

Abstract
The GCN5-related N-acetyltransferases family (GNAT) is an important family of proteins that includes more than 100000 members among eukaryotes and prokaryotes. Acetylation appears as a major regulatory post-translational modification and is as widespread as phosphorylation. N-Acetyltransferases transfer an acetyl group from acetyl-CoA to a large array of substrates, from small molecules such as aminoglycoside antibiotics to macromolecules. Acetylation of proteins can occur at two different positions, either at the amino-terminal end (αN-acetylation) or at the ε-amino group (εN-acetylation) of an internal lysine residue. GNAT members have been classified into different groups on the basis of their substrate specificity, and in spite of a very low primary sequence identity, GNAT proteins display a common and conserved fold. This Current Topic reviews the different classes of bacterial GNAT proteins, their functions, their structural characteristics, and their mechanism of action.
Graphical abstract

Histone acetylation and aminoglycoside resistance caused by acetylation were both discovered 50 years ago.1–3 Even though both discoveries were reported within a year of each other, most early work focused on histone acetylation and its consequences with respect to gene regulation.4,5 With the development of high-resolution mass spectrometry and genome sequencing, it has become clear that acetylation of proteins has emerged as a major modiffer of their activities in eukaryotes and prokaryotes.6 Acetylation can, however, occur in both non-enzymatic and enzyme-catalyzed processes. This is due to the extremely exothermic nature of the acetylation reaction. Acetyl-CoA (AcCoA) has an acetyl transfer potential higher than the phosphoryl transfer potential of ATP, and the acetylated amide product is extremely stable and considerably more so than the phosphomonoester product of the kinases.7 Thus, the high intracellular concentration of acyl-CoA’s, including AcCoA, will nonenzymatically react with any protein lysine residue with a reduced pK value. It is thus important in defining the “acetylome” to ensure that the acetylated protein be generated enzymatically by the action of an acetyltransferase and deacetylated by an appropriate deacetylase and that there be a catalytic or structural phenotype associated with the modification. Without this, the likelihood is that the protein simply contains a lysine residue that is accessible and is in an environment that reduces its pK value.
The use of the term GNAT to describe N-acetyltransferases was first derived from the yeast GCN5, which was shown to be a histone acetyltransferase.4,8 However, the first GNAT member was described by Okamoto in 1965 as a bacterial aminoglycoside acetyltransferase conferring kanamycin and gentamicin antibiotic resistance.3 Many aminoglycoside acetyltransferases have since been characterized and are clinically one of the most common reasons for aminoglycoside resistance.9 The GNAT superfamily currently contains more than 100000 members found in eukaryotes, prokaryotes, and archea.10
Despite the fact that all the members of the GNAT superfamily possess a very low primary sequence identity, their structures display a particularly conserved fold.11,12 The core secondary elements of all GNAT structures consist of six or seven β-strands and four α-helices (β0–β1–α1–α2–β2–β3–β4–α3–-β5–α4–β6) (Figure 1). The loop connecting β4 to α3, the pyrophosphate binding loop, appears as a characteristic structural feature of the GNAT superfamily, and the sequence is highly conserved between members (R/Q-X-X-G-X-A/G).13 The pyrophosphate binding loop plays a central role in AcCoA binding because it forms several hydrogen bonds between a number of amide backbone nitrogens and the pyrophosphate group of AcCoA.11,12 Another distinctive feature found in most structures of the GNAT members is a “β-bulge” located on β4, breaking apart parallel β-strands β4 and β5, resulting in a V-like shape that serves as a binding site for AcCoA. The pantothenate “arm” of AcCoA interacts with the C-terminal end of β4, while there are few interactions observed between the adenine ring and the active site. Typically, GNAT proteins function as dimers in solution, although we will discuss examples in which a monomeric unit is composed of two fused GNAT domains.
Figure 1.
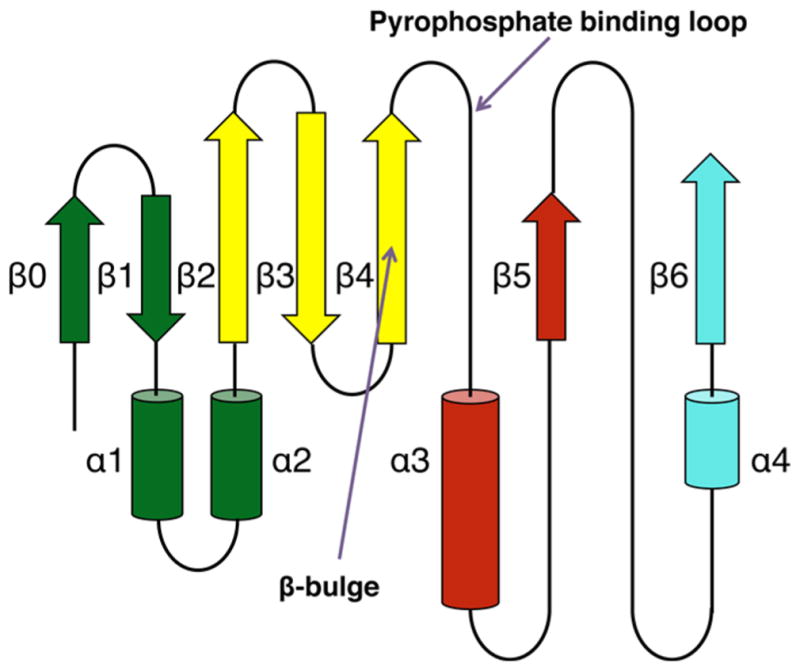
Topology scheme of the GNAT proteins. Starting at the N-terminal end, the secondary structure elements are colored dark green (β0, β1, α1, and α2), yellow (β2–β4), red (α3 and β5), and cyan (α4 and β6).
All members of the GNAT superfamily catalyze bireactant reactions, and the acetyltransferase reaction can proceed via either a ping-pong or sequential mechanism.12 There is a single example of a reaction occurring via a ping-pong kinetic mechanism. The yeast ESA1 is a member of the MYST subfamily of histone acetyltransferases, and there are compelling structural and mutagenesis data that support the kinetic necessity of an acetylated enzyme intermediate (C304 in the case of ESA1).14 For all other GNAT superfamily members, sequential kinetic mechanisms have been observed. Once both substrates are bound, a residue located in the active site, typically an aspartate or glutamate, acts as a general base and deprotonates the amine prior to nucleophilic attack on the thioester carbonyl. Residues acting as a general base have been identified by site-directed mutagenesis studies, and many ternary complex structures have been determined in recent years.11,12 In some cases, the deprotonation step involves an intervening water molecule connecting the side chain of a general base to the amine via hydrogen bonding.15,16 Thus, with the exception of the MYST subfamily of histone acetyltransferases or those GNATs with an appropriately positioned active site cysteine, the sequential mechanism involving the formation of a ternary complex is observed in a majority of the cases.11,12
GNAT members can acetylate the amino group of a large range of substrates and so have been classified into three groups: (1) small molecule acetyltransferases, such as aminoglycosides and mycothiol; (2) peptide acetyltransferases such as the peptidoglycan that is part of the bacterial cell wall; and (3) protein acetyltransferases, for instance, the histone family. In this Current Topic, we will focus on the classes of GNAT commonly found in bacteria, because the histone acetyltransferases have been reviewed in detail.17–19
1. SMALL MOLECULES
1.1. Aminoglycosides N-Acetyltransferases
Amino-glycosides are broad-spectrum antibacterial compounds containing typically one aminocyclitol ring (the most common being 2-deoxystreptamine) linked by a glycosidic bond to one or more amino sugars (Figure 2). The first aminoglycoside to be discovered was streptomycin; it was isolated from Streptomyces griseus and originally used for the treatment of tuberculosis.20 Typically, aminoglycosides work synergistically with β-lactams or other antibiotics that enhance their uptake.21 More recently, aminoglycosides have also been investigated for the treatment of certain genetic disorders, for instance, cystic fibrosis22,23 or Duchenne muscular dystrophy,24,25 as well as HIV therapies.26–28 Aminoglycosides inhibit protein translation in bacteria by binding to the 16S rRNA of the 30S ribosome.29,30 The site of binding and mechanism of inhibition has been significantly clarified with the elucidation of the structures of 16S rRNA fragments and the 30S ribosome subunit with various bound AGs.31–37
Figure 2.

General structures of various aminoglycosides. The amino-cyclitol (2-deoxystreptamine) is colored blue. Arrows indicate the positions that are commonly modified by AACs. (A) 4,6-Disubstituted deoxystreptamine aminoglycoside. Examples cited in this review are butirosin, paromomycin, and neomycin. (B) 4,5-Disubstituted deoxystreptamine aminoglycoside. Position 2′ can be either an amino group (kanamycin B, gentamicin C1a and C1, etc.) or a hydroxyl group (kanamycin A, amikacin, etc.).
Drug resistance to aminoglycosides emerged many decades ago. Several mechanisms have been identified, including mutation or methylation of certain 16S rRNA nucleotides involved in aminoglycoside binding, decreased aminoglycoside uptake, and chemical modification of aminoglycosides by aminoglycoside-modifying enzymes.38,39 Enzymatic modification is the most common and clinically relevant, although a combination of different mechanisms is sometimes observed.38–40 Three different classes of enzymes modify amino-glycosides: ATP-dependent phosphotransferases (O-phosphorylation, APH), ATP-dependent nucleotidyltransferases (O-nucleotidylation, ANT), and acetyl-CoA-dependent N-acetyl-transferases (N-acetylation, AAC), which are members of the GNAT family.
Aminoglycoside N-acetyltransferases were the first members of the GNAT family to be identified.3 Shaw et al. have established a nomenclature for describing the different aminoglycoside N-acetyltransferases.41 The enzymes are abbreviated by a three-letter code, AAC, followed by a number in parentheses that indicates the position of the acetylated amino group (Figure 2). A Roman numeral distinguishes between the different resistance profiles, and a lowercase letter specifically identifies the associated gene. Four types of AACs have been identified to date depending on the site of modification: AAC(1) when the 1-NH2 is acetylated, AAC(3), AAC(2′), and AAC(6′). Examples in each category of AAC will be provided in this Current Topic.
