

Abstract
Impaired nitric oxide (NO˙)-cyclic guanosine 3', 5'-monophosphate (cGMP) signaling has been observed in many cardiovascular disorders, including heart failure and pulmonary arterial hypertension. There are several enzymatic determinants of cGMP levels in this pathway, including soluble guanylyl cyclase (sGC) itself, the NO˙-activated form of sGC, and phosphodiesterase(s) (PDE). Therapies for some of these disorders with PDE inhibitors have been successful at increasing cGMP levels in both cardiac and vascular tissues. However, at the systems level, it is not clear whether perturbation of PDE alone, under oxidative stress, is the best approach for increasing cGMP levels as compared with perturbation of other potential pathway targets, either alone or in combination. Here, we develop a model-based approach to perturbing this pathway, focusing on single reactions, pairs of reactions, or trios of reactions as targets, then monitoring the theoretical effects of these interventions on cGMP levels. Single perturbations of all reaction steps within this pathway demonstrated that three reaction steps, including the oxidation of sGC, NO˙ dissociation from sGC, and cGMP degradation by PDE, exerted a dominant influence on cGMP accumulation relative to other reaction steps. Furthermore, among all possible single, paired, and triple perturbations of this pathway, the combined perturbations of these three reaction steps had the greatest impact on cGMP accumulation. These computational findings were confirmed in cell-based experiments. We conclude that a combined perturbation of the oxidatively-impaired NO˙-cGMP signaling pathway is a better approach to the restoration of cGMP levels as compared with corresponding individual perturbations. This approach may also yield improved therapeutic responses in other complex pharmacologically amenable pathways.
Author Summary
Developing drugs for a well-defined biochemical or molecular pathway has conventionally been approached by optimizing the inhibition (or activation) of a single target by a single pharmacologic agent. On occasion, drug combinations have been used that generally target multiple pathways affecting a common phenotype, again by optimizing the extent of inhibition of individual targets, semi-empirically adjusting their doses to minimize toxicities as they are manifest. Here, we present a computational approach for identifying optimal combinations of agents that can affect (inhibit) a well-defined biochemical pathway, doing so at minimal combined concentrations, thereby potentially minimizing dose-dependent toxicities. This approach is illustrated computationally and experimentally with a well-known pathway, the nitric oxide-cyclic GMP pathway, but is readily generalizable to rational polypharmacy.
Introduction
Signal transduction via the nitric oxide (NO˙)-cyclic guanosine 3', 5'-monophosphate (cGMP) pathway is involved in multiple and diverse biological responses, including smooth muscle relaxation, inhibition of platelet aggregation, and neural communication [1–6]. This pathway is composed of several molecular species acting in two opposing limbs, the cGMP-synthetic limb and the cGMP-degradative limb (see Fig 1). The proper function of these two limbs is crucial in controlling these biological responses. Within the cGMP-synthetic limb, NO˙ binds to soluble guanylyl cyclase (sGC) to catalyze the production of cGMP from guanosine-5'-triphosphate (GTP), whereas in the cGMP-degradative limb, cyclic nucleotide phosphodiesterase (PDE) converts cGMP to GMP. Impaired function of either or both limbs of the NO˙-cGMP signaling pathway has been reported in many cardiovascular disorders, including heart failure and pulmonary arterial hypertension.
Fig 1. Steps for modeling NO˙-cGMP signaling pathway.
