

Abstract
The architecture of the cotton plant, including fruit branch formation and flowering pattern, is the most important characteristic that directly influences light exploitation, yield and cost of planting. Nulliplex branch is a useful phenotype to study cotton architecture. We used RNA sequencing to obtain mRNA and miRNA profiles from nulliplex- and normal-branch cotton at three developmental stages. The differentially expressed genes (DEGs) and miRNAs were identified that preferentially/specifically expressed in the pre-squaring stage, which is a key stage controlling the transition from vegetative to reproductive growth. The DEGs identified were primarily enriched in RNA, protein, and signalling categories in Gossypium barbadense and Gossypium hirsutum. Interestingly, during the pre-squaring stage, the DEGs were predominantly enriched in transcription factors in both G. barbadense and G. hirsutum, and these transcription factors were mainly involved in branching and flowering. Related miRNAs were also identified. The results showed that fruit branching in cotton is controlled by molecular pathways similar to those in Arabidopsis and that multiple regulated pathways may affect the development of floral buds. Our study showed that the development of fruit branches is closely related to flowering induction and provides insight into the molecular mechanisms of branch and flower development in cotton.
Cotton is one of the most important economic crops for natural textile fibre and oilseed and has been widely planted in the United States, China, India, Pakistan, Australia, Uzbekistan and the other countries1,2. Cotton architecture is primarily determined based on shoot branching patterns and flowering patterns that directly influence light exploitation, yield, planting area, the efficiency of harvest mechanisation and the cost of planting2,3,4,5. In cotton, the floral bud’s forming indicates the beginning of reproductive growth on the basis of prior vegetative growth, and most cultivated cotton varieties produce lateral branch from the leaf axils, then the lateral branch develops and differentiates into vegetative branches and fruit branches. Vegetative buds often arise from the lower leaf axils, around the first five nodes of the stem, whereas the fruiting branch arises from the upper leaf axils. Shortly after the pre-squaring stage, visible triangular buds (approximately 3 mm) appear on the axil. The main shoot and the vegetative branch display monopodial growth, but the fruit branches display sympodial growth, typically with more than two nodes5,6,7. Floral bud bursting and fruit branching are major events in the development of cotton architecture and are among the most important productivity-related agronomic traits considered in breeding and cultivation programs8,9.
The prominent characteristics of cotton are the extensive overlap between vegetative growth and reproductive growth and the requirement for manual/mechanical pruning or artificial chemical regulation to perform alterations of the cotton architecture, which are important determinants of cotton yield10,11. Thus, it is important to balance vegetative growth and reproductive growth by improving the architecture, flowering and branching pattern of cotton to obtain higher yields and a lower cost of planting.
In recent years, the molecular mechanisms involving plant architecture, including floral transition, branching, shoot apical meristem development, and branch angle formation, have been extensively studied in rice etc., including the central roles of Branched1 (BRC1) in strigolactones, branching and Tiller Angle Control 1 (TAC1) in plant architecture12,13,14,15,16. Previous studies have mapped quantitative trait loci (QTLs) of cotton architectural traits using molecular markers, including fruit branch internode length and fruit branch angle8,9,17,18,19. The molecular mechanisms involved in the development of cotton architecture traits have been rarely reported.
There is a type of cotton branching mutation called nulliplex branch, for which most of the flowers arise directly from the leaf axils on the main shoot, and these individuals typically do not have a fruiting branch20,21,22. There are 1–3 flowers on the leaf axils on the main shoot and the fruiting branches seem to disappear in this branch mutation cotton. A nulliplex branch is an atypical branch that is genetically determined and is considered a cluster branch trait20,21,22. The nulliplex branching trait is controlled by the recessive gene gb_nb15. Cotton varieties with nulliplex branches have been planted in areas including Xinjiang, China, and Uzbekistan, and their unique plant architecture is suitable for high-density and mechanised planting, without needing pruning and chemical regulation, and with relatively short growing stages5. Chen et al. found that the nulliplex branch gene is localised to a 600 kb genome fragment and suggested that the trait may be related to an Arabidopsis thaliana centroradialis homologue (the ATC gene), which acts as a floral suppressor in Arabidopsis5,23. However, the molecular mechanisms of the development of nulliplex- and normal-branch cotton remain unknown.
The novel, high-throughput, deep-sequencing transcriptome approach known as RNA-seq has allowed for the generation of large-scale libraries of expressed sequence tags and has improved the speed of gene discovery in recent years. Beyond identifying and sequencing mRNA and small RNA transcriptomes, the Illumina RNA-seq platform allows researchers to examine the expression patterns of transcripts in tissues of interest. This approach has been successfully applied to a variety of plant species, including cotton24.
Small RNAs are short non-coding RNAs that play crucial roles in a wide range of biological processes, including cell proliferation, developmental timing, floral transition, and stress responses25,26,27,28. MicroRNAs (miRNAs) regulate a large number of genes in plants by binding to the 3′ UTR or other regions of target mRNAs leading to degradation or translational repression, which is common during development29. Previous studies in cotton have chiefly focused on miRNA expression in the ovules, during fibre development, and in the stress response30,31.
Recently, the genome of an allotetraploid cotton (Gossypium hirsutum) was published after the diploid species Gossypium raimondii and Gossypium arboreum were sequenced1,32,33,34. G. hirsutum is mainly cultivated as upland cotton, and the genomic data provides more comprehensive molecular information on production traits than genomic data for diploid species.
In this study, we performed high-throughput next-generation sequencing to identify the molecular mechanisms of the development of the fruit branch and floral bud in cotton. The square forming period of nulliplex and normal branches was used to simultaneously measure mRNA and miRNA expression profiles. Integration analysis of mRNA and miRNA expression and elucidation of the regulatory relationship between miRNA and their corresponding mRNA targets are crucial for understanding the development of fruit branch during the floral bud forming stage. Our results demonstrated that the differentially expressed genes (DEGs) between the nulliplex and normal branches in the pre-squaring stage of G. barbadense and G. hirsutum were primarily related to floral transition and branching development. These results provide a molecular basis for understanding the cotton plant’s architecture and imply that the development of the fruit branch may be associated with flowering transition.
Results
Transcriptome sequencing of mRNA libraries
To identify transcripts that are differentially expressed during the differentiation stage of flower and floral buds between nulliplex- and normal-branch cotton, samples from plants at three different developmental stages were used: the seedling stage, the pre-squaring stage, and the squaring stage. Four lines (varieties) were used to construct RNA-seq libraries (Fig. 1). The experiments were performed with three biological replicates, resulting in 36 RNA-seq libraries for sequencing. A total of 2,022,339,010 raw reads were obtained. After low-quality reads were filtered out, a total of 1,915,778,530 clean reads were selected for further analysis (See supplemental Table S1).
Figure 1. Plant appearance of bud on branch or main shoot in diffenrent cotton varieties (lines).

Red arrows indicate bud on main shoot (Nulliplex-branch) or normal branch (wild type). Nulliplex-branch: 3798 (G. hirsutum) and Xinhai 16 (G.barbadense), normal branch(wild type):Huazhong94-3130 (G. hirsutum) and Hai1(G.barbadense).
DEGs between nulliplex- and normal-branch cotton
Based on these expression analyses, we identified 422 upregulated and 307 downregulated DEGs in G. barbadense and 123 upregulated and 92 downregulated DEGs in G. hirsutum during the seedling stage. In G. barbadense and G. hirsutum, 2,538 upregulated and 2,419 downregulated and 339 upregulated and 304 downregulated DEGs, respectively, were identified during the pre-squaring stage and 1,069 upregulated and 943 downregulated and 1,344 upregulated and 990 downregulated DEGs, respectively, were identified during the squaring stage (Fig. 2, see supplemental Data set S1 to S6 online). There were more DEGs at the pre-squaring stage in G. barbadense than the other two stages, whereas the greatest number of DEGs was identified in the squaring stage in G. hirsutum.
Figure 2. Numbers of different expressed genes in each comparison.
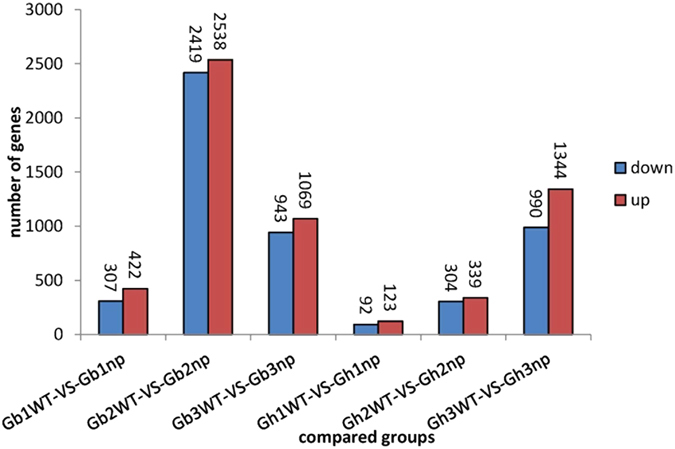
The numbers on column showed quantity of up-regulated (red) and down-regulated (blue) genes.
Functional category enrichment of DEGs
All DEGs of the six groups were assigned to Mapman functional categories. During the seedling stage, the DEGs were almost equally distributed in each category. During the second and third stages, the DEGs were mainly enriched for RNA, protein, signalling, and transport in both types of cotton (Fig. 3A,B). Regarding protein metabolism categories, the DEGs were mainly associated with ribosomal protein, posttranslational modification, and degradation at the second stage of G. barbadense. The third stage was mainly enriched with genes for posttranslational modification and degradation. However, the DEGs were considerably enriched in genes regulating degradation at the second stage and enriched in genes involved in posttranslational modification and degradation at the third stage (Fig. 3C). Regarding RNA categories, the DEGs were mainly enriched for transcription factor families, including homeobox, MYB, AP2/EREBP, WRKY, C2H2, and bHLH at the second stage in G. barbadense and enriched for MYB at the squaring stage in G. hirsutum (Fig. 3D).
Figure 3. Enrichment of functional categories for DEGs in six compared groups.

(A) Enrichment of functional categories for DEG lists. (B) Enrichment of protein subcategories. (C) Enrichment of transcription factor subcategories. (D) the percentage of each functional categories for DEGs in six compared groups. Gray, not significantly enriched. The bar showed the log2 value of enrich gene number.
