

Abstract
Background
The Gag polyprotein is a multifunctional regulator of retroviral replication and major structural component of immature virions. The nucleic acid chaperone (NAC) activity is considered necessary to retroviral Gag functions, but so far, NAC activity has only been confirmed for HIV-1 and RSV Gag polyproteins. The nucleocapsid (NC) domain of Gag is proposed to be crucial for interactions with nucleic acids and NAC activity. The major function of matrix (MA) domain is targeting and binding of Gag to the plasma membrane but MA can also interact with RNA and influence NAC activity of Gag. Here, we characterize RNA binding properties and NAC activity of HIV-2 MA and Gag, lacking p6 domain (GagΔp6) and discuss potential contribution of NC and MA domains to HIV-2 GagΔp6 functions and interactions with RNA.
Results
We found that HIV-2 GagΔp6 is a robust nucleic acid chaperone. HIV-2 MA protein promotes nucleic acids aggregation and tRNALys3 annealing in vitro. The NAC activity of HIV-2 NC is affected by salt which is in contrast to HIV-2 GagΔp6 and MA. At a physiological NaCl concentration the tRNALys3 annealing activity of HIV-2 GagΔp6 or MA is higher than HIV-2 NC. The HIV-2 NC and GagΔp6 show strong binding to the packaging signal (Ψ) of HIV-2 RNA and preference for the purine-rich sequences, while MA protein binds mainly to G residues without favouring Ψ RNA. Moreover, HIV-2 GagΔp6 and NC promote HIV-2 RNA dimerization while our data do not support MA domain participation in this process in vitro.
Conclusions
We present that contrary to HIV-1 MA, HIV-2 MA displays NAC activity and we propose that MA domain may enhance the activity of HIV-2 GagΔp6. The role of the MA domain in the NAC activity of Gag may differ significantly between HIV-1 and HIV-2. The HIV-2 NC and MA interactions with RNA are not equivalent. Even though both NC and MA can facilitate tRNALys3 annealing, MA does not participate in RNA dimerization in vitro. Our data on HIV-2 indicate that the role of the MA domain in the NAC activity of Gag differs not only between, but also within, retroviral genera.
Electronic supplementary material
The online version of this article (doi:10.1186/s12977-016-0245-1) contains supplementary material, which is available to authorized users.
Keywords: HIV-2, Gag, Matrix, Nucleocapsid, Nucleic acid chaperone, tRNALys3 annealing, RNA binding proteins, RNA dimerization, RNA structure
Background
The retroviral Gag polyprotein is not only a major structural component of immature virions, but also acts as multifunctional regulator of virus replication [1–4]. The Gag polyproteins of human immunodeficiency viruses type 1 and 2 (HIV-1 and HIV-2) share 50 % sequence homology and each contains the N-terminal matrix (MA) domain, which is responsible for Gag targeting and binding to the plasma membrane [5, 6], the capsid domain (CA), which facilitates Gag multimerization [4, 7], and the nucleocapsid (NC) domain that interacts with viral and cellular nucleic acids (NA) [2, 3, 8]. Gag also contains two spacer regions (SP1, SP2) and the p6 domain located at the C-terminus, which is necessary for virus release from the infected cell [9]. Before or shortly after virion release, the Gag polyprotein is cleaved in a highly ordered manner by viral protease into freestanding MA, CA, and NC proteins [1, 10]. In addition to the structural function in mature virions [4], the NC protein plays an important role in the facilitation of nucleic acid strand transfers during reverse transcription [11]. Mature MA remains bound to the viral membrane [4], but it was proposed that a fraction of HIV-1 MA also enters newly infected cells, associates with the pre-integration complex (PIC), and affects proviral DNA circularization and integration [12, 13].
At a late stage in the HIV replication cycle, Gag may be responsible for the annealing of tRNALys3 to an 18-nt primer binding sequence (PBS) localized in the 5′UTR of the viral RNA, where tRNALys3 primes reverse transcription [2, 14]. The NAC activity is considered necessary to anneal tRNALys3 but, so far, it has only been confirmed for HIV-1 and Rous sarcoma virus (RSV) Gag proteins in vitro [15–18]. Chaperone proteins can facilitate folding and formation of the most thermodynamically favoured structures of nucleic acids [19]. The NAC activity of HIV-1 Gag has been shown to depend on the NC domain, which is required for tRNALys3/viral RNA complex formation [14, 16, 17], whereas the MA domain can inhibit this process via RNA binding [17]. Numerous lines of evidence support the nucleic-acid-binding properties of retroviral MA [15, 20–27], but significant differences in the role of MA domain in the overall NAC activity of Gag were observed between retroviral genera. In contrast to HIV-1, the chaperone activity of alpharetrovirus RSV Gag is independent of the MA domain [15]. Moreover, HIV-1 and RSV NC display robust chaperone activity [15–17, 28], whereas MA proteins from both viruses do not promote annealing of primer tRNA in vitro [15–17]. The RNA-binding properties and chaperone activity of HIV-2 Gag have not been studied and the contribution of NC and MA domains remains undefined. We recently reported that HIV-2 NC is an effective NAC, but its activity is limited by the structural stability of the nucleic acid molecule to a much greater degree than that of HIV-1 NC [29].
As a nucleic acid chaperone, Gag binds NA non-specifically [2, 3], but is also engaged in highly specific recognition of cis-acting dimerization and packaging (Ψ) signals within the 5′UTR of the viral genomic RNA [30]. Although HIV-1 Gag binds GU-rich sequences in the cytoplasm, its binding specificity changes to A-rich RNA motifs during virion assembly [31]. The HIV-2 Gag-binding sites within the viral RNA remain uncharacterized and only limited information on HIV-2 NC binding to isolated domains of HIV-2 5′UTR in vitro is available [32, 33]. Similar packaging mechanisms are suggested for both viruses [34], but HIV-1 Gag is able to package HIV-2 RNA, whereas HIV-2 Gag cannot package HIV-1 RNA [35, 36]. The NC domains of HIV-1 and HIV-2 uncleaved Gag polyproteins are proposed to be crucial for the selection, dimerization, and packaging of viral RNA [8, 36–39]. In contrast, both NC and MA domains play direct roles in viral RNA packaging in deltaretroviruses [bovine leukaemia virus (BLV) and human T cell leukaemia virus type 2 (HTLV-2)] [22, 25, 26]. Interestingly, participation of the MA domain of HIV-1 Gag in these steps of viral replication was also suggested [23, 40, 41]. Moreover, an intriguing link between a mutation in the MA domain of HIV-2 Gag and viral RNA dimerization has been recently shown [42].
The three-dimensional structure of the entire HIV-1 and HIV-2 Gag is unknown, but the structures of their freestanding NC and MA have been presented [32, 43–46]. Moreover, the structure of the non-myristoylated HIV-1 Gag fragment (MA-CA-SP1-NC) was recently resolved by NMR spectroscopy [27]. The mature NC proteins of HIV-1 and HIV-2 are small basic proteins, containing two zinc finger domains (ZFs). The ZFs are proposed to be crucial for specific interactions of NC with nucleic acids, whereas basic residues from the disordered N-terminus play a role in non-specific interactions and NAC activity [8, 11, 29, 47]. Despite the limited sequence similarity between HIV-1 MA and HIV-2 MA, both proteins are composed of six α-helices and three β-sheet elements, and are myristoylated at the N-terminus [43, 44, 46]. The myristyl group and amino acid residues of HIV-1 and HIV-2 MA are engaged in PM binding [5, 43]. Importantly, some of those residues are located within the highly basic region (HBR) at the N-terminus of MA, which is proposed to be important for interactions with RNA in HIV-1 [21, 48–50]. RNA binding to the MA domain ensures the specificity of HIV-1 Gag interactions with PM phospholipids [6, 21, 48, 51]. Whether the MA domain of HIV-2 Gag is involved in RNA binding is not known.
Within this work we characterized the RNA-binding properties and nucleic acid chaperone activity of recombinant HIV-2 GagΔp6, NC and MA proteins. We identified binding sites of HIV-2 GagΔp6 and isolated NC and MA domains within the 5′UTR of HIV-2 RNA. Both HIV-2 NC and GagΔp6 show strong binding to the packaging signal and preference for the purine-rich sequences, while HIV-2 MA binds mainly to G residues without favouring Ψ RNA. Moreover, HIV-2 NC promotes HIV-2 RNA dimerization while this process is not supported by HIV-2 MA, suggesting that MA domain is dispensable for HIV-2 GagΔp6-promoted dimerization in vitro. We found that HIV-2 GagΔp6 is a robust nucleic acid chaperone and we propose that both NC and MA domains contribute to nucleic acids aggregation and tRNALys3 annealing in vitro. The NAC activity of HIV-2 NC is affected by salt in contrast to that of HIV-2 GagΔp6 and MA.
