

Abstract
Motivation: Nowadays, knowledge extraction methods from Next Generation Sequencing data are highly requested. In this work, we focus on RNA-seq gene expression analysis and specifically on case–control studies with rule-based supervised classification algorithms that build a model able to discriminate cases from controls. State of the art algorithms compute a single classification model that contains few features (genes). On the contrary, our goal is to elicit a higher amount of knowledge by computing many classification models, and therefore to identify most of the genes related to the predicted class.
Results: We propose CAMUR, a new method that extracts multiple and equivalent classification models. CAMUR iteratively computes a rule-based classification model, calculates the power set of the genes present in the rules, iteratively eliminates those combinations from the data set, and performs again the classification procedure until a stopping criterion is verified. CAMUR includes an ad-hoc knowledge repository (database) and a querying tool.
We analyze three different types of RNA-seq data sets (Breast, Head and Neck, and Stomach Cancer) from The Cancer Genome Atlas (TCGA) and we validate CAMUR and its models also on non-TCGA data. Our experimental results show the efficacy of CAMUR: we obtain several reliable equivalent classification models, from which the most frequent genes, their relationships, and the relation with a particular cancer are deduced.
Availability and implementation: dmb.iasi.cnr.it/camur.php
Contact: emanuel@iasi.cnr.it
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Among next generation sequencing experiments, RNA-seq gene expression profiling stands out as the process of quantifying the transcriptome abundance by counting the RNA fragments (reads) that are aligned on a reference genome (Wang etal., 2009).
In this work, we propose a new method for classifying RNA-seq case–control samples, which is able to compute multiple human readable classification models. We call this method and its software implementation CAMUR – Classifier with Alternative and MUltiple Rule-based models. Although RNA-seq data analysis tools (Howe etal., 2011; Kuehn etal., 2008) are widely used in case–control studies, the novelty of CAMUR consists in the extraction of several alternative and equivalent rule-based models, which represent relevant sets of genes related to the case and control samples. CAMUR extracts multiple classification models by adopting a feature elimination technique and by iterating the classification procedure.
CAMUR is based on the supervised learning approach (also called classification (Mehta etal., 1996)), the task of inferring a function from labeled training data (Tan etal., 2005b). Two data sets are required: (i) the training set, which consists of a group of training labeled samples, and hence each sample is a pair consisting of an input object – that can be a vector of features (attributes) – and its associated class label; (ii) the test set, which is used to classify new samples, after the inferred function is built; the test data may consists of a set of samples, whose class is known, but hidden and used only for verification purpose. Starting from the training set, a supervised machine learning algorithm builds the classification model based on the general hypotheses inferred from the features. Then, through this model, the classifier is able either to evaluate the model reliability on the test set, or to make predictions on new data. In other words, we can describe the classification problem as the process through which a system learns a mapping function (also called model) that assigns a sample to a class (Tan etal., 2005b). A classifier is the output of a supervised machine learning algorithm. There are many different state of the art classification algorithms: decision trees (Quinlan, 1993), rule-based (Boros etal., 2005; Cohen, 1995; Felici and Truemper, 2002; Frank and Witten, 1998; Gaines and Compton, 1995), ensembles (Bagging, Boosting, Random forest) (Dietterich, 2000), k-Nearest Neighbour (Dasarathy, 1990), linear regression (Seber and Lee, 2012), Naive Bayes (McCallum etal., 1998), neural networks (Haykin etal., 2009), Perceptrons (Riedmiller, 1994), Support Vector Machines (SVM) (Vapnik, 1998) and Relevance Vector Machine (RVM) (Tipping, 2001). For further details about the supervised learning paradigm and the algorithms the reader may refer to (Weitschek etal., 2014). Classification algorithms are frequently used in gene expression profiles analysis (Golub etal., 1999; Li etal., 2004; Nogueira etal., 2003; Park etal., 2014; Pirooznia etal., 2008; Shaik and Ramakrishna, 2014; Shipp et al., 2002; Tan and Gilbert, 2003; Tothill etal., 2015), in particular for experimental samples classification, i.e. the automatic assignment of each sample to its belonging class (e.g. case–control) after examining its profile. Rule-based classification algorithms are widespread for analyzing gene expression profiles (Dennis and Muthukrishnan, 2014; Geman etal., 2004; Hvidsten etal., 2003; Tan etal., 2005a; Weitschek etal., 2015; Zhou etal., 2003). These types of algorithms produce a classification model composed of logic formulas that provide an immediate relationship between the class and one or more features (genes). The assignment of a given class to each sample is performed by taking into account the satisfiability of the rules. In particular, the classifier uses logic propositional formulas in disjunctive (or conjunctive) normal form (‘if then rules’) for classifying the given records. Each classification rule (r) can be represented as: ri: Antecedent → Consequent (e.g. ). The antecedent contains a conjunction of attribute tests, each one known as literal (e.g. ), the consequent represents the covered class (e.g. control). Examples of rule-based classifiers are RIPPER (Cohen, 1995), LSQUARE (Felici and Truemper, 2002), LAD (Boros etal., 2005), RIDOR (Gaines and Compton, 1995) and PART (Frank and Witten, 1998).