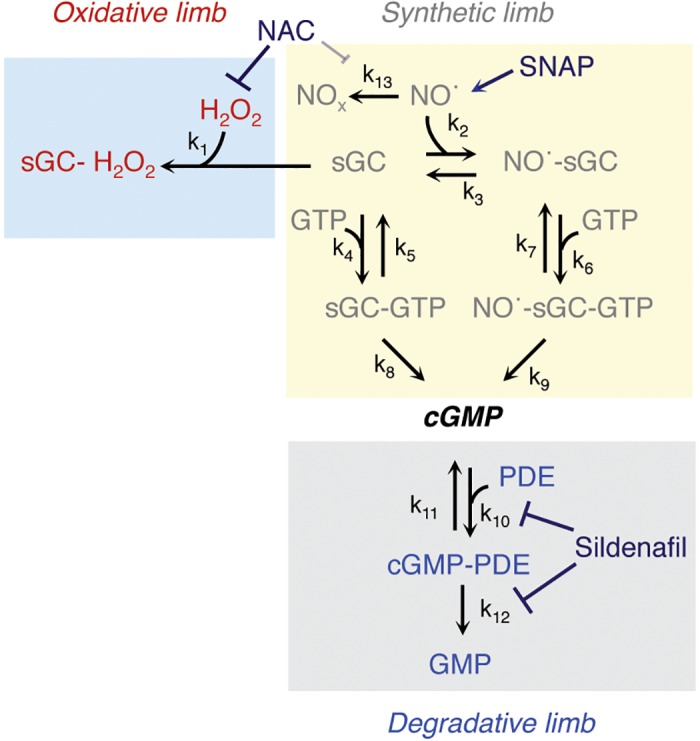
The reaction schema: NO˙ is synthesized in a generator cell and freely diffuses, either within the same cell or to a target cell, to activate sGC; sGC is oxidized by H2O2 and inactivated. Either sGC or NO˙-sGC can convert GTP to cGMP, which is degraded by PDE to GMP. Note that oxidative stress drives the system toward the oxidative limb, and that the goal of pharmacological modulation of this pathway is to reverse the adverse effects of oxidative stress and to minimize PDE inhibition in order to optimize cGMP levels. NAC can be used as an antioxidant (impairing hydrogen peroxide-dependent oxidation of SGC and, perhaps, oxidation of NO˙), sildenafil as a PDE5 inhibitor, and SNAP as an NO˙ donor to modulate this pathway experimentally. Abbreviations: cGMP, cyclic guanosine 3', 5'-monophosphate; GMP, guanosine-5'-monophosphate; GTP, guanosine-5'-triphosphate; H2O2, hydrogen peroxide; NAC, N-acetylcysteine; NO˙, nitric oxide; NOx, oxidized (inactive) nitrogen oxides; PDE, phosphodiesterase; SNAP, S-Nitroso-n-nacetylpenicillamine; sGC, soluble guanylyl cyclase.
Importantly, increased oxidative stress associated with malfunction of the NO˙-cGMP signaling pathway has been implicated in the pathobiology of several diseases [7, 8]. During oxidative stress, the pathway’s unresponsiveness can be explained by several mechanisms, among which sGC insensitivity to NO˙ (tolerance) is decisive. Elevated reactive oxygen species (ROS) may promote sGC insensitivity through either non-heme (cysteine) oxidation of sGC [9–14], S-nitrosation of sGC [15], heme oxidation of sGC [16], or oxidation of NO˙, such as via enhanced peroxynitrite (ONOO-) formation [17]. Potentially, there are several determinants of this oxidatively-adapted pathway, including oxidatively inactivated sGC itself, oxidized NO˙, and PDE. The pharmacological challenge is how best to deploy potential therapeutic options that focus on these determinants under increased oxidative stress in a way that optimizes restoration of the function of this pathway.
Investigating the complexity of biological systems using combinatorial perturbations is a rational strategy for predicting function and phenotype [18], understanding network mechanisms [19–22], and identifying new and more promising therapeutic targets for human diseases [23, 24]. In theory, using a combination of drugs that can perturb different components of a system could be a more effective strategy than treating a disease with a single drug [25]. Indeed, the most complex diseases, such as cardiovascular diseases, cancer, diabetes mellitus, neurodegenerative diseases, and asthma, are multifactorial diseases. Systems-based interventions using multi-component drug combinations have been used increasingly to treat these complex diseases, although these approaches have largely been developed empirically in the clinical setting. The main purpose of model-based drug discovery is to revisit classical pharmacology logically in order to replace the one-gene, one-protein, and one-mechanism perspective with a systems-oriented paradigm to improve the therapeutic index of potential drugs targeting these complex diseases [26, 27].
Relevant principles have emerged from different studies of combination therapies that do not always yield predicted outcomes. For example, the combination of niacin (vitamin B3) with a statin [5-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase] leads to an incremental decrease in low-density lipoprotein (LDL) cholesterol concentration and an increase in high-density lipoprotein (HDL) cholesterol concentration [28]. Combinations of drugs that perturb five different targets in the HIV life cycle have turned AIDS from a lethal infection into a manageable chronic disease [29]. Another interesting combination is that of nitroglycerin and N-acetylcysteine (NAC), which can potentiate the effects of nitroglycerin in the treatment of acute myocardial ischemia [30]. The combination of β2-adrenergic receptor activators with muscarinic receptor blockers is useful for the treatment of chronic obstructive pulmonary disease [31].
