4D, quantitation of Supplementary Fig. S4D) and thymidine/nocodazole assay (Supplementary Fig. S4E) indicated that Twist1 protein but not mRNA was degraded during S to G2/M phase. Moreover, downregulation of β-TrCP expression stabilized Twist1, suggesting that β-TrCP is an authentic E3 ligase that regulates Twist1 degradation at S phase.
Figure 4. AKT1 induces β-TrCP-mediated Twist1 degradation.
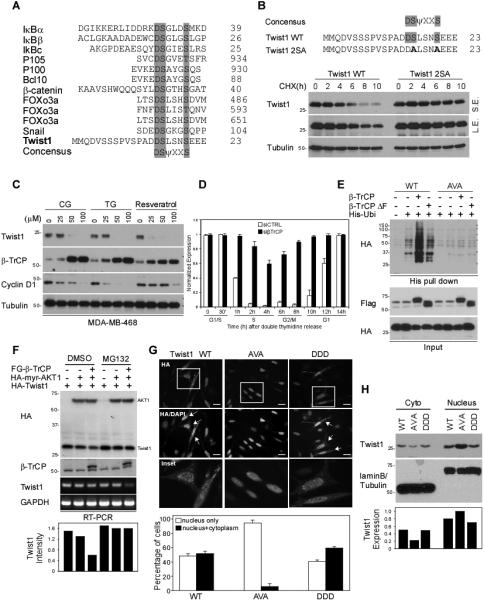
(A) Sequence alignment of β-TrCP destruction motif. Twist1 contains one putative β-TrCP recognition motifs (DSΨXXS). D, Aspartic acid; S, Serine; Ψ, hydrophobic; X, any amino acid.
(B) Pulse-chase experiment of Twist1 WT and 2SA protein stability using cycloheximide.
(C) MDA-MB-468 cells were treated with CG, TG and resveratrol at the indicated concentration for 48 h and subjected to Western blotting with the indicated antibodies.
(D) Quantification of Twist1 expression during cell cycle progression. HeLa cells carrying siCTRL or siβ-TrCP were synchronized using double thymidine block. Protein expression was measured by Western blot and quantified by densitometer. The full figure is presented in Figure S4D.
(E) Covalently conjugated His-ubiquitin of Twist1 was pulled down by Ni2+ agarose beads under denaturing condition and analyzed by Western blot with indicated antibodies.
(F) Western blot analysis of Twist1 degradation in HEK-293T cells.
(G) Subcellular localization of Twist1 WT, Twist1 AVA and Twist1 DDD. HA-Twist1 WT, AVA and DDD were transiently expressed in HeLa cells. The localization of Twist1 variants was imaged under a fluorescent microscope.
(H) Cell fractionation analysis of Twist1 variants.
To further validate how AKT1 is involved in β-TrCP-mediated Twist1 destabilization, we ectopically expressed β-TrCP WT and β-TrCP ΔF (a β-TrCP variant that lacks the F-box domain) and examined their effect on Twist1 ubiquitination. Expression of β-TrCP WT but not β-TrCP ΔF induced specific ubiquitination of Twist1 WT. Twist1 AVA was not ubiquitinated (Fig. 4E) in either β-TrCP WT or β-TrCP ΔF-expressing cells. In addition, co-expression of β-TrCP facilitated AKT1-mediated Twist1 degradation (Fig. 4F). Because the degradation machinery is enriched in the cytoplasm, we next asked whether AKT1-mediated Twist1 phosphorylation regulated by subcellular localization. Non-phosphorylation-mimic Twist1 AVA, which is resistant to 26S proteasome-mediated degradation, primarily localized to the nucleus (Fig. 4G). Likewise, expression of myr-AKT1 DN (dominant negative), which blocks endogenous AKT1 activity, led to an accumulation of non-phosphorylated Twist1 WT in the nucleus (Fig. 4G and Supplementary Fig. S5A). Cellular fractionation assay also demonstrated the nuclear translocation of non-phosphorylated Twist1 AVA variant (Fig. 4H). Our findings suggest that AKT1-mediated phosphorylation of Twist1 is required for its translocation from the nucleus to the cytoplasm where degradation occurs.