AAC(1) was first identified in Escherichia coli (E. coli) and Actinomycete strains.42,43 On the basis of high-performance liquid chromatography analysis, Lovering et al. demonstrated that E. coli AAC(1) could catalyze the transfer of one acetyl group from AcCoA to the N1 position of apramycin, lividomycin, paromomycin, and butirosin.42 Additionally, the enzyme could diacetylate neomycin and ribostamycin. However, the enzyme does not appear to be clinically relevant. Later, Sunada et al. identified another AAC(1) from an actinomycetes strain that predominantly acetylates paromomycin at the 1-NH2 position; however, the activity of the antibiotic was not significantly affected by this modification.43
The AAC(3) family includes nine subclasses of enzymes (I–X), but subclass V was later excluded after DNA analysis revealed that the genes encoding AAC(3)-II and -V were identical and conferred resistance to the same antibiotics.41 The subclass I group can be subdivided into five groups (a–e) exhibiting resistance to gentamicin, sisomicin, and fortimicin.41 Gentamicin acetyltransferase from E. coli catalyzing the acetylation of gentamicin at the 3-NH2 position was the first purified and kinetically characterized aminoglycoside-modifying enzyme.44–46 Kinetic analysis using a spectrophotometric assay revealed that the enzyme utilizes a random bi-bi mechanism. Serratia marcescens (S. marcescens) AAC(3)-Ia was the first aminoglycoside acetyltransferase GNAT structure to be determined, simultaneously with the first structure of the histone acetyltransferase, Hat1.47,48 The enzyme harboring a C-terminal truncation was cocrystallized with CoA, and the structure was determined to 2.3 Å by multiwavelength anomalous dispersion.47 The structure exhibits the typical GNAT fold that includes four α-helices and six β-strands (Figure 3A). The pyrophosphate group of CoA was found to form hydrogen bonds with the conserved motif R/Q-X-X-G-X-A/G, while the pantothenate group interacts with the end of strand β4. Although a dimer was observed in the asymmetric unit, the existence of a dimer in solution was not conclusively determined. AAC(3)-III enzymes confer resistance to gentamicin, kanamycin, neomycin, paromomycin, and tobramycin.41 Recently, one enzyme of this subclass, AAC(3)-IIIb from Pseudomonas aeruginosa, has been extensively characterized thermodynamically and kinetically to provide insights into the binding mode of CoA and aminoglycosides.49–51
Figure 3.

Structures of aminoglycoside N-acetyltransferases. The secondary structure elements are colored like the topology scheme presented in Figure 1. The substrates or products bound are rendered as sticks with white carbons. (A) Structure of S. marcescens AAC(3)-Ia (PDB entry 1BO4). (B) Structure of M. tuberculosis AAC(2′)-Ic in complex with CoA and kanamycin A (KAN) (PDB entry 1M4I). (C) Superimposition of the S. enterica AAC(6′)-Ib structure in complex with CoA and its variant AAC(6′)-Ib11 (PDB entries 2PRB and 2PR8, respectively). The α-helical flap (highlighted with an arrow in each structure) located above the aminoglycoside binding site is colored purple in the AAC(6′)-Ib structure and orange in the AAC(6′)-Ib11 structure.
The AAC(2′) family includes only one subclass. The enzymes generally promote the acetylation of dibekacin, gentamicin, kanamycin, netilmicin, and tobramycin.41 Initially, AAC(2′)-Ia was identified in Providencia stuartii in which overexpression of the acetyltransferase is observed in the presence of aminoglycosides.52,53 The other AAC(2′) enzymes are found in only mycobacteria, including AAC(2′)-Ib in Mycobacterium fortuitum,54 AAC(2′)-Ic in Mycobacterium tuberculosis (M. tuberculosis), AAC(2′)-Id in Mycobacterium smegmatis (M. smegmatis), and AAC(2′)-Ie in Mycobacterium leprae.55 AAC(2′)-Ic from M. tuberculosis has been very well characterized, both enzymatically and structurally.56,57 Kinetic analysis reveals that the enzyme can acetylate the amino group at position 2′ of a broad range of AGs.56 An interesting feature of AAC(2′)-Ic specificity is the demonstration that the enzyme can also perform O-acetylation and acetylate AGs such as kanamycin A or amikicin, each of which contains a 2′-hydroxyl group. Dead-end inhibition studies indicate that the acetylation reaction, like other AACs, follows a sequential kinetic mechanism in which AcCoA binds first, promoting the binding of the AG. The crystal structure of AAC(2′)-Ic was determined in an apo form and in complex with CoA and various aminoglycosides (kanamycin A, ribostamycin, and tobramycin) (Figure 3B).57 The overall fold of the enzyme, in addition to the presence of the characteristic “β-bulge” (G83) and pyrophosphate binding loop, confirms that it belongs to the GNAT family. As observed for other GNAT proteins, AAC(2′)-Ic forms a dimer. The binding mode of CoA is similar to that observed in GNAT structures. However, this was the first reported structure of an aminoglycoside N-acetyltransferase in a ternary complex with CoA and aminoglycoside bound. The different aminoglycosides bind within a proximal site to the CoA binding pocket, in a region containing acidic residues and a tryptophan residue (W181) that are conserved among the AAC(2′) enzymes. Most of the interactions occur with the amino and hydroxyl groups from the 2-deoxystreptamine ring, which explains the specificity of the AAC(2′) enzymes for aminoglycosides containing this ring. The 2′-substituent, either an amino group or a hydroxyl group, is located 3.6–4.1 Å from the thiol group of CoA, supporting the direct nucleophilic attack mechanism.
AAC(6′) is the most common class of aminoglycoside N-acetyltransferases because the 6′-substituent is important in the binding of aminoglycosides to the 30S ribosome subunit.31,33,36 Two different classes of enzymes have been determined on the basis of their ability to confer resistance, and both can acetylate netilmicin, tobramycin, and 2′-N-ethylnetilmicin. However, type I enzymes confer resistance to amikacin and gentamicin C1a and C2 but not to gentamicin C1, while type II enzymes exhibit activity with all types of gentamicin, but not amikacin.58 AAC(6′)-I is the most common aminoglycoside N-acetyltransferase,38 and an extensive list of enzymes belonging to the AAC(6′) family has recently been reviewed by Ramirez et al.39 AAC(6′)-Ii from Enterococcus faecium59–62 and AAC(6′)-Iy from Salmonella enterica (S. enterica)63–65 were the first AAC(6′) enzymes to be extensively characterized, both kinetically and structurally. Interesting examples among the members of the AAC(6′) family are AAC(6′)-Ib and its two variants AAC(6′)-Ib-cr, a bifunctional enzyme that confers resistance to both aminoglycosides and fluoroquinolones,66 and AAC(6′)-Ib11, an enzyme that displays activity with both amikacin and gentamicin.67 This enzyme is the most widespread AG-modifying enzyme and is found in more than 70% of Gram-negative clinical isolates that produce AAC(6′)-I enzymes.68 Kinetic analysis and dead-end inhibition studies demonstrate that both AAC(6′)-Ib and AAC(6′)-Ib-cr use an ordered sequential kinetic mechanism in which AcCoA binds first, followed by the addition of either aminoglycoside or fluoroquinolone.69 Both enzymes exhibit a bell-shaped pH–activity profile, suggesting that they use a catalytic base and a catalytic acid in the reaction. In the case of AAC(6′)-Ib-cr, the pH optima are different depending on the substrate used, either aminoglycoside or fluoroquinolone. Both AAC(6′)-Ib and AAC(6′)-Ib11 structures have been determined.69,70 Both structures exhibit a characteristic GNAT fold. Surprisingly, AAC(6′)-Ib and AAC(6′)-Ib-cr are monomeric in solution, while AAC(6′)-Ib11 is in a monomer/dimer equilibrium.70 Variant AAC(6′)-Ib11 possesses two mutations compared to wild-type AAC(6′)-Ib (Q106L and L107S). These two mutations are located in the vicinity of the binding pocket of AcCoA, more specifically where the pantothenate arm is positioned, and lead to a severe structural change compared to the AAC(6′)-Ib structure (Figure 3C).70 The central β-strand is disrupted, and the α-helical flap, which forms a lid above the aminoglycoside binding site in AAC(6′)-Ib, is more flexible. This structural difference between the wild-type enzyme and its variant could explain the substrate specificity of both enzymes. Minor differences in the binding interactions were observed depending on the AG bound, either kanamycin or 4,5-disubstituted aminoglycosides (ribostamycin and paromomycin).69 On the basis of the positions of the substrates, the structures of AAC(6′)-Ib ternary complexes suggest a direct nucleophilic attack with D115 and Y164 acting as the proposed catalytic base and acid.
It should also be noted that AAC enzymes can be fused into bifunctional enzymes and can be associated with either another AAC enzyme [e.g., AAC(3)-Ib/AAC(6′)-Ib′]71 or another AG-modifying enzyme [e.g., AAC(6′)/APH(2′′)].72,73
1.2. Mycothiol Transferase
Mycothiol (MSH), whose function is analogous to that of glutathione in eukaryotes and eubacteria, is found only in actinomycetes. MSH is the major low-molecular weight thiol and serves as a cellular antioxidant to protect the bacteria against oxidative damage and electrophilic toxins.74,75 High levels of MSH are found in mycobacteria, especially in M. tuberculosis.76 Four unique enzymes conduct the biosynthesis of MSH in five steps (Figure 4A).77 First, N-acetylglucosamine is transferred from UDP-N-acetylglucosamine to 1-L-myo-inositol-1-phosphate, leading to the formation of 3-phospho-1-D-myo-inositol-2-acetamido-2-deoxy-α-D-glucopyranoside (GlcNAc-Ins-P). This step is catalyzed by the glycosyltransferase MshA.78 The intermediate is then dephosphorylated by a phosphatase, MshA2, yet to be identified.79 The zinc-dependent metalloprotein MshB subsequently deacetylates the intermediate to generate 1-D-myo-inositol-2-amino-2-deoxy-α-D-glucopyranoside (GlcN-Ins).80 MshC catalyzes the ATP-dependent ligation between L-cysteine and the 2-amino group from GlcN-Ins, yielding 1-D-myo-inosityl-2-L-cysteinylamino-2-deoxy-α-D-glucopyranoside (Cys-GlcN-Ins).81 Finally, Cys-GlcN-Ins is acetylated by MshD at the cysteinyl amine, resulting in the formation of MSH.82
Figure 4.

Role and structure of MshD in mycothiol biosynthesis. (A) Biosynthetic pathway of mycothiol. The box highlights the reaction catalyzed by MshD. (B) Structure of M. tuberculosis MshD in complex with CoA and DAM (PDB entry 2C27). The secondary structure elements are colored like the topology scheme presented in Figure 1. The substrates or products bound are rendered as sticks with white carbons. (C) Superimposition of MshD in complex with CoA and DAM (PDB entry 2C27, gray) and MshD in complex with AcCoA (PDB entry 1OZP, beige). Upon substrate binding, the N-terminal domain rotates closer to the C-terminal domain to allow a narrower binding cavity for DAM as highlighted by the arrow.