Opposing and independent regulatory mechanisms within the NO˙-cGMP pathway determine the biological level of cGMP in the steady-state. Model-based approaches have facilitated our understanding of these regulatory mechanisms for cGMP formation [32–34]. Thus far, modeling has been used to study two distinct limbs of the NO˙-cGMP signaling pathway separately [33–39]; however, here, we will build this model as an integrated system that also includes oxidative inactivation. We then pose the question of whether a combination of two or three agents with orthogonal therapeutic actions (and toxicities), used at lower concentrations than when used alone, will enhance cGMP formation beyond that of single agents in the presence of oxidative stress.
In this study, we pursued this question using a dynamical model of the NO˙-cGMP signaling pathway in the presence of hydrogen peroxide. Impaired activation of NO˙-cGMP signaling has been observed in several cardiovascular disorders, including heart failure [40] and pulmonary arterial hypertension [41], due, in part, to excess oxidants. Current treatments for these disorders that involve this pathway includes nitrovasodilators and phosphodiesterase inhibitors. Thus, this theoretical approach, were it to demonstrate benefit, may offer initial strategies for optimal drug combinations for the treatment of these (and other) disorders in which NO˙-cGMP signaling is dysfunctional using approved agents.
Previous studies have used models of cell signaling networks to evaluate the action of drug pairs as compared with corresponding individual drugs [42]. Modeling drug action using ordinary differential equations could be challenging without sufficient information about the integrated network kinetics of drug action. To address this challenge, Araujo and colleagues investigated an interesting concept, perturbation simulation, on the epidermal growth factor receptor (EGFR) signaling network. They found that pairwise perturbations of reaction rates was more effective at restoring optimal function of the network than individual perturbation of corresponding reaction rates [43].
We expanded this approach to a practically remediable pathway and examined how hydrogen peroxide (H2O2)-induced oxidant stress affects the key reaction steps of the NO˙-cGMP signaling pathway to diminish cGMP levels, and then developed a combinatorial approach to perturb the oxidatively-impaired NO˙-cGMP signaling pathway and restore cGMP levels toward normal. In addition, in contrast to [43], we also examined the consequences of lesser degrees of inhibition in combination modeling to infer lower dose-dependent toxicity. Lastly, in contrast to [43], we performed cell-based experiments to validate the modeling strategy. To do so, all rate constants were perturbed individually, in pairs, or in trios by step-wise ten-fold changes in their values to their original values, which is analogous to concentration-dependent inhibition of a given reaction by a specific inhibitor. Our goal was to identify an optimal perturbation that augments the cGMP levels toward normal during oxidative stress.
Using a single perturbation, we found that the potential therapeutic targets, including the oxidation of sGC, NO˙ dissociation from sGC, and cGMP degradation by PDE, had a profound effect on enhancing cGMP accumulation as compared with other reaction steps. Using combined perturbations, we were able to identify an optimal triple perturbation that increases cGMP levels beyond that observed with the corresponding individual or paired perturbations that comprise it. Importantly, these theoretical findings were confirmed in cell-based experiments in which a combination of a nitric oxide donor (S-nitroso-N-penicillamine), an antioxidant (N-acetyl-L-cysteine), and a phosphodiesterase type 5-inhibitor (sildenafil) significantly improved the cyclic GMP output of the pathway in the setting of oxidant stress (hydrogen peroxide) in pulmonary artery vascular smooth muscle cells.
Results
Elevated reactive oxygen species (ROS) can affect both the cGMP-synthetic limb [9, 10] and the cGMP-degradative limb [44] of the NO˙-cGMP signaling pathway. In order to assess the combinatorial effects of different pharmacological modulators on dynamical pathway behavior (as determined by cGMP output), we first modeled the dynamical behavior and then assessed the effects of optimal combinations of pharmacological inhibitors in cell-based assays using pulmonary artery vascular smooth muscle cells.