We chose to analyze RNA-seq data with rule-based algorithms, because of their human readability, i.e. the investigator is provided with a list of meaningful features (genes) that appear in the rules. Specifically, among the state of the art classifiers we implement our method relying on the Repeated Incremental Pruning to Produce Error Reduction – RIPPER algorithm, because it is a robust and effective rule-based approach that provides reliable case–control models in terms of classification rates and computational performances (Lehr etal., 2011). In RNA-seq, rule-based algorithms may provide a low number of features (genes) into the resulting rules. For example, in a binary classification problem the classifier can build a model made of only two rules, with two or three features (e.g. ). Although this fact does not affect the classification performances, many other features that have discriminant power may not be present in the classification model. Therefore, our aim is to extract a comprehensive amount of knowledge from the analyzed data composed of equivalent and alternative classification models (i.e. rules). For example, to maximize the knowledge extraction in RNA-seq samples classification, we aim to detect all the genes that are implied with the analyzed disease, i.e. the discriminant genes that appear in alternative classification models. For extracting multiple classification solutions, one approach is presented in (Deb and Reddy, 2003), where the authors found 352 different three-gene combinations providing a 100% correct classification to the Leukemia gene expression profile data available at (Golub etal., 1999), by extending a genetic algorithm (Deb etal., 2002) into a multi-objectives evolutionary one that finds multiple and multimodal solutions in one single run (Miettinen, 1999). Those are defined as solutions that have identical objective values, but they differ in their format. Furthermore, another classification approach is presented in (Gholami etal., 2012) and relies on a feature elimination method, which consists of choosing features and then, removing those that do not match an assumption criteria. The deletion is performed in order to obtain a smaller set of features that can perform as well as the larger one, and hence the computational overhead is reduced. However, the authors aim is not to extract alternative and equivalent classification models. Conversely, we aim to obtain more than one reliable classification model by performing an iterative feature elimination without implementing an optimization method.
2 Materials and methods
First, the terminology adopted in the paper is introduced. We collect n samples, each one described by its m features (gene expression profiles) and labeled with a class (condition), e.g. normal – tumoral (We adopt The Cancer Genome Atlas terminology (i.e. normal – tumoral), where normal corresponds to a healthy sample (control) and tumoral to a diseased one (case).). The ith sample of the data set is represented by the vector , where and . Therefore, the vectors compose the data matrix, whose rows correspond to the samples and whose columns to their features. The reader may refer to Table 1 for an example.
Table 1.
Example of the breast cancer RNA-seq data matrix extracted from The Cancer Genome Atlas (TCGA)
| SampleID | ANO8 | C1orf27 | TRPM6 | Class | |
|---|---|---|---|---|---|
| A8-A09D | 2.64 | 5.42 | 0.38 | Breast cancer | |
| BH-A0DH | 1.46 | 6.47 | 0.76 | Normal | |
| GM-A2DC | 2.22 | 22.50 | 0.53 | Breast cancer | |
| GM-A2D9 | 3.13 | 14.21 | 0.61 | Breast cancer | |
| ⋯ | ⋯ | ⋯ | |||
| GM-A2DB | 3.86 | 5.15 | 0.59 | Breast cancer |
The rows correspond to the samples and the columns to their features (gene expression profiles). The cells contain the gene expression measure Reads Per Kilobase per Million mapped reads (RPKM) explained in Section 2.3.
2.1 CAMUR: classifier with alternative and multiple rule-based models
In this section, we describe CAMUR, a method and a software designed to find alternative and equivalent solutions for a classification problem. CAMUR is based on: In brief, the method iteratively computes a rule-based classification model through the supervised RIPPER algorithm, calculates the power set (or a partial combination) of the features present in the rules, iteratively eliminates those combinations from the data set, and performs again the classification procedure until a stopping criterion is verified.
a rule-based classifier (i.e. in this work RIPPER);
an iterative feature elimination technique;
a repeated classification procedure;
an ad-hoc storage structure for the classification rules (CAMUR database).