The enzyme MshD that catalyzes the final acetylation step in MSH biosynthesis is a GNAT protein. This enzyme was first identified in M. smegmatis and M. tuberculosis.82 A comparison of the MshD sequence with other acetyltransferase proteins revealed that MshD possesses twice the number of amino acids compared to other GNAT proteins and appeared to include two GNAT motifs in its sequence. Because the C-terminal region displays the highest degree of similarity to acetyltransferases, Koledin and co-workers hypothesized that MshD is the result of gene duplication and the C-terminal region is involved in the acetylation reaction while the N-terminal region is inactive.82
M. tuberculosis MshD (Rv0819) was crystallized in the presence of both AcCoA and CoA.83 The structure of a ternary complex of MshD cocrystallized with CoA and desacetylmycothiol (DAM) was also determined (Figure 4B).16 The structure confirms the presence of two GNAT motifs with the N-terminal domain (residues 1–140) and the C-terminal domain (residues 151–315) linked by a random coil. While most members of the GNAT family form dimers in solution, dynamic light scattering experiments and gel filtration revealed that MshD is a monomer in solution.83 Typically, the terminal β-strands (β6) of each subunit of the dimer exchange across the dimer interface to establish a continuous β-sheet. The same pattern is observed in the MshD structure. The two domains exhibit very similar structures as highlighted by a low root-mean-square (rms) displacement value for the Cα atom positions (1.7 Å) when superimposing one domain onto the other. However, the two domains appear to utilize different binding modes for AcCoA. In fact, the acetyl moiety of AcCoA is found buried in a hydrophobic pocket and is not correctly positioned to donate its acetyl group to Cys-GlcN-Ins in the N-terminal domain.83 Additionally, the βbulge is lacking in the latter domain. These two structural observations support the hypothesis that only the C-terminal domain is catalytically active. The structure of the ternary complex confirmed this hypothesis.16 In the binary complexes, a large cavity was observed between the two domains, and it was considered that upon binding of CoA and the acetyl acceptor, a conformational change between the two motifs might occur.83 In the ternary complex structure, the N-terminal domain is rotated toward the C-terminal domain, resulting in a narrower central cavity where DAM is bound.16 The amine moiety from DAM is oriented toward CoA bound in the C-terminal domain. Residues from both N-terminal and C-terminal domains participate in specific interactions with DAM. Interestingly, although only CoA and DAM were added to MshD prior to crystallization, an AcCoA molecule was found in the N-terminal domain, suggesting that AcCoA remained bound to the C-terminal domain during purification. The pH dependence of the maximal velocity suggested that MshD uses both a catalytic base and a catalytic acid. On the basis of their positions in the ternary complex structure, E234 (deprotonates a water molecule, which in turn accepts the proton from the amino group of DAM) and Y294 are believed to play those roles.16
1.3. Other Small Molecule Acetyltransferases
In addition to the aminoglycoside N-acetyltransferases and the mycothiol transferase that have been extensively studied, many other types of small molecule acetyltransferases have been partially or fully characterized. To give the reader a taste of acetyltransferase substrate diversity, a short list of diverse bacterial acetyltransferases is compiled in Table 1. This table highlights acetyltransferases involved in peptidoglycan recycling,84 detoxification pathways,85–88 production of virulence factor,89,90 iron acquisition,91,92 and redox balance.93
Table 1.
Examples of GNAT Enzymes and Their Targets
| names | targets | notes |
|---|---|---|
| PA4794148 | potential protein | acetyltransferase |
| ApxC89 | prototoxin HlyA | acyltransferase |
| GAT, SaGNAT149,150 | unknown small molecule | acetyltransferase, herbicide |
| CBG151 | clavaminic acid | acetyltransferase, tandem GNAT |
| MccE90 | antibiotic microcin C7 | acetyltransferase, bidomain |
| PseH84 | UDP-4-amino-4,6-dideoxy-β-L-AltNAc | acetyltransferase |
| SpeG/PaiA85,86 | spermidine | acetyltransferase, dodecamer GNAT |
| WecD152 | 4-amido-4,6-dideoxy-D-galactose | acetyltransferase |
| MddA/ADP187,88 | methionine sulfoxide, methionine sulfone | acetyltransferase |
| MbtK91,92 | mycobactin | acyltransferase |
| PhnO153 | aminoalkylphosphonic acid | acetyltransferase |
| Rv0802c154 | unknown | succinyl transferase |
| GlmA93 | glucosamine | acetyltransferase |
2. PEPTIDES
2.1. Fem Family
Members of the family of Fem amino-acyltransferases catalyze the addition of amino acids to the nascent peptidoglycan using aminoacylated tRNA as a substrate to form an elongated, branched cell wall cross-link.94 Fem family members are found primarily in Gram-positive bacteria such as Staphylococcus, Streptococcus, Lactobacillus, Clostridium, and Streptomyces and in few Gram-negative pathogenic spirochetes, including Borrelia burgdorferi and Treponema pallidum. The peptidoglycan, a critical component of the cell wall, is composed of alternating N-acetylglucosamine and N-acetylmuramic acid, on which the pentapeptide is linked via a lactyl group (Figure 5).95 In organisms containing the Fem enzymes, the pentapep-ide (L-Ala)-(D-Glu)-X-(D-Ala)-(D-Ala) is further modified by the addition of up to five amino acids at position X, where X is either lysine, ornithine, or the D,L-diamino acid, meso-diaminopimelic acid (Dap) (Table 2). The enzymes that synthesize the interchain peptide were first discovered in a methicillin-resistant Staphylococcus aureus (S. aureus) strain (pentaglycine extension at position X) and were initially believed to be factors essential for methicillin resistance, from which the name Fem is derived.96 In the 1990s, three fem genes were identified by insertional mutagenesis in S. aureus: femX (also known as fmhB), femA, and femB. femX was shown to be an essential gene in S. aureus that catalyzed the addition of the first glycine substituent onto the peptidoglycan precursor associated with the membrane (lipid II).97 On the other hand, femA and femB insertional mutants were not detrimental and lead to the formation of a one-glycine extended branched peptide and a three-glycine extended branched peptide, respectively.98 These results suggested that FemX adds the first glycine, FemA the second and third, and FemB the last two glycines. In the early 2000s, the first Fem was successfully expressed and assayed.99 FemX from Lactobacillus viridescens [also known as Weissella viridescens (W. viridescens)] was shown to catalyze the amino acyl transfer of glycine from charged tRNAGly to a soluble UDP-N-acetylmuramyl pentapeptide (Figure 5).100 In the literature, several Fem homologues have been reported. In Streptococcus pneumonia, MurM adds L-alanine or L-serine to ε-L-lysine, and subsequently, MurN adds a second L-alanine residue.101,102 In Enterococcus faecalis, BppA1 and BppA2 transfer L-alanine from Ala-tRNA to the first and second positions of the interchain peptide, respectively.103 All Fem enzymes and homologues use an amino-acylated tRNA as an acyl donor. S. aureus encodes three tRNAGly isoacceptors that bind poorly to elongation factor EF-Tu, thus preventing their use in ribosomal protein synthesis, and two “adequate” tRNAGly molecules suitable for protein synthesis.104 Specific nucleotide pairing changes or additional base pairing in the isoacceptor tRNA conserved loops led to the distortion of the anticodon loop and the inability to bind to the translational machinery.105 Because FemX is essential for bacterial survival, the enzyme constitutes an ideal drug target. To date, analogues of nonribosomal tRNA containing an oxadiazole group mimicking the 3′-amino acyl ester of the tRNA and peptidyl-RNA bisubstrate analogues have been developed and shown to be effective FemX inhibitors in vitro.106,107 The three-dimensional structures of two Fem enzymes have been determined, FemA from S. aureus and FemX from W. viridescens (Figure 6).108,109 The FemA structure revealed a globular domain composed of two GNAT subdomains and a pair of antiparallel α-helix domains located between β2′ and β3′ of the second GNAT subdomain. The α-helical coiled coil arm was suggested to play a role in the binding of Gly-tRNAGly during glycine transfer based on similar structural features found for seryl-tRNA synthetases.110 The main difference between the FemX structure complexed with UDP-MurNac-pentapeptide and the FemA structure is the absence of the coiled coil domain, which is replaced by a tight five-residue loop. FemX is also composed of two GNAT domains, separated by a cleft where the UDP-MurNac-pentapeptide is interacting primarily with the second GNAT domain.109 A newer FemX structure in complex with a bisubstrate analogue revealed crucial mechanistic information.111 The bisubstrate analogue is composed of a peptidoglycan precursor analogue linked by a 1,4-triazole ring to a RNA molecule mimicking the acceptor arm of tRNAAla. Superimposition of the FemX-UDP-MurNac-pentapeptide and FemX-peptidyl-RNA structures gave a rms displacement value of 0.4 Å for the Cα atom positions, indicating almost no change in the protein conformation. The only exception is the rearrangement of the loop between strands β5 and β6 (residues 136–140) toward the catalytic cavity, which is critical for maintaining the position of the pentapeptide lysine, which is ε-aminoacylated. The triazole ring of the pentapeptide is in the proximity of K305 and was proposed to be the residue participating in the chemical step. K305 and F304 are highly conserved in Fem enzymes, and mutation of those residues to alanine decreased the rate of turnover of FemX by 1.3 × 104- and 16-fold, respectively. On the basis of those results, a structure-based mechanism for substrate-assisted catalysis was proposed.
Figure 5.

Scheme of peptidoglycan layer formation. Fem enzymes act at different stages of the peptide bridge formation depending on the bacterial species. In W. viridescens, FemX adds only the first amino acid. In S. aureus, FemX adds the first glycine residue whereas FemA adds the second and third glycine residues and FemB the last two glycine residues. For the sake of simplicity, only three of five amino acids in the peptide bridge are represented in the figure. After peptide bridge formation, the terminal Gly5 α-NH2 group of the peptide bridge is cross-linked to the penultimate amino acid of the neighboring pentapeptide stem by transpeptidation leading to the release of the terminal amino acid from the stem peptide.
Table 2.
Interpeptide Bridge Composition of Various Bacterial Species
| species | interpeptide bridge composition | position X of the pentapeptide |
|---|---|---|
| Lactobacillus coprophilus | (L-Ala)2 | ε-lysine155 |
| S. aureus | (Gly)5 | ε-lysine156 |
| Staphylococcus epidermidis | Gly-L-Ala | ε-lysine157 |
| W. viridescens | L-Ala-L-Ser | ε-lysine100 |
| Streptococcus pneumoniae | L-Ser-(L-Ala)2 | ε-lysine158 |
| Borrelia burgdorferi | Gly | δ-L-ornithine159 |
| Treponema palladium | Gly | δ-L-ornithine159 |
| Clostridium perfringens | Gly | ω-L,L-diaminopimelic acid160 |
| M. tuberculosis | Gly | ω-L,L-diaminopimelic acid161 |
| B. subtilis | none | ω-L,L-diaminopimelic acid162 |
| Streptomyces coelicolor | Gly | ω-L,L-diaminopimelic acid163 |
Figure 6.