Results
Nucleic acid-binding properties of HIV-2 GagΔp6, MA, and NC
Interactions of HIV-1 Gag polyprotein as well as NC and MA proteins with nucleic acids are documented (reviewed in Ref. [3]). However, little information, and limited to freestanding NC [32, 33], is available for HIV-2. Therefore, the first step of our study was to characterize the binding of non-myristoylated HIV-2 Gag polyprotein lacking the p6 domain (referred to as GagΔp6) and freestanding HIV-2 NC and MA proteins to RNA. We performed salt-dependent filter-binding assays to examine interactions with three RNA molecules that represent functional domains of 5′UTR of HIV-2 genomic RNA: TARpA, PBS, and Ψ (Fig. 1) [33, 52, 53].
Fig. 1.
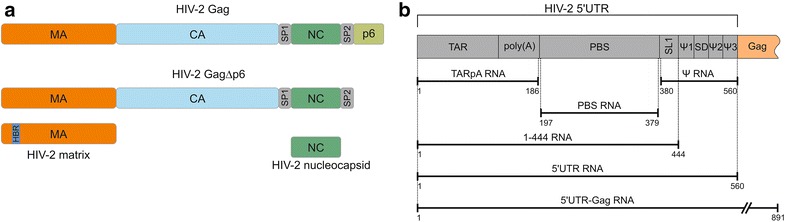
Schematic representation of HIV-2 proteins (a) and RNAs (b) used in this study
At physiological salt concentration (150 mM NaCl), the highest binding affinity was observed for Ψ RNA and HIV-2 GagΔp6 or NC (15 and 71 nM, respectively) (Table 1). However, GagΔp6 binding was salt-independent up to 250 mM NaCl, while NC affinity to Ψ RNA decreased nine-fold within a 100–250 mM NaCl range. At physiological salt concentration GagΔp6 displayed slightly lower affinity to PBS and TARpA than to Ψ RNA. A similar but more apparent trend was observed for HIV-2 NC protein, whereas HIV-2 MA bound PBS, TARpA and Ψ RNA with comparable affinities. Both HIV-2 NC and MA bound non-Ψ RNA molecules with similar affinities, but MA binding to Ψ RNA was significantly weaker than that of NC. Contrary to HIV-2 NC, GagΔp6 and MA consistently showed resistance to increasing salt within a 100–250 mM range, independently of the RNA substrate used. Importantly, even at 500 mM NaCl, HIV-2 GagΔp6 remained tightly bound to Ψ (Kd ≈ 125 nM) and PBS (Kd ≈ 314 nM) RNAs and only GagΔp6/TARpA interaction was reduced (Kd ≈ 965 nM). Both HIV-2 NC and MA proteins showed significantly lower affinity to RNA at 500 mM NaCl and only NC/Ψ RNA interaction was somewhat tighter.
Table 1.
Calculated dissociation constants for HIV-2 NC, MA, and GagΔp6 complexes with selected RNA
| NaCl (mM) | Kd (nM) | ||
|---|---|---|---|
| NC | MA | GagΔp6 | |
| TARpA RNA | |||
| 50 | 207 ± 9 | 431 ± 88 | 49 ± 5 |
| 100 | 285 ± 21 | 417 ± 74 | 52 ± 5 |
| 150 | 385 ± 16 | 430 ± 46 | 53 ± 5 |
| 200 | 541 ± 6 | 538 ± 68 | 65 ± 4 |
| 250 | 679 ± 16 | 593 ± 76 | 97 ± 7 |
| 500 | 1432 ± 22 | 2377 ± 209 | 965 ± 83 |
| PBS RNA | |||
| 50 | 36 ± 3 | 335 ± 28 | 31 ± 1 |
| 100 | 117 ± 6 | 289 ± 4 | 31 ± 3 |
| 150 | 196 ± 9 | 290 ± 5 | 30 ± 2 |
| 200 | 277 ± 26 | 340 ± 25 | 31 ± 2 |
| 250 | 392 ± 37 | 382 ± 25 | 41 ± 3 |
| 500 | 2036 ± 212 | 2072 ± 98 | 314 ± 29 |
| Ψ RNA | |||
| 50 | 7 ± 2 | 413 ± 52 | 27 ± 1 |
| 100 | 25 ± 1 | 300 ± 41 | 17 ± 1 |
| 150 | 71 ± 1 | 312 ± 29 | 15 ± 1 |
| 200 | 135 ± 4 | 357 ± 21 | 24 ± 2 |
| 250 | 240 ± 10 | 374 ± 19 | 26 ± 3 |
| 500 | 979 ± 26 | 1514 ± 210 | 125 ± 5 |
Hydroxyl radical (HR) footprinting was used to further characterize HIV-2 GagΔp6, NC, and MA interactions with HIV-2 5′UTR. Overall reactivity profiles for RNA in the presence or absence of HIV-2 GagΔp6, NC, or MA were compared to reveal RNA sequences protected when proteins were present. At the highest protein concentration, almost the entire HIV-2 leader RNA was protected from HR cleavage for all proteins studied. The strongest decreases in HR cleavage were observed in the presence of HIV-2 GagΔp6 and NC proteins, while lesser effects were observed at the same concentration of HIV-2 MA, which supports the RNA-binding analysis (Table 1). A strong decrease in susceptibility to HR was noted for all studied proteins within TAR, PBS, and domains within the 3′ end of the 5′UTR (Fig. 2a). Several sites within the PBS domain that were protected from cleavage in the presence of HIV-2 GagΔp6 were also protected by MA but not NC, while reduced reactivities for some sequences within the 3′ end of the 5′UTR that were noted in the presence of HIV-2 GagΔp6 were also observed in the presence of NC but not MA. HIV-2 GagΔp6 or NC protected mostly purine residues, whereas mostly G residues displayed reduced cleavage in the presence of MA (Additional file 1). The protection patterns were detected in both single and double-stranded regions. Analysis of the composition of protected sites and their vicinity revealed a prevalence of G residues in the case of MA, A residues for NC, while mixed nucleotide residue composition was observed for HIV-2 GagΔp6 (Fig. 2b).
Fig. 2.
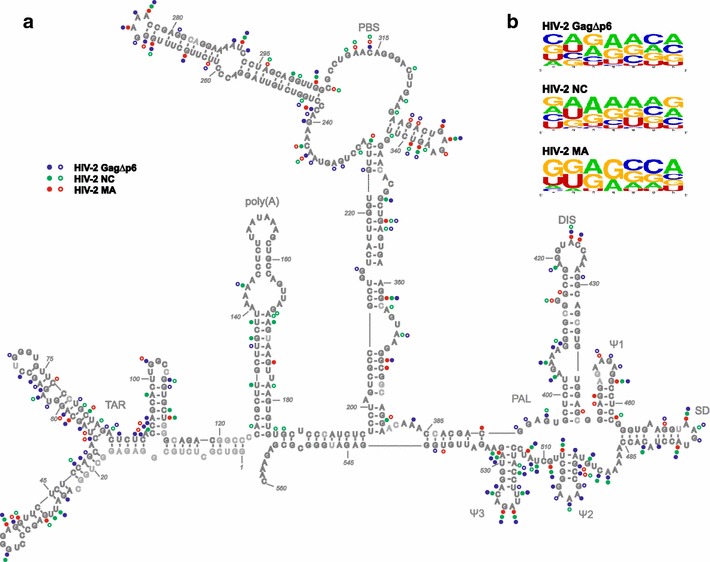
Protein binding to HIV-2 5′UTR. a Secondary structure model of +1–560 nt region of HIV-2 RNA [33] with the positions protected from hydroxyl radical cleavage in the presence of the HIV-2 GagΔp6, NC, and MA proteins marked. The level of protection increases with protein concentration. Closed circles indicate residues protected starting at lowest protein concentration; open circles indicate residues protected starting at intermediate protein concentration. Light grey annotates lack of data. b Sequences within binding sites and their vicinities were compared using http://weblogo.berkeley.edu/ [68]
HIV-2 MA displays high TAR annealing activity
The capability to facilitate the annealing of HIV-1 TAR RNA and TAR(−) DNA hairpins serves as a basic test of the in vitro NAC activity of proteins [54]. This assay resembles the annealing of (–) ssDNA to 3′UTR of viral RNA during the first strand transfer of reverse transcription. HIV-1 NC and Gag were found to be excellent nucleic acid chaperones, whereas HIV-1 MA displays little, if any, NAC activity [16, 17, 25]. We have recently shown that HIV-2 NC displays robust annealing activity of HIV-1 TAR hairpins [29], but the NAC activity of HIV-2 MA and Gag has not been reported yet. Therefore, the concentration and time course TAR RNA/TAR(−) DNA gel-shift annealing assays were performed to characterize and compare the NAC activity of HIV-2 MA, NC, and GagΔp6. A comparison of the proportion of annealed product obtained at increasing concentrations of HIV-2 MA, NC, or GagΔp6 showed that all analysed HIV-2 proteins effectively facilitated the annealing reaction (Fig. 3a). At 0.2 µM concentration (1 protein per 3.9 nt) of HIV-2 GagΔp6, NC, or MA, over 75 % of the TAR(−) DNA was annealed. However, on a molar basis, HIV-2 GagΔp6 is a more effective chaperone than NC or MA, since a twofold lower concentration of GagΔp6 was sufficient for maximal percentage of TAR annealing. Moreover, in the presence of 0.05 µM HIV-2 GagΔp6, the annealing was ~60 %, whereas no significant amount of annealed products was observed at the same concentration of HIV-2 NC or MA. Similarly to the observations made previously for HIV-1 GagΔp6 [16], we found that a further increase in HIV-2 GagΔp6 concentration led to a decrease in annealed products.