In greater details, CAMUR executes at first the RIPPER algorithm, which extracts from a training set the classification model that contains rules with a number of features (i.e. genes) and their values (i.e. quantification levels). Accuracy and F – measure (see Eq. 1) are used on a test set to evaluate the extracted classification model. Then, CAMUR stores the classification model and the results into a database and extracts the features from the generated model. We call this set of features St (where t is the current iteration) and we define the list where those features are memorized as FL. After that, CAMUR computes the power set of the features Pt by storing all the combinations into the main memory. In the following, we refer to the Original Data Set of features as ODS. Starting from Pt, the software performs a feature elimination by deleting from ODS one combination of features at time (i.e. an item of the power set) and executes the RIPPER classification algorithm on the new data set with (ptj is the jth element of Pt and ). All the results of the elimination and classification steps are memorized in the CAMUR database. These operations are iterated on the new generated data sets with where k < t and , updating FL at each iteration. We highlight that the power set () generation on the new feature sets is performed by not taking into account duplicate combinations that occurred in previous power sets Pk with . CAMUR terminates the execution when one of the following conditions is satisfied: At the end of this procedure, we have a collection of alternative classification models composed of several features that are able to distinguish the samples with high reliability. For evaluating the classification models and consequently to terminate the procedure, we adopt the accuracy and the F – measure (refer to Eq. 1). Given True Positives (TP), objects of that class recognized in the same class; False Positives (FP), objects not belonging to that class recognized in that class; True Negatives (TN), objects not belonging to that class and not recognized in that class; False Negatives (FN), objects belonging to that class and not recognized in that class, the measures are defined as follows:
| (1) |
where is the Precision and is the Recall.
the reliability of the classification models is below a given threshold, e.g. F – measure (see Eq. 1) lower than 0.85;
the list of features FL has been completely processed;
the maximum number of iterations has been reached.
In the following, we provide an execution example of the algorithm. Given a data set composed of 10 features (genes) and 10 samples – 5 tumoral and 5 normal –, CAMUR extracts through the first execution of RIPPER a classification model composed of a set of rules (e.g. ). The rules contain a set of three features , gene2, which is stored in the features list FL. Starting from S1 the power set (except the empty set) P1 is computed: . The first item of the power set is eliminated from the data set and the classification procedure is performed, which provides a new set of features, e.g. . The first power set P1 is completely processed, generating a number of feature sets S, which are stored in FL. After the processing of P1, the power set P2 from S2 is computed and the classification is performed. The algorithm continues until one of the stopping criteria is verified. To speed up the procedure, it is worth noting that the next power set is computed and processed only when the current power set has been completely examined.
The computational time depends on: (i) the size of the power sets, which are related to the size of the feature sets Si – if the cardinality of the feature set is equal to m (), then the power set generation requires in the worst-case ; (ii) the worst-case complexity of RIPPER, i.e. with n number of samples in the training set. Therefore, the total complexity of CAMUR is . We highlight that usually the number of features present in rule-based classification models is limited, especially when dealing with two-class classification problems, as case–control studies.
Additionally, we investigate the possibility to iterate the feature elimination in different ways, and hence our algorithm can be executed as follows: .
In the loose feature elimination mode, the algorithm performs a combined iterative feature elimination. As above-mentioned, this execution mode takes the model and the results from the first classification and builds the power set of the found features, whose combinations are iteratively eliminated from the data set. A classification step follows each elimination of the feature combinations. The new extracted features that are present in the current model are added to the features list FL and are going to be processed in the next iterations.
In the strict feature elimination mode, the algorithm performs a single iterative feature elimination. First, a classification with the RIPPER algorithm is performed, the features that appear into the rules are extracted, and then eliminated one by one. The classification is iterated after each elimination on the resulting data set. In contrast to the , once a feature is eliminated, it is never inserted again into the data set. Referring to the example given above, in the the execution is straightforward. Starting from the above-mentioned feature set S1, CAMUR proceeds with the elimination of gene1 from the original data set ODS and performs the classification on the new data set, obtaining . Then, it eliminates gene2 from and performs the classification again, obtaining S3. It finishes to process S1, and then all the other ones contained in FL if a proper stopping criteria is satisfied.
The is faster than the , this can be explained by the two main differences: (i) the does not compute the power set, so there are less classification procedures to run; (ii) on each classification run one discriminating feature is eliminated from the original data matrix. Therefore, the accuracy of the models may decrease faster. Conversely, the extracts more knowledge but is slower, because the computed power set has a sets and for each one CAMUR runs the classification algorithm again.
Finally, it is possible to execute both the strict and the through the . This execution mode performs first the strict and then the , storing all the models and results into the CAMUR database.
2.2 The CAMUR software package
The CAMUR software package is composed of two distinct parts: The MSE corresponds to the implementation of the CAMUR algorithm described in Section 2.1. In brief, it performs the iterative classification and feature elimination procedures and fills the database with the results and models.
Multiple Solutions Extractor (MSE);
Multiple Solutions Analyzer (MSA).
The MSE is organized in following modules:
InputManager, which manages the user interactions and the input;
CamurLauncher, which executes the iterative CAMUR classification algorithm;
DataElaborator, which is responsible for the data set to classify and performs feature elimination;
FeaturesManager, which manages the feature lists and power sets;
ResultsElaborator, which processes the classification results and models;
DataAccessObject(DAO), which has the responsibility to communicate with the database.
The component diagram of the software is shown in Figure 1.
Fig. 1.