Twist1 AVA is a potent inducer of EMT
Because Twist1 is a key mediator during EMT progression, we also investigated the effect of AKT1-mediated Twist1 phosphorylation in EMT using Twist1 phosphorylation-deficient (Twist1 AVA) and phosphorylation-mimic (Twist1 DDD) mutants. To this end, we established stable transfectants with empty vector (EV), Twist1 WT, and AVA and DDD Twist1 mutants in MCF7 breast cancer cells, in which the basal level of endogenous Twist1 WT is low. Of the 35 clones screened, more than 15 clones expressed Twist1 WT with little or no effect on E-cadherin expression and no apparent morphological changes that resemble EMT. Likewise, the Twist1 DDD stable transfectants did not induce any E-cadherin loss or EMT phenotype. Interestingly, half of the 20 neomycin-resistant clones of Twist1 AVA transfectants underwent morphological changes that resembled EMT. Stable transfectants with similar Twist1 expression levels were used for comparison (Figs. 5A, 5B, and Supplementary Figs. S5B and S5C). To exclude the random effect from stable transfectant selection, we infected MDCK cells with replication-incompetent retroviruses expressing Flag-Twist1 WT, AVA, and DDD. In both MCF7 and MDCK cells, Twist1 AVA induced significantly downregulated E-cadherin and upregulated N-cadherin expression (Figs. 5A, 5B and Supplementary Fig. S5D). Vimentin was also selectively expressed in Twist1 AVA-expressing cells (Fig. 5B, and Supplementary Fig. S5C). RT-PCR analysis indicated that E-cadherin mRNA level was partially reduced in MCF7-Twist1 WT cells but completely abrogated in MCF7-Twist1 AVA cells (Fig. 5C). These results suggest that AKT1-mediated phosphorylation of Twist1 suppresses expression of several EMT markers such as N-cadherin and vimentin and derepresses Twist1-repressed E-cadherin. Blocking Twist1 phosphorylation as exemplified by the Twist1 AVA mutant induced expression of EMT markers.
Figure 5. Twist1 AVA mutant induces EMT.
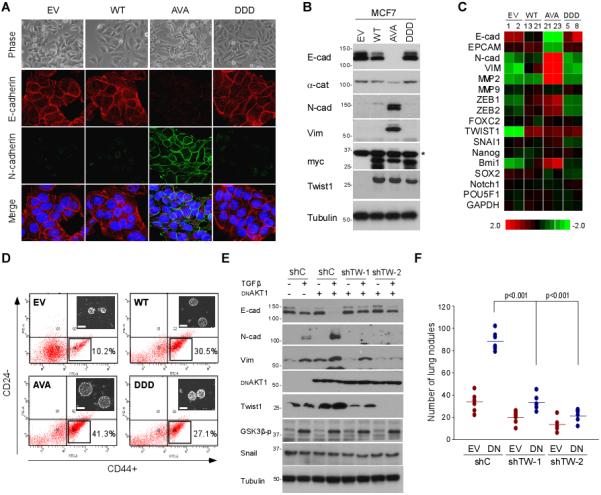
(A) Morphological changes of Twist1 stable clones. Phase contrast imaging of the morphological changes of each stable clone (upper panel; 20x magnification). Expression of EMT markers was detected using E-cadherin, N-cadherin and vimentin antibodies by confocal microscopy (lower panel). Cell nuclei were stained with DAPI.
(B) Western blot analysis of MCF7 Twist1 stable clones.
(C) qPCR analysis of Twist1 stable clones. Heatmap was generated using TreeView.
(D) Cancer stem cell side-population of Twist1 stable clones. Flow cytometric analysis showing the percentage of CD24− and CD44+ cells in MCF7 stable clones. Inset: tumor initiation ability measured by mammosphere formation.
(E) Western blot analysis myr-AKT1 DN overexpression clones in MCF10A cells.