Fig. 3.
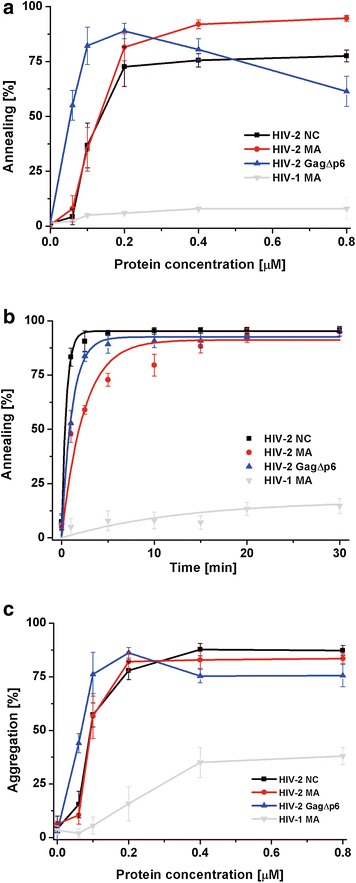
Comparison of HIV-2 GagΔp6, MA, NC and HIV-1 MA nucleic acid chaperone activity. a TAR annealing assays in the presence of increasing concentrations of protein. b Time course TAR annealing assays using 0.2 µM of each protein (1 protein per 3.9 nt). The curves are single-exponential fits to average data. Representative gels are included in Additional file 2. c TAR(−) DNA aggregation by the different proteins
Subsequently, time course TAR annealing at 0.2 µM protein concentration (1 protein per 3.9 nt) was performed to compare the annealing rates of HIV-2 NC, MA, and GagΔp6 (Fig. 3b). We found that the annealing rate of HIV-2 NC is higher than that of GagΔp6 and MA (twofold and threefold, respectively) (Table 2). However, the final levels of TAR hairpins annealed in the presence of each analysed HIV-2 protein were similar and close to 95 %. To directly compare HIV-2 and HIV-1 MA proteins, the ability to facilitate TAR annealing was assayed for HIV-1 MA (Fig. 3a, b). In agreement with previous study [25], low level of TAR annealing (~16 %) was measured in the presence of HIV-1 MA.
Table 2.
Annealing parameters of HIV-2 NC, MA, GagΔp6
| HIV-2 protein (μM) | Annealing rate | NA annealed (%) |
|---|---|---|
| TAR annealing (20 mM NaCl) | ||
| NC (0.2) | 2.0 ± 0.40 | 95.3 ± 0.7 |
| MA (0.2) | 0.6 ± 0.05 | 91.1 ± 2.4 |
| GagΔp6 (0.2) | 0.9 ± 0.11 | 92.5 ± 1.3 |
| tRNALys3 annealing (20 mM NaCl) | ||
| NC (1.5) | 1.87 ± 0.44 | 95.1 ± 1.8 |
| MA (1.5) | 0.17 ± 0.08 | 37.8 ± 5.0 |
| GagΔp6 (1.5) | 0.18 ± 0.06 | 85.1 ± 5.2 |
| tRNALys3 annealing (150 mM NaCl) | ||
| NC (1.5) | 0.07 ± 0.03 | 35.8 ± 4.9 |
| MA (1.5) | 0.21 ± 0.06 | 48.1 ± 4.9 |
| GagΔp6 (1.5) | 0.26 ± 0.03 | 82.1 ± 3.4 |
| NC (3) | 0.11 ± 0.02 | 43 ± 5.2 |
| MA (3) | 0.22 ± 0.07 | 60 ± 4.9 |
| GagΔp6 (3) | 0.37 ± 0.07 | 90 ± 3.7 |
| RNA dimerization (150 mM NaCl) | ||
| NC (6) | 0.48 ± 0.10 | 49.2 ± 1.6 |
| GagΔp6 (6) | 0.15 ± 0.01 | 45.7 ± 2.2 |
HIV-2 MA effectively aggregates nucleic acids
The ability to sequence non-specific aggregation of NA is considered an important characteristic of NAC proteins [55]. We directly compared the NA-aggregation properties of analysed HIV-2 proteins and HIV-1 MA using sedimentation assays (Fig. 3c). In this assay, the 32P-labelled HIV-1 TAR(−) DNA and TAR RNA were incubated with increasing concentrations of protein, centrifuged, and the amount of non-aggregated NA was measured. We found that HIV-2 NC, MA, and GagΔp6 are effective NA-aggregating agents, since ~80 % of NA aggregation was detected at a 0.2 µM concentration of each protein. The observed NA aggregation at a given protein concentration was similar for HIV-2 NC and MA, but significantly greater for HIV-2 GagΔp6, since a two-fold lower concentration of GagΔp6 was sufficient for the maximal NA aggregation. HIV-1 MA aggregated NA much weaker than HIV-2 proteins since only up to ~40 % of aggregation was detected at the highest protein concentration used (0.8 µM).
NC and MA domains may contribute to the tRNALys3 annealing activity of HIV-2 Gag
The HIV-1 Gag and NC, via their NAC activity, mediate tRNALys3 annealing in vitro and in vivo [14, 17, 36]. On the contrary, HIV-1 MA does not promote the tRNALys3 annealing in vitro even when high MA concentrations and long reaction times were employed [17]. The aggregation and TAR annealing assays demonstrated that HIV-2 MA displays high NAC activity in vitro (Fig. 3). To further characterize and compare the activity of the analysed HIV-2 proteins, gel-shift tRNALys3 annealing assays were performed, using the in vitro transcribed, unmodified tRNALys3, and a 560-nt RNA, corresponding to the 5′UTR of HIV-2 genomic RNA (Fig. 1b). To determine the influence of salt on the NAC activity of proteins, the annealing reactions were conducted at low (20 or 50 mM) and physiological (150 mM) NaCl concentrations.
The annealing assays showed that HIV-2 GagΔp6 displays high tRNALys3 annealing activity and, on a molar basis, is an even better chaperone than HIV-2 NC (Fig. 4a, b). Moreover, in contrast to HIV-2 NC, GagΔp6 activity is not influenced by NaCl concentration. In the presence of 1 µM HIV-2 GagΔp6, the annealing was ~70 % at 20 and 150 mM NaCl. At the same concentration of HIV-2 NC, only 25 % of annealing was measured at 20 mM NaCl, whereas no activity was observed at 150 mM NaCl. Furthermore, at 1.5 µM concentration (1 protein per 3.9 nt) of HIV-2 NC the presence of 150 mM NaCl reduced the annealing activity by 80 % and, even at 3 µM NC (1 protein per 1.8 nt), reduction by 30 % was still observed. Interestingly, in the presence of 0.5 µM HIV-2 MA, ~25 and ~37 % annealing of tRNALys3 was achieved at 20 and 150 mM NaCl, respectively (Fig. 4a, b). These results suggest that an increase in salt concentration stimulates to some extent the activity of HIV-2 MA. Although HIV-2 MA promoted tRNALys3 annealing at lower concentrations than HIV-2 NC, in contrast to NC, a further increase in its concentration up to 3 µM (1 protein per 1.8 nt) did not lead to an increase in annealing. In our hands, and in agreement with previous results [17], HIV-1 MA did not promote tRNALys3 annealing even at high protein concentration and during an extended period of reaction (Fig. 4).
Fig. 4.