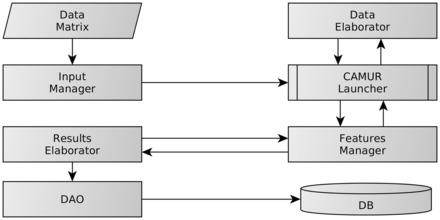
Component diagram of the MSE part of the CAMUR software package
The workflow of the software is as follows: the InputManager processes the user input data (data matrix) and the parameters (e.g. maximum number of iterations, execution mode), input data are taken by the CamurLauncher and managed through the DataElaborator. Then, CamurLauncher performs the iterative classification by managing the feature eliminations and combinations through the FeaturesManager. The ResultsElaborator stores the classification models and results in the database with the aid of the DataAccessObject(DAO).
On the other hand, the MSA is a support tool dedicated to the analysis and interpretation of the obtained results, it extracts knowledge from the database by running predefined queries. The following queries have been included in the software:
Q1 Genes list: Which are all the genes that are able to distinguish tumoral samples from normal ones in a given RNA-seq experiment? And how many times do they occur in all the obtained classification models?
In this query, we extract the list of genes and their occurrences in all the extracted rule-based classification models.
Q2 Literals and conjunctions list: Which are the most relevant literals (e.g. ) and conjunctions (e.g. ) and their related correctly classified instances?
Through this query, we identify the conjunctions of one or more rule literals (e.g. ) optionally linked with a logic . For each conjunction, we report: (i) the number of correctly (incorrectly) classified instances; (ii) the percentage of correctly (incorrectly) classified instances.
Q3 Rules list: Which are the classification rules and how is their reliability?
In this query, we extract the rule disjunctions (i.e. conjunctions linked with a logic ), their measures of reliability, i.e. F – measure, accuracy (refer to Eq. 1 of Section 2.1).
Q4 Literals statistic: Which are the literals (e.g. ) that more frequently occur within a specific range?
Such a query provides the gene name, the literal operator (e.g. <, >), its minimum and maximum value, the values average () and their standard deviation (σ), the number of occurrences of each literal with the same operator, and finally the coefficient of variation measure defined as: .
Q5 Gene pairs: Which are all the pairs of genes that appear within a same rule and how many are their occurrences?
This query extracts all the couples of genes that are present in a same rule and counts how many times these two genes appear together.
The MSA is organized in the following modules: The MSA software is released with a graphic interface, which enables to choose a predefined query and to set additional parameters. It provides the real knowledge in terms of gene lists, gene interactions, expression thresholds, classification results and models. A screenshot of the graphic interface is depicted in Figure 2. The CAMUR software package composed of the MSE and the MSA and described above is implemented in JAVA for linux, windows and mac-os operating systems under a GPL license and is available at dmb.iasi.cnr.it/camur.php. A comprehensive user guide is provided as supplementary data S1.
Fig. 2.
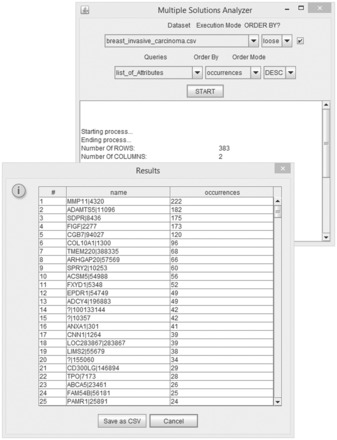
Screenshot of the MSA part of the CAMUR software package: it displays the initial parameters configuration available to the user
, which is responsible for user interactions and for showing the results of the queries;
, which executes the query and collects the results;
, which builds a query according to the user input;
, which processes the query by retrieving all the information from the database;
, which has the responsibility to communicate with the database.
CAMUR database
CAMUR stores the classification models and the results of the procedure into an ad-hoc storage structure, called CAMUR database. It permits the execution of the MSA queries for knowledge extraction. This database has a total of 16 relationships and 13 entities, the main ones are described below: CAMUR database is implemented with the open source software MySql (www.mysql.com).
, which contains information about the execution of the MSE;
, which represents an execution of the classification procedure and stores its results;
, which consists of the whole set of disjunctions that predict a class;
that is a set of conjunctions;
that is composed by a feature, an operator (i.e. >, <, ) and a value;
that represents the set of features extracted from the rules;
that is the set of features eliminated before an experiment execution.
2.3 Experimental data
In this work, we test our method on RNA-seq experimental data extracted from The Cancer Genome Atlas (TCGA) (Weinstein etal., 2013). Additionally, we validate our method on non-TCGA data.
The Cancer Genome Atlas is a project that aims to offer a comprehensive overview of genomic changes involved in human cancer. A data portal available at www.tcga.org offers access to a large number of genomic and clinical experiments related to more than 10 000 patients affected by 33 different tumor types. In addition, it provides a collection of diverse metadata (e.g. clinical health records) associated to the patients. The TCGA portal contains clinical information, genomic characterization data, and high level sequence analysis of the tumor genomes.