Modeling the NO˙-cGMP Signaling Pathway
Once NO˙ is generated in source cells (endothelial cells) in the vasculature, it diffuses into vascular smooth muscle cells and binds to sGC, a ferrous iron hemoprotein receptor, to generate the NO˙-sGC complex. Either sGC alone or the NO˙-sGC complex, whose specific activity is ~200 times greater than sGC alone [45], can convert GTP to the second messenger molecule, cGMP, which is degraded by cyclic nucleotide PDE(s) to GMP. However, under oxidative stress conditions, sGC is also oxidized by H2O2 and thereby desensitized (Fig 1). The biological reactions comprising this system were modeled using ordinary differential equations and mass action kinetics involving 12 molecular species and 13 rate constants (S1 Table).
The simulation time intervals were selected to monitor cGMP dynamics between 0 and 200 seconds (based on the cGMP dynamics in S1A Fig for pulmonary artery vascular smooth muscle cells). Next, an oxidant (500 μM H2O2) was added to the system to alter the dynamics of all molecular species, including cGMP, as compared with control. One prior experimental study showed that both cGMP-degrading enzymes and sGC desensitization cooperatively accounted for the diverse patterns of cGMP responses to NO˙. Two different temporal dynamic signatures of cGMP were reported within platelets and astrocytes that have high and low levels of PDEs, respectively [32, 46, 47]. In our system under control conditions, the cGMP concentrations increased abruptly to a peak concentration within 40 seconds, and then decreased to baseline within 200 seconds (mirroring the experimental dynamics of S1A Fig). When the system was exposed to H2O2, the cGMP levels decreased by ~6-fold (Fig 2A). In this model, we proposed that H2O2 impairs activation of sGC and its generation of cGMP [48]; however, conflicting results have been reported by others [49].
Fig 2. Monitoring cGMP levels after single perturbation of oxidatively impaired NO˙-cGMP pathway.

(A) The effect of H2O2 on cGMP dynamics. H2O2 caused ~6-fold reduction in cGMP levels. (B and C) Reducing either k3 or k10 during oxidative stress was not an effective perturbation for restoring cGMP to the normal level. The value of k3 or k10 was reduced to 10% of their original values (S1 Table), with perturbation of k3 or k10 denoted as ρk3 and ρk10, respectively. While these perturbations can be effective strategies in enhancing cGMP levels in the absence of H2O2, they are not effective in restoring cGMP levels to normal in the presence of H2O2. (D) All single perturbations of the NO˙-cGMP pathway in the presence of H2O2. The cGMP levels were monitored in the absence of H2O2. Subsequently, in the presence of H2O2, the system was perturbed by reducing one of thirteen rate constants (k1‒k13) to 10% of their original values, denoted as ρks, and then the cGMP levels calculated. Note that ρk1, ρk3, ρk10, and ρk12 markedly increased the cGMP levels beyond other perturbations.
Relative Contributions of the Determinants of cGMP Synthesis and Degradation to the NO˙-cGMP Signaling Pathway
To restore cGMP generation to the normal level, there are several potential therapeutic targets, including oxidatively inactivated sGC, the NO˙-activated form of sGC, and PDE. Therapies for some diseases with PDE inhibitors have been successful at increasing cGMP levels in both cardiac and vascular tissues. However, to predict which one of these potential targets would be most effective at increasing cGMP levels, we perturbed either the synthetic limb (k3) or the degradative limb (k10) of the pathway in the absence or the presence of H2O2, and then evaluated cGMP dynamics. We found that targeting these two reaction steps can significantly increase the cGMP levels as compared with control if there is sufficient unoxidized sGC available. However, under significant oxidative stress, targeting these two reaction steps cannot be an effective strategy for restoring cGMP levels to normal (Fig 2B and 2C).
To evaluate more systematically the role of any given reaction in cGMP formation, we compared cGMP dynamics by reducing each of the thirteen rate constants to 10% of its original value (simulating significant reaction inhibition) in the presence of H2O2. We found that the cGMP levels were not restored toward control levels by decreasing k1 (oxidizing sGC), k3 (desensitizing sGC), or k10 (degrading cGMP) to 10% of their original values (Fig 2D). This observation suggested that under oxidative stress, targeting either the synthetic limb or the degradative limb of the pathway alone is not an effective approach for restoring cGMP to normal levels.