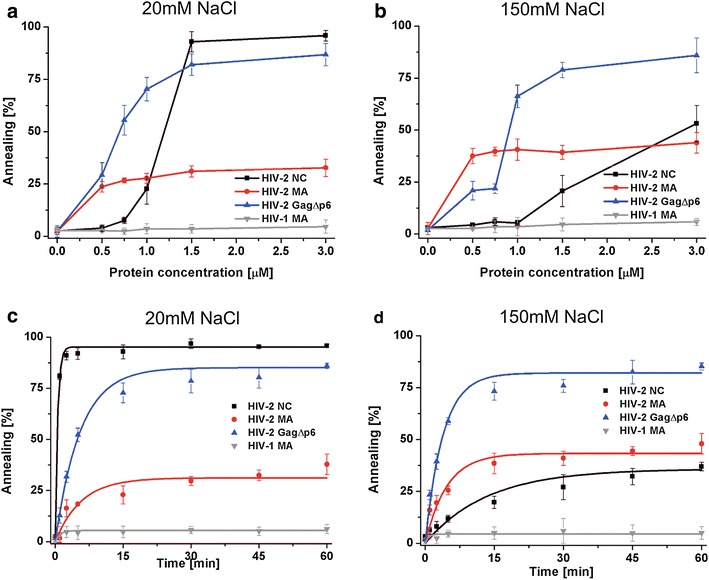
Protein induced tRNALys3 annealing in the presence of varying NaCl concentrations. a and b Concentration course annealing assays. c and d Time course annealing assays using 1.5 µM of each protein (1 protein per 3.9 nt), the curves are single-exponential fits to average data. Representative gels are included in Additional file 2
The time course tRNALys3 annealing assays performed at 1.5 µM protein concentration (1 protein per 3.9 nt), revealed important differences between the annealing rates of HIV-2 NC, MA, and GagΔp6 at 20 mM and 150 mM NaCl (Fig. 4c, d). At low NaCl, HIV-2 NC displayed a significantly higher (~11-fold) annealing rate than HIV-2 GagΔp6 or MA. The observed final percentages of tRNALys3 annealed were similar for HIV-2 NC and GagΔp6 (~95 and ~85 %, respectively), and ~38 % in the presence of HIV-2 MA. At physiological NaCl concentration, the annealing rates and final percentages of annealing in the presence of HIV-2 MA and GagΔp6 did not change significantly. Interestingly, HIV-2 NC exhibited an almost 27-fold lower annealing rate and the final percentage of tRNALys3 annealed was reduced to ~38 %. The increase in HIV-2 proteins concentration to 3 µM (1 protein per 1.8 nt) did not change the observed trend and inhibitory effect of 150 mM NaCl was still observed for HIV-2 NC but not for HIV-2 MA or GagΔp6 (Table 2).
HIV-2 MA does not promote HIV-2 RNA dimerization
HIV-2 genomic RNA is packaged as a dimer into the virions and, as in other retroviruses, Gag is involved in genome dimerization and packaging [35, 37, 38]. It was presented that, similarly to HIV-1, the intact NC domain within the uncleaved HIV-2 Gag confers specific binding of dimerization and Ψ signals via its two zinc finger motifs [37]. Our results showed that freestanding HIV-2 MA binds several sequences of HIV-2 RNA that are important for dimerization and packaging (Fig. 2a). To elucidate the role of the MA domain in HIV-2 RNA dimerization, in vitro assays in the presence of HIV-2 GagΔp6, MA, and NC were performed. In contrast to HIV-1, HIV-2 RNA forms tight dimers in vitro inefficiently, so the +1–444 transcript derived from the 5′UTR of HIV-2 RNA was chosen, since it forms dimers in vitro more efficiently than the entire (560 nt) 5′UTR [56]. The +1–444 transcript contains sequences proposed to be involved in HIV-2 genome dimerization: palindrome of SL1, called DIS (dimer initiation site), Ψ sequence with palindrome pal, and TAR domain [33, 38, 42, 56, 57]. We observed that HIV-1 MA and HIV-2 MA were unable to induce dimerization in vitro, whereas both HIV-2 NC and GagΔp6 facilitated the dimerization of +1–444 RNA (Fig. 5). In comparison to annealing assays, a significantly lower concentration of HIV-2 NC or GagΔp6 (1 protein per 30 nt) was needed to obtain the maximum percentage of RNA dimer (Fig. 5a). This observation is consistent with the hypothesis that, at the initial steps of packaging, a limited number of Gag is engaged in genome selection and dimerization [37]. Surprisingly, regardless of the presence of 150 mM NaCl in dimerization assays, HIV-2 NC was more effective on a molar basis than HIV-2 GagΔp6 and exhibited a three-fold higher dimerization rate (Fig. 5a; Table 2).
Fig. 5.
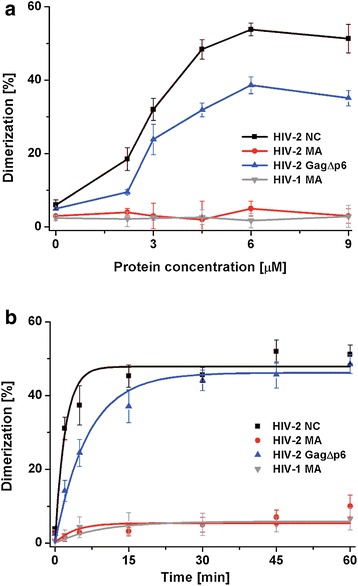
Protein-induced dimerization of HIV-2 RNA. a +1–444 RNA dimerization in the presence of increasing concentrations of proteins. b Time course dimerization in the presence of 6 µM protein (1 protein per 30 nt). The curves in panel b are single-exponential fits to average data. Representative gels are included in Additional file 2
Discussion and conclusions
In this work, we investigated the chaperone activity of HIV-2 GagΔp6, MA, and NC proteins, their binding specificity, and interactions with HIV-2 RNA. We also included HIV-1 MA in chaperone assays for direct comparison. The results of NA aggregation, TAR, or tRNALys3 annealing assays showed that, on a molar basis, HIV-2 GagΔp6 is a more robust nucleic acid chaperone than NC. Moreover, at a physiological salt concentration, the rate and final percentage of annealed tRNALys3 were significantly higher in the presence of HIV-2 GagΔp6 than HIV-2 NC (Fig. 4b, c; Table 2). The salt-dependent binding assays revealed that HIV-2 GagΔp6 binds to RNA with higher affinity than freestanding HIV-2 NC (Table 1). These observations suggest that domains other than NC contribute to the NAC activity of HIV-2 Gag. Indeed, we found that HIV-2 MA binds RNA and displays high NAC activity in vitro, since it effectively aggregated NA and facilitated the annealing of TAR hairpins (Fig. 3). This is in contrast to HIV-1 MA, which displays very poor NA aggregation and TAR annealing activity (Fig. 3) [25]. In addition HIV-1 MA does not chaperone tRNALys3 annealing in vitro while in the presence of HIV-2 MA, up to ~50 % of tRNALys3 annealing was measured (Fig. 4). In low salt concentration HIV-2 MA displays reduced tRNALys3 annealing activity compared to that of HIV-2 NC and GagΔp6. However the difference in activity is less evident in TAR annealing assays, suggesting that NAC activity of HIV-2 MA is limited by substrates length and stability to a greater degree than that of HIV-2 NC or GagΔp6.
We observed that HIV-2 GagΔp6 and MA binding to RNA is salt-independent in the range from 50 to 250 mM NaCl (Table 1). Consistently with these results, the tRNALys3 annealing activity of HIV-2 GagΔp6 and MA was not sensitive to monovalent salt at 20–150 mM (Fig. 4). Contrary to our data on HIV-2 MA, the increase in monovalent salt concentration from 50 to 150 mM significantly decreased the RNA-binding affinity of HIV-1 MA [25]. The RNA-binding properties and NAC activity of HIV-2 NC are highly salt-sensitive. At a physiological NaCl concentration, the extent and rate of tRNALys3 annealing in the presence of HIV-2 NC were lower than in the presence of HIV-2 MA (Fig. 4; Table 2). The comparison of the HIV-2 NC and MA chaperone activity at different salt concentrations supports involvement of both RNA-binding domains of HIV-2 GagΔp6 in tRNALys3 annealing.
Based on the presented results, we propose that both NC and MA domains contribute to the chaperone activity of HIV-2 Gag. Although HIV-2 NC is an effective chaperone, its activity is lower than that of HIV-1 NC [29]. Additionally to the NC domain, the MA domain via interactions with RNA and its NAC activity may enhance the activity of HIV-2 Gag. However we cannot exclude influence of other domains or multimerization on the NAC activity of HIV-2 Gag. The available data indicate that for HIV-1 the NC domain is primarily responsible for the overall NAC activity of HIV-1 Gag [16, 17], whereas the MA domain via RNA binding inhibits the NAC activity of HIV-1 Gag [17]. Interestingly, the MA domain does not influence the NAC activity of RSV Gag (alpharetrovirus) [15], while the HTLV-2 MA protein (deltaretrovirus) displays significantly higher chaperone activity than HTLV-2 NC [25]. Our data on HIV-2 indicate that the role of the MA domain in the NAC activity of Gag may differ not only between, but also within, retroviral genera.