(F) 4T1-Luc cells with myr-AKT1 DN and two shTwist1 stable clones were injected into female nude mice via the tail vein. Error bars represent the mean ± SD of five mice.
To further validate the association between Twist1 AVA mutant and EMT phenotype, we performed several EMT related functional assays using MCF7 stable transfectants. Although cell proliferation rate of Twist1 variants remained similar (Supplementary Figs. S5E and S5F), cell invasion ability by a Transwell assay was significantly enhanced in MCF7 Twist1 AVA transfectants. MCF7-Twist1 AVA cells also displayed accelerated migration by wound healing assay compared to the WT and DDD mutant under time-lapse microscopy (Supplementary Fig. S6A). Twist1 AVA demonstrated aggressive phenotype as indicated by the high percentage of migrating cells (Supplementary Fig. S6B). To further validate the migration ability was due to the loss of E-cadherin, we restored E-cadherin into two independent Twist1 AVA clones to revert Twist1 AVA-induced EMT (Supplementary Fig. S6C). Because EMT is usually accompanied by an increase in cancer stem cell properties, we also conducted stem cell analysis and found that CD24−/CD44+ population (Fig. 5D) and mammosphere formation (Fig. 5D, inset) was higher in cells expressing Twist1 AVA compared those expressing WT or DDD mutant Twist. Moreover, the percentage of Twist1 AVA-expressing cells that underwent adriamycin-induced apoptosis was significantly less compared with that of Twist1 S42A and VA (Supplementary Fig. S6D). Taken together, blocking AKT1-mediated Twist1 phosphorylation enhances EMT phenotypic changes that resemble AKT1-induced EMT repression.
To consolidate the role of Twist1 in AKT1-mediated EMT repression, we knocked down Snail and Twist1 in shAKT1 clone and found that downregulation of Twist1 but not Snail abolished shAKT1-induced EMT (Supplementary Fig. S6E). We also stably expressed myr-AKT1 DN in MCF 10A (shCTRL) and two Twist1 knockdown stable clones (shTW-1 and shTW-2) (13). Myr-AKT1 DN increased Twist1 expression and enhanced TGFβ-induced EMT in MCF 10A cells as measured by downregulation of E-cadherin and upregulation of vimentin and N-cadherin expression. However, knocking down Twist1 inhibited TGFβ-induced EMT in AKT1 DN expressing cells, whereas cells that received shControl (shC) showed no effect (Fig. 5E). Thus, expression of Twist1 in MCF 10A cells is required for TGFβ/myr-AKT1 DN-induced EMT, establishing a role of Twist1 in the AKT1-mediated EMT repression.
Next, we asked whether AKT1 DN increases the metastatic potential of breast cancer in vivo, and if so, whether the AKT1 DN-induced Twist1 expression plays a role in this process. Results from an in vivo experimental metastasis assay by a lung colonization xenograft model showed that myr-AKT1 DN expression in 4T1-Luc cells enhanced metastatic potential as measured by the number of lung colonization, and knockdown of Twist1 expression antagonized myr-AKT1 DN-induced metastasis (Fig. 5F).