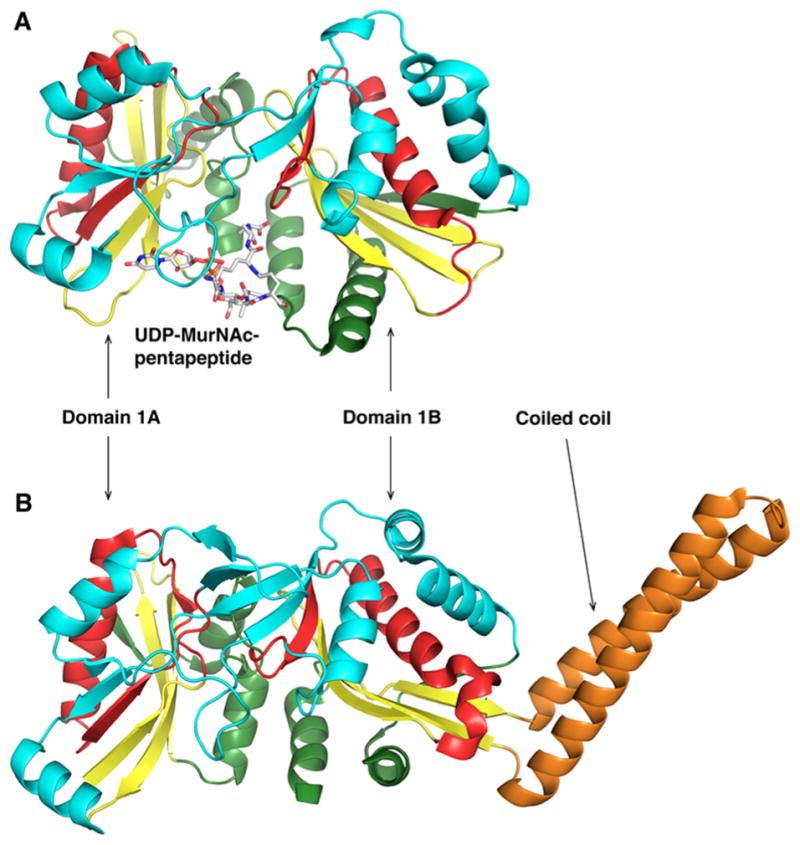
Structural comparison of W. viridescens FemX and S. aureus FemA. The GNAT secondary structure elements are colored like the topology scheme presented in Figure 1. The bisubstrate is rendered as sticks with white carbons. (A) The W. viridescens FemX consists of two similar GNAT domains, 1A and 1B (PDB entry 1P4N). (B) The S. aureus FemX structure also displays domains 1A and 1B and contains an additional coiled coil segment that is colored orange (PDB entry 1LRZ).
3. PROTEINS
GNAT enzymes also acetylate proteins, either at the N-terminus (αN-acetylation) or on the side chain of internal lysine residues (εN-acetylation). The largest family of protein N-acetyltransferases is the histone acetyltransferase family found in eukaryotes, which acetylate histones predominantly at the N-terminus.11 Because this Current Topic is focused on GNAT proteins in bacteria, the histone acetyltransferase family will not be discussed here.
3.1. Rim Proteins
αN-Acetylation of proteins is a common post-translational modification in eukaryotes, but there are relatively few examples in prokaryotes.112 Three ribosomal proteins, S5, S18, and L12, are known to be αN-acetylated in bacteria.113 Using nitrosoguanine mutagenesis followed by two-dimensional gel electrophoresis, Isono and co-workers identified the three proteins responsible for the αN-acetylation of these ribosomal proteins: RimI, RimJ, and RimL acetylate S18, S5, and L12, respectively.113–116 The function of αN-acetylation of these proteins is not fully understood.
The acetylation of L12 leads to the formation of L7, an acetylated version of L12. L12 and L7 are dimers that interact with the ribosome through an adaptor ribosomal protein, L10.117 Contrary to other ribosomal proteins, multiple copies of L12/L7 are present in the ribosome.118 More recently, Gordiyenko et al. demonstrated that the αN-acetylation of L12 allows for tighter binding between L12/L7 and L10, stabilizing the ribosomal stalk complex.119 Three RimL structures from different organisms have been determined to date: RimL from Salmonella typhimurium LT2 (S. typhimurium),120 RimL from Thermus thermophilus (T. thermophilus),121 and YdaF (a homologue of RimL) from Bacillus subtilis (B. subtilis).122 Although the YdaF structure exhibits a typical GNAT fold, its acetyltransferase activity has not been confirmed. Experiments including gel filtration and dynamic light scattering suggest that S. typhimurium RimL forms a dimer in solution.120 In the RimL–CoA complex, CoA is bound between the splayed β4 and β5 strands, and few specific interactions are observed between the adenine/ribose moieties and the protein (Figure 7A). Additionally, backbone amides from the pyrophosphate binding loop, as well as a conserved water molecule, interact with the pyrophosphate moiety through hydrogen bonds while the pantothenate “arm” interacts with residues located at the C-terminal end of strand β4. A conformational change involving the loop linking α1 to α2 is observed upon the binding of CoA. One characteristic structural feature of the GNAT family is the presence of a “β-bulge” on β4, responsible for the splaying of strands β4 and β5, but this structural feature is not observed in the S. typhimurium RimL structure. A cysteine (C134) proximal to the active site is involved in a disulfide bond with CoA in one of the S. typhimurium RimL–CoA structures, suggesting that the reaction catalyzed by RimL could exploit a ping-pong mechanism. However, this residue does not appear to be conserved among the other RimL enzymes. The mutant enzyme (C134A) displays kinetic properties similar to those of the wild-type enzyme, indicating that the enzyme utilizes a direct nucleophilic attack. S141 and E160 are hypothesized as the general acid and base, respectively. Finally, the cleft formed at the dimer interface could be the binding site for L12 as it is also observed in the T. thermophilus RimL and B. subtilis YdaF structures.121,122
Figure 7.
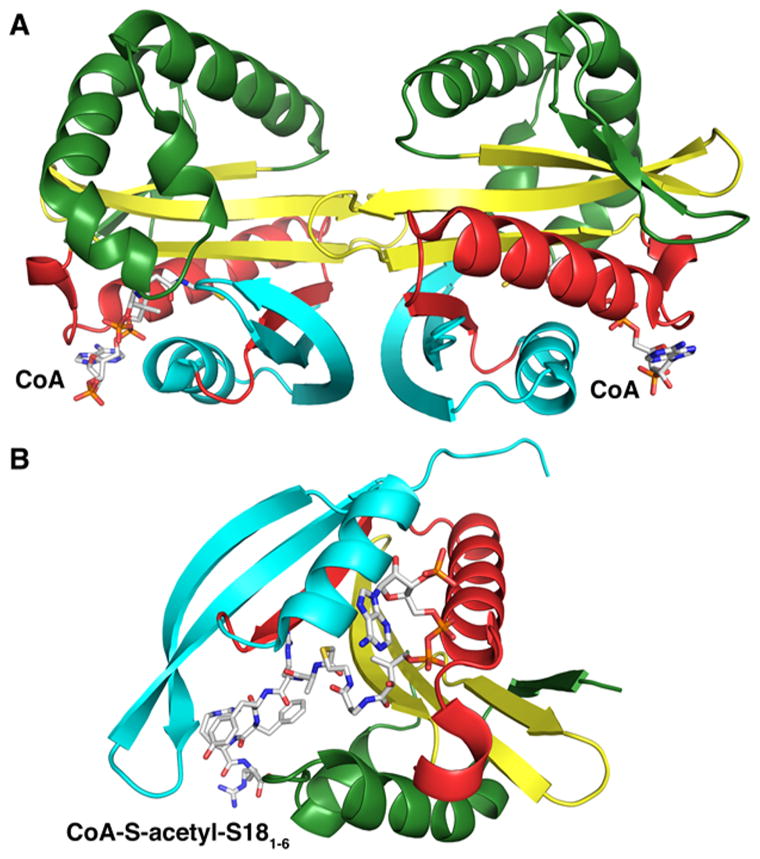
Structures of Rim proteins. The secondary structure elements are colored like the topology scheme presented in Figure 1. CoA and the bisubstrate are rendered as sticks with white carbons. (A) Structure of S. tphimurium LT2 RimL in complex with CoA (PDB entry 1S7N). (B) Structure of S. typhimurium LT2 RimI in complex with a bisubstrate CoA-S-acetyl-S181–6 (PDB entry 2CNM).
The S. typhimurium RimI that acetylates S18 has been extensively characterized, both kinetically and structurally.123,124 RimI utilizes an ordered kinetic mechanism involving the binding of AcCoA first, followed by the binding of S18.123 Because full-length S18 from S. typhimurium was not successfully expressed, a peptide representing the first six amino acids of S18 was used for the kinetic and inhibition studies (abbreviated as S181–6). S. typhimurium RimI was crystallized in the presence of AcCoA, CoA, and a bisubstrate inhibitor.124 The structures exhibit all the characteristic features of the GNAT fold. However, the enzyme appears to be a monomer in solution. The cocrystal structure with a bisubstrate inhibitor (CoA-S-acetyl-S181–6) reveals how the inhibitor binds to the active site (Figure 7B). The peptide from the bisubstrate inhibitor binds perpendicularly to the pantothenate group of CoA. Several residues of the active site are specifically interacting with the peptide portion through hydrogen bonds, as well as van der Waals interactions. Several conformational changes are observed upon the binding of the peptide compared to the structure containing CoA, the most significant involving the orientation of the loop connecting β6 and β7.
The structure of a putative RimJ protein from Vibrio fischeri has been determined (PDB entry 3IGR) and exhibits a structural fold similar to that of the RimI and RimL structures. In addition to acetylating S5, RimJ has been proposed to be involved in ribosome assembly125 and to acetylate thymosin α1, an immunomodulating peptide.126
3.2. Protein Acetyltransferase, Pat
Acetylation at the ε-amino group of a lysine residue is a major post-translational protein regulation mechanism found in all kingdoms of life. Since the discovery of εN-lysine-acetylated AcCoA synthase (ACS) in S. enterica127 and acetylome studies in E. coli128,129 and S. enterica,130 εN-lysine acetylation appears to be widespread in prokaryotes. The first εN-lysine acetyltransferase was identified in S. enterica as SePat (formerly yhiQ) and is responsible for ACS acetylation leading to enzyme inhibition.131 The SePat enzyme is composed of two domains: a GNAT acetyltransferase domain at the C-terminus and a NDP-forming AcCoA synthetase domain at the N-terminus, which is 96% identical to E. coli PatZ.132 However, the NDP-forming AcCoA domain is unable to produce AcCoA from acetate, ATP, and CoA because the catalytic histidine, present in PatZ, is replaced with an asparagine (N114) in SePat. SePat has been reported to acetylate several metabolic enzymes, including ACS,131 glyceraldehyde-3-phosphate dehydrogenase (GapA), isocitrate lyase (AceA), and isocitrate dehydrogenase kinase (aceK),130 and to propionylate propionyl-CoA synthetase (PprE).133 It has been proposed that Pat acetylates AcCoA synthase at high intracellular concentrations of AcCoA to prevent further increases in its concentration, maintain the acetate pool, and prevent unnecessary ATP hydrolysis.134
Enzyme acetylation is reversed by deacetylase enzymes, including the NAD+-dependent sirtuin-like deacetylases, which allows for the rapid response and adaptation to new metabolic needs or physiological changes. In B. subtilis, ACS activity is also post-translationally modified by AcuA, a GNAT protein acetyltransferase.135 The ACS gene and AcuABC operon are adjacent to each other, with AcuA functioning as the acetylase and AcuC as an NAD+-independent deacetylase. A second NAD+-dependent deacetylase, CobB, is also able to deacetylate ACS, suggesting that depending on growth conditions, two independent acetylases could be differentially used.135 Reversible ACS acetylation by Pat enzymes has been reported in many organisms, including Rhodopseudomonas palutris,136 Streptomyces coelicolor,137 Streptomyces lividans (S. lividans),138 M. smegmatis,139 and M. tuberculosis.140 The recently determined structure of an active catalytic complex between the S. lividans Pat GNAT domain and S. enterica ACS revealed key determinants for protein substrate recognition and subsequent acetylation.141 In addition to the conserved PX4GK motif on the C-terminus of the ACS protein substrate, a trio of arginines located after the PX4GK motif also conserved in ACS homologues was shown to interact with a negative patch on Pat. Those complementary ionic interactions contribute to Pat substrate specificity. A unique feature of M. tuberculosis Pat and M. smegmatis Pat acetyltransferase structures is the fusion of an N-terminal cyclic nucleotide binding domain to the GNAT domain.142,143 This cyclic nucleotide binding domain specifically recognizes cAMP, allowing allosteric activation of Pat. Following macrophage phagocytosis of M. tuberculosis, a dramatic increase (50-fold) in cAMP concentration was observed in the macrophage cytoplasm as well as within the bacteria.144 cAMP is a key regulatory molecule in mycobacterial physiological adaptation during infection and influences the host response. The M. tuberculosis Pat crystal structure reveals that after cAMP binds to the N-terminal domain, a conformational change causes M. tuberculosis Pat to switch from an autoinhibited state to a cAMP-activated state (Figure 8). Activated M. tuberculosis Pat acetylates and inhibits the activity of multiple downstream enzymes such as ACS, fatty acyl-CoA ligase FadD13, and fatty acyl-AMP ligase FadD33, which regulate fatty acid metabolism and siderophore biosynthesis, respectively.140,145,146
Figure 8.