We extract RNA-seq experiment data related to Breast (BRCA), Head and Neck (HNSC) and Stomach (STAD) Cancers. The data set characteristics are summarized in Table 2. For each data set, we take into account the Reads Per Kilobase per Million mapped reads (RPKM) value of each gene expression measure (Mortazavi etal., 2008), which normalizes the gene raw counts by considering the length of the gene and the total number of the fragments:
| (2) |
where R is the number of mapped reads onto the gene exons, Nr is the total number of mapped reads, and L is the feature length that corresponds to the number of nucleotides of the exonic region of the gene. For each tumor, we build a unique matrix of RPKM values, where the rows correspond to the samples, the columns to genes, and the cells to the RPKM values. The matrix given as input to CAMUR is similar to that one depicted in Table 1 of Section 2. An ad-hoc software ‘Tcga2Camur’ that converts the TCGA RNA-seq data sets into the CAMUR data matrix has been developed and is available at dmb.iasi.cnr.it/camur.php.
Table 2.
Summary of the analyzed data sets
| Cancer | Tissues | Tumoral | Normal | Genes | [MB] |
|---|---|---|---|---|---|
| BRCA | 884 | 783 | 101 | 20532 | 292 |
| HNSC | 295 | 264 | 31 | 20532 | 92 |
| STAD | 271 | 238 | 33 | 29699 | 56 |
The three data sets are extracted from The Cancer Genome Atlas. The numbers refer to the sequenced tissues, belonging to tumoral and normal classes (first three columns). It is worth noting that for each data set the number of analyzed samples corresponds to the number of tumoral tissues (third column). The last two columns refer to the number of genes and the size of the three data sets.
It is worth noting that CAMUR can be applied also to gene expression data processed by other normalization methods, such as RSEM (RNA-seq by Expectation Maximixation) (Li and Dewey, 2011). RSEM guesses how many ambiguously mapping reads belong to a transcript/gene (i.e. raw count value of the TCGA data) and estimates the frequency of the gene/transcript among the sequenced transcripts (i.e. scaled estimate value of the TCGA data). RSEM provides an accurate transcript quantification without requiring a reference genome. In particular, we test CAMUR on RNA-seq data of BRCA extracted from TCGA and normalized with the RSEM method.
Moreover, we validate CAMUR on a non-TCGA data set: the Wilms Tumor (WT) (Walz etal., 2015) among the Kidney Tumors of the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project.
Finally, we evaluate CAMUR classification models on non-TCGA BRCA data sets downloaded from Gene Expression Omnibus (GEO) with accession numbers GSE56022 and GSM1308330.
3 Results and discussion
In this section, we provide an overview of the extracted knowledge from the analyzed data, including statistics of the performed experiments, and a more specific discussion of the obtained results.
We analyzed the TCGA data sets of Breast, Head and Neck, and Stomach Cancers with both the loose and the of CAMUR. CAMUR ran a total of 1486 classification procedures with a percentage split sampling schema (80% training, 20% test) (Tan etal., 2005b). The classification procedures stopped either for the decreasing of the classification performances, or because the maximum number of iterations was reached. On average, the obtained precision, recall and F – measure values are greater than 99%. Within the generated rules CAMUR found 904 different genes, each gene is found on average 23.34 times (the occurrences of each gene range from 1 to 900). CAMUR computed 8182 sets of combinations (736 in and 7446 in ), each one composed of 1–7 genes (average 2.57). The amount of removed sets is 1480 (364 in , 1116 in ), which represent the removed genes from a data set. The number of genes within these sets is on average 2.025 and the values range from 1 to 6.
The CAMUR analysis (strict and ) on the Breast, the Head and Neck and the Stomach Cancer data set provides 513, 218 and 272 different genes, respectively. The corresponding gene expressions allow the scientist to distinguish normal samples from tumoral ones and they are potential markers for the diseases.
The total amount of gene pairs identified in all rule sets are 20139, 610 and 272, for the Breast, the Head and Neck and the Stomach Cancer, respectively. Among those genes pairs, 2212 for Breast, 256 for Head and Neck, 137 for Stomach Cancer have been found into rule sets containing exactly a pair of genes.
We show the execution times of CAMUR in Table 3. It is worth noting that the processing of the Breast Cancer data set requires longer time, because of the large number of samples and of the rules size.
Table 3.
CAMUR execution times
| Cancer | Total time [h] | Loose mode time [h] | Strict mode time [h] |
|---|---|---|---|
| BRCA | 6 h:56 m | 6 h:17 m | 0 h:39 m |
| HNSC | 0 h:33 m | 0 h:26 m | 0 h:7 m |
| STAD | 0 h:27 m | 0 h:17 m | 0 h:10 m |
The execution times for Breast (BRCA), Head and Neck (HNSC), Stomach (STAD) Cancer. Times are reported in hours.
In the following, we report the extracted knowledge related to the Breast cancer by discussing the obtained results of each query described in Section 2.1. The results related to the other data sets can be found in supplementary data S2.
With query 1 (features list), we extract 383 genes and their occurrences found by CAMUR during the classification experiments. We show in Table 4 an example of the results of this query.
Table 4.