AKT1 controls the molecular switch of Twist1 by T121 and S123 phosphorylation
To differentiate AKT2-induced EMT via Twist1 S42 phosphorylation, a series of functional analyses was performed to distinguish the isoform-specific regulation in the context of Twist1. Twist1 S42A exhibited similar protein stability (Supplementary Fig. S7A) and ubiquitination (Supplementary Fig. S7B) compared with the wild-type Twist1. Interestingly, mutation of the second motif on Twist1 S42A (Twist1 AVA) robustly stabilized Twist1 by blocking ubiquitination. These results suggest that T121 and S123 phosphorylation by AKT1 favor β-TrCP recognition for protein degradation. We performed qChIP (Supplementary Fig. S7C) and luciferase assay (Supplementary Fig. S7D) to evaluate the transcriptional activities of the Twist1 variants on the E-cadherin promoter. We showed that Twist1 S42A by transient transfection reduced its DNA binding ability and derepressed the transcriptional activity of the E-Cadherin promoter, which is in line with prior studies showing AKT2 phosphorylates Twist1 at S42 to induce EMT (28). Neither DNA binding nor transcriptional repression of the E-cadherin promoter was affected by the Twist1 VA (T121V/S123A) mutant, which suggests that T121/S123 phosphorylation per se does not affect Twist1 binding affinity. Results from functional assays using Twist MCF7 stable transfectants by retrovirus, however, indicated that Twist1 S42A lost its EMT potential whereas Twist1 AVA induced EMT phenotypic change (Supplementary Fig. S7E) as well as cell invasion (Supplementary Fig. S7F). The half-life of Twist1 S42A mutant was much shorter than that of the AVA mutant (Supplementary Fig. S7A). The additional phosphorylation at T121/S123 in Twist1 S42A likely enhances its degradation by β-TrCP in the stable transfectants, reducing its ability to induce EMT. Altogether, we propose a model (Supplementary Fig. S7G) as follows: AKT2 phosphorylates Twist1 at S42, which reduces E-cadherin expression to induce EMT whereas AKT1 catalyzes two additional phosphorylation at Twist1 T121 and S123, allowing β-TrCP to degrade Twist1, which leads to inhibition of EMT.
Correlation of AKT1, β-TrCP, Twist1, and E-cadherin in human tumor tissues
To validate the pathological relevance of the identified mechanism, we studied the expression of AKT1, β-TrCP, Twist1, and E-cadherin in 104 human metastatic breast tumor specimens by IHC staining. Twist1 was detected in 13 of the 39 specimens with high AKT1 expression but in 39 of the 45 specimens with low AKT1 expression, indicating that there is an inverse correction between AKT1 and Twist1 expression. Consistent with this finding, we found that AKT1 expression correlated with β-TrCP and E-cadherin expression (Supplementary Fig. S8 and Table S1).
Inhibition of AKT1 induces Twist1 upregulation and metastatic potential
Multiple AKT inhibitors have been tested in clinical trials as anti-cancer agents (29). The model shown in Supplementary Fig. S7G raised the concern that the use of AKT inhibitors as anti-cancer agents could potentially enhance EMT/metastasis while suppressing tumor growth. To this end, we tested this potential adverse effect of a clinically used AKT inhibitor, MK-2206, which possesses differential inhibition toward specific AKT isoform (selective inhibitor of AKT1, AKT2 and AKT3 with IC50 of 5 nM, 12 nM, and 65 nM, respectively) (30). Since breast cancer cells with lower ratios of AKT1/AKT2 are less sensitive to MK-2206 (31), we sought to determine whether AKT1 inhibition by MK-2206 has potential to induce EMT via Twist1 upregulation in breast cancer cells. MCF10A cells were treated with 0.2 μM MK-2206 which enhanced TGFβ-induced EMT (Fig. 6A, lane 4, 8 and 12; and Supplementary Fig. S9A). MK-2206 alone also induced EMT in MCF 10A cells but required a longer stimulation than TGFβ treatment (Fig. 6A, lane 3, 7 and 11). Continuous treatment with MK-2206 alone to passage 3 (3 days/passage) led to late EMT morphological changes similar to those by TGFβ. In fact, MK-2206 mediated EMT primarily through inhibition of AKT1 as knockdown of AKT2 and AKT3 had no effect on the expression of EMT markers (Supplementary Fig. S9B). Consistent with our results from AKT1 knockdown cells (Fig. 3C), inhibition of AKT1 by MK-2206 increases Twist1 stability by blocking Twist1 degradation (Fig. 6A). To determine whether stabilization of Twist1 is required for MK-2206-mediated EMT, MCF 10A cells carrying shCTRL or shTwist1 (shTW) were treated with MK-2206 for 10 days. Downregulation of Twist1 inhibited MK2206-mediated cell migration, invasion, and acquisition of EMT phenotype (Fig. 6B), suggesting an indispensable role of Twist1 in MK-2206-mediated phenotypic changes associated with EMT.