Activation of M. tuberculosis Pat via cAMP binding (left structure, PDB entry 4AVA; right structure, PDB entry 4AVC). The N-terminal regulatory domain (orange) is linked to C-terminal catalytic GNAT domain. Upon cAMP binding, the Pat protein undergoes a conformational rearrangement allowing protein–substrate binding in the catalytic domain active site. The GNAT secondary structure elements are colored like the topology scheme presented in Figure 1. The substrates and activators bound are rendered as sticks with white carbons.
4. CONCLUSION
It has been 50 years since the discovery of the AcCoA-dependent N-acetyltransferases, and our understanding of their roles in antibiotic resistance, metabolism, and biosynthesis and most recently control and regulation of central carbon metabolism has improved dramatically. Their kinetic and chemical mechanisms have been dissected, and numerous structures of the different “classes” of GNATs have been reported. Although originally thought to use exclusively AcCoA, there are examples shown in this review in which the acyl transfer substrate can be an aminoacylated tRNA molecule and other examples in which the nature of the fatty acyl substituent is proprionyl, and longer acyl chains (e.g., yeast myristoyltransferase,147 also a monomeric enzyme composed of two GNAT domains). The size and nature of the eubacterial “acetylome” are largely unknown and, in some reported cases, unlikely to be correct due to nonenzymatic acetylation reactions. Today, a majority of the identifiable bacterial GNATs are of unknown biochemical function, and further efforts are necessary to unravel the full picture of a promising field of research.
Acknowledgments
Funding
This work was supported by Grant RO1 AI 060899 to J.S.B.
ABBREVIATIONS
- AAC
aminoglycoside N-acetyltransferase
- AcCoA
acetyl-coenzyme A
- ANT
aminoglycoside nucleotidyltransferase
- APH
aminoglycoside phosphotransferases
- CoA
coenzyme A
- Cys-GlcN-Ins
1-D-myo-inosityl-2-L-cysteinylamino-2-deoxy-αD-glucopyranoside
- DAM
desacetylmycothiol
- Dap
diaminopimelic acid
- GlcNAc-Ins-P
3-phospho-1-D-myo-inositol-2-acet-amido-2-deoxy-α-D-glucopyranoside
- GlcN-Ins
1-D-myo-inositol-2-amino-2-deoxy-α-D-glucopyranoside
- GNAT
GCN5-related N-acetyltransferase
- MSH
mycothiol
- PDB
Protein Data Bank
- rms
root-mean-square
Footnotes
The authors declare no competing financial interest.
References
- 1.Phillips DM. The presence of acetyl groups of histones. Biochem J. 1963;87:258–263. doi: 10.1042/bj0870258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okamoto S, Suzuki Y. Chloramphenicol-, dihydrostreptomycin-, and kanamycin-inactivating enzymes from multiple drug-resistant Escherichia coli carrying episome ‘R’. Nature. 1965;208:1301–1303. doi: 10.1038/2081301a0. [DOI] [PubMed] [Google Scholar]
- 4.Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 5.Parthun MR, Widom J, Gottschling DE. The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell. 1996;87:85–94. doi: 10.1016/s0092-8674(00)81325-2. [DOI] [PubMed] [Google Scholar]
- 6.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 7.Cleland WW, Hengge AC. Mechanisms of phosphoryl and acyl transfer. FASEB J. 1995;9:1585–1594. doi: 10.1096/fasebj.9.15.8529838. [DOI] [PubMed] [Google Scholar]
- 8.Horiuchi J, Silverman N, Pina B, Marcus GA, Guarente L. ADA1, a novel component of the ADA/GCN5 complex, has broader effects than GCN5, ADA2, or ADA3. Mol Cell Biol. 1997;17:3220–3228. doi: 10.1128/mcb.17.6.3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies J, Wright GD. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol. 1997;5:234–240. doi: 10.1016/S0966-842X(97)01033-0. [DOI] [PubMed] [Google Scholar]
- 10.Xie L, Zeng J, Luo H, Pan W, Xie J. The roles of bacterial GCN5-related N-acetyltransferases. Crit Rev Eukaryotic Gene Expression. 2014;24:77–87. doi: 10.1615/critreveukaryotgeneexpr.2014007988. [DOI] [PubMed] [Google Scholar]
- 11.Vetting MW, de Carvalho LPS, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neuwald AF, Landsman D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci. 1997;22:154–155. doi: 10.1016/s0968-0004(97)01034-7. [DOI] [PubMed] [Google Scholar]
- 14.Yan Y, Harper S, Speicher DW, Marmorstein R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat Struct Biol. 2002;9:862–869. doi: 10.1038/nsb849. [DOI] [PubMed] [Google Scholar]
- 15.Rojas JR, Trievel RC, Zhou J, Mo Y, Li X, Berger SL, Allis CD, Marmorstein R. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature. 1999;401:93–98. doi: 10.1038/43487. [DOI] [PubMed] [Google Scholar]
- 16.Vetting MW, Yu M, Rendle PM, Blanchard JS. The substrate-induced conformational change of Mycobacterium tuberculosis mycothiol synthase. J Biol Chem. 2005;281:2795–2802. doi: 10.1074/jbc.M510798200. [DOI] [PubMed] [Google Scholar]
- 17.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 18.Marmorstein R, Roth SY. Histone acetyltransferases: function, structure, and catalysis. Curr Opin Genet Dev. 2001;11:155–161. doi: 10.1016/s0959-437x(00)00173-8. [DOI] [PubMed] [Google Scholar]
- 19.Berndsen CE, Denu JM. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr Opin Struct Biol. 2008;18:682–689. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schatz A, Bugie E, Waksman SA, Hanssen AD, Patel R, Osmon DR. Streptomycin, a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria. 1944. Clin Orthop Relat Res. 2005:3–6. doi: 10.1097/01.blo.0000175887.98112.fe. [DOI] [PubMed] [Google Scholar]
- 21.Labby KJ, Garneau-Tsodikova S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med Chem. 2013;5:1285–1309. doi: 10.4155/fmc.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 23.Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- 24.Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside-induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000;48:164–169. [PubMed] [Google Scholar]
- 25.Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, Atkins JF, Flanigan KM. Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol. 2004;55:422–426. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- 26.Lapidot A, Berchanski A, Borkow G. Insight into the mechanisms of aminoglycoside derivatives interaction with HIV-1 entry steps and viral gene transcription. FEBS J. 2008;275:5236–5257. doi: 10.1111/j.1742-4658.2008.06657.x. [DOI] [PubMed] [Google Scholar]
- 27.Venkataraman N, Cole AL, Ruchala P, Waring AJ, Lehrer RI, Stuchlik O, Pohl J, Cole AM. Reawakening retrocyclins: ancestral human defensins active against HIV-1. PLoS Biol. 2009;7:e1000095. doi: 10.1371/journal.pbio.1000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranjan N, Kumar S, Watkins D, Wang D, Appella DH, Arya DP. Recognition of HIV-TAR RNA using neomycin-benzimidazole conjugates. Bioorg Med Chem Lett. 2013;23:5689–5693. doi: 10.1016/j.bmcl.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies J, Gorini L, Davis BD. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol Pharmacol. 1965;1:93–106. [PubMed] [Google Scholar]
- 30.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 31.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 32.Fourmy D, Recht MI, Puglisi JD. Binding of neomycin-class aminoglycoside antibiotics to the A-site of 16 S rRNA. J Mol Biol. 1998;277:347–362. doi: 10.1006/jmbi.1997.1552. [DOI] [PubMed] [Google Scholar]
- 33.Fourmy D, Yoshizawa S, Puglisi JD. Paromomycin binding induces a local conformational change in the A-site of 16 S rRNA. J Mol Biol. 1998;277:333–345. doi: 10.1006/jmbi.1997.1551. [DOI] [PubMed] [Google Scholar]
- 34.Yoshizawa S, Fourmy D, Puglisi JD. Structural origins of gentamicin antibiotic action. EMBO J. 1998;17:6437–6448. doi: 10.1093/emboj/17.22.6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brodersen DE, Clemons WM, Jr, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 36.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 37.Francois B, Russell RJ, Murray JB, Aboulela F, Masquida B, Vicens Q, Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005;33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magnet S, Blanchard JS. Molecular insights into aminoglycoside action and resistance. Chem Rev. 2005;105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 39.Ramirez MS, Tolmasky ME. Aminoglycoside modifying enzymes. Drug Resist Updates. 2010;13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright GD. Molecular mechanisms of antibiotic resistance. Chem Commun (Cambridge, U K) 2011;47:4055–4061. doi: 10.1039/c0cc05111j. [DOI] [PubMed] [Google Scholar]
- 41.Shaw KJ, Rather PN, Hare RS, Miller GH. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev. 1993;57:138–163. doi: 10.1128/mr.57.1.138-163.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lovering AM, White LO, Reeves DS. AAC(1): a new aminoglycoside-acetylating enzyme modifying the Cl aminogroup of apramycin. J Antimicrob Chemother. 1987;20:803–813. doi: 10.1093/jac/20.6.803. [DOI] [PubMed] [Google Scholar]
- 43.Sunada A, Nakajima M, Ikeda Y, Kondo S, Hotta K. Enzymatic 1-N-acetylation of paromomycin by an actinomycete strain #8 with multiple aminoglycoside resistance and paromomycin sensitivity. J Antibiot. 1999;52:809–814. doi: 10.7164/antibiotics.52.809. [DOI] [PubMed] [Google Scholar]
- 44.Williams JW, Northrop DB. Purification and properties of gentamicin acetyltransferase I. Biochemistry. 1976;15:125–131. doi: 10.1021/bi00646a019. [DOI] [PubMed] [Google Scholar]
- 45.Williams JW, Northrop DB. Substrate specificity and structure-activity relationships of gentamicin acetyltransferase I. The dependence of antibiotic resistance upon substrate Vmax/Km values. J Biol Chem. 1978;253:5908–5914. [PubMed] [Google Scholar]
- 46.Williams JW, Northrop DB. Kinetic mechanisms of gentamicin acetyltransferase I. Antibiotic-dependent shift from rapid to nonrapid equilibrium random mechanisms. J Biol Chem. 1978;253:5902–5907. [PubMed] [Google Scholar]