The residues important for the chaperone activity of deltaretroviral MA proteins are highly conserved, including the presence of positively charged amino acid residues, and are located in α-helix II [25]. In case of HIV-1 MA two basic residues within the corresponding region (α-helix II) are also conserved between the HIV-1 and related SIV subtypes (Fig. 6a) but are not sufficient to confer NA chaperone activity. However, substitution of E40R/E42L/N47K in HIV-1 α-helix II resulting in the basic character mimicking HTLV-2 MA increased HIV-1 MA NAC activity to the level comparable to that of HTLV-2 [25]. In case of HIV-2 and related SIV subtypes this region is less positively charged than that of HIV-1 MA (Fig. 6a). Interestingly, calculated [58] isoelectric point is higher for the full length MA protein of HIV-2 than HIV-1 MA (9.68 and 9.10, respectively). The observed differences in the NAC activity of MA proteins may result from the more basic character of HIV-2 MA relative to HIV-1 MA. This is further supported by the analysis of electrostatic potential surfaces of HIV-1 and HIV-2 MA (Additional files 3 and 4). The amino acid residues of HBR were shown to be responsible for the RNA binding in HIV-1 MA [48]. The sequence alignment of HIV-1, HIV-2 and related SIV isolates demonstrated high level of conservation of the HBR between those retroviruses (Fig. 6b). Since HIV-1 MA do not promote tRNALys3 annealing it is unlikely that HBR region is a major determinant of HIV-2 MA chaperone activity. Analysis of the electrostatic potential surfaces of HIV-2 and HIV-1 MA proteins reveals regions other than α-helix II or HBR that might be considered important for HIV-2 MA NAC activity (Additional files 3 and 4). The difference between electrostatic potential surfaces of the two proteins was observed e.g. in the C terminus. The C terminus of MA proteins is disordered [43, 46]. Unstructured regions containing basic residues able to nonspecifically interact with nucleic acids are suggested to be important for the proteins’ NAC activity [19, 29, 47, 59]. On the other hand, relatively high salt resistance (up to 250 mM) of HIV-2 MA (Table 1) may suggest that some specific interactions contribute to HIV-2 MA/RNA binding.
Fig. 6.
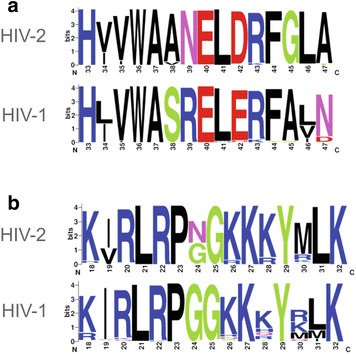
Sequence alignment of a MA α-helix II and b highly basic region (HBR); from HIV and related SIV subtypes. Sequences were taken from HIV Sequence Database (http://hiv.lanl.gov) and at least hundred sequences were align for each group. Alignments were visualized using http://weblogo.berkeley.edu/ [68]
A recent study has shown that HIV-1 Gag exhibits significant differences in salt-dependent binding to diverse HIV-1 RNA fragments and binds non-Ψ RNA with low specificity via its NC and MA domains, whereas binding to Ψ RNA is highly specific and only the NC domain is engaged [60]. Such a binding model is not common to all retroviral Gag polyproteins, since, in deltaretroviruses (HTLV-2), the MA domain binds RNA more specifically than NC and plays a dominant role in the initial recognition of the Ψ signal in genomic RNA [25]. Our results demonstrate that HIV-2 GagΔp6 binds both Ψ and non-Ψ RNAs with high specificity, which was manifested by a negligible change in the dissociation constants within the 50–250 mM NaCl range (Table 1). Moreover, even at 500 mM NaCl, HIV-2 GagΔp6 interacted with Ψ RNA and non-Ψ (PBS) RNA with strong affinities (Kd ≈ 125 and 314 nM, respectively). HIV-2 GagΔp6 binding to Ψ RNA was similar to that presented for HIV-1 GagΔp6, but HIV-1 GagΔp6 did not bind non-Ψ RNA (TARpA) at 500 mM NaCl [60]. HIV-2 GagΔp6 bound TARpA at 500 mM NaCl but with affinity (Kd ≈ 965 nM) lower than PBS. At low ionic strength (50 mM NaCl), HIV-2 NC binding to Ψ RNA was comparable to that of HIV-2 GagΔp6, but highly susceptible to salt concentration. HIV-2 NC and MA bound non-Ψ RNA with comparable affinity, but MA binding to Ψ RNA was notably weaker than that of NC or GagΔp6. Taken together, our results suggest contributions of both the NC and MA domains to the interactions of HIV-2 Gag with RNA, but the NC domain plays a major role in recognizing the Ψ signal. Our results showing that HIV-2 GagΔp6 and NC, but not MA, occupy some of the sites within the Ψ region of 5′UTR may further support this notion. Although the majority of HIV-2 GagΔp6, NC, and MA binding sites cluster within the Ψ region of HIV-2 5′UTR, extensive interactions were also detected within the TAR and PBS domains (Fig. 2). A recent study demonstrated that the RNA-binding specificity of HIV-1 Gag changes during viral replication [31]. Only a few regions of the viral RNA interacted with HIV-1 Gag in cytosol, including the 5′UTR, while the entire RNA was covered within the virus particles. Interestingly, the binding of HIV-1 Gag to TAR observed in the cytosol was HIV-1 subtype-dependent. Several lines of evidence suggested that TAR might be important for HIV dimerization and packaging [61, 62]. Moreover, TAR stability is considered important in the strand transfer during reverse transcription [63] and may also influence the Gag translation efficiency [64]. Our in vitro conditions are likely in favour of the detection of the high-affinity binding sites, but without differentiation of the replication stage. Binding of HIV-2 GagΔp6, NC, and MA to PBS in the vicinity of the tRNALys3 binding regions may support their involvement in primer annealing. Indeed, all proteins promoted tRNALys3 annealing to the 5′UTR (Fig. 4). Interestingly, findings that HIV-1 Gag has a strong preference for G-rich binding sites in cells and A-rich in virions [31] is reflected in our in vitro binding studies, showing a high purine content within the HIV-2 GagΔp6 binding sites.
In HIV-2 RNA, packaging and dimerization signals overlap, and the NC domain of HIV-2 Gag is proposed to be required for viral genome dimerization and packaging in vivo [37, 38, 56]. Indeed, we found that HIV-2 NC and GagΔp6 effectively promote in vitro dimerization of HIV-2 RNA containing DIS and pal dimerization signals (Fig. 5). For HIV-1 Gag, numerous lines of evidence support the involvement of the NC domain in the genomic RNA selection, dimerization, and packaging, but several observations suggest a contribution of the MA domain to these processes [41]. It was found that the presence of either the NC or the MA domain is required for genome packaging during HIV-1 particle assembly [23, 40]. However, a recent study revealed that the MA domain of HIV-1 Gag binds almost exclusively to specific cellular tRNAs [31]. Our data suggest that the MA domain is dispensable for HIV-2 GagΔp6-promoted dimerization, since this process is not supported by HIV-2 MA in vitro. On the other hand, HIV-2 MA binds some cis-acting dimerization and packaging sequences in 5′UTR of HIV-2 RNA (Fig. 2). Therefore, we cannot exclude the participation of the MA domain in HIV-2 genome selection and dimerization in the cell.
Methods
Cloning, expression and purification of recombinant proteins
The HIV-2ROD NC protein was obtained using pGEX-4T-3-NCp8 as described previously [33]. Sequences encoding HIV-2 MA, GagΔp6 (HIV-2ROD isolate) and HIV-1 MA (HIV-1NL4-3 isolate) were PCR amplified from HIV-2 pROD10-EVA232 and pNL4-3-ARP2006 (National Institute for Biological Standards and Control, Centre for AIDS Reagents, UK). PCR products were digested, purified using PureLink® spin columns (Invitrogen), and cloned into a pGEX-4T-3 expression vector. The sequence of each construct was confirmed by DNA sequencing. The glutathione S-transferase (GST) fusion HIV-2 NC, MA, and GagΔp6 and HIV-1 MA recombinant proteins were expressed in One Shot® BL21(DE3)pLysS E. coli (Invitrogen) and purified by affinity chromatography on Glutathione Sepharose (GE Healthcare) as described in the Additional file 5. The GST tag was removed by thrombin cleavage. The purity of proteins was assessed by SDS–PAGE and estimated to be above 90 %. Protein concentrations were determined by their absorption spectrum and protein samples were aliquoted and stored at −80 °C.