Figure 6. Twist1 is required for AKT1-mediated EMT repression.
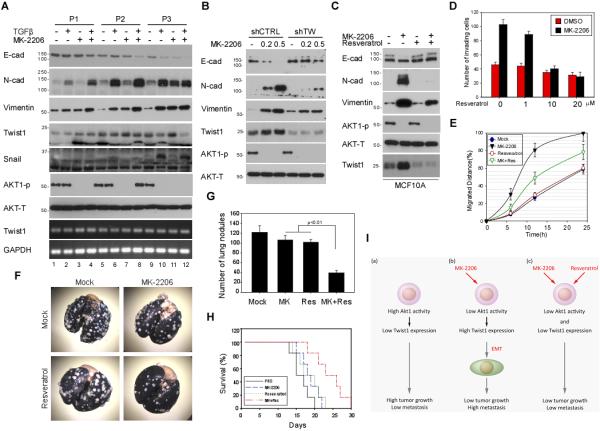
(A) MCF 10A cells were continuously treated with or without TGFβ or MK-2206. Phenotypic changes were examined by Western blot with the indicated antibodies.
(B) MCF 10A cells harboring shCTRL and shTwist1 were treated with 0.2 μM or 0.5 μM MK-2206 for 10 days and subjected to Western blotting with the indicated antibodies.
(C) Resveratrol reduces MK-2206-induced EMT. MCF 10A cells were continuously treated with or without MK-2206 or Resveratrol and subjected to Western blotting with the indicated antibodies.
(D and E) The number of migrating cells and their distance of migration (arbitrary unit) calculated from three independent views.
(F) Representative photograph of metastatic lung nodules. 4T1 cells (1 × 105 cells/0.1 ml) were injected into the lateral tail vein of Balb/c female mice. MK-2206 and resveratrol were then administered orally at a dosage of 200 mg/kg (n = 6/group) per day for 21 days. Mice were sacrificed, and the lungs were stained with India ink and resected for imaging.
(G) Quantification of the lung nodules. Error bars represent ± SD for n = 6.
(H) The survival time of mice in (F). Survival curve was plotted by Kaplan-Meier analysis.
(I) Schematic diagram showing the combinatorial strategy of utilizing resveratrol to overcome MK-2206-induced metastatic potential.
Since the protein stability of Twist1 is tightly regulated by β-TrCP (Fig. 4C and Supplementary Fig. S4C), we asked whether destabilization of Twist1 by β-TrCP inducer, resveratrol, would reduce MK-2206-mediated EMT. We first treated MCF 10A cells with MK-2206 to induce EMT followed by resveratrol (10 μM) for 2 days. Resveratrol downregulated Twist1 and reverted MK-2206-mediated phenotypic changes associated with EMT (Fig. 6C). At a concentration of 10 μM, resveratrol inhibited MK-2206-mediated cell invasion (Fig. 6D) and migration ability (Fig. 6E). Similar results were observed in 4T1 cells (Supplementary Fig. S9C). The possibility that resveratrol decreases the MK-2206-mediated aggressiveness was further explored by investigating the effects of resveratrol on tumor metastasis in animal model. We inoculated 4T1 cells in Balb/c mice by tail vein injection (13). Tumor-bearing mice were then treated with or without MK-2206 or/and resveratrol. The metastatic nodules in the lungs were resected from each mouse and quantified by a dissecting microscope (Fig. 6F). The number of lung metastasis was significantly reduced in mice co-treated with both MK-2206 and resveratrol but not in mice that received each agent alone (Fig. 6G). In addition, the combined therapy of MK-2206 and resveratrol also prolonged mice survival by 20% (Fig. 6H). These observations suggest that Twist1 is a key factor in MK-2206-induced EMT, cell migration/invasion, and lung colonization in breast cancer and that destabilization of Twist1 by inducing β-TrCP through resveratrol enhances the therapeutic efficacy of MK-2206 for breast cancer treatment (Fig. 6I).