DEGs in flower and floral bud development
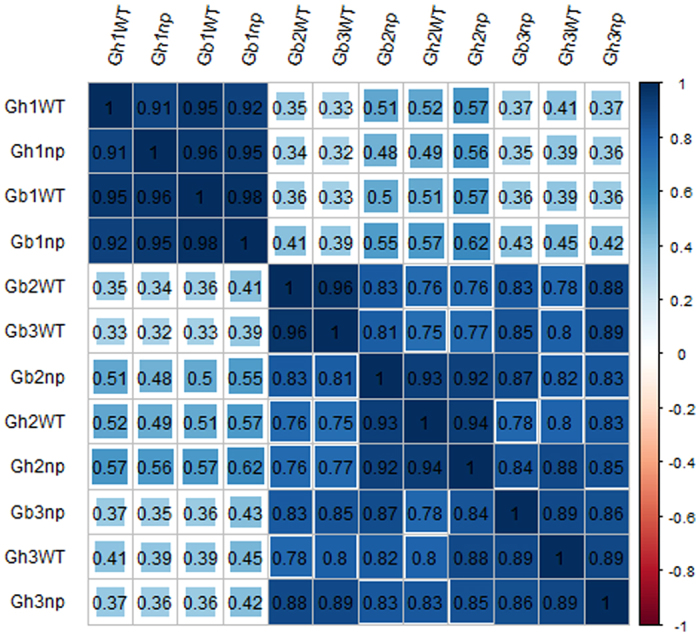
For interlibrary comparison, read numbers were normalised to relative abundance as reads per kilobase transcriptome per million mapped reads (RPKM). The RPKM value (mean value of three biological replicates) of each gene was further used to compute the related coefficients between each sample. The expression correlations of genes showed accordance between the same development stages in both types of cotton, excluding Gb3WT. The correlations were substantially lower between the seedling stage and squaring stage and/or pre-squaring stages (Fig. 4). Partial correlations between pre-squaring stage samples and squaring stage samples were lower than same-stage samples, such as that between Gb2WT and Gb2np with a coefficient of 0.83, which was slightly lower than 0.96 for Gb3WT (Fig. 4). This finding suggests that the development of the pre-squaring stage and squaring stage may be closely associated with integral processes.
Figure 4. Correlation coefficients between gene expression data sets form three biological duplicates.
To study the molecular mechanisms of cotton fruit branch initiation and flower development, we analysed the stage-specific DEGs at three stages in each type of cotton. There were 187, 3,825, and 845 DEGs in the seedling stage, pre-squaring stages and squaring stages of G. barbadense between nulliplex and normal branches, respectively (Fig. 5A). In G. hirsutum, there were 66, 353, and 2,047 specific DEGs in the three stages between the branching types, respectively (Fig. 5B).
Figure 5.

Venn diagram analyses of stage-specific expression genes in G.barbadense (A) G. hirsutum (B).
DEGs specifically expressed in the pre-squaring stage
Flowers and floral buds are forming in the pre-squaring stage; genes specifically expressed during this stage may play a key role in the early regulation of cotton flower and floral bud formation. Gene ontology (GO) enrichment analyses of DEGs specifically expressed during the pre-squaring stage revealed a major enrichment of oxidoreductase activity in both G. barbadense and G. hirsutum (see supplemental Tables S2 and S3; correction p < 0.05). In addition, DEGs were primarily enriched for transmembrane transporter activity and hydrolase activity and acting on glycosyl bonds in G. barbadense and ion binding in G. hirsutum (Fig. 6A, Tables 1 and 2). We further analysed the DEGs collectively expressed in the pre-squaring stage, and the results revealed 68 DEG candidates (See supplemental Fig. S1 and S2).
Figure 6. Enrichment of GO and Mapman functional categories for stage specific DEG in square initiating stage.
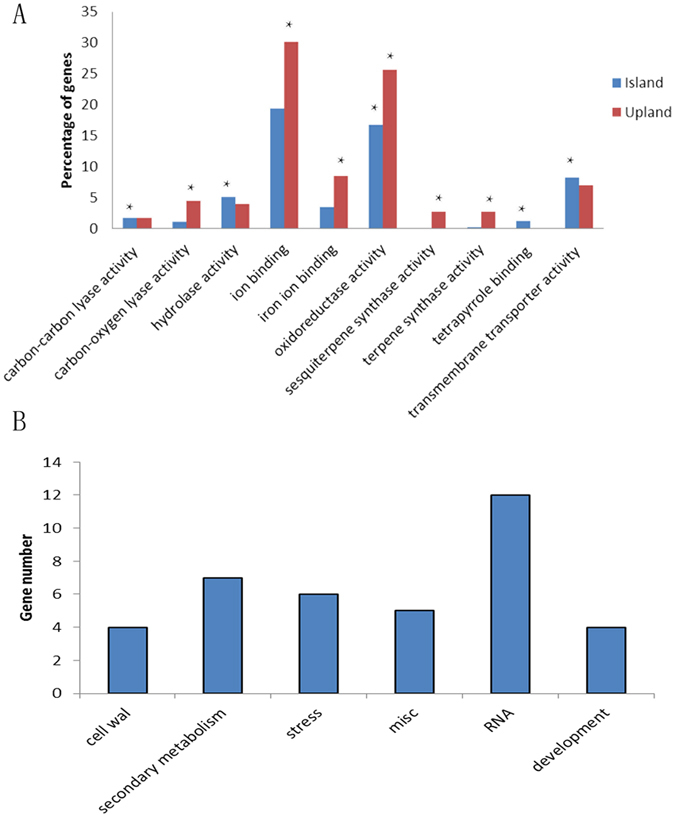
(A) Go enrichment of the DEG between nulliplex branch and normal branch in G. hirsutum and G.barbadense in square initiating stage. (B) Enrichment of functional categories for DEG both in G. hirsutum and G.barbadense. Asterisk indicates p < 0.05.
Table 1. Pathway annotation to KEGG of pre-squaring stage-specific DEG in G.barbadense.
| Pathway | Gene number(2243) | Pvalue | Qvalue | Pathway ID | |
|---|---|---|---|---|---|
| 1 | Photosynthesis-antenna proteins | 27 (1.2%) | 3.36E-20 | 9.69E-18 | ko00196 |
| 2 | Metabolic pathways | 768 (34.24%) | 7.32E-11 | 1.05E-08 | ko01100 |
| 3 | DNA replication | 49 (2.18%) | 4.77E-10 | 4.58E-08 | ko03030 |
| 4 | Amino sugar and nucleotide sugar metabolism | 77 (3.43%) | 1.83E-08 | 1.32E-06 | ko00520 |
| 5 | Starch and sucrose metabolism | 117 (5.22%) | 4.13E-08 | 2.38E-06 | ko00500 |
| 6 | Biosynthesis of secondary metabolites | 413 (18.41%) | 2.61E-07 | 1.25E-05 | ko01110 |
| 7 | Plant hormone signal transduction | 224 (9.99%) | 1.56E-06 | 6.40E-05 | ko04075 |
| 8 | Plant-pathogen interaction | 256 (11.41%) | 5.72E-06 | 2.06E-04 | ko04626 |
| 9 | Fructose and mannose metabolism | 119 (5.31%) | 7.83E-05 | 2.51E-03 | ko00051 |
| 10 | Alanine, aspartate and glutamate metabolism | 43 (1.92%) | 0.000155339 | 4.47E-03 | ko00250 |
| 11 | Tyrosine metabolism | 110 (4.9%) | 0.000375324 | 9.83E-03 | ko00350 |
| 12 | Cyanoamino acid metabolism | 32 (1.43%) | 0.000818251 | 1.96E-02 | ko00460 |
| 13 | Other glycan degradation | 35 (1.56%) | 0.001132179 | 2.19E-02 | ko00511 |
| 14 | Apoptosis | 81 (3.61%) | 0.001144075 | 2.19E-02 | ko04210 |
| 15 | Chloroalkane and chloroalkene degradation | 25 (1.11%) | 0.001226188 | 2.19E-02 | ko00625 |
| 16 | Cysteine and methionine metabolism | 35 (1.56%) | 0.001249362 | 2.19E-02 | ko00270 |
| 17 | Mismatch repair | 29 (1.29%) | 0.001290759 | 2.19E-02 | ko03430 |
| 18 | Carotenoid biosynthesis | 35 (1.56%) | 0.001746863 | 2.79E-02 | ko00906 |
| 19 | Pentose phosphate pathway | 35 (1.56%) | 0.002632152 | 3.08E-02 | ko00030 |
| 20 | NF-kappa B signaling pathway | 78 (3.48%) | 0.002674192 | 3.08E-02 | ko04064 |
| 21 | Drug metabolism-cytochrome P450 | 22 (0.98%) | 0.002785508 | 3.08E-02 | ko00982 |
| 22 | Chlorocyclohexane and chlorobenzene degradation | 8 (0.36%) | 0.002830841 | 3.08E-02 | ko00361 |
| 23 | Toll-like receptor signaling pathway | 82 (3.66%) | 0.002855844 | 3.08E-02 | ko04620 |
| 24 | Atrazine degradation | 5 (0.22%) | 0.002867008 | 3.08E-02 | ko00791 |
| 25 | Neurotrophin signaling pathway | 89 (3.97%) | 0.002874946 | 3.08E-02 | ko04722 |
| 26 | Lipopolysaccharide biosynthesis | 11 (0.49%) | 0.002949529 | 3.08E-02 | ko00540 |
| 27 | Microbial metabolism in diverse environments | 225 (10.03%) | 0.003075122 | 3.08E-02 | ko01120 |
| 28 | Pertussis | 84 (3.74%) | 0.00308791 | 3.08E-02 | ko05133 |
| 29 | Glutathione metabolism | 33 (1.47%) | 0.003273129 | 3.08E-02 | ko00480 |
| 30 | Neuroactive ligand-receptor interaction | 2 (0.09%) | 0.003312447 | 3.08E-02 | ko04080 |
| 31 | Leishmaniasis | 81 (3.61%) | 0.003312899 | 3.08E-02 | ko05140 |
| 32 | Pyrimidine metabolism | 166 (7.4%) | 0.004005977 | 3.61E-02 | ko00240 |
| 33 | Photosynthesis | 29 (1.29%) | 0.004622528 | 4.03E-02 | ko00195 |
| 34 | Chagas disease (American trypanosomiasis) | 83 (3.7%) | 0.005105472 | 4.32E-02 | ko05142 |
| 35 | Nitrogen metabolism | 26 (1.16%) | 0.005286608 | 4.35E-02 | ko00910 |
| 36 | D-Alanine metabolism | 3 (0.13%) | 0.005595 | 4.38E-02 | ko00473 |
| 37 | Benzoate degradation | 87 (3.88%) | 0.005774368 | 4.38E-02 | ko00362 |
| 38 | Insulin signaling pathway | 46 (2.05%) | 0.005838026 | 4.38E-02 | ko04910 |
| 39 | Ascorbate and aldarate metabolism | 30 (1.34%) | 0.005933246 | 4.38E-02 | ko00053 |
| 40 | Metabolism of xenobiotics by cytochrome P450 | 21 (0.94%) | 0.00619936 | 4.46E-02 | ko00980 |
| 41 | Measles | 82 (3.66%) | 0.006586086 | 4.58E-02 | ko05162 |
| 42 | Tuberculosis | 98 (4.37%) | 0.006679158 | 4.58E-02 | ko05152 |
Table 2. Pathway annotation to KEGG of pre-squaring stage-specific DEG in G. hirsutum.