DNA and RNA substrates
TAR(−) DNA, corresponding to the trans activation response (TAR) sequences of HIV-1MAL, was 32P-labelled at the 5′-end with [γ-32P]ATP using T4 polynucleotide kinase (Fermentas) and purified using NucAway Spin Columns (Life Technologies). HIV-1 TAR RNA and unmodified human tRNALys3 (referred to here as tRNALys3) were obtained using a PCR-generated template (Additional file 6) and Ambion T7-MEGAshortscript. Transcripts were purified by denaturing gel electrophoresis (8 M urea) in 1 × TBE, followed by elution and ethanol precipitation. The tRNALys3 was 3′-end labelled using [α-32P]pCp and T4 RNA ligase (Fermentas) and purified on G50 columns (GE Healthcare). Templates for in vitro transcription of HIV-2 RNA molecules were obtained by PCR amplification of fragments from the HIV-2 plasmid pROD10-EVA232 using a forward primer containing a T7 promoter sequence (Additional file 6). The RNA molecules were as follows: TARpA (nt +1–188), PBS (nt +197–379), Ψ (nt +380–560), RNA +1–444, 5′UTR (nt +1–560) and RNA +1–891. RNAs were synthesized using T7-MEGAscript (Ambion) and purified using Direct-zol™ RNA MiniPrep (Zymo Research). The integrity of the RNAs was assessed by agarose gel electrophoresis under denaturing conditions. Purified RNA was stored at −20 °C. For some assays, RNA was 3′-end labelled using [α-32P]pCp and T4 RNA ligase (Fermentas) following purification using Direct-zol™ RNA MiniPrep (Zymo Research).
Filter-binding assay
Equilibrium-binding experiments were performed as described previously [59] with the following modifications. Reactions were carried out in binding buffer (20 mM HEPES–KOH pH 7.5, 1 mM MgCl2, 10 µM TCEP, 5 mM β-mercaptoethanol, 10 µM ZnCl2, and 50–500 mM NaCl). The final concentration of RNA was 0.2 nM. The binding reactions were incubated for 25 min at room temperature and then 50 µl of each reaction was filtered and washed with 200 µl of binding buffer containing 50 mM NaCl. After filtration, the membranes were dried and exposed to a phosphoimager screen. Data were analysed using Multigauge (Fuji) and Origin (OriginLab) software.
Hydroxyl radical footprinting and detection of RNA cleavage products
RNA +1–891 was used for footprinting experiments (Additional file 6) and the secondary structure of the 5′UTR within this RNA was confirmed previously [33]. RNA samples (5 pmol) were heated at 95 °C for 1 min and slowly cooled to 4 °C. Subsequently, buffer was added to the final concentration of 40 mM Tris–HCl, pH 8.0, 130 mM KCl, 0.5 mM EDTA, and 5 mM MgCl2, and samples were incubated for 25 min at 37 °C. Folded RNA samples were diluted 20-fold with 20 mM Tris–HCl, pH 8.0, followed by addition of NC, MA, or GagΔp6 (6 μl of 3 µM, 6 µM, or 12 µM protein in the buffer containing 50 mM Tris–HCl, pH 8.0, 1 M NaCl, 6.7 mM β-mercaptoethanol, 2.5 mM DTT, 0.1 mM ZnCl2) to a 70 μl reaction. RNA/protein complexes were formed at 0 °C for 20 min. Footprinting reactions were initiated by applying on the wall of the tube 1 μl of 2.5 mM (NH4)Fe(SO4)2, 50 mM sodium ascorbate, 1.5 % H2O2, and 2.75 mM EDTA, and centrifugating. After 15 s at 24 °C, reactions were quenched by the addition of 20 μl of stop solution containing 0.1 M thiourea and 0.2 M EDTA. RNA were purified using Direct-zol RNA MiniPrep Kit (Zymo Research). For the reverse transcription reactions, a total of 1.5 pmols of RNA was mixed with 2 µl of fluorescently labelled primer 186, 540, or 787 (Additional file 6) [4 μM Cy5 (with reagent) and 6 uM Cy5.5 (without reagent)] and 12 μl of primer-template solutions were incubated at 85 °C for 3 min, 60 °C for 5 min, 35 °C for 5 min, and 50 °C for 2 min. Reverse transcription and sample processing were carried out as previously described [65]. Sequencing ladders were prepared using a Thermo Sequenase Cycle Sequencing Kit (Affymetrix) according to the manufacturer’s protocol. Samples and sequencing ladders were purified using a ZR DNA Sequencing Clean-up Kit (Zymo Research) and analysed on a GenomeLab GeXP Analysis System (Beckman-Coulter). Three to nine repetitions were obtained for each read. Electropherogram peaks were converted to reactivity values using Shapefinder software [66]. Reverse transcription stops in the control reaction were identified as outlying high peaks in the plotted background area. To normalize the data, peak intensity for each nucleotide was divided by the average intensity of the 8 % most reactive peaks excluding outliers. The outliers were defined as greater than 1.5 times the interquartile difference above the 3rd quartile [67]. Normalized data were averaged and nucleotide positions corresponding to reverse transcription stops were excluded from further analysis. Differences between reactivity values for reaction without protein and containing protein were calculated. The consistent drop in reactivity with increasing protein concentration larger than at least 20 % of reactivity value was regarded as a possible binding site.
To estimate number of binding sites within different domains of 5′UTR for HIV-2 GagΔp6, NC and MA, number of residues protected from hydroxyl radical cleavage in the presence of only GagΔp6 and NC were compared to the number of those protected only in the presence of GagΔp6 and MA.
TAR annealing assay
32P-labelled HIV-1 TAR(−) DNA (1 nM) and unlabelled HIV-1 TAR RNA (6 nM) were heat denaturated and folded separately in buffer containing 20 mM Tris–HCl, pH 7.5, 30 mM NaCl, 0.1 mM MgCl2, 10 µM ZnCl2, and 5 mM DTT. Then, the mixture of both oligonucleotides was incubated with increasing concentrations of each protein (0–0.8 µM) at 37 °C for 5 min. The time course assays were conducted at 37 °C using 0.2 µM protein and the samples were removed at the indicated time points. All reactions were quenched with 0.5 volume of stop solution (20 % glycerol, 20 mM EDTA pH 8.0, 0.1 % SDS, 0.25 % bromophenol blue, and 0.4 mg/ml yeast tRNA). Samples were analysed by native PAGE (8 %) in 0.5 × TBE at 4 °C.
Sedimentation assays
32P-labelled HIV-1 TAR(−) DNA (1 nM) was combined with complementary unlabelled TAR RNA in a buffer containing 50 mM Tris pH 7.5, 20 mM NaCl, and 0.2 mM MgCl2. Reactions (10 µl) were incubated with increasing protein concentrations (0–0.8 µM) at 37 °C for 5 min. Subsequently, the mixtures were centrifuged at 11,400 rpm for 20 min. Supernatants (2 µl) were collected and subjected to scintillation counting.
tRNALys3 annealing assay
32P-labelled tRNALys3 (2 nM) and unlabelled +1–560 HIV-2 RNA (10 nM) were refolded in 50 mM Tris–HCl, pH 7.5 by heating at 95 °C for 1 min and slow cooling to 37 °C, followed by addition of MgCl2 to 10 mM and placement on ice. The annealing buffer contained 50 mM Tris–HCl pH 7.5, 5 mM DTT, and 1 mM MgCl2, and different NaCl concentrations of 20, 50, or 150 mM. The mixture was incubated at 37 °C for 10 min, followed by addition of protein and further incubation for 10 min. The time course assays were conducted at 37 °C using 1.5 or 3 µM protein and the aliquots were removed at the indicated time points. All reactions were quenched by incubation with 1 % (w/v) SDS at room temperature for 5 min. The samples were phenol/chloroform-extracted, mixed with loading buffer (50 % glycerol with dyes), and separated on 1.4 % SDS-agarose gel in 1 × TBE at room temperature.
RNA dimerization
The unlabelled +1–444 HIV-2 (400 nM) spiked with the trace amount of the same 32P-labelled transcript was heat denatured in 50 mM Tris–HCl pH 7.5, 40 mM KCl, 150 mM NaCl, and 0.1 mM MgCl2. The mixture was slowly cooled to 37 °C and placed on ice, followed by addition of different concentrations of proteins. The dimerization was allowed to proceed at 37 °C for 30 min. The time course dimerization assays were conducted at 37 °C using 6 µM protein and the aliquots were removed at the indicated time points. All dimerization reactions were quenched by incubation with 1 % (w/v) SDS at room temperature for 5 min, phenol/chloroform-extracted, and mixed with loading buffer (50 % glycerol with dyes). The products were separated on 1 % agarose gel in 1 × TBE at room temperature.
All gels were autoradiographed and quantitatively analysed by phosphorimaging using a FLA-5100 phosphorimager with MultiGaugeV 3.0 software (FujiFilm). The obtained data were analysed using Origin (OriginLab) software. All graphs represent averaged data from three or more independent experiments with standard deviations indicated. In all cases, at least three independent experiments were performed, and the data presented are representative of the whole.
Authors’ contributions
KPW designed the study. KPW and KJP performed the experiments, analysed and interpreted the data and wrote the manuscript. LB and MB performed the experiments, analysed the data and helped to draft the manuscript. RWA provided important suggestions to the manuscript and helped to secure funding. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by National Science Centre Poland [2011/01/D/NZ1/03478 to KPW, 2012/06/A/ST6/00384 to RWA] and Ministry of Science and Higher Education Poland [0397/IP1/2011/71 to KPW, 0492/IP1/2013/72 to KJP], and European Union Regional Development Fund and the Polish Ministry of Science and Higher Education, under the Leading National Research Centre (KNOW) Program. Funding for open access charge National Science Centre Poland [2012/06/A/ST6/00384].