- 47.Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley SK. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell. 1998;94:439–449. doi: 10.1016/s0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 48.Dutnall RN, Tafrov ST, Sternglanz R, Ramakrishnan V. Structure of the histone acetyltransferase Hat1: a paradigm for the GCN5-related N-acetyltransferase superfamily. Cell. 1998;94:427–438. doi: 10.1016/s0092-8674(00)81584-6. [DOI] [PubMed] [Google Scholar]
- 49.Norris AL, Serpersu EH. Interactions of coenzyme A with the aminoglycoside acetyltransferase (3)-IIIb and thermodynamics of a ternary system. Biochemistry. 2010;49:4036–4042. doi: 10.1021/bi1001568. [DOI] [PubMed] [Google Scholar]
- 50.Norris AL, Ozen C, Serpersu EH. Thermodynamics and kinetics of association of antibiotics with the aminoglycoside acetyltransferase (3)-IIIb, a resistance-causing enzyme. Biochemistry. 2010;49:4027–4035. doi: 10.1021/bi100155j. [DOI] [PubMed] [Google Scholar]
- 51.Norris AL, Nickels J, Sokolov AP, Serpersu EH. Protein dynamics are influenced by the order of ligand binding to an antibiotic resistance enzyme. Biochemistry. 2014;53:30–38. doi: 10.1021/bi401635r. [DOI] [PubMed] [Google Scholar]
- 52.Rather PN, Orosz E, Shaw KJ, Hare R, Miller G. Characterization and transcriptional regulation of the 2′-N-acetyltransferase gene from Providencia stuartii. J Bacteriol. 1993;175:6492–6498. doi: 10.1128/jb.175.20.6492-6498.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Macinga DR, Rather PN. The chromosomal 2′-N-acetyltransferase of Providencia stuartii: physiological functions and genetic regulation. Front Biosci, Landmark Ed. 1999;4:D132–D140. doi: 10.2741/macinga. [DOI] [PubMed] [Google Scholar]
- 54.Ainsa JA, Martin C, Gicquel B, Gomez-Lus R. Characterization of the chromosomal aminoglycoside 2′-N-acetyltrans-ferase gene from Mycobacterium fortuitum. Antimicrob Agents Chemother. 1996;40:2350–2355. doi: 10.1128/aac.40.10.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ainsa JA, Perez E, Pelicic V, Berthet FX, Gicquel B, Martin C. Aminoglycoside 2′-N-acetyltransferase genes are universally present in mycobacteria: characterization of the aac(2′)-Ic gene from Mycobacterium tuberculosis and the aac(2′)-Id gene from Mycobacterium smegmatis. Mol Microbiol. 1997;24:431–441. doi: 10.1046/j.1365-2958.1997.3471717.x. [DOI] [PubMed] [Google Scholar]
- 56.Hegde SS, Javid-Majd F, Blanchard JS. Overexpression and mechanistic analysis of chromosomally encoded aminoglycoside 2′-N-acetyltransferase (AAC(2′)-Ic) from Mycobacterium tuberculosis. J Biol Chem. 2001;276:45876–45881. doi: 10.1074/jbc.M108810200. [DOI] [PubMed] [Google Scholar]
- 57.Vetting MW, Hegde SS, Javid-Majd F, Blanchard JS, Roderick SL. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat Struct Biol. 2002;9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 58.Rather PN, Munayyer H, Mann PA, Hare RS, Miller GH, Shaw KJ. Genetic analysis of bacterial acetyltransferases: identification of amino acids determining the specificities of the aminoglycoside 6′-N-acetyltransferase Ib and IIa proteins. J Bacteriol. 1992;174:3196–3203. doi: 10.1128/jb.174.10.3196-3203.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wright GD, Ladak P. Overexpression and characterization of the chromosomal aminoglycoside 6′-N-acetyltransferase from Enterococcus faecium. Antimicrob Agents Chemother. 1997;41:956–960. doi: 10.1128/aac.41.5.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DiGiammarino EL, Draker KA, Wright GD, Serpersu EH. Solution studies of isepamicin and conformational comparisons between isepamicin and butirosin A when bound to an aminoglycoside 6′-N-acetyltransferase determined by NMR spectroscopy. Biochemistry. 1998;37:3638–3644. doi: 10.1021/bi972778b. [DOI] [PubMed] [Google Scholar]
- 61.Wybenga-Groot LE, Draker K, Wright GD, Berghuis AM. Crystal structure of an aminoglycoside 6′-N-acetyltransferase: defining the GCN5-related N-acetyltransferase superfamily fold. Structure. 1999;7:497–507. doi: 10.1016/s0969-2126(99)80066-5. [DOI] [PubMed] [Google Scholar]
- 62.Burk DL, Ghuman N, Wybenga-Groot LE, Berghuis AM. X-ray structure of the AAC(6′)-Ii antibiotic resistance enzyme at 1.8 A resolution; examination of oligomeric arrangements in GNAT superfamily members. Protein Sci. 2003;12:426–437. doi: 10.1110/ps.0233503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Magnet S, Lambert T, Courvalin P, Blanchard JS. Kinetic and mutagenic characterization of the chromosomally encoded Salmonella enterica AAC(6′)-Iy aminoglycoside N-acetyltransferase. Biochemistry. 2001;40:3700–3709. doi: 10.1021/bi002736e. [DOI] [PubMed] [Google Scholar]
- 64.Hegde SS, Dam TK, Brewer CF, Blanchard JS. Thermodynamics of aminoglycoside and acyl-coenzyme A binding to the Salmonella enterica AAC(6′)-Iy aminoglycoside N-acetyltransferase. Biochemistry. 2002;41:7519–7527. doi: 10.1021/bi020190l. [DOI] [PubMed] [Google Scholar]
- 65.Vetting MW, Magnet S, Nieves E, Roderick SL, Blanchard JS. A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem Biol. 2004;11:565–573. doi: 10.1016/j.chembiol.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 66.Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Hye Park C, Bush K, Hooper DC. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med. 2006;12:83–88. doi: 10.1038/nm1347. [DOI] [PubMed] [Google Scholar]
- 67.Casin I, Hanau-Bercot B, Podglajen I, Vahaboglu H, Collatz E. Salmonella enterica serovar Typhimurium bla(PER-1)-carrying plasmid pSTI1 encodes an extended-spectrum aminoglycoside 6′-N-acetyltransferase of type Ib. Antimicrob Agents Chemother. 2003;47:697–703. doi: 10.1128/AAC.47.2.697-703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vakulenko SB, Mobashery S. Versatility of aminoglycosides and prospects for their future. Clin Microbiol Rev. 2003;16:430–450. doi: 10.1128/CMR.16.3.430-450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vetting MW, Park CH, Hegde SS, Jacoby GA, Hooper DC, Blanchard JS. Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry. 2008;47:9825–9835. doi: 10.1021/bi800664x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maurice F, Broutin I, Podglajen I, Benas P, Collatz E, Dardel F. Enzyme structural plasticity and the emergence of broad-spectrum antibiotic resistance. EMBO Rep. 2008;9:344–349. doi: 10.1038/embor.2008.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim C, Villegas-Estrada A, Hesek D, Mobashery S. Mechanistic characterization of the bifunctional aminoglycoside-modifying enzyme AAC(3)-Ib/AAC(6′)-Ib′ from Pseudomonas aeruginosa. Biochemistry. 2007;46:5270–5282. doi: 10.1021/bi700111z. [DOI] [PubMed] [Google Scholar]
- 72.Daigle DM, Hughes DW, Wright GD. Prodigious substrate specificity of AAC(6′)-APH(2″), an aminoglyco-side antibiotic resistance determinant in enterococci and staphylococci. Chem Biol. 1999;6:99–110. doi: 10.1016/S1074-5521(99)80006-4. [DOI] [PubMed] [Google Scholar]
- 73.Smith CA, Toth M, Weiss TM, Frase H, Vakulenko SB. Structure of the bifunctional aminoglycoside-resistance enzyme AAC(6′)-Ie-APH(2″)-Ia revealed by crystallographic and small-angle X-ray scattering analysis. Acta Crystallogr, Sect D: Biol Crystallogr. 2014;70:2754–2764. doi: 10.1107/S1399004714017635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Newton GL, Fahey RC, Cohen G, Aharonowitz Y. Low-molecular-weight thiols in streptomycetes and their potential role as antioxidants. J Bacteriol. 1993;175:2734–2742. doi: 10.1128/jb.175.9.2734-2742.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newton GL, Bewley CA, Dwyer TJ, Horn R, Aharonowitz Y, Cohen G, Davies J, Faulkner DJ, Fahey RC. The structure of U17 isolated from Streptomyces clavuligerus and its properties as an antioxidant thiol. Eur J Biochem. 1995;230:821–825. doi: 10.1111/j.1432-1033.1995.0821h.x. [DOI] [PubMed] [Google Scholar]
- 76.Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J Bacteriol. 1996;178:1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fan F, Vetting MW, Frantom PA, Blanchard JS. Structures and mechanisms of the mycothiol biosynthetic enzymes. Curr Opin Chem Biol. 2009;13:451–459. doi: 10.1016/j.cbpa.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Newton GL, Ta P, Bzymek KP, Fahey RC. Biochemistry of the initial steps of mycothiol biosynthesis. J Biol Chem. 2006;281:33910–33920. doi: 10.1074/jbc.M604724200. [DOI] [PubMed] [Google Scholar]
- 79.Newton GL, Buchmeier N, Fahey RC. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol Mol Biol Rev. 2008;72:471–494. doi: 10.1128/MMBR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Newton GL, Av-Gay Y, Fahey RC. N-Acetyl-1-D-myo-inosityl-2-amino-2-deoxy-alpha-D-glucopyranoside deacetylase (MshB) is a key enzyme in mycothiol biosynthesis. J Bacteriol. 2000;182:6958–6963. doi: 10.1128/jb.182.24.6958-6963.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rawat M, Newton GL, Ko M, Martinez GJ, Fahey RC, Av-Gay Y. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother. 2002;46:3348–3355. doi: 10.1128/AAC.46.11.3348-3355.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koledin T, Newton GL, Fahey RC. Identification of the mycothiol synthase gene (mshD) encoding the acetyltransferase producing mycothiol in actinomycetes. Arch Microbiol. 2002;178:331–337. doi: 10.1007/s00203-002-0462-y. [DOI] [PubMed] [Google Scholar]