Discussion
Some discrepancies observed in AKT1's role in breast cancer metastasis have been reported. For instance, overexpression of AKT1 suppresses tumor invasion but accelerates ErbB2-mediated mammary tumorigenesis (7). In other studies, AKT1 knockout impaired lung metastasis in MMTV-ErbB2/Neu and MMTV-PyMT transgenic mice (32,33). While this may be attributed to the differences between the transgenic models tested, one possibility could be that knockout of AKT1 already compromised the ErbB2- and PyMT-induced primary tumor formation, which may in turn impair AKT1-mediated EMT and metastasis inhibition.
Consistent with the literature (4-6,34,35), our study provides a comprehensive pathological and mechanistic understanding of the complexity of AKT isoform regulation in breast cancer EMT. Multiple dataset analysis identified the inhibitory nature of AKT1 in EMT, which was further validated by patient sample analysis in which a correlation between AKT1 expression and improved patient survival in invasive breast cancer cohorts was observed. In vitro and in vivo studies using the Twist1 AVA mutant demonstrated that AKT1 mediates EMT inhibition in breast cancer cells, further supporting an inverse relationship between AKT1 expression and EMT from gene expression analysis. It is worthwhile to mention that stable clones of Twist1 variant were selected with equal expression level; however, Twist1 AVA still exhibited obvious EMT, suggesting other function of Twist1 AVA may exist to stimulate EMT in addition to its ability to increase Twist1 protein stability. Although AKT1 inhibits EMT in breast cancer, AKT1 was shown to induce EMT in squamous cancer and sarcoma (36-38). Thus, it is of importance to understand how cell-type-specific AKT isoform expression, localization, and activities elicit diverse functions in cancer cells.
AKT2 phosphorylates Twist1 at S42 to enhance Twist1-mediated E-cadherin suppression (23). Since AKT1 also phosphorylates Twist1 at S42, AKT1 may exert dual and contradictory influences on Twist1. According to our data, the complexity may be minimal as Twist1 protein expression is downregulated as long as AKT1 is expressed in breast cancer. These results are also in agreement with many published research showing that AKT1 inhibits EMT in breast cancer (4-6,34,35). Hence, we believe that Twist1 S42-mediated EMT induction by AKT1 may exist but is outpaced by AKT1-mediated negative regulation through T121 and S123 phosphorylation in breast cancer.
In summary, we have identified a novel AKT1/β-TrCP/Twist1/E-cadherin signaling axis in breast cancer cells (Supplementary Table S1). The AKT1- and AKT2-specific EMT regulation of Twist1 phosphorylation appear to have differential effects depending on the cellular context. Dissecting the effect of each AKT isoform, in this case, AKT1, enabled us to further understand how breast cancer cells undergo EMT and reach metastatic potential. Moreover, resveratrol may provide therapeutic benefits by limiting the metastatic potential when administering MK-2206.
Supplementary Material
Acknowledgments
This work was partially supported by the following: National Institutes of Health (CA109311, CA099031, and CCSG CA16672); Susan G. Komen Foundation; Patel Memorial Breast Cancer Endowment Fund; Breast Cancer Research Foundation; The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund; Ministry of Science and Technology, International Research-intensive Centers of Excellence in Taiwan (I-RiCE; MOST 104-2911-I-002-302); Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW104-TDU-B-212-124-002); Center for Biological Pathways.