Combined Perturbation of the Oxidized NO˙-cGMP Pathway
The relative involvement of both synthetic and degradative components of the NO˙-cGMP signaling pathway led us to propose that these components could exert autonomous effects on cGMP accumulation. This concept raised the possibility that combined perturbations may have more profound effects on cGMP levels than single perturbations. Addressing this concept, the NO˙-cGMP pathway was perturbed using all possible single, paired, or triple perturbations in the presence of H2O2, and then the time-integrated cGMP (cGMPT) levels were calculated (Fig 3). We found that the simultaneous perturbation (ρ) of several rate processes along with the perturbation of k1 (ρk1) yielded the highest cGMPT levels relative to other perturbations. This finding suggested that targeting the primary driver of pathway dysfunction (ρk1) along with other potential therapeutic targets might be a better approach for increasing cGMP levels (even) beyond control levels under oxidative stress.
Fig 3. Relative cGMPT levels after single, paired, and triple kinetic perturbations of the oxidatively impaired NO˙-cGMP pathway.

The relative level of cGMPT as a function of all possible single (13 brown bars), paired (78 gray bars), and triple (286 green bars) perturbations is shown. Values of the rate constants were reduced to 10% of their original values (S1 Table). Each bar shows the relative integrated cGMP levels over the period of simulation, or . Note that some of the optimal perturbations are highlighted in this figure.
Evaluation of the Effects of Combined Perturbations on cGMP Responses
We next perturbed the proposed rate constants, including ρk1, ρk3, ρk10, or all possible combinations of these three rate constants, in the presence of H2O2. Modeling a fractional linear reduction of values for these rate constants, we created a vector of eleven different values for each wherein the maximum value for each rate constant was its original value (S1 Table) and the minimum value was 0.1, 0.3, or 0.5 of its original value for a rate constant in single, paired, or triple perturbations, respectively (lesser minimal values were used with greater combined perturbations to attempt to demonstrate efficacy at combined doses that might limit dose-dependent toxicities). Rate constants that were not varied under each set of modeling conditions were maintained at their full values. The cGMP dynamics was then calculated using the range of rate constants (Fig 4). These results suggested that under oxidative stress, decreasing dissociation of NO˙ from the NO˙-sGC complex (ρk3) is the most sensitive reaction step for increasing cGMP levels as compared with the use of an anti-oxidant (ρk1) and PDE inhibitor (ρk10). Furthermore, perturbation of k1 (ρk1) is the best strategy by which to increase cGMP levels beyond perturbation of either k3 (ρk3) or k10 (ρk10).
Fig 4. The effects of linear reduction of values for three individual rate constants on cGMP dynamics.

Using the linear reduction of values for k1, k3, and k10, we created a vector of eleven different values for each of these rate constants. We next generated three linearly spaced vectors for each of the rate constants by fractionally reducing each decrementally. Using these vectors of rate constants, cGMP dynamics were calculated. The cGMP levels after serial reduction of (A) k1, (B) k2, and (C) k3. (D) The relative cGMP levels. Data are shown as mean (dashed lines) ± S.E.M (shaded lines) of 11 simulated replicates. Note: k3 was the most sensitive parameter in cGMP accumulation as compared with k1 and k10. In addition, the highest cGMP levels were achieved at ~40 seconds.
Subsequently, we used the Bliss model [50] (which, based on probability theory, assumes two inhibitors work through independent mechanisms of action, and assumes that the two inhibitors do not interfere or compete with each other) to evaluate the power of paired perturbations. Under oxidative stress, optimal parameters were perturbed either individually or in pairs in order to compare the effects of perturbations on cGMP levels. The effects of single perturbations on cGMP levels were used to calculate the Bliss model, as indicated by eq (15). As depicted in Fig 5, paired perturbations of optimized single parameters increased cGMP levels beyond the Bliss model predictions. To assess whether these differences are additive or non-additive, we used an isobologram analysis. We examined the combined effects on cGMPT when two or three rate constants were perturbed simultaneously. The isobologram (contour plot) [51–56] was used to quantify the combined effects (Fig 6), wherein we observed that combined perturbations act additively to increase cGMP levels in this system.
Fig 5. Pairwise perturbation of optimal rate constants increased cGMP levels beyond that predicted by the Bliss model.