- 83.Vetting MW, Roderick SL, Yu M, Blanchard JS. Crystal structure of mycothiol synthase (Rv0819) from Mycobacterium tuberculosis shows structural homology to the GNAT family of N-acetyltransferases. Protein Sci. 2003;12:1954–1959. doi: 10.1110/ps.03153703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ud-Din AI, Liu YC, Roujeinikova A. Crystal structure of Helicobacter pylori pseudaminic acid biosynthesis N-acetyltransferase PseH: implications for substrate specificity and catalysis. PLoS One. 2015;10:e0115634. doi: 10.1371/journal.pone.0115634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Filippova EV, Kuhn ML, Osipiuk J, Kiryukhina O, Joachimiak A, Ballicora MA, Anderson WF. A novel polyamine allosteric site of SpeG from Vibrio cholerae is revealed by its dodecameric structure. J Mol Biol. 2015;427:1316–1334. doi: 10.1016/j.jmb.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Filippova EV, Shuvalova L, Minasov G, Kiryukhina O, Zhang Y, Clancy S, Radhakrishnan I, Joachimiak A, Anderson WF. Crystal structure of the novel PaiA N-acetyltransferase from Thermoplasma acidophilum involved in the negative control of sporulation and degradative enzyme production. Proteins: Struct, Funct, Genet. 2011;79:2566–2577. doi: 10.1002/prot.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davies AM, Tata R, Snape A, Sutton BJ, Brown PR. Structure and substrate specificity of acetyltransferase ACIAD1637 from Acinetobacter baylyi ADP1. Biochimie. 2009;91:484–489. doi: 10.1016/j.biochi.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 88.Hentchel KL, Escalante-Semerena JC. In Salmonella enterica, the Gcn5-related acetyltransferase MddA (formerly YncA) acetylates methionine sulfoximine and methionine sulfone, blocking their toxic effects. J Bacteriol. 2015;197:314–325. doi: 10.1128/JB.02311-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Greene NP, Crow A, Hughes C, Koronakis V. Structure of a bacterial toxin-activating acyltransferase. Proc Natl Acad Sci U S A. 2015;112:E3058–3066. doi: 10.1073/pnas.1503832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Agarwal V, Metlitskaya A, Severinov K, Nair SK. Structural basis for microcin C7 inactivation by the MccE acetyltransferase. J Biol Chem. 2011;286:21295–21303. doi: 10.1074/jbc.M111.226282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Card GL, Peterson NA, Smith CA, Rupp B, Schick BM, Baker EN. The crystal structure of Rv 1347c, a putative antibiotic resistance protein from Mycobacterium tuberculosis, reveals a GCN5-related fold and suggests an alternative function in siderophore biosynthesis. J Biol Chem. 2005;280:13978–13986. doi: 10.1074/jbc.M413904200. [DOI] [PubMed] [Google Scholar]
- 92.Frankel BA, Blanchard JS. Mechanistic analysis of Mycobacterium tuberculosis Rv1347c, a lysine Nepsilon-acyltransferase involved in mycobactin biosynthesis. Arch Biochem Biophys. 2008;477:259–266. doi: 10.1016/j.abb.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reith J, Berking A, Mayer C. Characterization of an N-acetylmuramic acid/N-acetylglucosamine kinase of Clostridium acetobutylicum. J Bacteriol. 2011;193:5386–5392. doi: 10.1128/JB.05514-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dare K, Ibba M. Roles of tRNA in cell wall biosynthesis. Wiley Interdiscip Rev RNA. 2012;3:247–264. doi: 10.1002/wrna.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schleifer KH, Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev. 1972;36:407–477. doi: 10.1128/br.36.4.407-477.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Berger-Bachi B, Barberis-Maino L, Strassle A, Kayser FH. FemA, a host-mediated factor essential for methicillin resistance in Staphylococcus aureus: molecular cloning and characterization. Mol Gen Genet. 1989;219:263–269. doi: 10.1007/BF00261186. [DOI] [PubMed] [Google Scholar]
- 97.Rohrer S, Ehlert K, Tschierske M, Labischinski H, Berger-Bachi B. The essential Staphylococcus aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation. Proc Natl Acad Sci U S A. 1999;96:9351–9356. doi: 10.1073/pnas.96.16.9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ehlert K, Schroder W, Labischinski H. Specificities of FemA and FemB for different glycine residues: FemB cannot substitute for FemA in staphylococcal peptidoglycan pentaglycine side chain formation. J Bacteriol. 1997;179:7573–7576. doi: 10.1128/jb.179.23.7573-7576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hegde SS, Shrader TE. FemABX family members are novel nonribosomal peptidyltransferases and important pathogen-specific drug targets. J Biol Chem. 2001;276:6998–7003. doi: 10.1074/jbc.M008591200. [DOI] [PubMed] [Google Scholar]
- 100.Hegde SS, Blanchard JS. Kinetic and mechanistic characterization of recombinant Lactobacillus viridescens FemX (UDP-N-acetylmuramoyl pentapeptide-lysine N6-alanyltransferase) J Biol Chem. 2003;278:22861–22867. doi: 10.1074/jbc.M301565200. [DOI] [PubMed] [Google Scholar]
- 101.Filipe SR, Tomasz A. Inhibition of the expression of penicillin resistance in Streptococcus pneumoniae by inactivation of cell wall muropeptide branching genes. Proc Natl Acad Sci U S A. 2000;97:4891–4896. doi: 10.1073/pnas.080067697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Pascale G, Lloyd AJ, Schouten JA, Gilbey AM, Roper DI, Dowson CG, Bugg TD. Kinetic characterization of lipid II-Ala:alanyl-tRNA ligase (MurN) from Streptococcus pneumoniae using semisynthetic aminoacyl-lipid II substrates. J Biol Chem. 2008;283:34571–34579. doi: 10.1074/jbc.M805807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bouhss A, Josseaume N, Severin A, Tabei K, Hugonnet JE, Shlaes D, Mengin-Lecreulx D, Van Heijenoort J, Arthur M. Synthesis of the L-alanyl-L-alanine cross-bridge of Enterococcus faecalis peptidoglycan. J Biol Chem. 2002;277:45935–45941. doi: 10.1074/jbc.M207449200. [DOI] [PubMed] [Google Scholar]
- 104.Giannouli S, Kyritsis A, Malissovas N, Becker HD, Stathopoulos C. On the role of an unusual tRNAGly isoacceptor in Staphylococcus aureus. Biochimie. 2009;91:344–351. doi: 10.1016/j.biochi.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 105.Villet R, Fonvielle M, Busca P, Chemama M, Maillard AP, Hugonnet JE, Dubost L, Marie A, Josseaume N, Mesnage S, Mayer C, Valery JM, Etheve-Quelquejeu M, Arthur M. Idiosyncratic features in tRNAs participating in bacterial cell wall synthesis. Nucleic Acids Res. 2007;35:6870–6883. doi: 10.1093/nar/gkm778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chemama M, Fonvielle M, Villet R, Arthur M, Valery JM, Etheve-Quelquejeu M. Stable analogues of aminoacyl-tRNA for inhibition of an essential step of bacterial cell-wall synthesis. J Am Chem Soc. 2007;129:12642–12643. doi: 10.1021/ja0749946. [DOI] [PubMed] [Google Scholar]
- 107.Fonvielle M, Mellal D, Patin D, Lecerf M, Blanot D, Bouhss A, Santarem M, Mengin-Lecreulx D, Sollogoub M, Arthur M, Etheve-Quelquejeu M. Efficient access to peptidyl-RNA conjugates for picomolar inhibition of non-ribosomal FemX(Wv) aminoacyl transferase. Chem - Eur J. 2013;19:1357–1363. doi: 10.1002/chem.201201999. [DOI] [PubMed] [Google Scholar]
- 108.Benson TE, Prince DB, Mutchler VT, Curry KA, Ho AM, Sarver RW, Hagadorn JC, Choi GH, Garlick RL. X-ray crystal structure of Staphylococcus aureus FemA. Structure. 2002;10:1107–1115. doi: 10.1016/s0969-2126(02)00807-9. [DOI] [PubMed] [Google Scholar]
- 109.Biarrotte-Sorin S, Maillard AP, Delettre J, Sougakoff W, Arthur M, Mayer C. Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: insights into FemABX family substrates recognition. Structure. 2004;12:257–267. doi: 10.1016/j.str.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 110.Cusack S, Berthet-Colominas C, Hartlein M, Nassar N, Leberman R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 A. Nature. 1990;347:249–255. doi: 10.1038/347249a0. [DOI] [PubMed] [Google Scholar]
- 111.Fonvielle M, Li de La Sierra-Gallay I, El-Sagheer AH, Lecerf M, Patin D, Mellal D, Mayer C, Blanot D, Gale N, Brown T, van Tilbeurgh H, Etheve-Quelquejeu M, Arthur M. The structure of FemX(Wv) in complex with a peptidyl-RNA conjugate: mechanism of aminoacyl transfer from Ala-tRNA(Ala) to peptidoglycan precursors. Angew Chem, Int Ed. 2013;52:7278–7281. doi: 10.1002/anie.201301411. [DOI] [PubMed] [Google Scholar]
- 112.Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem Sci. 1998;23:263–267. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 113.Isono S, Isono K, Hirota Y. Mutations affecting the structural genes and the genes coding for modifying enzymes for ribosomal proteins in Escherichia coli. Mol Gen Genet. 1978;165:15–20. doi: 10.1007/BF00270371. [DOI] [PubMed] [Google Scholar]
- 114.Cumberlidge AG, Isono K. Ribosomal protein modification in Escherichia coli. I A mutant lacking the N-terminal acetylation of protein S5 exhibits thermosensitivity. J Mol Biol. 1979;131:169–189. doi: 10.1016/0022-2836(79)90072-x. [DOI] [PubMed] [Google Scholar]
- 115.Isono K, Isono S. Ribosomal protein modification in Escherichia coli. II Studies of a mutant lacking the N-terminal acetylation of protein S18. Mol Gen Genet. 1980;177:645–651. doi: 10.1007/BF00272675. [DOI] [PubMed] [Google Scholar]
- 116.Isono S, Isono K. Ribosomal protein modification in Escherichia coli. III Studies of mutants lacking an acetylase activity specific for protein L12. Mol Gen Genet. 1981;183:473–477. doi: 10.1007/BF00268767. [DOI] [PubMed] [Google Scholar]
- 117.Pettersson I, Hardy SJ, Liljas A. The ribosomal protein L8 is a complex L7/L12 and L10. FEBS Lett. 1976;64:135–138. doi: 10.1016/0014-5793(76)80267-0. [DOI] [PubMed] [Google Scholar]
- 118.Subramanian AR. Copies of proteins L7 and L12 and heterogeneity of the large subunit of Escherichia coli ribosome. J Mol Biol. 1975;95:1–8. doi: 10.1016/0022-2836(75)90330-7. [DOI] [PubMed] [Google Scholar]
- 119.Gordiyenko Y, Deroo S, Zhou M, Videler H, Robinson CV. Acetylation of L12 increases interactions in the Escherichia coli ribosomal stalk complex. J Mol Biol. 2008;380:404–414. doi: 10.1016/j.jmb.2008.04.067. [DOI] [PubMed] [Google Scholar]
- 120.Vetting MW, de Carvalho LP, Roderick SL, Blanchard JS. A novel dimeric structure of the RimL Nalpha-acetyltransferase from Salmonella typhimurium. J Biol Chem. 2005;280:22108–22114. doi: 10.1074/jbc.M502401200. [DOI] [PubMed] [Google Scholar]
- 121.Sakamoto K, Murayama K, Oki K, Iraha F, Kato-Murayama M, Takahashi M, Ohtake K, Kobayashi T, Kuramitsu S, Shirouzu M, Yokoyama S. Genetic encoding of 3-iodo-L-tyrosine in Escherichia coli for single-wavelength anomalous dispersion phasing in protein crystallography. Structure. 2009;17:335–344. doi: 10.1016/j.str.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 122.Brunzelle JS, Wu R, Korolev SV, Collart FR, Joachimiak A, Anderson WF. Crystal structure of Bacillus subtilis YdaF protein: a putative ribosomal N-acetyltransferase. Proteins: Struct, Funct, Genet. 2004;57:850–853. doi: 10.1002/prot.10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yu M, Magalhães ML, Cook PF, Blanchard JS. Bisubstrate inhibition: Theory and application to N-acetyltransferases. Biochemistry. 2006;45:14788–14794. doi: 10.1021/bi061621t. [DOI] [PubMed] [Google Scholar]