A portion from the output results of the ‘list of attributes’ query
| Gene | Occurrences |
|---|---|
| ADAMTS511096 | 109 |
| MMP114320 | 102 |
| FIGF2277 | 84 |
| SDPR8436 | 82 |
| COL10A11300 | 51 |
The extracted genes are sorted by their occurrences, which may point to a relation with the disease. In Table 5, we provide a list of the most frequent 12 genes extracted during the execution of CAMUR.
Table 5.
List of the most common 12 genes (row-wise) extracted by CAMUR
| MMP114320 | ADAMTS511096 | SDPR8436 |
| FIGF2277 | CGB794027 | COL10A11300 |
| TMEM220388335 | ARHGAP2057569 | SPRY210253 |
| ACSM554988 | FXYD15348 | EPDR154749 |
With query 2 (conjunctions list), we extract 1708 conjunctions and the values of the correctly (incorrectly) classified samples. For example, we extracted ‘(SDPR8436 11.6) (ANXA1301 161.3) Normal’ that classified correctly all the 87 instances of the test set. Query 3 (disjunctions list) extracts 1564 classification rules, e.g. ‘(SPRY210253 14.4) (C20orf160140706 1.8) (COL10A11300 0.7) (AASS10157 1.6) Normal’, which provides an accuracy of 100%. Through query 4 (literal statistics), we extract 397 most frequent genes and we may capture if they show comparable expression values. An interesting example for the output interpretation of query 3 in Breast Cancer is: gene TMEM220388335 occurs 33 times, and its attribute value is , and hence provides a strong and stable signal. Query 5 (pairs of features) displays 2212 pairs of genes and a counter of how many times they appear together. The pairs that appear mostly are depicted in Table 6. Additionally, it is worth noting that the user can define personalized queries and run them directly on the database.
Table 6.
An example of the output for query 5
| Gene 1 | Gene 2 | Occurrences |
|---|---|---|
| FIGF2277 | MMP114320 | 100 |
| CGB794027 | ADAMTS511096 | 73 |
| SDPR8436 | ANXA1301 | 37 |
| EPDR154749 | MMP114320 | 34 |
Furthermore, among the gene lists extracted by CAMUR, we found 3 genes (i.e. ACOT711332, ADAR103 and GLT25D179709) shared by Breast, Head and Neck and Stomach Cancer set: in panel a of Figure 3 we show all the overlaps among the three sets of genes through an Eulero-Venn diagram. A preliminary functional analysis on the human protein atlas (Uhlén etal., 2015) confirms the relation of those genes with the three above-mentioned cancer types.
Fig. 3.
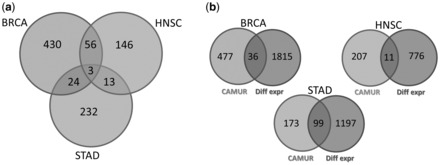
Eulero-Venn diagram of the CAMUR gene lists for BRCA, HNSC and STAD: (a) diagram of overlapped genes extracted by CAMUR; (b) diagram of the overlapped genes between the CAMUR gene lists and the differential expressed ones
In order to strengthen CAMUR, we performed the following tests. Since we have not found other state of the art classification algorithms that implement multiple models extraction, a direct comparison of our method is not feasible. Therefore, we compared CAMUR with respect to a standard wide-spread technique that relies on the differential expression analysis (Storey and Tibshirani, 2003) and that provides a list of statistically significant genes related to case–control samples, by applying Benjamini-Hochberg correction (Benjamini and Hochberg, 1995) to estimate a False Discovery Rate (FDR)-adjusted p-value. We extracted a list of 1851, 787, 1296 genes with a P-value for BRCA, HNSC and STAD, respectively. The above-mentioned lists were compared with those extracted by CAMUR. We found 36 for BRCA, 11 for HNSC, 99 for STAD genes that are shared in both lists (panel b of Fig. 3). The lists of shared genes are available as supplementary data S3. It is worth noting that the size of the lists extracted by CAMUR is smaller, and hence our approach allows to focus on few core genes related to the investigated disease. Additionally, most of those genes are not selected by the differential expression analysis enhancing the novelty of our approach.
Additionally, we ran several tests to validate CAMUR, its classification models, and its performances. The detailed results are available as supplementary data S4. First, we randomly selected ten BRCA rules extracted by CAMUR and verified them on two external breast cancer RNA-seq data sets of GEO (GSE56022 and GSM1308330). Most of the rules succeed in the identification of the diseased samples confirming the validity of our method: 9 out of 10 correctly cover the GSM1308330 samples, 7 out of 10 the GSE56022 ones (but we remark that 2 of the not successful rules cannot be applied because a gene is not present in the data set). Second, we tested CAMUR on a non-TCGA data set: the Wilms Tumor (WT) (Walz etal., 2015) of the (TARGET) project. It consists of 94 tissues (82 tumoral, 12 normal) and 58450 mRNA gene expression values normalized with the RPKM method. CAMUR performed 320 runs (212 loose and 108 in ) finding 231 different genes with an average F-measure of 0.98. Third, we validated CAMUR on RNA-seq data of BRCA normalized with the RSEM method. CAMUR executed 2048 classification experiments (1895 loose and 153 in ) and extracted 986 different genes with an average F-measure of 0.99. Finally, we performed a comparative analysis of CAMUR with respect to the SVM classifier by computing the same number of classification runs: both methods reached high reliable results (average F-measure of 0.97 for SVM, 0.99 for CAMUR) on all data sets. We remark that SVM outputs just a single classification model that cannot be easily interpreted by human experts.