Comparison of cGMP levels after individual and pairwise perturbation of (A) k1 + k3, (B) k1 + k10, and (C) k3 + k10 with the Bliss model [generated using the perturbation of corresponding individual rate constants and applying eq (15) in the Supplement]. Single perturbations were used to predict paired perturbation signatures. The simulated combination produced cGMP levels that were greater than those predicted by the Bliss model.
Fig 6. Pairwise or triple perturbations of three agents additively enhance the cGMPT levels.

(A-C) The cGMPT dose matrix-responses to all possible combined perturbations of three rate constants. We iteratively reduced the rate constants by fractional decrements with ρ defined as the (normalized) perturbation ratio with values either 0.3 or 0.5 for pairwise or triple perturbations, respectively. For pairwise perturbations, two vectors of rate constants were combined in 11×11 matrices in which the value of each rate constant was fractionally reduced by linear decrements along each axis. The combined effects of (A) k1 plus k3, (B) k1 plus k10, and (C) k3 plus k10 on cGMPT levels. (D-E) The contour plots illustrate that the pairwise perturbations exerted additive effects on cGMPT levels. (G-I) The cGMP dose matrix responses to the triple perturbation of k1, k3, and k10. Three vectors of rate constants were combined in 11×11×11 matrices in which the value of each rate constant was linearly reduced along each axis. (J-L) The contour plots reveal additive augmentation of cGMPT levels.
cGMP Dynamics in the Absence and Presence of H2O2
We studied cGMP dynamics using human embryonic kidney (HEK) 293 cells and human pulmonary artery vascular smooth muscle (PAVSM) cells. PAVSM contain abundant PDE5 compared with HEK293 cells (35), thereby ensuring that both the cGMP-synthetic and degradative limbs determine the cGMP levels (S1 Fig). Thus, in PAVSM, the rapid accumulation of cGMP is followed by its equally rapid reduction (S1A Fig). In HEK293 cells, which contain lower amounts of PDE5 (35) as compared with PAVSM cells, the cGMP-synthetic limb of the pathway primarily determines the cGMP levels (S1B Fig). The cells were pretreated with either H2O2 or buffer for 30 minutes. Time points were selected to capture cGMP dynamics. When the HEK293 cells were exposed to H2O2 at 500 μM, we found that NO˙-stimulated cGMP production was significantly reduced as a function of time (S1B Fig). This result suggested that H2O2 blocked the cGMP-synthetic limb of the pathway, which plays a predominant role in determining the cGMP levels in HEK293 cells (as confirmed by the absence of a biphasic response in cGMP dynamics compared with the PAVSM cells in S1 Fig).
cGMP Dynamics in the Presence of Pharmacological Modulators of the cGMP Pathway
In order to determine the validity of the combinatorial modeling described above, we measured cGMP in PAVSM cells treated with various combinations of agents that act on different steps in the pathway of Fig 1. Agents were chosen because they have been used in human studies, and because they affect each of the limbs of the pathway in Fig 1. As shown in Fig 7A, we first showed that the addition of a NO.-donor, S-acetyl-N-penicillamine (SNAP), increased the cGMP produced by 58% over vehicle-treated control cells; hydrogen peroxide treatment, however, abrogated this increase. When cells were treated with hydrogen peroxide and the reducing agent, N-acetyl-L-cysteine (NAC), cGMP levels increased to ~2-fold above control. The addition of sildenafil, a PDE5 inhibitor (the primary PDE isoform found in PAVSM responsible for cGMP degradation), to SNAP and NAC in the presence of hydrogen peroxide further increased cGMP levels to ~3.2-fold above vehicle-treated control cells. With these baseline measurements, we next explored key comparative combinations of agents that mimicked the optimal modeled combinations, as shown in Fig 7B. Here, cGMP responses are reported as the % of the maximal response (to sildenafil and SNAP in the absence of hydrogen peroxide) owing to variation from experiment to experiment. We observed that the combinations of NAC and SNAP, sildenafil and SNAP, and sildenafil, NAC, and SNAP each increased cGMP in the presence of hydrogen peroxide, and that the relative magnitude of the increases was consistent with the modeled data in Fig 3. The use of NAC inhibits reactions 1 (and possibly 13), the use of SNAP ‘inhibits’ reaction 3 indirectly by driving reaction 2, and the use of sildenafil inhibits reaction 10 as a competitive inhibitor of PDE5 and indirectly inhibits reaction 12 by limiting the formation of the catalytic complex and hence substrate turn-over. Thus, as in Fig 3, the magnitude of increase in cGMP was of the following order: inhibition of reactions 1 + 3 < inhibition of reactions 3 + 10 (or 12) < inhibition of reactions 1 + 3 + 10, which is similar to the experimental reaction order we observed in the data of Fig 7.