Competing interests
The authors declare that they have no competing interests.
Additional files
10.1186/s12977-016-0245-1 Analysis of the nucleotides identity at the HIV-2 GagΔp6, NC and MA binding sites within HIV-2 5′UTR RNA.
10.1186/s12977-016-0245-1 The representative electrophoretic analyses of the TAR RNA/TAR(−) DNA and tRNALys3/HIV-2 5′UTR annealing assays, and +1–444 RNA dimerization assays in the presence of HIV-2 GagΔp6, NC, MA and HIV-1 MA.
10.1186/s12977-016-0245-1 MA proteins of HIV-1 (PDB id. 2HMX) and HIV-2 (PDB id. 2K4E) represented by their molecular surfaces.
10.1186/s12977-016-0245-1 Movie file. 3D view of MA proteins of HIV-1 (PDB id. 2HMX) and HIV-2 (PDB id. 2K4E) represented by their molecular surfaces.
10.1186/s12977-016-0245-1 Detailed description of HIV-2 NC, MA, GagΔp6 and HIV-1 MA proteins expression and purification.
10.1186/s12977-016-0245-1 Table containing sequences of the primers used in this study.
Contributor Information
Katarzyna Pachulska-Wieczorek, Phone: +48 61 8528503, Email: kasiapw@ibch.poznan.pl.
Leszek Błaszczyk, Email: blaszcz@ibch.poznan.pl.
Marcin Biesiada, Email: biesiada@ibch.poznan.pl.
Ryszard W. Adamiak, Email: adamiakr@ibch.poznan.pl
Katarzyna J. Purzycka, Phone: +48 61 8528503, Email: purzycka@ibch.poznan.pl
References
- 1.Bell NM, Lever AM. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol. 2013;21(3):136–144. doi: 10.1016/j.tim.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Rein A. Nucleic acid chaperone activity of retroviral Gag proteins. RNA Biol. 2010;7(6):700–705. doi: 10.4161/rna.7.6.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rein A, Datta SA, Jones CP, Musier-Forsyth K. Diverse interactions of retroviral Gag proteins with RNAs. Trends Biochem Sci. 2011;36(7):373–380. doi: 10.1016/j.tibs.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganser-Pornillos BK, Yeager M, Sundquist WI. The structural biology of HIV assembly. Curr Opin Struct Biol. 2008;18(2):203–217. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci USA. 2006;103(30):11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chukkapalli V, Ono A. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J Mol Biol. 2011;410(4):512–524. doi: 10.1016/j.jmb.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayo K, Huseby D, McDermott J, Arvidson B, Finlay L, Barklis E. Retrovirus capsid protein assembly arrangements. J Mol Biol. 2003;325(1):225–237. doi: 10.1016/S0022-2836(02)01176-2. [DOI] [PubMed] [Google Scholar]
- 8.Muriaux D, Darlix JL. Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol. 2010;7(6):744–753. doi: 10.4161/rna.7.6.14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell S, Rein A. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J Virol. 1999;73(3):2270–2279. doi: 10.1128/jvi.73.3.2270-2279.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swanstrom R, Wills JW. Synthesis, assembly, and processing of viral proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- 11.Levin JG, Guo J, Rouzina I, Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog Nucleic Acid Res Mol Biol. 2005;80:217–286. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 12.Mannioui A, Nelson E, Schiffer C, Felix N, Le Rouzic E, Benichou S, et al. Human immunodeficiency virus type 1 KK26-27 matrix mutants display impaired infectivity, circularization and integration but not nuclear import. Virology. 2005;339(1):21–30. doi: 10.1016/j.virol.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Bukrinskaya A. HIV-1 matrix protein: a mysterious regulator of the viral life cycle. Virus Res. 2007;124(1–2):1–11. doi: 10.1016/j.virusres.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Guo F, Saadatmand J, Niu M, Kleiman L. Roles of Gag and NCp7 in facilitating tRNA(Lys)(3) Annealing to viral RNA in human immunodeficiency virus type 1. J Virol. 2009;83(16):8099–8107. doi: 10.1128/JVI.00488-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rye-McCurdy TD, Nadaraia-Hoke S, Gudleski-O’Regan N, Flanagan JM, Parent LJ, Musier-Forsyth K. Mechanistic differences between nucleic acid chaperone activities of the Gag proteins of Rous sarcoma virus and human immunodeficiency virus type 1 are attributed to the MA domain. J Virol. 2014;88(14):7852–7861. doi: 10.1128/JVI.00736-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu T, Datta SA, Mitra M, Gorelick RJ, Rein A, Levin JG. Fundamental differences between the nucleic acid chaperone activities of HIV-1 nucleocapsid protein and Gag or Gag-derived proteins: biological implications. Virology. 2010;405(2):556–567. doi: 10.1016/j.virol.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones CP, Datta SA, Rein A, Rouzina I, Musier-Forsyth K. Matrix domain modulates HIV-1 Gag’s nucleic acid chaperone activity via inositol phosphate binding. J Virol. 2011;85(4):1594–1603. doi: 10.1128/JVI.01809-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng YX, Campbell S, Harvin D, Ehresmann B, Ehresmann C, Rein A. The human immunodeficiency virus type 1 Gag polyprotein has nucleic acid chaperone activity: possible role in dimerization of genomic RNA and placement of tRNA on the primer binding site. J Virol. 1999;73(5):4251–4256. doi: 10.1128/jvi.73.5.4251-4256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajkowitsch L, Chen D, Stampfl S, Semrad K, Waldsich C, Mayer O, et al. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007;4(3):118–130. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- 20.Alfadhli A, McNett H, Tsagli S, Bachinger HP, Peyton DH, Barklis E. HIV-1 matrix protein binding to RNA. J Mol Biol. 2011;410(4):653–666. doi: 10.1016/j.jmb.2011.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alfadhli A, Still A, Barklis E. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol. 2009;83(23):12196–12203. doi: 10.1128/JVI.01197-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katoh I, Kyushiki H, Sakamoto Y, Ikawa Y, Yoshinaka Y. Bovine leukemia virus matrix-associated protein MA(p15): further processing and formation of a specific complex with the dimer of the 5′-terminal genomic RNA fragment. J Virol. 1991;65(12):6845–6855. doi: 10.1128/jvi.65.12.6845-6855.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ott DE, Coren LV, Gagliardi TD. Redundant roles for nucleocapsid and matrix RNA-binding sequences in human immunodeficiency virus type 1 assembly. J Virol. 2005;79(22):13839–13847. doi: 10.1128/JVI.79.22.13839-13847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purohit P, Dupont S, Stevenson M, Green MR. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA. 2001;7(4):576–584. doi: 10.1017/S1355838201002023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun M, Grigsby IF, Gorelick RJ, Mansky LM, Musier-Forsyth K. Retrovirus-specific differences in matrix and nucleocapsid protein-nucleic acid interactions: implications for genomic RNA packaging. J Virol. 2014;88(2):1271–1280. doi: 10.1128/JVI.02151-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Norris KM, Mansky LM. Involvement of the matrix and nucleocapsid domains of the bovine leukemia virus Gag polyprotein precursor in viral RNA packaging. J Virol. 2003;77(17):9431–9438. doi: 10.1128/JVI.77.17.9431-9438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshmukh L, Ghirlando R, Clore GM. Conformation and dynamics of the Gag polyprotein of the human immunodeficiency virus 1 studied by NMR spectroscopy. Proc Natl Acad Sci USA. 2015;112(11):3374–3379. doi: 10.1073/pnas.1501985112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart-Maynard KM, Cruceanu M, Wang F, Vo MN, Gorelick RJ, Williams MC, et al. Retroviral nucleocapsid proteins display nonequivalent levels of nucleic acid chaperone activity. J Virol. 2008;82(20):10129–10142. doi: 10.1128/JVI.01169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pachulska-Wieczorek K, Stefaniak AK, Purzycka KJ. Similarities and differences in the nucleic acid chaperone activity of HIV-2 and HIV-1 nucleocapsid proteins in vitro. Retrovirology. 2014;11:54. doi: 10.1186/1742-4690-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Souza V, Summers MF. How retroviruses select their genomes. Nat Rev Microbiol. 2005;3(8):643–655. doi: 10.1038/nrmicro1210. [DOI] [PubMed] [Google Scholar]
- 31.Kutluay SB, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, et al. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell. 2014;159(5):1096–1109. doi: 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsui T, Tanaka T, Endoh H, Sato K, Tanaka H, Miyauchi E, et al. The RNA recognition mechanism of human immunodeficiency virus (HIV) type 2 NCp8 is different from that of HIV-1 NCp7. Biochemistry. 2009;48(20):4314–4323. doi: 10.1021/bi802364b. [DOI] [PubMed] [Google Scholar]