- 124.Vetting MW, Bareich DC, Yu M, Blanchard JS. Crystal structure of RimI from Salmonella typhimurium LT2, the GNAT responsible for N(alpha)-acetylation of ribosomal protein S18. Protein Sci. 2008;17:1781–1790. doi: 10.1110/ps.035899.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roy-Chaudhuri B, Kirthi N, Kelley T, Culver GM. Suppression of a cold-sensitive mutation in ribosomal protein S5 reveals a role for RimJ in ribosome biogenesis. Mol Microbiol. 2008;68:1547–1559. doi: 10.1111/j.1365-2958.2008.06252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fang H, Zhang X, Shen L, Si X, Ren Y, Dai H, Li S, Zhou C, Chen H. RimJ is responsible for N(alpha)-acetylation of thymosin alpha1 in Escherichia coli. Appl Microbiol Biotechnol. 2009;84:99–104. doi: 10.1007/s00253-009-1994-8. [DOI] [PubMed] [Google Scholar]
- 127.Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- 128.Yu BJ, Kim JA, Moon JH, Ryu SE, Pan JG. The diversity of lysine-acetylated proteins in Escherichia coli. J Microbiol Biotechnol. 2008;18:1529–1536. [PubMed] [Google Scholar]
- 129.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics. 2008;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Starai VJ, Escalante-Semerena JC. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J Mol Biol. 2004;340:1005–1012. doi: 10.1016/j.jmb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 132.Thao S, Escalante-Semerena JC. Biochemical and thermodynamic analyses of Salmonella enterica Pat, a multidomain, multimeric N(epsilon)-lysine acetyltransferase involved in carbon and energy metabolism. mBio. 2011;2:e00216–11. doi: 10.1128/mBio.00216-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem. 2007;282:30239–30245. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- 134.Chan CH, Garrity J, Crosby HA, Escalante-Semerena JC. In Salmonella enterica, the sirtuin-dependent protein acylation/deacylation system (SDPADS) maintains energy homeostasis during growth on low concentrations of acetate. Mol Microbiol. 2011;80:168–183. doi: 10.1111/j.1365-2958.2011.07566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gardner JG, Grundy FJ, Henkin TM, Escalante-Semerena JC. Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD(+) involvement in Bacillus subtilis. J Bacteriol. 2006;188:5460–5468. doi: 10.1128/JB.00215-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Crosby HA, Heiniger EK, Harwood CS, Escalante-Semerena JC. Reversible N epsilon-lysine acetylation regulates the activity of acyl-CoA synthetases involved in anaerobic benzoate catabolism in Rhodopseudomonas palustris. Mol Microbiol. 2010;76:874–888. doi: 10.1111/j.1365-2958.2010.07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mikulik K, Felsberg J, Kudrnacova E, Bezouskova S, Setinova D, Stodulkova E, Zidkova J, Zidek V. CobB1 deacetylase activity in Streptomyces coelicolor. Biochem Cell Biol. 2012;90:179–187. doi: 10.1139/o11-086. [DOI] [PubMed] [Google Scholar]
- 138.Tucker AC, Escalante-Semerena JC. Acetoacetyl-CoA synthetase activity is controlled by a protein acetyltransferase with unique domain organization in Streptomyces lividans. Mol Microbiol. 2013;87:152–167. doi: 10.1111/mmi.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hayden JD, Brown LR, Gunawardena HP, Perkowski EF, Chen X, Braunstein M. Reversible acetylation regulates acetate and propionate metabolism in Mycobacterium smegmatis. Microbiology. 2013;159:1986–1999. doi: 10.1099/mic.0.068585-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu H, Hegde SS, Blanchard JS. Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry. 2011;50:5883–5892. doi: 10.1021/bi200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tucker AC, Taylor KC, Rank KC, Rayment I, Escalante-Semerena JC. Insights into the specificity of lysine acetyltransferases. J Biol Chem. 2014;289:36249–36262. doi: 10.1074/jbc.M114.613901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee HJ, Lang PT, Fortune SM, Sassetti CM, Alber T. Cyclic AMP regulation of protein lysine acetylation in Mycobacterium tuberculosis. Nat Struct Mol Biol. 2012;19:811–818. doi: 10.1038/nsmb.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Podobnik M, Siddiqui N, Rebolj K, Nambi S, Merzel F, Visweswariah SS. Allostery and conformational dynamics in cAMP-binding acyltransferases. J Biol Chem. 2014;289:16588–16600. doi: 10.1074/jbc.M114.560086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bai G, Schaak DD, McDonough KA. cAMP levels within Mycobacterium tuberculosis and Mycobacterium bovis BCG increase upon infection of macrophages. FEMS Immunol Med Microbiol. 2009;55:68–73. doi: 10.1111/j.1574-695X.2008.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nambi S, Gupta K, Bhattacharyya M, Ramakrishnan P, Ravikumar V, Siddiqui N, Thomas AT, Visweswariah SS. Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J Biol Chem. 2013;288:14114–14124. doi: 10.1074/jbc.M113.463992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vergnolle O, Xu H, Blanchard JS. Mechanism and regulation of mycobactin fatty acyl-AMP ligase FadD33. J Biol Chem. 2013;288:28116–28125. doi: 10.1074/jbc.M113.495549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bhatnagar RS, Futterer K, Farazi TA, Korolev S, Murray CL, Jackson-Machelski E, Gokel GW, Gordon JI, Waksman G. Structure of N-myristoyltransferase with bound myristoyl-CoA and peptide substrate analogs. Nat Struct Biol. 1998;5:1091–1097. doi: 10.1038/4202. [DOI] [PubMed] [Google Scholar]
- 148.Majorek KA, Kuhn ML, Chruszcz M, Anderson WF, Minor W. Structural, functional, and inhibition studies of a Gcn5-related N-acetyltransferase (GNAT) superfamily protein PA4794: a new C-terminal lysine protein acetyltransferase from pseudomonas aeruginosa. J Biol Chem. 2013;288:30223–30235. doi: 10.1074/jbc.M113.501353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Siehl DL, Castle LA, Gorton R, Keenan RJ. The molecular basis of glyphosate resistance by an optimized microbial acetyltransferase. J Biol Chem. 2007;282:11446–11455. doi: 10.1074/jbc.M610267200. [DOI] [PubMed] [Google Scholar]
- 150.Srivastava P, Khandokar YB, Swarbrick CM, Roman N, Himiari Z, Sarker S, Raidal SR, Forwood JK. Structural characterization of a Gcn5-related N-acetyltransferase from Staphylococcus aureus. PLoS One. 2014;9:e102348. doi: 10.1371/journal.pone.0102348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Iqbal A, Arunlanantham H, Brown T, Jr, Chowdhury R, Clifton IJ, Kershaw NJ, Hewitson KS, McDonough MA, Schofield CJ. Crystallographic and mass spectrometric analyses of a tandem GNAT protein from the clavulanic acid biosynthesis pathway. Proteins: Struct, Funct, Genet. 2010;78:1398–1407. doi: 10.1002/prot.22653. [DOI] [PubMed] [Google Scholar]
- 152.Hung MN, Rangarajan E, Munger C, Nadeau G, Sulea T, Matte A. Crystal structure of TDP-fucosamine acetyltransferase (WecD) from Escherichia coli, an enzyme required for enterobacterial common antigen synthesis. J Bacteriol. 2006;188:5606–5617. doi: 10.1128/JB.00306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Errey JC, Blanchard JS. Functional annotation and kinetic characterization of PhnO from Salmonella enterica. Biochemistry. 2006;45:3033–3039. doi: 10.1021/bi052297p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vetting MW, Errey JC, Blanchard JS. Rv0802c from Mycobacterium tuberculosis: the first structure of a succinyl-transferase with the GNAT fold. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2008;64:978–985. doi: 10.1107/S1744309108031679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Holzapfel W, Kandler O. Taxonomy of the species Lactobacillus Beijerinck. VI Lactobacillus coprophilus subsp confusus nov subsp., a new variety of the subspecies Betabacterium. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg. 1969;123:657–666. [PubMed] [Google Scholar]
- 156.Schneider T, Senn MM, Berger-Bachi B, Tossi A, Sahl HG, Wiedemann I. In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol Microbiol. 2004;53:675–685. doi: 10.1111/j.1365-2958.2004.04149.x. [DOI] [PubMed] [Google Scholar]
- 157.Tipper DJ, Berman MF. Structures of the cell wall peptidoglycans of Staphylococcus epidermidis Texas 26 and Staphylococcus aureus Copenhagen. I Chain length and average sequence of cross-bridge peptides. Biochemistry. 1969;8:2183–2192. doi: 10.1021/bi00833a060. [DOI] [PubMed] [Google Scholar]
- 158.Filipe SR, Severina E, Tomasz A. Functional analysis of Streptococcus pneumoniae MurM reveals the region responsible for its specificity in the synthesis of branched cell wall peptides. J Biol Chem. 2001;276:39618–39628. doi: 10.1074/jbc.M106425200. [DOI] [PubMed] [Google Scholar]
- 159.Yanagihara Y, Kamisango K, Yasuda S, Kobayashi S, Mifuchi I, Azuma I, Yamamura Y, Johnson RC. Chemical compositions of cell walls and polysaccharide fractions of spirochetes. Microbiol Immunol. 1984;28:535–544. doi: 10.1111/j.1348-0421.1984.tb00706.x. [DOI] [PubMed] [Google Scholar]
- 160.Takumi K, Kawata T. Quantitative chemical analyses and antigenic properties of peptidoglycans from Clostridium botulinum and other clostridia. Jpn J Microbiol. 1976;20:287–292. doi: 10.1111/j.1348-0421.1976.tb00990.x. [DOI] [PubMed] [Google Scholar]
- 161.Lavollay M, Arthur M, Fourgeaud M, Dubost L, Marie A, Veziris N, Blanot D, Gutmann L, Mainardi JL. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J Bacteriol. 2008;190:4360–4366. doi: 10.1128/JB.00239-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Magnet S, Arbeloa A, Mainardi JL, Hugonnet JE, Fourgeaud M, Dubost L, Marie A, Delfosse V, Mayer C, Rice LB, Arthur M. Specificity of L,D-transpeptidases from gram-positive bacteria producing different peptidoglycan chemotypes. J Biol Chem. 2007;282:13151–13159. doi: 10.1074/jbc.M610911200. [DOI] [PubMed] [Google Scholar]
- 163.Hong HJ, Hutchings MI, Hill LM, Buttner MJ. The role of the novel Fem protein VanK in vancomycin resistance in Streptomyces coelicolor. J Biol Chem. 2005;280:13055–13061. doi: 10.1074/jbc.M413801200. [DOI] [PubMed] [Google Scholar]