Fig 7. Increases in cGMP in PAVSM cells in the presence and absence of pharmacological agents that affect the cGMP pathway.

In (A) and (B), PAVSM cells were pre-incubated for 90 min with buffer, 10 mM NAC (N-acetylcysteine), or 100 nM sildenafil (sild), followed by 30 min incubation with 500 μM hydrogen peroxide. Cells were then incubated with 100 μM SNAP for 10 min prior to harvesting for cGMP determination. Panel (A) shows a representative experiment, with three biological replicates under each condition. Shown in (B) are the averages of 3–4 separate experiments with each condition tested in duplicate or triplicate. Sildenafil, NAC, H2O2, plus SNAP condition was not significantly different from the sildenafil plus SNAP condition; these two conditions (* indicates p<0.05 by ANOVA followed by a Newman-Keuls test) were significantly different from all other conditions.
Discussion
Impaired activation of NO˙-cGMP signaling pathway has been observed in cardiovascular disorders and other common disease states. There are multiple enzymatic determinants of cGMP production in this pathway, including sGC itself, the oxidatively inactivated form of sGC, the NO˙-activated form of sGC, enzymatic sources of NO˙, and PDE. Therapies for these disorders with PDE inhibitors have been successful at enhancing cGMP levels in cardiac and vascular tissue with attendant improvement in lusitropy and vasodilation, respectively. However, PDE is only one of the enzymatic determinants of cGMP formation. In this systems-level approach, we used all possible single, paired, or triple perturbations to propose a combined perturbation that was more effective in cGMP accumulation than any single perturbation. The optimal number of the perturbations was three owing to there being only three key processes that determine independently the cGMP levels, i.e., cGMP synthesis, cGMP degradation, and oxidative inactivation of sGC. By having this modeled information, one can improve experimental design, curb cost, and save time in performing the experiments necessary for gaining useful results. Alternatively, one could randomly target any given component of this pathway either individually or in combination with other components of the pathway. Yet another approach is the maximal damage targeting strategy [57], theoretically a better approach relative to the random targeting of a pathway. However, using either the random targeting or the maximal damage targeting approach, we might overlook the optimal perturbations among many combinations that may never have been tested.
PDEs are essential enzymes within normal cells that degrade the phosphodiester bond in the second messenger molecules cAMP and cGMP. PDEs are, therefore, important regulators of signal transduction mediated by these second messenger molecules. As with many drugs that affect molecular pathways involved in (many) different signaling pathways, the side effects of PDE inhibitors are dose-dependent [58]; thus, to reduce the dose of a PDE inhibitor and then combine it with other potential drugs that have non-overlapping mechanisms of action and toxicities may significantly improve the overall therapeutic index of the treatment strategy.
One of the rationales for using combination therapy is to block redundant pathways that exist extensively within the molecular networks whose functions are modified in human diseases. To overlook this network property may limit the potential for reformulating existing drugs that can be used in combination with higher efficacy and fewer toxicities. Our results show how the combined perturbations of the NO˙-cGMP signaling pathway represent a useful strategy for increasing cGMP levels. A model-based analysis suggests that the combinatorial perturbation of biological networks is a promising approach by which to identify drug combinations with higher efficacy and perhaps lower toxicity (rational polypharmacy) [42]. Further work on other specific pathways will be required to validate the general approach.