4 Conclusion
In this work, we presented CAMUR, a new method for multiple solutions extraction in rule-based classification problems. We showed that the amount of knowledge extracted by our algorithm is higher than a standard supervised classification. We described the two parts of CAMUR software package: MSE that performs the classification procedure and MSA that analyzes the obtained results. Additionally, we designed and developed a database for an effective and comprehensive knowledge extraction. We proved the efficacy of our algorithm on large sets of RNA-seq data, focusing on Breast, Head and Neck and Stomach Cancer from TCGA, and validating it on external data sets from TARGET and GEO. To conclude, CAMUR results as a reliable technique for solving classification problems by extracting many alternative and equally accurate solutions.
In future, we intend to test our method on other RNA-seq data sets in order to build a large knowledge repository of classification models related to a particular disease. The extracted genes may then be analyzed by domain experts with functional and enrichment analyses (D’Andrea etal., 2013). It would be also interesting to perform a simulation study for evaluating the performance of CAMUR under different scenarios in a quantitative manner. Additionally, we plan to integrate in our software other rule-based classifiers, as well as to enrich the software with new functions and higher performances. Finally, we plan to extend the analysis to other biological data sets as sequences classification, e.g. DNA-Barcoding.
Supplementary Material
Acknowledgements
We wish to thank Prof. Riccardo Torlone and Paolo Atzeni for supporting this work during the master of science in computer engineering and during the big data course. The authors’ contributions can be found in supplementary data.
Funding
This work was supported by the Italian PRIN ‘GenData 2020’ [2010RTFWBH], the FLAGSHIP ‘InterOmics’ [PB.P05] project, and The Epigenomics Flagship Project ‘EPIGEN’ [PB.P01].
Conflict of Interest: none declared.
References
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological), 57, 289–300. [Google Scholar]
- Boros E., et al. (2005) Logical Analysis of Data. In: Wang J. (ed), Encyclopedia of Data Warehousing and Mining, Hershey, PA, USA: Idea Group Reference, pp. 689–692. [Google Scholar]
- Cohen W.W. (1995) Fast effective rule induction. In Proceedings of the Twelfth International Conference on Machine Learning, pp. 115–123. [Google Scholar]
- D’Andrea D., et al. (2013) Fidea: a server for the functional interpretation of differential expression analysis. Nucleic Acids Res., 41, W84–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasarathy B.V. (1990) Nearest Neighbor (NN) Norms: NN Pattern Classification Techniques. IEEE Computer Society Press, 2001 L Street N.W., Suite 700 Washington, USA. [Google Scholar]
- Deb K., Reddy A.R. (2003) Reliable classification of two-class cancer data using evolutionary algorithms. BioSystems, 72, 111–129. [DOI] [PubMed] [Google Scholar]
- Deb K., et al. (2002) A fast and elitist multiobjective genetic algorithm: Nsga-ii. IEEE Trans. Evol. Comput., 6, 182–197. [Google Scholar]
- Dennis B., Muthukrishnan S. (2014) Agfs: adaptive genetic fuzzy system for medical data classification. Appl. Soft Comput., 25, 242–252. [Google Scholar]
- Dietterich T.G. (2000) Ensemble methods in machine learning. In: Multiple classifier systems. Springer; New York, USA, pp. 1–15. [Google Scholar]
- Felici G., Truemper K. (2002) A minsat approach for learning in logic domains. INFORMS J. Comput., 13, 1–17. [Google Scholar]
- Frank E., Witten I.H. (1998) Generating accurate rule sets without global optimization. In Proceedings of the 15th International Conference on Machine Learning Morgan Kaufmann. [Google Scholar]
- Gaines B.R., Compton P. (1995) Induction of ripple-down rules applied to modeling large databases. J. Intell. Inf. Syst., 5, 211–228. [Google Scholar]
- Geman D., et al. (2004) Classifying gene expression profiles from pairwise mrna comparisons. Stat. Appl. Genet. Mol. Biol., 3, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholami B., et al. (2012) Recursive feature elimination for brain tumor classification using desorption electrospray ionization mass spectrometry imaging. In Engineering in Medicine and Biology Society (EMBC), 2012 Annual International Conference of the IEEE, pp. 5258–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub T.R., et al. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science, 286, 531–537. [DOI] [PubMed] [Google Scholar]
- Haykin S.S., et al. (2009) Neural networks and learning machines. Vol. 3 Pearson Education, Upper Saddle River, NJ, USA. [Google Scholar]
- Howe E.A., et al. (2011) RNA-seq analysis in mev. Bioinformatics, 27, 3209–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvidsten T.R., et al. (2003) Learning rule-based models of biological process from gene expression time profiles using gene ontology. Bioinformatics, 19, 1116–1123. [DOI] [PubMed] [Google Scholar]