| Pathway | Gene number (40) | Pvalue | Qvalue | Pathway ID | |
|---|---|---|---|---|---|
| 1 | Photosynthesis-antenna proteins | 2 (5%) | 0.001319016 | 0.09855362 | ko00196 |
| 2 | Isoquinoline alkaloid biosynthesis | 2 (5%) | 0.00323993 | 0.09855362 | ko00950 |
| 3 | Biosynthesis of secondary metabolites | 13 (32.5%) | 0.003478363 | 0.09855362 | ko01110 |
| 4 | Benzoxazinoid biosynthesis | 2 (5%) | 0.00914341 | 0.19429746 | ko00402 |
| 5 | Sesquiterpenoid and triterpenoid biosynthesis | 2 (5%) | 0.01309328 | 0.22258576 | ko00909 |
| 6 | Glycolysis/Gluconeogenesis | 3 (7.5%) | 0.01931683 | 0.27001173 | ko00010 |
| 7 | Chloroalkane and chloroalkene degradation | 2 (5%) | 0.02223626 | 0.27001173 | ko00625 |
| 8 | Fatty acid metabolism | 2 (5%) | 0.03182014 | 0.31785701 | ko00071 |
| 9 | Pentose and glucuronate interconversions | 3 (7.5%) | 0.03903229 | 0.31785701 | ko00040 |
| 10 | Retinol metabolism | 1 (2.5%) | 0.04813431 | 0.31785701 | ko00830 |
| 11 | Cell cycle-yeast | 3 (7.5%) | 0.04846708 | 0.31785701 | ko04111 |
| 12 | Glycerolipid metabolism | 2 (5%) | 0.04915537 | 0.31785701 | ko00561 |
| 13 | Two-component system | 2 (5%) | 0.04991741 | 0.31785701 | ko02020 |
| 14 | Tyrosine metabolism | 4 (10%) | 0.05235292 | 0.31785701 | ko00350 |
| 15 | Diterpenoid biosynthesis | 2 (5%) | 0.0561666 | 0.3182774 | ko00904 |
Then, we classified the genes that were specifically expressed during the pre-squaring stage using the Mapman program and found that these genes fell into six major groups: cell wall-related processes, secondary processes, stress response, miscellaneous, RNA processes, and development (Fig. 6B). More interestingly, 12 DEGs (approximately one-fifth of the tissue-specific genes) in the pre-squaring stage were mainly enriched in RNA processes, including key flower development genes such as short vegetative phase (SVP, CotAD_47245), late elongated hypocotyl (LHY, CotAD_32839), and pseudo-response regulator 5 (PRR5, CotAD_14148), and were related to ABI3/VP1 1 (RAV1, CotAD_22031), suggesting that the pre-squaring stage specifically expressed genes that tend to promote flower development. In addition, genes specifically expressed during bud and shoot apical meristem development included branched 1 (BRC1, CotAD_02759), breast cancer associated RING 1 (BARD1, CotAD_54816), and indole-3-acetic acid inducible 18 (IAA18, CotAD_00235), which implied that these genes control the growth and development of floral buds to form the cotton architecture (Table 3).
Table 3. The Mapman function annotation of DEG at square initiating stage both in G. hirsutum and G.barbadense.
| Gene id | BinName | Description (homology gene in Arabidopsis) |
|---|---|---|
| CotAD_58304 | PS.lightreaction.other electron carrier (ox/red).ferredoxin | (at1g02180) ferredoxin-related |
| CotAD_35943 | major CHO metabolism.degradation.starch.starch cleavage.beta amylase | (at4g15210) ATBETA-AMY |
| CotAD_16567 | minor CHO metabolism.trehalose.potential TPS/TPP | (at4g17770) TPS5 |
| CotAD_00103 | glycolysis.cytosolic branch.non-phosphorylating glyceraldehyde 3-phosphate dehydrogenase (NPGAP-DH) | (at2g24270)aldehyde dehydrogenase 11A3 (ALDH11A3) |
| CotAD_10000 | fermentation.ADH | (at1g77120) alcohol dehydrogenase 1 (ADH1) |
| CotAD_07955 | TCA/org transformation.carbonic anhydrases | (at3g01500) carbonic anhydrase 1 (CA1) |
| CotAD_48130 | mitochondrial electron transport/ATP synthesis.alternative oxidase | (at5g64210)alternative oxidase 2 (AOX2) |
| CotAD_71865 | cell wall.modification | (at4g38210) expansin A20 (EXPA20) |
| CotAD_72788 | cell wall.modification | (at4g17030) expansin-like B1 (EXLB1) |
| CotAD_27981 | cell wall.modification | (at3g29030) expansin A5 (EXPA5) |
| CotAD_16307 | cell wall.pectin*esterases.PME | (at1g02810) Plant invertase/pectin methylesterase inhibitor superfamily |
| CotAD_12986 | metal handling | (at5g27690) Heavy metal transport/detoxification superfamily protein |
| CotAD_07016 | secondary metabolism.isoprenoids.non-mevalonate pathway.geranylgeranyl pyrophosphate synthase | (at4g38460) geranylgeranyl reductase (GGR) |
| CotAD_24175 | secondary metabolism.phenylpropanoids.lignin biosynthesis.F5H | (at5g07990) TRANSPARENT TESTA 7 (TT7) |
| CotAD_56508 | secondary metabolism.sulfur-containing.glucosinolates | (at4g03070) AOP1 |
| CotAD_44293 | secondary metabolism.sulfur-containing.glucosinolates.synthesis.aliphatic.sulfotransferase | (at1g74090) desulfo-glucosinolate sulfotransferase 18 (SOT18) |
| CotAD_44293 | secondary metabolism.sulfur-containing.glucosinolates.synthesis.indole.indole-3-methyl-desulfoglucosinolate sulfotransferase | (at1g74090) desulfo-glucosinolate sulfotransferase 18 (SOT18) |
| CotAD_15307 | secondary metabolism.flavonoids.dihydroflavonols | (at5g24530) DOWNY MILDEW RESISTANT 6 (DMR6) |
| CotAD_24175 | secondary metabolism.flavonoids.dihydroflavonols.flavonoid 3”-monooxygenase | (at5g07990)TRANSPARENT TESTA 7 (TT7) |
| CotAD_49345 | hormone metabolism.abscisic acid.induced-regulated-responsive-activated | (at5g59220) highly ABA-induced PP2C gene 1 (HAI1) |
| CotAD_56508 | hormone metabolism.gibberelin.synthesis-degradation | (at4g03070) AOP1 |
| CotAD_12787 | tetrapyrrole synthesis.unspecified | (at1g17100) SOUL heme-binding family protein |
| CotAD_40314 | stress.biotic | (at3g12500) basic chitinase (HCHIB) |
| CotAD_73916 | stress.biotic.PR-proteins | (at1g61180) LRR and NB-ARC domains-containing disease resistance protein |
| CotAD_52530 | stress.biotic.PR-proteins.proteinase inhibitors.trypsin inhibitor | (at1g17860) Kunitz family trypsin and protease inhibitor protein |
| CotAD_22468 | stress.biotic.PR-proteins.proteinase inhibitors.trypsin inhibitor | (at1g17860) Kunitz family trypsin and protease inhibitor protein |
| CotAD_41277 | stress.abiotic.drought/salt | (at1g30360) early-responsive to dehydration 4 (ERD4) |
| CotAD_68015 | stress.abiotic.unspecified | (at5g66590) CAP superfamily protein |
| CotAD_24175 | misc.cytochrome P450 | (at5g07990)TRANSPARENT TESTA 7 (TT7) |
| CotAD_65900 | misc.short chain dehydrogenase/reductase (SDR) | (at4g11410) NAD(P)-binding Rossmann-fold superfamily protein |
| CotAD_44293 | misc.sulfotransferase | (at1g74090)desulfo-glucosinolate sulfotransferase 18 (SOT18) |
| CotAD_09729 | misc.GDSL-motif lipase | (at5g40990) GDSL lipase 1 (GLIP1) |
| CotAD_26978 | misc.GDSL-motif lipase | (at5g40990) GDSL lipase 1 (GLIP1) |
| CotAD_22031 | RNA.regulation of transcription.AP2/EREBP, APETALA2/Ethylene-responsive element binding protein family | (at1g13260) related to ABI3/VP1 1 (RAV1) |
| CotAD_21647 | RNA.regulation of transcription.CCAAT box binding factor family, HAP5 | (at5g43250) “nuclear factor Y, subunit C13” (NF-YC13) |
| CotAD_47245 | RNA.regulation of transcription.MADS box transcription factor family | (at2g22540) SHORT VEGETATIVE PHASE (SVP) |
| CotAD_32839 | RNA.regulation of transcription.MYB-related transcription factor family | (at1g01060) LATE ELONGATED HYPOCOTYL (LHY) |
| CotAD_30985 | RNA.regulation of transcription.MYB-related transcription factor family | (at1g18330) EARLY-PHYTOCHROME-RESPONSIVE1 (EPR1) |
| CotAD_02759 | RNA.regulation of transcription.TCP transcription factor family | (at3g18550) BRANCHED 1 (BRC1) |
| CotAD_00235 | RNA.regulation of transcription.Aux/IAA family | (at1g51950) indole-3-acetic acid inducible 18 (IAA18) |
| CotAD_14148 | RNA.regulation of transcription.Psudo ARR transcription factor family | (at5g24470) pseudo-response regulator 5 (PRR5) |
| CotAD_54816 | RNA.regulation of transcription.putative transcription regulator | (at1g04020)breast cancer associated RING 1 (BARD1) |
| CotAD_04662 | RNA.regulation of transcription.putative transcription regulator | (at2g29570)proliferating cell nuclear antigen 2 (PCNA2) |
| CotAD_55265 | RNA.regulation of transcription.SET-domain transcriptional regulator family | (at5g24330)ARABIDOPSIS TRITHORAX-RELATED PROTEIN 6 (ATXR6) |
| CotAD_00066 | RNA.regulation of transcription.unclassified | (at5g10770) Eukaryotic aspartyl protease family protein |
| CotAD_65745 | DNA.synthesis/chromatin structure | (at1g26840) origin recognition complex protein 6 (ORC6) |
| CotAD_49345 | protein.postranslational modification | (at5g59220) highly ABA-induced PP2C gene 1 (HAI1) |
| CotAD_00063 | protein.degradation.aspartate protease | (at5g10770) Eukaryotic aspartyl protease family protein |
| CotAD_64010 | protein.degradation.ubiquitin.E3.RING | (at4g01270) RING/U-box superfamily protein |
| CotAD_72041 | signalling.receptor kinases.leucine rich repeat VIII.VIII-2 | (at1g56130) Leucine-rich repeat transmembrane protein kinase |
| CotAD_58997 | signalling.phosphinositides.phosphatidylinositol-4-phosphate 5-kinase | (at1g71010) FORMS APLOID AND BINUCLEATE CELLS 1C (FAB1C) |
| CotAD_25160 | signalling.MAP kinases | (at3g51630) with no lysine (K) kinase 5 (WNK5) |
| CotAD_06239 | development.unspecified | (at3g59550)SYN3 |
| CotAD_07322 | development.unspecified | (at3g59550) SYN3 |
| CotAD_42150 | development.unspecified | (at4g19450) Major facilitator superfamily protein |
| CotAD_50063 | development.unspecified | (at1g69490) NAC-like, activated by AP3/PI (NAP) |
| CotAD_07846 | transport.amino acids | (at5g40780) LHT1 (lysine histidine transporter) |
| CotAD_40809 | transport.unspecified cations | (at5g64560)magnesium transporter 9 (MGT9) |
| CotAD_61138 | transport.misc | (at5g52450) MATE efflux family protein |
| CotAD_54187 | not assigned.no ontology.hydroxyproline rich proteins | (at4g01050)thylakoid rhodanese-like (TROL) |
| CotAD_43563 | not assigned.unknown | no original description |
| CotAD_18636 | not assigned.unknown | (at5g40460) unknown protein |
| CotAD_17275 | not assigned.unknown | (at3g48490) unknown protein |
| CotAD_29961 | not assigned.unknown | (at5g05250) unknown protein |
| CotAD_23730 | not assigned.unknown | (at5g56850) unknown protein |
| CotAD_26649 | not assigned.unknown | (at2g38640)unknown function (DUF567) |
| CotAD_34939 | not assigned.unknown | (at3g01680)unknown protein |
| CotAD_46956 | not assigned.unknown | (gnl|cdd|36433) no description available |
| CotAD_56138 | not assigned.unknown | (at3g56870) unknown protein |
| CotAD_48331 | not assigned.unknown | (gnl|cdd|36433) no description available |
| CotAD_20876 | not assigned.unknown | (at4g28310) unknown protein |
| CotAD_22049 | not assigned.unknown | (at3g18440) aluminum-activated malate transporter 9 (ALMT9) |
| CotAD_74231 | not assigned.unknown | (at4g28310) unknown protein |
| CotAD_40867 | not assigned.unknown | (at5g21940) unknown protein |
SVP, RAV1, and IAA18 were substantially downregulated and NF-YC13, PCNA2, BARD1, and ATXRP were upregulated during the pre-squaring stage compared to the other two stages in both G. barbadense and G. hirsutum. Moreover, EPR1, PPR5, and LHY were upregulated in G. barbadense but downregulated in G. hirsutum at the second stage. For BRC1 and CotAD_00066, the tendency to change was in the opposite direction of the latter three genes (Fig. 7, see supplemental Fig. S2).