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- 1.Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8(16):2502–8. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A. 2001;98(20):10983–5. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phung TL, Du W, Xue Q, Ayyaswamy S, Gerald D, Antonello Z, et al. Akt1 and akt3 exert opposing roles in the regulation of vascular tumor growth. Cancer Res. 2015;75(1):40–50. doi: 10.1158/0008-5472.CAN-13-2961. [DOI] [PubMed] [Google Scholar]
- 4.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171(6):1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Molecular cell. 2005;20(4):539–50. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 6.Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, et al. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Science signaling. 2009;2(92):ra62. doi: 10.1126/scisignal.2000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004;64(9):3171–8. doi: 10.1158/0008-5472.can-03-3465. [DOI] [PubMed] [Google Scholar]
- 8.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nature reviews Cancer. 2002;2(6):442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 9.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17(5):548–58. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, et al. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelialmesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67(19):9066–76. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, et al. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. The Journal of biological chemistry. 2008;283(21):14665–73. doi: 10.1074/jbc.M707429200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-mesenchyme transition induced by TNF-a requires NF-kB mediated transcriptional upregulation of Twist1. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130(3):440–55. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 15.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. The New England journal of medicine. 2002;347(25):1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 17.Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC genomics. 2006;7:96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 Promotes Expansion of Breast Tumor Initiating Cells through Activation of RAF1-beta-Catenin Signaling. Cancer cell. 2010;19(1):86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer cell. 2006;10(6):515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 22.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15(23):6541–51. [PMC free article] [PubMed] [Google Scholar]
- 23.Vichalkovski A, Gresko E, Hess D, Restuccia DF, Hemmings BA. PKB/AKT phosphorylation of the transcription factor Twist-1 at Ser42 inhibits p53 activity in response to DNA damage. Oncogene. 2010 doi: 10.1038/onc.2010.115. [DOI] [PubMed] [Google Scholar]
- 24.Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, et al. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell. 2003;4(6):799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 25.Margottin-Goguet F, Hsu JY, Loktev A, Hsieh HM, Reimann JD, Jackson PK. Prophase destruction of Emi1 by the SCF(betaTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev Cell. 2003;4(6):813–26. doi: 10.1016/s1534-5807(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 26.Wei S, Lin LF, Yang CC, Wang YC, Chang GD, Chen H, et al. Thiazolidinediones modulate the expression of beta-catenin and other cell-cycle regulatory proteins by targeting the F-box proteins of Skp1-Cul1-F-box protein E3 ubiquitin ligase independently of peroxisome proliferator-activated receptor gamma. Mol Pharmacol. 2007;72(3):725–33. doi: 10.1124/mol.107.035287. [DOI] [PubMed] [Google Scholar]
- 27.Wei S, Chuang HC, Tsai WC, Yang HC, Ho SR, Paterson AJ, et al. Thiazolidinediones mimic glucose starvation in facilitating Sp1 degradation through the up-regulation of beta-transducin repeat-containing protein. Mol Pharmacol. 2009;76(1):47–57. doi: 10.1124/mol.109.055376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, et al. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov. 2012;2(3):248–59. doi: 10.1158/2159-8290.CD-11-0270. [DOI] [PubMed] [Google Scholar]
- 29.Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert opinion on investigational drugs. 2010;19(11):1355–66. doi: 10.1517/13543784.2010.520701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29(35):4688–95. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 31.Sangai T, Akcakanat A, Chen H, Tarco E, Wu Y, Do KA, et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res. 2012;18(20):5816–28. doi: 10.1158/1078-0432.CCR-12-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, et al. Akt1 governs breast cancer progression in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(18):7438–43. doi: 10.1073/pnas.0605874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007;67(1):167–77. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- 34.Dillon RL, Muller WJ. Distinct biological roles for the akt family in mammary tumor progression. Cancer Res. 2010;70(11):4260–4. doi: 10.1158/0008-5472.CAN-10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong KO, Kim JH, Hong JS, Yoon HJ, Lee JI, Hong SP, et al. Inhibition of Akt activity induces the mesenchymal-to-epithelial reverting transition with restoring E-cadherin expression in KB and KOSCC-25B oral squamous cell carcinoma cells. J Exp Clin Cancer Res. 2009;28:28. doi: 10.1186/1756-9966-28-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, van Roy F, Lee-Kwon W, et al. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003;63(9):2172–8. [PubMed] [Google Scholar]
- 37.Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26(53):7445–56. doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- 38.Zhu QS, Rosenblatt K, Huang KL, Lahat G, Brobey R, Bolshakov S, et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene. 2011;30(4):457–70. doi: 10.1038/onc.2010.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.