- Kuehn H., et al. (2008) Using genepattern for gene expression analysis. Current Protocols in Bioinformatics, 22, 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehr T., et al. (2011) Rule based classifier for the analysis of gene–gene and gene–environment interactions in genetic association studies. BioData Min., 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. (2011) Rsem: accurate transcript quantification from rna-seq data with or without a reference genome. BMC Bioinformatics, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., et al. (2004) A comparative study of feature selection and multiclass classification methods for tissue classification based on gene expression. Bioinformatics, 20, 2429–2437. [DOI] [PubMed] [Google Scholar]
- McCallum A., et al. (1998) A comparison of event models for naive bayes text classification. In AAAI-98 Workshop on Learning for Text Categorization, Vol. 752, pp. 41–48. Citeseer. [Google Scholar]
- Mehta M., et al. (1996) Sliq: a fast scalable classifier for data mining. In Advances in Database Technology-EDBT’96, pp. 18–32. Springer. [Google Scholar]
- Miettinen K. (1999) Nonlinear multiobjective optimization, Vol. 12 Springer, New York, USA. [Google Scholar]
- Mortazavi A., et al. (2008) Mapping and quantifying mammalian transcriptomes by rna-seq. Nat. Methods, 5, 621–628. [DOI] [PubMed] [Google Scholar]
- Nogueira F.T., et al. (2003) RNA expression profiles and data mining of sugarcane response to low temperature. Plant Physiol., 132, 1811–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C., et al. (2014) Integrative gene network construction to analyze cancer recurrence using semi-supervised learning. PLoS One, 9, e86309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia M., et al. (2008) A comparative study of different machine learning methods on microarray gene expression data. BMC Genomics, 9, S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan J.R. (1993) C4.5: Programs for Machine Learning (Morgan Kaufmann Series in Machine Learning), 1st edn.Morgan Kaufmann, San Francisco, California, USA. [Google Scholar]
- Riedmiller M. (1994) Advanced supervised learning in multi-layer perceptronsfrom backpropagation to adaptive learning algorithms. Comput. Stand. Interfaces, 16, 265–278. [Google Scholar]
- Seber G.A., Lee A.J. (2012) Linear regression analysis, Vol. 936 John Wiley & Sons, Wiley, Hoboken, NJ, USA. [Google Scholar]
- Shaik R., Ramakrishna W. (2014) Machine learning approaches distinguish multiple stress conditions using stress-responsive genes and identify candidate genes for broad resistance in rice. Plant physiology, 164, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipp M.A., et al. (2002) Diffuse large b-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med., 8, 68–74. [DOI] [PubMed] [Google Scholar]
- Storey J.D., Tibshirani R. (2003) Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA, 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A.C., Gilbert D. (2003) Ensemble machine learning on gene expression data for cancer classification. In Proceedings of New Zealand Bioinformatics Conference, Te Papa, Wellington, New Zealand University of Glasgow. [PubMed] [Google Scholar]
- Tan A.C., et al. (2005a) Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics, 21, 3896–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P., et al. (2005b) Introduction to Data Mining. Addison Wesley, Oxford University Press, Oxford, UK. [Google Scholar]
- Tipping M.E. (2001) Sparse Bayesian learning and the relevance vector machine. J. Mach. Learn. Res., 1, 211–244. [Google Scholar]
- Tothill R.W., et al. (2015) Development and validation of a gene expression tumour classifier for cancer of unknown primary. Pathol. J. RCPA, 47, 7–12. [DOI] [PubMed] [Google Scholar]
- Uhlén M., et al. (2015) Tissue-based map of the human proteome. Science, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Vapnik V.N. (1998) Statistical Learning Theory. Wiley, Hoboken, NJ, USA. [Google Scholar]
- Walz A.L., et al. (2015) Recurrent dgcr8, drosha, and six homeodomain mutations in favorable histology wilms tumors. Cancer Cell, 27, 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., et al. (2009) RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet., 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J.N., et al. (2013) The cancer genome atlas pan-cancer analysis project. Nat. Genet., 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitschek E., et al. (2014) Supervised DNA barcodes species classification: analysis, comparisons and results. BioData Min., 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitschek E., et al. (2015) Gela: a software tool for the analysis of gene expression data. In Database and Expert Systems Applications (DEXA), BIOKDD, pp. 31–35. IEEE. [Google Scholar]
- Zhou C., et al. (2003) Evolving accurate and compact classification rules with gene expression programming. IEEE Trans. Evol. Comput., 7, 519–531. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.