Figure 7. Expression profiles for transcription factors in square initiating stage-specific DEGs both in upland and G.barbadense.

The bar showed log2 value of fold change.
DEGs of different developmental stages
The DEGs were analysed among the different developmental stages of each cotton variety, and the results showed that the number of DEGs in the squaring stage compared to the seedling stage and pre-squaring stage compared to seedlings in G. barbadense was much higher than in G. hirsutum in both nulliplex- and normal-branch cotton (Fig. 8A). The pre-squaring stage may be controlled by the differentiation of the fruit branch tissue, whereas the squaring stage is mainly controlled by the growth of the fruit branches in normal-branch cotton. The results revealed 395 DEGs between the pre-squaring stage and seedling stage and 435 DEGs between the squaring stage and seedling stage in normal-branch cotton (Fig. 8B). In nulliplex branches, there were 328 DEGs between the pre-squaring stage and the seedling stage and 535 DEGs between the squaring stage and the seedling stage.
Figure 8. The distribution of DEG in different development stages.

(A) Numbers of different expressed genes in each comparison. The numbers on column showed quantity of up-regulated (red) and down-regulated (blue) genes. (B,C) Venn diagram analyses of different stages in normal branch cotton and nulliplex branch cotton. (D) Enrichment of function categories for DEG. Gray, not significantly enriched. The bar mean the p value, p < 0.05 were significantly enriched.
These DEGs were enriched for functional categories, as determined using the Mapman software. The results showed that the DEGs of the nulliplex branches (Gb1np-vs-Gb2np, Gb1np-vs-Gb3np, Gh1np-vs-Gh2np, and Gh1np-vs-Gh3np) were substantially enriched in signalling categories, and the DEGs of the normal branches (Gb1WT-vs-Gb2WT, Gb1WT-vs-Gb3WT, Gh1WT-vs-Gh2WT, and Gh1WT-vs-Gh3WT) were mainly enriched in transport categories (Fig. 8D). In addition, the DEGs of the normal branches in the floral bud’s forming stage and seedling stage (Gb1WT-vs-Gb3WT and Gh1WT-vs-Gh3WT) were considerably enriched in development categories. These results suggest that nulliplex-branch and normal-branch cotton may have different developmental patterns.
Sequencing of small RNA libraries
Twelve small RNA samples (each sample was the same as above with three biological repeats) yielded a total of 422,920,856 reads and 175,305,470 unique reads. There were more than 11 million reads and more than 4 million unique reads for each library. The reads of each library were mapped back to the cotton genome and accounted for 75% to 88% of the total reads, representing 67% to 81% of their unique read counterparts (See supplemental Table S4). In both types of cotton, rRNA in the squaring stage were substantially different between nulliplex and normal branches, showing 0.56% unique reads in normal branches and 1.36% unique reads in nulliplex branches for G. barbadense (miGb3WT vs. miGb3np) and 0.38% unique reads in the normal branches and 0.85% unique reads in nulliplex branches for G. hirsutum (miGh3WT vs. miGh3np). However, the other RNA families had similar proportions, including miRNA (0.5–0.7% for unique reads), snRNA (0.02–0.05% for unique reads), snoRNA (approximately 0.02% for unique reads), and tRNA (approximately 0.15% for unique reads) (See supplemental Table S4). For all sRNA libraries, the major distribution of read lengths was between 21 and 24 nt.
Identification of known conserved miRNA families
A total of 712 known conserved miRNAs were identified in 12 samples (three biological replications). Of these, some miRNAs were highly and stably expressed in all samples, such as miR157a-5p, miR166a-3p, and miR3954 (See supplemental data set S7 online). However, other miRNA counts diverged sharply. For example, miRNA167h had more than 35 k reads in miGb3WT and miGb2np and no reads in miGb3np and miGh2np (See supplemental data set S7 online).
Differentially expressed miRNA (DEM) in three stages of cotton
To study the regulation of miRNA in development of floral buds and fruit branch in cotton, we further analysed conserved DEMs using similar strategies as that used for RNA-seq. There were 17 (9 downregulated and 8 upregulated), 31 (17 downregulated and 14 upregulated), and 43 (28 downregulated and 15 upregulated) DEMs in the seedling stage, pre-squaring stage, and squaring stage of G. barbadense between nulliplex and normal branches, respectively (Fig. 9A, See supplemental data set 8–10 online). In G. hirsutum, there were 24 (11 downregulated and 13 upregulated), 28 (18 downregulated and 10 upregulated), and 43 (31 downregulated and 12 upregulated) DEMs, respectively (Fig. 9A, See supplemental data set S11–S13 online).
Figure 9. The statistics of differentially expressed miRNA.
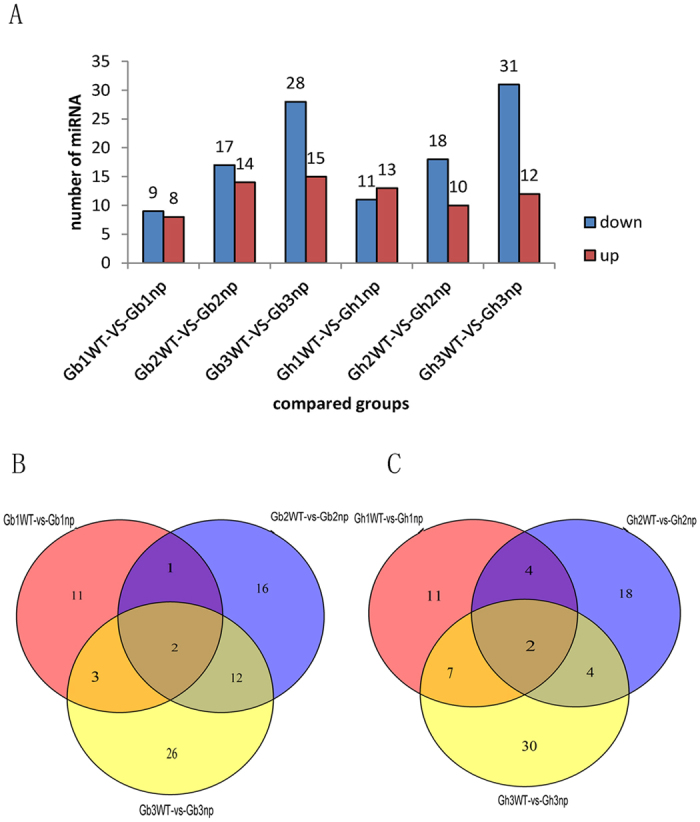
(A) Numbers of different expressed miRNA in each comparison. The numbers on column showed quantity of up-regulated (red) and down-regulated (blue) genes. Venn diagram analyses of stage-specific expression miRNAs in G.barbadense (B) G. hirsutum (C).
Among these DEMs, there were 11, 16, and 26 stage-specific DEMs in the G. barbadense seedling stage, pre-squaring stage, and squaring stage, respectively (Fig. 9B). In G. hirsutum, we identified 11, 18, and 30 stage-specific DEMs in the seedling stage, pre-squaring stage, and squaring stage, respectively (Fig. 9C). In G. barbadense, the uniquely expressed miRNAs at the pre-squaring stage, which included miR156c-3p, miR156i-3p, and miR171c of the miR156 and miR171 family may be related to floral transition (See supplemental Table S5)27. In G. hirsutum, the specifically expressed miRNAs at the pre-squaring stage including miR166c, a member of the miR166 family, were also involved in floral organ polarity and shoot apical meristem formation (See supplemental Table S6)27,35. In addition, at the pre-squaring stage of G. barbadense, the DEMs included miR167 and miR166 family members and the stage-specific DEMs noted above (See supplemental data set S9 online).
Novel miRNAs were identified and the results showed that of these 3,350 novel miRNAs, 13, 8, and 13 were DEMs in the seedling stage, pre-squaring stage, and squaring stage of G. barbadense and 11, 9, and 13 were DEMs in the three stages of G. hirsutum, respectively (See supplemental Fig. S3A and S3B).
DEM target identification at three stages
The DEM target pairs above were identified from G. hirsutum genome sequences based on negative regulation between miRNA and their targets by combining the results of DEG analysis with RNA-seq. Among the three stages, 80 targets of the specifically expressed miRNAs were screened out. In the pre-squaring stage of G. barbadense, 11 DEMs targeting 32 DEGs were identified, including CotAD_35367 (MADS box protein, a homologue of sepallata3 (SEP3), which is involved in flower development)36 and CotAD_75643 (a homologue of HB8, which regulates post-embryonic meristem initiation and is also bound by SVP in vegetative tissue in Arabidopsis) (Table 4)37,38.