- 33.Purzycka KJ, Pachulska-Wieczorek K, Adamiak RW. The in vitro loose dimer structure and rearrangements of the HIV-2 leader RNA. Nucleic Acids Res. 2011;39(16):7234–7248. doi: 10.1093/nar/gkr385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ni N, Nikolaitchik OA, Dilley KA, Chen J, Galli A, Fu W, et al. Mechanisms of human immunodeficiency virus type 2 RNA packaging: efficient trans packaging and selection of RNA copackaging partners. J Virol. 2011;85(15):7603–7612. doi: 10.1128/JVI.00562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dilley KA, Ni N, Nikolaitchik OA, Chen J, Galli A, Hu WS. Determining the frequency and mechanisms of HIV-1 and HIV-2 RNA copackaging by single-virion analysis. J Virol. 2011;85(20):10499–10508. doi: 10.1128/JVI.05147-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaye JF, Lever AM. Nonreciprocal packaging of human immunodeficiency virus type 1 and type 2 RNA: a possible role for the p2 domain of Gag in RNA encapsidation. J Virol. 1998;72(7):5877–5885. doi: 10.1128/jvi.72.7.5877-5885.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaye JF, Lever AM. Human immunodeficiency virus types 1 and 2 differ in the predominant mechanism used for selection of genomic RNA for encapsidation. J Virol. 1999;73(4):3023–3031. doi: 10.1128/jvi.73.4.3023-3031.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffin SD, Allen JF, Lever AM. The major human immunodeficiency virus type 2 (HIV-2) packaging signal is present on all HIV-2 RNA species: cotranslational RNA encapsidation and limitation of Gag protein confer specificity. J Virol. 2001;75(24):12058–12069. doi: 10.1128/JVI.75.24.12058-12069.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berkowitz RD, Ohagen A, Hoglund S, Goff SP. Retroviral nucleocapsid domains mediate the specific recognition of genomic viral RNAs by chimeric Gag polyproteins during RNA packaging in vivo. J Virol. 1995;69(10):6445–6456. doi: 10.1128/jvi.69.10.6445-6456.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ott DE, Coren LV, Chertova EN, Gagliardi TD, Nagashima K, Sowder RC, 2nd, et al. Elimination of protease activity restores efficient virion production to a human immunodeficiency virus type 1 nucleocapsid deletion mutant. J Virol. 2003;77(10):5547–5556. doi: 10.1128/JVI.77.10.5547-5556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parent LJ, Gudleski N. Beyond plasma membrane targeting: role of the MA domain of Gag in retroviral genome encapsidation. J Mol Biol. 2011;410(4):553–564. doi: 10.1016/j.jmb.2011.04.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.L’Hernault A, Weiss EU, Greatorex JS, Lever AM. HIV-2 genome dimerization is required for the correct processing of Gag: a second-site reversion in matrix can restore both processes in dimerization-impaired mutant viruses. J Virol. 2012;86(10):5867–5876. doi: 10.1128/JVI.00124-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saad JS, Ablan SD, Ghanam RH, Kim A, Andrews K, Nagashima K, et al. Structure of the myristylated human immunodeficiency virus type 2 matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J Mol Biol. 2008;382(2):434–447. doi: 10.1016/j.jmb.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci USA. 1996;93(7):3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF. Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science. 1998;279(5349):384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 46.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol. 1994;244(2):198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 47.Mirambeau G, Lyonnais S, Gorelick RJ. Features, processing states, and heterologous protein interactions in the modulation of the retroviral nucleocapsid protein function. RNA Biol. 2010;7(6):724–734. doi: 10.4161/rna.7.6.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chukkapalli V, Oh SJ, Ono A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci USA. 2010;107(4):1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Datta SA, Zhao Z, Clark PK, Tarasov S, Alexandratos JN, Campbell SJ, et al. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J Mol Biol. 2007;365(3):799–811. doi: 10.1016/j.jmb.2006.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inlora J, Collins DR, Trubin ME, Chung JY, Ono A. Membrane binding and subcellular localization of retroviral Gag proteins are differentially regulated by MA interactions with phosphatidylinositol-(4,5)-bisphosphate and RNA. MBio. 2014;5(6):e02202. doi: 10.1128/mBio.02202-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chukkapalli V, Inlora J, Todd GC, Ono A. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J Virol. 2013;87(12):7155–7159. doi: 10.1128/JVI.00075-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pachulska-Wieczorek K, Purzycka KJ, Adamiak RW. New, extended hairpin form of the TAR-2 RNA domain points to the structural polymorphism at the 5′ end of the HIV-2 leader RNA. Nucleic Acids Res. 2006;34(10):2984–2997. doi: 10.1093/nar/gkl373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dirac AM, Huthoff H, Kjems J, Berkhout B. Regulated HIV-2 RNA dimerization by means of alternative RNA conformations. Nucleic Acids Res. 2002;30(12):2647–2655. doi: 10.1093/nar/gkf381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rajkowitsch L, Semrad K, Mayer O, Schroeder R. Assays for the RNA chaperone activity of proteins. Biochem Soc Trans. 2005;33(Pt 3):450–456. doi: 10.1042/BST0330450. [DOI] [PubMed] [Google Scholar]
- 55.Stoylov SP, Vuilleumier C, Stoylova E, De Rocquigny H, Roques BP, Gerard D, et al. Ordered aggregation of ribonucleic acids by the human immunodeficiency virus type 1 nucleocapsid protein. Biopolymers. 1997;41(3):301–312. doi: 10.1002/(SICI)1097-0282(199703)41:3<301::AID-BIP5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 56.Lanchy JM, Ivanovitch JD, Lodmell JS. A structural linkage between the dimerization and encapsidation signals in HIV-2 leader RNA. RNA. 2003;9(8):1007–1018. doi: 10.1261/rna.5590603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dirac AM, Huthoff H, Kjems J, Berkhout B. The dimer initiation site hairpin mediates dimerization of the human immunodeficiency virus, type 2 RNA genome. J Biol Chem. 2001;276(34):32345–32352. doi: 10.1074/jbc.M103462200. [DOI] [PubMed] [Google Scholar]
- 58.Bjellqvist B, Basse B, Olsen E, Celis JE. Reference points for comparisons of 2-dimensional maps of proteins from different human cell-types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis. 1994;15(3–4):529–539. doi: 10.1002/elps.1150150171. [DOI] [PubMed] [Google Scholar]
- 59.Nishida Y, Pachulska-Wieczorek K, Blaszczyk L, Saha A, Gumna J, Garfinkel DJ, et al. Ty1 retrovirus-like element Gag contains overlapping restriction factor and nucleic acid chaperone functions. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Webb JA, Jones CP, Parent LJ, Rouzina I, Musier-Forsyth K. Distinct binding interactions of HIV-1 Gag to Psi and non-Psi RNAs: implications for viral genomic RNA packaging. RNA. 2013;19(8):1078–1088. doi: 10.1261/rna.038869.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andersen ES, Contera SA, Knudsen B, Damgaard CK, Besenbacher F, Kjems J. Role of the trans-activation response element in dimerization of HIV-1 RNA. J Biol Chem. 2004;279(21):22243–22249. doi: 10.1074/jbc.M314326200. [DOI] [PubMed] [Google Scholar]
- 62.Song R, Kafaie J, Laughrea M. Role of the 5′ TAR stem–loop and the U5-AUG duplex in dimerization of HIV-1 genomic RNA. Biochemistry. 2008;47(10):3283–3293. doi: 10.1021/bi7023173. [DOI] [PubMed] [Google Scholar]
- 63.Heilman-Miller SL, Wu T, Levin JG. Alteration of nucleic acid structure and stability modulates the efficiency of minus-strand transfer mediated by the HIV-1 nucleocapsid protein. J Biol Chem. 2004;279(42):44154–44165. doi: 10.1074/jbc.M401646200. [DOI] [PubMed] [Google Scholar]
- 64.Soto-Rifo R, Limousin T, Rubilar PS, Ricci EP, Decimo D, Moncorge O, et al. Different effects of the TAR structure on HIV-1 and HIV-2 genomic RNA translation. Nucleic Acids Res. 2012;40(6):2653–2667. doi: 10.1093/nar/gkr1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Purzycka KJ, Legiewicz M, Matsuda E, Eizentstat LD, Lusvarghi S, Saha A, et al. Exploring Ty1 retrotransposon RNA structure within virus-like particles. Nucleic Acids Res. 2013;41(1):463–473. doi: 10.1093/nar/gks983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vasa SM, Guex N, Wilkinson KA, Weeks KM, Giddings MC. ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA. 2008;14(10):1979–1990. doi: 10.1261/rna.1166808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Low JT, Weeks KM. SHAPE-directed RNA secondary structure prediction. Methods. 2010;52(2):150–158. doi: 10.1016/j.ymeth.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]