Table 4. Identified targets for known miRNAs in cotton.
| miR-name | log2Ratio | TargetID | log2Ratio | type | Target annotation | |
|---|---|---|---|---|---|---|
| Gb1np/Gb1WT | miR9484 | 18.30964 | CotAD_21580 | −1.13888 | up-down | unkown |
| CotAD_34848 | −2.5911 | up-down | LRR family protein | |||
| miR9674a-3p | −19.9987 | CotAD_56347 | 1.333495 | down-up | EMBRYO DEFECTIVE 976 (EMB976) | |
| Gb2np/Gb2WT | miR5291a | −16.6434 | CotAD_63772 | 1.518275 | down-up | Glycosyl hydrolase family protein |
| miR5537 | 19.1318 | CotAD_07870 | −1.05164 | up-down | chloride channel A (CLC-A) | |
| CotAD_32730 | −1.01925 | up-down | Tetratricopeptide repeat (TPR)-like superfamily protein | |||
| CotAD_72047 | −1.34026 | up-down | HXXXD-type acyl-transferase family protein | |||
| CotAD_73186 | −2.15217 | up-down | HXXXD-type acyl-transferase family protein | |||
| miR5641 | −18.4987 | CotAD_30697 | 3.752667 | down-up | glycosyl hydrolase 9B18 (GH9B18) | |
| CotAD_35367 | 2.500502 | down-up | SEPALLATA3 (SEP3) | |||
| miR4393b | −19.5048 | CotAD_41814 | 1.089024 | down-up | cellulose-synthase like D2 (CSLD2) | |
| miR1077-5p | 22.57754 | CotAD_07688 | −1.3105 | up-down | unkown | |
| CotAD_35213 | −1.28959 | up-down | unkown | |||
| CotAD_47342 | −1.17882 | up-down | unkown | |||
| CotAD_48531 | −1.29591 | up-down | RRNA intron-encoded homing endonuclease | |||
| CotAD_53140 | −1.34414 | up-down | unkown | |||
| CotAD_70473 | −1.13899 | up-down | unkown | |||
| miR156i-3p | 17.38975 | CotAD_31099 | −2.04149 | up-down | unkown | |
| CotAD_54581 | −2.46349 | up-down | galacturonosyltransferase 12 (GAUT12) | |||
| miR166i | 21.68899 | CotAD_39504 | −2.67928 | up-down | unkown | |
| CotAD_75643 | −1.8728 | up-down | homeobox gene 8 (HB-8) | |||
| miR9484 | 21.06244 | CotAD_01114 | −1.16244 | up-down | NB-ARC domain-containing disease resistance protein | |
| CotAD_16057 | −1.32682 | up-down | unkown | |||
| CotAD_21416 | −1.28186 | up-down | Late embryogenesis abundant protein, LEA-5 | |||
| CotAD_25778 | −1.15489 | up-down | pleiotropic drug resistance 5 (PDR5) | |||
| CotAD_26434 | −1.74762 | up-down | nitrate transporter 1:2 (NRT1:2) | |||
| CotAD_34848 | −2.29922 | up-down | LRR family protein | |||
| CotAD_40632 | −1.10811 | up-down | allantoate amidohydrolase (AAH) | |||
| CotAD_44160 | −1.96883 | up-down | receptor like protein 35 (RLP35) | |||
| CotAD_47999 | −1.29278 | up-down | pleiotropic drug resistance 5 (PDR5) | |||
| CotAD_68929 | −2.75838 | up-down | unkown | |||
| miR862-3p | −16.5445 | CotAD_40927 | 1.124491 | down-up | unkown | |
| miR482d | 17.53683 | CotAD_34619 | −5.55857 | up-down | RECOGNITION OF PERONOSPORA PARASITICA 8 (RPP8) | |
| miR5298d | −16.4595 | CotAD_45973 | 2.497785 | down-up | unkown | |
| CotAD_67595 | 4.281629 | down-up | Cysteine/Histidine-rich C1 domain family protein | |||
| Gb3np/Gb3WT | miR7502 | −4.33643 | CotAD_35035 | 1.416654 | down-up | FRAGILE FIBER 1 (FRA1) |
| miR482b | 19.36951 | CotAD_59175 | −1.5279 | up-down | NB-ARC domain-containing disease resistance protein | |
| miR167a-5p | 25.12849 | CotAD_21735 | −1.82028 | up-down | GDSL-like Lipase/Acylhydrolase superfamily protein | |
| CotAD_24173 | −2.20715 | up-down | Flavonoid 3′-monooxygenase | |||
| miR829-5p | −17.6664 | CotAD_20201 | 3.869985 | down-up | Disease resistance protein (TIR-NBS-LRR class) family | |
| miR5641 | −18.9006 | CotAD_30080 | 1.385631 | down-up | SABRE (SAB) | |
| CotAD_37128 | 1.089237 | down-up | unkown | |||
| CotAD_38231 | 1.789472 | down-up | PARVUS (PARVUS) | |||
| CotAD_65515 | 1.039363 | down-up | P-loop containing nucleoside triphosphate hydrolases superfamily protein | |||
| miR1159.1 | 21.12174 | CotAD_28336 | −14.1677 | up-down | unkown | |
| miR157a-5p | −3.41905 | CotAD_19197 | 5.586874 | down-up | O-methyltransferase family protein | |
| Gh1np/Gh1WT | miR9484 | −18.1864 | CotAD_52129 | 1.875941 | down-up | probable E3 ubiquitin-protein ligase LOG2-like |
| Gh3np/Gh3WT | miR5537 | 18.61224 | CotAD_28668 | −3.57269 | up-down | wall associated kinase-like 1 (WAKL1) |
| miR6103-3p | −20.0962 | CotAD_05723 | 1.008805 | down-up | cysteine-rich RLK (RECEPTOR-like protein kinase) 25 (CRK25) | |
| CotAD_15382 | 1.28365 | down-up | guanylyl cyclase 1 (GC1) | |||
| CotAD_57423 | 3.435504 | down-up | cysteine-rich RLK (RECEPTOR-like protein kinase) 26 (CRK26) | |||
| CotAD_57424 | 1.427739 | down-up | cysteine-rich RLK (RECEPTOR-like protein kinase) 10 (CRK10) | |||
| CotAD_75962 | 1.142944 | down-up | phragmoplast orienting kinesin 2 (POK2) | |||
| miR2950 | −3.81938 | CotAD_23640 | 1.161408 | down-up | hydroxyproline-rich glycoprotein family protei | |
| miR5021 | −17.16 | CotAD_02433 | 2.113803 | down-up | MYB | |
| CotAD_05722 | 1.87195 | down-up | cysteine-rich RLK (RECEPTOR-like protein kinase) 25 (CRK25) | |||
| CotAD_08430 | 1.194253 | down-up | Vacuolar import/degradation, Vid27-related protein | |||
| CotAD_08496 | 1.074783 | down-up | DREB1C | |||
| CotAD_13093 | 1.070816 | down-up | lectin protein kinase family protein | |||
| CotAD_13340 | 1.687647 | down-up | AAA-ATPase 1 (AATP1) | |||
| CotAD_19878 | 1.284197 | down-up | senescence-related gene 3 (SRG3) | |||
| CotAD_28123 | 1.288063 | down-up | early nodulin-like protein 1 (ENODL1) | |||
| CotAD_31169 | 2.538615 | down-up | sucrose-proton symporter 2 (SUC2) | |||
| CotAD_35030 | 1.129006 | down-up | WRKY transcription factor 26 | |||
| CotAD_38978 | 2.753387 | down-up | dehydrin 2 | |||
| CotAD_68811 | 2.057678 | down-up | plastid developmental protein DAG | |||
| CotAD_72901 | 1.577081 | down-up | FERONIA (FER) | |||
| miR156a-5p | −20.6349 | CotAD_28172 | 1.611284 | down-up | PAS domain-containing protein tyrosine kinase family protein | |
| miR414 | −18.2483 | CotAD_05693 | 2.220604 | down-up | unkown | |
| CotAD_08496 | 1.074783 | down-up | DREB1C | |||
| CotAD_20312 | 1.280126 | down-up | plant intracellular ras group-related LRR 2 | |||
| CotAD_35408 | 1.248969 | down-up | global transcription factor group E4 (GTE4) | |||
| CotAD_39897 | 4.054389 | down-up | ABA REPRESSOR1 (ABR1) | |||
| CotAD_40639 | 2.371314 | down-up | Calcium-binding EF-hand family protein | |||
| CotAD_41567 | 1.228057 | down-up | NBS-LRR resistance protein RGH2 | |||
| CotAD_42550 | 1.495769 | down-up | unkown | |||
| CotAD_53959 | 3.793863 | down-up | CYCLIN D1;1 (CYCD1;1) | |||
| CotAD_57315 | 4.286361 | down-up | unkown | |||
| CotAD_62266 | 2.179486 | down-up | receptor like protein 13 (RLP13) | |||
| CotAD_63349 | 4.788476 | down-up | formin homology5 (Fh5) |
In the seedling stage, there were two DEMs with three target DEGs in G. barbadense and one DEM and one target in G. hirsutum. At the squaring stage, 7 DEMs were found in 11 DEG target genes in G. barbadense, and in G. hirsutum, 6 DEMs were identified with a predicted 33 target genes (Table 4).
RNA-seq expression validation by qPCR
We used qPCR to confirm the reliability of the RNA-seq data. The results showed that among nine selected DEGs of the pre-squaring stages, approximately 90% were consistent with RNA-seq data (See supplemental Fig. S5).
Discussion
Plant architecture is fundamental to agricultural productivity and the artificial selection of desired growth habits. The regulation of cotton architecture has enormous potential applications for high-density planting, mechanised harvesting, decreasing the cost of cultivation and improving yield. In the present study, to investigate the intricate molecular mechanisms of fruit branch and flower development in cotton plants, RNA-seq technology was utilized to examine the global gene expression profile of shoot apexes by generating both mRNA and miRNA libraries from the shoot apex of four cotton varieties (lines), including two with nulliplex branches and two with normal branches in three developmental stages with the aim of subtracting the unrelated differences in genotype, applying the newly published allotetraploid genome of G. hirsutum as a reference genome, which provided us with more complete and informative results for credible data mining33,34.
While it would have been highly desirable to start with genetic material that differs only in the trait of interest (e.g., near-isogenic lines), it would have taken many years to establish such lines given that G. hirsutum and G. barbadense are allotetraploid.
In this study although the nulliplex branches and normal branches lines of G. hirsutum and G. barbadense did not share parentage with each other, the differentially expressed genes observed should include the result of both the np mutation and unrelated differences in genotype in each comparison of G. hirsutum or G. barbadense. In order to acquire the result of the np mutation and subtract the unrelated differences in genotype, our experimental design with three different developmental stages of the two cotton fruit branch types in G. hirsutum and G. barbadense can help to find the differentially expressed genes related to the development of fruit branch. Firstly, we screened the common DEGs among three stages (the seedling, pre-squaring and squaring stage) in two kinds of branch type (nulliplex-branch and normal branch) of G. barbadense and G. hirsutum lines, respectively. Secondly, the common DEGs between the above two groups of G. barbadense and G. hirsutum were selected and can be considered as candidate DEGs for the np mutation. Finally, in this way we have in reality obtained interesting results and selected 68 genes associated with development of the np mutation in the pre-squaring stage. Among them, we have acquired the key genes related to development of branch (Table 3), such as Branched1(GhBRC1), IAA 18 etc., which play central roles in the development of branch in plants13,39,40,41,42,43,44. The results showed that the nulliplex branch is a useful trait and model for the study of cotton architecture.
The results indicated that the number of DEGs was significantly higher in the second stage than in the third stage of development in G. barbadense, but the DEG numbers of two stages in G. hirsutum showed the opposite results. The correlation coefficient between Gh2np and Gh2WT was 0.93, significantly higher than the 0.89 correlation found between Gh3np and Gh3WT. The correlation coefficient between Gb2np and Gb2WT was 0.83, lower than the correlation coefficient of 0.84 between Gb3np and Gb3WT (Fig. 4).
Mapman software analysis indicated that each developmental stage of the two cotton plant types was enriched with DEGs. The four samples of the seedling stage were still in the early period of vegetative growth, and many biochemical pathways were not initiated; consequently there was a relatively small number of DEGs. The pre-squaring stage is a key stage that may control the transition from vegetative growth to reproductive growth, and both types of growth may occur simultaneously during the squaring stage. DEGs between nulliplex branches and normal branches were mainly enriched in RNA, protein, secondary metabolism, stress, and signalling processes in the pre-squaring stage and squaring stage (Fig. 3A). The results showed remarkable differences between nulliplex-branch and normal-branch cotton in the development of the shoot apex, especially in the RNA category, which contains a large number of transcription factors. Fruiting branch development and differentiation from the apical or lateral meristem may be regulated by a series of transcription factors in cotton. However, there were different proportions of DEGs in some functional categories between G. barbadense and G. hirsutum (Fig. 3B). In the hormone metabolism category, the proportion of DEGs was significantly higher in G. barbadense than in G. hirsutum. This difference indicates that the developmental process may be more closely regulated by plant hormones in G. barbadense than in G. hirsutum in these four varieties (lines).
Regarding the different stages, the DEGs of the normal-branch cotton plants in the squaring stage and seedling stage (Gb1WT-vs-Gb3WT and Gh1WT-vs-Gh3WT) were remarkably enriched in development categories. In normal-branch cotton, the fruit branch grows during the squaring stage (there are no fruit branches in nulliplex-branch cotton), which is in agreement with the RNA-seq results. The second stage is a key developmental stage that regulates the transition from vegetative growth to reproductive growth and fruit branch formation. We further examined specific-stage DEGs in the second stage, and 68 candidate genes were identified.
The screened DEGs included 12 transcription factors (Table 3). The gene CotAD_02759 (GhBRC1), a homologue of Arabidopsis branched1 (AtBRC1), was downregulated in nulliplex-branch G. barbadense and upregulated in nulliplex-branch G. hirsutum at the pre-squaring stage. BRC1 downregulation leads to branch outgrowth in Arabidopsis39. Based on mutants and expression analysis in Arabidopsis, BRC1 is likely downstream of the more axillary growth pathway and is required for auxin-induced apical dominance13,39,40. The cotton GhBRC1 gene may have the same function as AtBRC1, and the normal branch (fruit branch) is analogous to branch outgrowth in Arabidopsis to form axillary meristems. Cotton GhBRC1 was upregulated in Gh2np (nulliplex-branch, G. hirsutum), which may be repressed during fruit branch development. However, it remains unclear why GhBRC1 was downregulated in Gb2np, a nulliplex-branch (no fruit branch) variety of G. barbadense (Fig. 6). The homologue (CotAD_00235, GhIAA18) of the auxin-related transcription factor IAA18 (indole-3-acetic acid inducible 18) in Arabidopsis, which causes aberrant cotyledon placement in embryos apical patterning, has been shown to be downregulated in nulliplex-branch cotton of both G. barbadense and G. hirsutum41. This finding suggests that another regulated pathway of lateral development may play a role.
The axillary meristem differentiates into fruit branches to form floral buds. Based on our results, the homologous genes may also regulate the differentiation of apical meristems in addition to BRC1 modulation of florigen activity in the floral buds of the axillary meristems in Arabidopsis13. The gene CotAD_54816 (GhBARD1) is homologous with BARD1 in Arabidopsis (also named ROW1) and is a repressor of Wuschel145. BARD1 regulates shoot apical meristem organisation and maintenance by limiting WUS expression to the organising centre. Recently, BARD1 was shown to be essential for QC maintenance and stem cell niche development through the repression of WOX5 in the proximal meristem45,46. During the pre-squaring stage, GhBARD1 was substantially upregulated in Gb2np and Gh2np, which may repress downstream gene expression or the shoot apical meristem formation of floral buds, causing nulliplex-branch cotton.
In addition, GhBRC1 and GhBARD1 are involved in floral meristem determinacy. BRC1 interacts with Flowering Locus T to repress the floral transition, and BARD1 can limit WUS expression, which forms a negative feedback loop with agamous (AG) and is involved in floral determinacy and the specialisation of reproductive floral organs13,47,48,49. These results suggest that the formation of nulliplex-branch cotton is associated with lateral axillary meristem development and floral formations, similar to other flowering time genes.
Interestingly, in addition to GhBRC1 and GhBARD1, which may be involved in the flowering time network, there are four other Arabidopsis homologous genes among 12 transcription factors that may be related to the flowering time network, including CotAD_47245 (GhSVP, a homologue of short vegetative phase [SVP]), CotAD_32839 (GhLHY, a homologue of late elongated hypocotyl [LHY]), CotAD_30985 (GhEPR1, a homologue of early-phytochrome-responsive1 [EPR1), and CotAD_14148 (GhPPR5, a homologue of pseudo-response regulator 5 [PRR5]) (Table 3, Fig. 7).
In Arabidopsis, PRR5 can bind to LHY promoters and recruit transcriptional corepressors of the Groucho/tup1 corepressor family to repress LHY transcription in circadian clocks50,51. EPR1 is regulated by both phytochrome A and phytochrome B and is a component of a slave circadian oscillator in Arabidopsis52. To the best of our knowledge, the circadian clock gene and photoperiodic response can induce early flowering in Arabidopsis53,54,55. Based on our results, GhPRR5, GhLHY, and GhEPR1 genes were upregulated in Gb2np and downregulated in Gh2np (Fig. 7).The same expression trend implied that GhPRR5 might also bind to the GhLHY promoter and regulate the circadian clock of flowering time in cotton.
In addition, SVP is a negative regulator of the floral transition, which forms a flowering repressor complex together with FLC in Arabidopsis56,57,58,59. In our study, the SVP homologue GhSVP was downregulated in nulliplex-branch plants in both G. barbadense and G. hirsutum, which suggests that GhSVP may promote floral transition in nulliplex-branch cotton compared to normal-branch cotton in our four varieties (lines). Furthermore, the overexpression of RAV1, a homologue of cotton CotAD_22031 (GhRAV1) in Arabidopsis, slows the development of lateral roots and rosette leaves60,61,62. In addition, low expression causes an early flowering phenotype, implying that RAV1 may function as a negative regulator of growth and development60. In the present study, the GhRAV1 gene was downregulated in nulliplex-branch cotton at the pre-squaring stage in both G. barbadense and G. hirsutum, which implies that GhRAV1 may have accelerated the flowering speed of cotton varieties Xinhai16 and 3798.
Although there is no report on trehalose-6-phosphate synthase 5 (TPS5: the homology of cotton gene CotAD_16567, GhTPS5), TPS1 (a homology family number with TPS5) is essential for normal vegetative growth and transition to flowering in Arabidopsis63.
Similarity analyses of the above differentially expressed genes showed that many DEGs of the fruit branch and floral transition may be predominantly expressed in cotton similar to Arabidopsis, Theobroma cacao, Populus euphratica, and other species. However, most of the related evidence has been obtained from studies on Arabidopsis.
In the past decade, numerous studies have demonstrated that different miRNA families play important roles in regulating plant architecture and branching patterns42,64,65, although a high level of conservation of miRNAs and their regulatory pathways/target genes has been demonstrated in many species. In the present study, DEMs were analysed between nulliplex-branch and normal-branch plants. In the pre-squaring stage, stage-specific DEMs such as miR156 and miR171 family members were found in G. barbadense (See supplemental table S5) and miR166 was found in G. hirsutum (See supplemental table S6). The miR156 miRNA family promotes juvenile-to-adult phase transition and apical meristem formation64,66, and miR171 negatively regulates shoot branching35,67. In the pre-squaring stage, the miR166 family found in G. hirsutum regulates the shoot apical meristem and lateral organ formation in Arabidopsis66,68. In addition, these miRNA families have been confirmed to be closely related to flowering time26,60, in agreement with our RNA-seq results.
The majority of the miRNA families are expressed during several plant growth periods or developmental processes. The non-stage-specific DEMs may also be involved in branching and flowering processes. More members of mi156, miR166, and miR167 were differentially expressed in three developmental stages (See supplemental data set S8 to S13 online). Nevertheless, the target of the DEMs was predicted, and we could not identify the genes related to branching and flowering (Table 4). These results imply that there were a number of genes involved in branching and flowering that had not been previously identified, but the molecular mechanism of regulation remains unclear. This conclusion suggests that nulliplex branching may be related to a flower time-related candidate gene5.
In nulliplex-branch cotton, the floral bud directly arises from the leaf axils of the main shoot, whereas in normal-branch the floral bud arises from the node of the fruit branch. With the fruit branch growing on leaf axils, the floral bud is produced at the same time. Based on the above results, genes controlling the formation of nulliplex-branch cotton may also be involved in floral induction. Interestingly, in the four varieties (lines) used for sequencing in this study, the squaring stage of Xinhai16 (G. barbadense L., nulliplex branch) was significantly earlier than that of Hai1 (G. barbadense L., normal branch) and line 3,798 (G. hirsutum L., nulliplex-branch) was significantly earlier than Huazhong 94-3130 (G. hirsutum L., normal branch). The earlier squaring stage (flowering time) in the nulliplex-branch compared to the normal-branch cotton in both G. barbadense and G. hirsutum in the four varieties (lines) may coincide with branching control genes involved in floral induction.
Our results showed that multiple pathways, including genes related to fruit branch development, e.g., GhBRC1, may control nulliplex branching. At the same time, the flowering time genes may influence fruit branch growth. The different tendencies of DEGs in the island and G. hirsutum imply that multiple pathways may regulate branching. The KEGG pathway of the pre-squaring stage DEGs showed that the DEGs of the island and G. hirsutum were both enriched in circadian rhythm genes, which may be associated with flowering (See supplemental Tables S5 and S6). In the circadian rhythm pathway, all enriched DEGs were downregulated (Fig. 10A). However, DEGs such as GhLHY have different expression patterns in G. barbadense compared to G. hirsutum. Other DEGs homologous to GhLHY showed the same expression trend as GhLHY in G. hirsutum, such as CotAD_17426. In addition, other genes in the circadian rhythm pathway have the same condition, such as CCA1, PIF3, and APR7 (Fig. 10B). Furthermore, the plant hormone signal transduction pathway was substantially enriched in G. barbadense but not in G. hirsutum (See supplemental Tables S5–S7). This finding supports the hypothesis that other regulation pathways may be involved in G. barbadense, and the results showed that many DEGs were related to multi-hormone signal transduction pathways (Fig. 10C, See supplemental Table S7).
Figure 10. Schematic diagram in KEGG pathway of DEGs in square initiating stage by RNA-Seq data analysis and the model of bud outgrowth.

(A) and (B) DEGs enriched in circadian rhythm pathway of G. hirsutum and G.barbadense, respectively. (C) DEGs enriched of plant hormone signal transduction pathway in G.barbadense. Up-regulated genes are colored red, down-regulated genes are colored green. (D) Proposed model for cotton fruit branch.
In conclusion, GhBRC1 may play a key role in floral bud formation (flowering) and fruit branch development in cotton, which is similar to BRC1′s central role in the architecture of Arabidopsis42,43. GhBRC1 may also be associated with plant hormones and the flowering network, including the circadian rhythm genes that control fruit branching and floral transition, and thus is involved in elaborate regulatory mechanisms and networks.
Conclusions
Overall, our results imply that the molecular pathways similar to those in Arabidopsis may control fruit branching in cotton. Multiple regulated pathways may affect the development of floral buds on leaf axils in G. barbadense and G. hirsutum. The development of the fruit branch is closely related to the induction of flowering. Our results provide a basis for plant type research in crops and an approach for elucidating the molecular mechanism of the development of the nulliplex branch and the normal branch in the flowering and fruit branch forming stage for improving plant architecture and yield in cotton and other plants.
Materials and Methods
Plant material
The cotton varieties Xinhai16 (G. barbadense L., nulliplex-branch), Hai1 (G. barbadense L., normal branch), line 3798 (G. hirsutum L., nulliplex-branch), and line Huazhong 94-3130 (G. hirsutum L., normal branch, obtained from the Cotton Institute of the Chinese Academy of Agricultural Sciences of China) were grown in a natural field environment (Kaifeng, China) in 2014, all the materials are stable and pure lines or varieties which have been self-fertilized for at least eight generations before we used in the studies. Shoot apices (approximately 10 mm) from each individual were collected at three sampling stage and stored immediately in liquid nitrogen after washing with water (See supplemental Fig. 3). The three stages used for constructing RNA-seq libraries included the seedling stage with two leaves of Xinhai16 (Gb1np), Hai1 (Gb1WT), 3798 (Gh1np), and Huazhong 94-3130 (Gh1WT). The pre-squaring stage with five and six leaves for Xinhai16 (Gb2np), seven and eight leaves for Hai1 (Gb2WT), five leaves for 3798 (Gh2np), and six and seven leaves for Huazhong94-3130 (Gh2WT), respectively, all without any visible triangular floral bud, just in the forming stage of floral bud (flowering) and fruiting branch in the pre-squaring stage. The squaring stage with nine leaves for Xinhai16 (Gb3np), nine and ten leaves for Hai1 (Gb3WT), and eight to ten leaves for 3798 (Gh3np) and Huazhong94-3130 (Gh3WT), respectively, all with visible triangular floral buds in the field. These leaf numbers above refer to visual, expanded true leaves in cotton field69. The nodes of first fruiting branch we investigated for each line including Xinhai16, Hai1, 3798 and Huazhong 94-3130 were 6-7,8-9,5-6,7-8 true expanded leaves, respectively, in virtue of the different lines (varieties). The same RNA samples were used for sRNA-seq library construction, and we labelled the libraries with the prefix ‘mi’ as in ‘miGb1np’.
Sample preparation and sequencing library construction
The mRNA and small RNA libraries were constructed as suggested by Illumina and were sequenced on a HiSeq 2000 platform. Briefly, total RNA was isolated using a Plant RNA EASYspin Plus Kit (Aidlab, Peking, China) according to the manufacturer’s instructions. Then, the total RNA of each sample was sent to BGI (Shenzhen, China) for library construction and sequencing. Finally, an Agilent 2100 Bioanalyzer and ABI Step One Plus Real-Time PCR System was used for the quantification and qualification of the sample library, after which the library was sequenced using an Illumina HiSeq™ 2000 platform.
Alignment of RNA-seq reads to the reference genome
Reference genome sequences and annotations were downloaded from the Institute of cotton Research of CAAS (http://cgp.genomics.org.cn)33. Reads were aligned to the genome with no more than five mismatches using the SOAP aligner/SOAP2 software after low-quality reads (reads with unknown sequences ‘N’), adaptor sequence fragments, and empty reads were removed70. After passing the QC process of the alignment, the results were used for further analysis.
Differential gene expression analysis
RPKM was used to obtain the relative levels of expression71,72. Differential expression analysis was performed using R packages of DESeq for comparisons among samples with three biological replicates71,73. We used a false discovery rate of <0.001 and an absolute value of the log 2 ratio >1 as the threshold for judging the significance of the gene expression differences74. The DEGs were further analysed by GO-Term Finder and path_finder for GO and KEGG enrichment75. The calculated p-value was Bonferroni corrected, taking the corrected p-value < 0.05 as a threshold for GO annotation. After the GO annotation was obtained for each gene, WEGO software was used to classify the genes by function and to determine the distribution of gene functions in the species at the macro level76. The DEGs were annotated using the Mercator web tool77 and then loaded into Mapman software for function enrichment analysis78,79,80. The Venn diagrams in this study were prepared using the function Venn diagram in R based on the gene list for each tissue type.
Small RNA classification
After trimming adaptor sequences at the 5′ and 3′ ends of sequenced reads low-quality reads (reads with unknown sequences ‘N’), adaptor sequence fragments, and empty reads were filtered out. Reads were aligned to the genome using the SOAP aligner/SOAP2 software with parameter settings for perfect matches. Then, sRNA-seq reads were compared to miRNA databases including repeat RNA, miRBase (version 21, http://www.mirbase.org/ftp.shtml), GenBank (ftp://ftp.ncbi.nlm.nih.gov/genbank/), Rfam (version 11, http://rfam.janelia.org/), and snoRNA (http://www.mir2disease.org/) using the BlastN program (version 2.2.16). Mireap software (version 0.1) was used for novel miRNA prediction.
Differential expression analysis of small RNAs and target prediction
Differentially expressed miRNAs between samples were identified using DESeq software73. The false discovery rate of <0.001 and an absolute value of the log2 ratio > 1 were used as the threshold for determining the significance of miRNA expression differences. miRNA target gene prediction was performed using psRobot and TargetFinder81,82,83,84,85. miRNA function categorisation and pathway enrichment were based on the GO and KEGG databases.
Data validation by qPCR
Total RNA was extracted as described for DEG library preparation and sequencing. Total RNA (2 μg) from each sample was reverse transcribed in a 20 μL reaction using Reverse Transcriptase M-MLV (Takara). The sequences of the primers used are shown in Supplemental Table S8. The actin gene and/or UBQ7 gene of cotton (GeneBank accession No. is AY305733 and DQ116441) were used as internal control genes. qRT-PCR was performed using a SYBR Premix Ex Taq™ Kit (Takara) according to the manufacturer’s protocol. The 20 μL reaction volume consisted of forward and reverse primers (1 μL) SYBR premix Ex Taq II (10 μL), ddH2O (2.6 μL), cDNA (5 μL) and ROX Dye II (0.4 μL). The selected genes were verified using an ABI 7500 real-time PCR detection system with a cycling temperature of 60 °C and a single peak on the melting curve to ensure a single product. Relative transcript levels for each sample were obtained using the 2−△△Ct method. At least three replicates were tested per sample.
Additional Information
Accession Numbers: All the RNA-seq and small RNA raw data and processed data used in this study have been deposited at NCBI Gene Expression Omnibus under accession number GSE69025.
How to cite this article: Sun, Q. et al. To Be a Flower or Fruiting Branch: Insights Revealed by mRNA and Small RNA Transcriptomes from Different Cotton Developmental Stages. Sci. Rep. 6, 23212; doi: 10.1038/srep23212 (2016).
Supplementary Material
Acknowledgments
We acknowledge the Beijing Genomics institute at Shenzhen for its assistance in original data processing and related bioinformatics analysis. Thanks to the suggestion of Professor Yonghong Wang, Siyi Guo, Ling Bai and Guangzhou Gene de novo Biotechnology Co., Ltd. This work was supported by the Natural Science Foundation of China (NSFC grant No. 31571724, 31071461, 31470354), the National Key Basic Special Funds (2012CB1143001), 2014 Henan outstanding talent project (154200510006) and Chongqing Natural Science Foundation (cstc2012jjA80037, cstc2015jA80006, cstc2013jcyA10005) and Chongqing Municipal Commission of Education (KJ130502, KJ130520, KJ1400424, KJ1500420).
Footnotes
Author Contributions Y.F.C. and C.P.S. conceived the study. X.M.D., W.N.W., G.H.W., K.X.Z., S.P.G., L.L., X.L., C.Y., C.W.C., J.C.M., H.Z. and W.G. participated in experiment materials preparation. X.M.D. guided and provided the cotton materials, G.H.W., W.N.W. and S.Z. participated in RNA extraction. Q.S.,C.W.C. and P.Q. analysed the data. Q.S. and S.Z. performed qRT-PCR. Q.S., Y.F.C. and C.W.C. wrote the paper. C.M. guided the study. All authors read and approved the final manuscript.
References
- Paterson A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427 (2012). [DOI] [PubMed] [Google Scholar]
- Reinhardt D. & Kuhlemeier C. Plant architecture. EMBO Rep. 3, 846–851 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T. & Matsuoka M. Generating high-yielding varieties by genetic manipulation of plant architecture. Curr. Opin. Biotechnol. 15, 144–147 (2004). [DOI] [PubMed] [Google Scholar]
- Wang Y. & Li J. Molecular basis of plant architecture. Annu. Rev. Plant Biol. 59, 253–279 (2008). [DOI] [PubMed] [Google Scholar]
- Chen W. et al. Genetic mapping of the nulliplex-branch gene (gb_nb1) in cotton using next-generation sequencing. Theor. Appl. Genet. 128, 539–547 (2015). [DOI] [PubMed] [Google Scholar]
- Jenkins J. N., McCarty J. & Parrott W. Fruiting efficiency in cotton: Boll size and boll set percentage. Crop Sci. 30, 857–860 (1990). [Google Scholar]
- Boquet D. J., Moser E. B. & Breitenbeck G. A. Boll weight and within-plant yield distribution in field-grown cotton given different levels of nitrogen. Agron. J. 86, 20–26 (1994). [Google Scholar]
- Li C. Q. et al. QTL detection for node of first fruiting branch and its height in upland cotton (Gossypium hirsutum L.). Euphytica 188, 441–451 (2012). [Google Scholar]
- Guo Y. F., McCarty J., Jenkins J. N., An C. F. & Saha S. Genetic detection of node of first fruiting branch in crosses of a cultivar with two exotic accessions of upland cotton. Euphytica 166, 317–329 (2009). [Google Scholar]
- Pettigrew W. T. Source-to-sink manipulation effects on cotton lint yield and yield components. Agron. J. 86, 731–735 (1994). [Google Scholar]
- Reta-Sánchez D. G. & Fowler J. L. Canopy light environment and yield of narrow-row cotton as affected by canopy architecture. Agron. J. 94, 1317–1323 (2002). [Google Scholar]
- Blumel M., Dally N. & Jung C. Flowering time regulation in crops-what did we learn from Arabidopsis? Curr. Opin. Biotechnol. 32, 121–129 (2015). [DOI] [PubMed] [Google Scholar]
- Niwa M. et al. BRANCHED1 interacts with FLOWERING LOCUS T to repress the floral transition of the axillary meristems in Arabidopsis. Plant Cell 25, 1228–1242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels S. D. Flowering time regulation produces much fruit. Curr. Opin. Plant Biol. 12, 75–80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B. et al. TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J. 52, 891–898 (2007). [DOI] [PubMed] [Google Scholar]
- Busch B. L. et al. Shoot branching and leaf dissection in tomato are regulated by homologous gene modules. Plant Cell 23, 3595–3609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J. Z. et al. Mapping genomic loci for cotton plant architecture, yield components, and fiber properties in an interspecific (Gossypium hirsutum L. x G. barbadense L.) RIL population. Mol. Genet. Genomics 289, 1347–1367 (2014). [DOI] [PubMed] [Google Scholar]
- Dai T. C. & Wang Z. M. Molecular cloning and expression analysis of GhLOF genes in upland cotton (Gossypium hirsutum L.). Genet. Mol. Res. 14, 4575–4583 (2015). [DOI] [PubMed] [Google Scholar]
- McGarry R. C., Prewitt S. & Ayre B. G. Overexpression of FT in cotton affects architecture but not floral organogenesis. Plant Signal. Behav. 8, e23602 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney T. H. Short branch, another character of cotton showing monohybrid inheritance. Journal of Agricultural Research 41, 379–387 (1930). [Google Scholar]
- Winston J. Inheritance of cluster habit and its linkage relation with anthocyanin pigmentation in upland cotton. Journal of Agricultural Research 64, 105–119 (1942). [Google Scholar]
- Pathak R. S. & Singh R. B. Genetic analysis of the duplicate loci, cluster and short branch in Gossypium hirsutum L. Theor. Appl. Genet. 46, 281–287 (1975). [DOI] [PubMed] [Google Scholar]
- Huang N. C., Jane W. N., Chen J. & Yu T. S. Arabidopsis thaliana CENTRORADIALIS homologue (ATC) acts systemically to inhibit floral initiation in Arabidopsis. Plant J. 72, 175–184 (2012). [DOI] [PubMed] [Google Scholar]
- Sun Q. et al. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genomics 14, 852 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira T. H. et al. microRNAs associated with drought response in the bioenergy crop sugarcane (Saccharum spp.). PLoS ONE 7, e46703 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benlloch R., Berbel A., Serrano-Mislata A. & Madueno F. Floral initiation and inflorescence architecture: a comparative view. Ann. Bot. 100, 659–676 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Guo Z. & Li L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev. Biol. 380, 133–144 (2013). [DOI] [PubMed] [Google Scholar]
- Carrington J. C. & Ambros V. Role of microRNAs in plant and animal development. Science 301, 336–338 (2003). [DOI] [PubMed] [Google Scholar]
- Ambros V. review article The functions of animal microRNAs. Nature 431, 350–355 (2004). [DOI] [PubMed] [Google Scholar]
- Xie F., Sun G., Stiller J. W. & Zhang B. Genome-wide functional analysis of the cotton transcriptome by creating an integrated EST database. PLoS one 6, e26980 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X. et al. Identification of novel microRNAs in the Verticillium wilt-resistant upland cotton variety KV-1 by high-throughput sequencing. Springerplus 3, 564 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44, 1098–1103 (2012). [DOI] [PubMed] [Google Scholar]
- Li F. et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530 (2015). [DOI] [PubMed] [Google Scholar]
- Zhang T. et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537 (2015). [DOI] [PubMed] [Google Scholar]
- Teotia S. & Tang G. To bloom or not to bloom: role of microRNAs in plant flowering. Mol. Plant 8, 359–377 (2015). [DOI] [PubMed] [Google Scholar]
- Kaufmann K. et al. Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biol. 7, e1000090 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge M. J. et al. Class III homeodomain-leucine zipper gene family members have overlapping, antagonistic, and distinct roles in Arabidopsis development. Plant Cell 17, 61–76 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregis V. et al. Identification of pathways directly regulated by SHORT VEGETATIVE PHASE during vegetative and reproductive development in Arabidopsis. Genome Biol. 14, R56 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Martinez J. A., Poza-Carrion C. & Cubas P. Arabidopsis BRANCHED1 acts as an integrator of branching signals within axillary buds. Plant Cell 19, 458–472 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlayson S. A. Arabidopsis Teosinte Branched1-like 1 regulates axillary bud outgrowth and is homologous to monocot Teosinte Branched1. Plant Cell Physiol. 48, 667–677 (2007). [DOI] [PubMed] [Google Scholar]
- Ploense S. E., Wu M. F., Nagpal P. & Reed J. W. A gain-of-function mutation in IAA18 alters Arabidopsis embryonic apical patterning. Development 136, 1509–1517 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameau C. et al. Multiple pathways regulate shoot branching. Front. Plant Sci. 5, 741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann T. & Muhr M. Shaping plant architecture. Front. Plant Sci. 6, 233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychoudhry S., Del Bianco M., Kieffer M. & Kepinski S. Auxin controls gravitropic setpoint angle in higher plant lateral branches. Curr Biol. 23(15), 1497–504 (2013). [DOI] [PubMed] [Google Scholar]
- Han P. & Zhu Y. X. BARD1 may be renamed ROW1 because it functions mainly as a REPRESSOR OF WUSCHEL1. Plant Signal. Behav. 4, 52–54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Jiao Y., Liu Z. & Zhu Y.-X. ROW1 maintains quiescent centre identity by confining WOX5 expression to specific cells. Nature communications 6, 6003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanofsky M. F. et al. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 346, 35–39 (1990). [DOI] [PubMed] [Google Scholar]
- Lenhard M., Bohnert A., Jürgens G. & Laux T. Termination of Stem Cell Maintenance in Arabidopsis Floral Meristems by Interactions between WUSCHEL and AGAMOUS. Cell 105, 805–814 (2001). [DOI] [PubMed] [Google Scholar]
- Lohmann J. U. et al. A Molecular Link between Stem Cell Regulation and Floral Patterning in Arabidopsis. Cell 105, 793–803 (2001). [DOI] [PubMed] [Google Scholar]
- Xing H. et al. LNK1 and LNK2 recruitment to the evening element require morning expressed circadian related MYB-like transcription factors. Plant Signal. Behav. 10, e1010888 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Kim J. & Somers D. E. Transcriptional corepressor TOPLESS complexes with pseudoresponse regulator proteins and histone deacetylases to regulate circadian transcription. Proc Natl Acad Sci USA 110, 761–766 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno N. et al. The novel MYB protein EARLY-PHYTOCHROME-RESPONSIVE1 is a component of a slave circadian oscillator in Arabidopsis. Plant Cell 15, 2476–2488 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashino T. et al. Clock-controlled and FLOWERING LOCUS T (FT)-dependent photoperiodic pathway in Lotus japonicus II: characterization of a microRNA implicated in the control of flowering time. Bioscience, biotechnology, and biochemistry 77, 1179–1185 (2013). [DOI] [PubMed] [Google Scholar]
- Shim J. S. & Imaizumi T. Circadian clock and photoperiodic response in Arabidopsis: from seasonal flowering to redox homeostasis. Biochemistry 54, 157–170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H. et al. The circadian clock gene regulatory module enantioselectively mediates imazethapyr-induced early flowering in Arabidopsis thaliana. J. Plant Physiol. 171, 92–98 (2014). [DOI] [PubMed] [Google Scholar]
- Seo E. et al. Crosstalk between cold response and flowering in Arabidopsis is mediated through the flowering-time gene SOC1 and its upstream negative regulator FLC. Plant Cell 21, 3185–3197 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann U. et al. Molecular cloning of SVP: a negative regulator of the floral transition in Arabidopsis. Plant J. 21, 351–360 (2000). [DOI] [PubMed] [Google Scholar]
- Lee J. H. et al. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 21, 397–402 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos J. L. et al. Combinatorial activities of SHORT VEGETATIVE PHASE and FLOWERING LOCUS C define distinct modes of flowering regulation in Arabidopsis. Genome Biol. 16, 31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. X., Wang Y. X., Liu X. F. & Li J. Y. Arabidopsis RAV1 is down-regulated by brassinosteroid and may act as a negative regulator during plant development. Cell Res. 14, 8–15 (2004). [DOI] [PubMed] [Google Scholar]
- Kagaya Y., Ohmiya K. & Hattori T. RAV1, a novel DNA-binding protein, binds to bipartite recognition sequence through two distinct DNA-binding domains uniquely found in higher plants. Nucleic Acids Res. 27, 470–478 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanel E. A., Schrago C. G., Counago R. M., Russo C. A. & Alves-Ferreira M. Evolution of the B3 DNA binding superfamily: new insights into REM family gene diversification. PLoS one 4, e5791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijken A. J., Schluepmann H. & Smeekens S. C. Arabidopsis trehalose-6-phosphate synthase 1 is essential for normal vegetative growth and transition to flowering. Plant Physiol. 135, 969–977 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogale S. et al. MicroRNA156: A Potential Graft-Transmissible MicroRNA That Modulates Plant Architecture and Tuberization in Solanum tuberosum ssp. andigena. Plant physiol. 164, 1011–1027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz S., Grande A. V., Bujdoso N., Saedler H. & Huijser P. The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant molecular biology 67, 183–195 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G. K., Kubo M., Zhong R., Demura T. & Ye Z. H. Overexpression of miR165 affects apical meristem formation, organ polarity establishment and vascular development in Arabidopsis. Plant Cell Physiol. 48, 391–404 (2007). [DOI] [PubMed] [Google Scholar]
- Jin D., Wang Y., Zhao Y. & Chen M. MicroRNAs and their cross-talks in plant development. J. Genet. Genomics 40, 161–170 (2013). [DOI] [PubMed] [Google Scholar]
- Williams L., Grigg S. P., Xie M., Christensen S. & Fletcher J. C. Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 132, 3657–3668 (2005). [DOI] [PubMed] [Google Scholar]
- Huang J. Q. In China Cotton Cultivation (ed. Zhang J. R.) 198–199 (Press of China Agricultural Sciences and Technology, 1998). [Google Scholar]
- Li R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009). [DOI] [PubMed] [Google Scholar]
- Audic S. & Claverie J. M. The significance of digital gene expression profiles. Genome Res. 7, 986–995 (1997). [DOI] [PubMed] [Google Scholar]
- Mortazavi A., Williams B. A., McCue K., Schaeffer L. & Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008). [DOI] [PubMed] [Google Scholar]
- Anders S. & Huber W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. & Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Annals of statistics 29, 1165–1188 (2001). [Google Scholar]
- Kanehisa M. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–484 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–297 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M. et al. Mercator: a fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 37, 1250–1258 (2014). [DOI] [PubMed] [Google Scholar]
- Lohse M. et al. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 40, W622–627 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O. et al. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939 (2004). [DOI] [PubMed] [Google Scholar]
- Usadel B. et al. A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species, Maize. Plant Cell Environ 32, 1211–1229 (2009). [DOI] [PubMed] [Google Scholar]
- Wu H. J., Ma Y. K., Chen T., Wang M. & Wang X. J. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 40, W22–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bo X. & Wang S. TargetFinder: a software for antisense oligonucleotide target site selection based on MAST and secondary structures of target mRNA. Bioinformatics 21, 1401–1402 (2005). [DOI] [PubMed] [Google Scholar]
- Kielbasa S. M., Bluthgen N., Fahling M. & Mrowka R. Targetfinder.org: a resource for systematic discovery of transcription factor target genes. Nucleic Acids Res. 38, W233–238 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavorgna G., Guffanti A., Borsani G., Ballabio A. & Boncinelli E. TargetFinder: searching annotated sequence databases for target genes of transcription factors. Bioinformatics 15, 172–173 (1999). [DOI] [PubMed] [Google Scholar]
- Srivastava P. K., Moturu T. R., Pandey P., Baldwin I. T. & Pandey S. P. A comparison of performance of plant miRNA target prediction tools and the characterization of features for genome-wide target prediction. BMC Genomics 15, 348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.