Materials and Methods
cGMP Measurements
Enzyme immunoassay (EIA): human pulmonary artery vascular smooth muscle (PAVSM) cells and growth media were obtained from Lonza Inc. (Walkersville, MD., USA). Confluent cells were pre-treated with phenol-red free DMEM (supplemented with 10% fetal calf serum) in the presence of absence of 10 mM N-acetyl-L-cysteine (NAC), a thiol reducing agent and antioxidant, to reverse mildly oxidized critical sulfhydryl groups in sGC [14] and, possibly, to prevent the oxidation of NO to NOx; and/or 100 nM sildenafil, a PDE5 inhibitor, for 90 min followed by 500 μM H2O2 for 30 min. Cells were next treated with either phosphate-buffered saline or 100 μM S-nitroso-N-acetylpenicillamine (SNAP), a NO.-donor, for 10 min. PAVSM cells were rinsed in ice-cold phosphate buffered saline and then solubilized in ice-cold 6% trichloroacetic acid. Samples were stored at -80° until the day of the assay. Samples were processed and cGMP and protein were measured as previously described [14]. cGMP formation was measured by immunoassay according to the cGMP Assay (Cayman Chemical Co., Ann Arbor, MI). H2O2, trichloroacetic acid, NAC, and sildenafil were purchased from Sigma-Aldrich (St. Louis, MO). Phenol-red-free DMEM was obtained from Gibco, Life Technologies, Grand Island, NY and fetal calf serum was from Atlanta biologicals, Flowery Branch, GA.
Constructing the Model
Assuming mass-action kinetics, the reaction scheme (Fig 2, S1 Table) was deconstructed into 12 ordinary differential equations (ODEs):
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
to simulate the dynamics of the molecular species within the NO˙-cGMP signaling pathway. The ODEs were solved using a numerical ODE solver (ode15s). All mathematical modeling and simulations were performed using the SimBiology toolbox in MatLab (Version 8, 2012b, MathWorks, Natick, MA). The parameter values for this model include 13 rate constants and 12 initial concentrations (S1 Table), which were chosen or estimated from the literature, as indicated in the Table. The system dynamics were assessed in the absence or presence of H2O2.
Model Perturbations
In the presence of H2O2 (500 μM), the NO˙-cGMP pathway was perturbed by assuming the presence of an effective inhibitor of a given reaction(s) sufficient to impair the reaction kinetics (ρk = 0.1k). We perturbed all possible individual (13), pairs (78), or trios (286) of reactions in the model. The total number of perturbations (up to triple perturbations) was computed by inserting the total number of rate constants (q = 13) and the maximum number of perturbations (p = 3) into following equation:
| (13) |
Thus, the total possible number of perturbations (for triple perturbations) is 13C3 = 377. To assess the relative role of each perturbation, cGMP dynamics were illustrated (Fig 3).
The time-integrated cumulative cGMP (cGMPT) level is defined as:
| (14) |
Comparing the simulated pairwise perturbations with the Bliss model
We next used the Bliss model [50] to predict the effects of paired perturbations using the product effect of corresponding single perturbations. The Bliss model was used to evaluate whether combined perturbations enhance cGMP levels beyond corresponding single perturbations. The combined perturbation of two rate constants (ρk) is computed as the product of the individual perturbations of the two rate constants, ρki and ρkj.
| (15) |
Evaluating the Combined Perturbations Using Contour and Dose Matrix Plots
Perturbation of k1, k3, or k10 alone can induce a dose-dependent cGMPT response in the presence of H2O2. We varied the rate constants by fractional linear decrements.
To depict the matrix response of cGMPT, two vectors of rate constant values were combined in 11×11 matrices where the value of each rate constant is depicted along each axis (Fig 6). The contour plots were used to evaluate additive and non-additive effects. Thus, the combined actions were either additive (Fig 6) or synergistic if the isobole is a straight line or a convex line, respectively. Likewise, 11×11×11 matrix of all model parameters was constructed for triple perturbations and three-dimensional contour plots analyzed accordingly.
Supporting Information
(DOCX)
(DOCX)
(TIF)
Acknowledgments
The authors thank Ms. Stephanie Tribuna for expert technical assistance.
Data Availability
All relevant data are within the paper and supporting information.
Funding Statement
This work was funded by National Institutes of Health grant numbers: R37 HL61795, P01 HL48743, U01 HL108630, U01 HL007690, and P50 GM 107618 (Sorger, PI) to JL. URL: http://www.nih.gov. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Furchgott RF. Introduction to EDRF research. J Cardiovasc Pharmacol. 1993;22 Suppl 7:S1–2. [PubMed] [Google Scholar]
- 2.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987. December;84(24):9265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977. August;74(8):3203–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992. December 18;258(5090):1898–902. [DOI] [PubMed] [Google Scholar]
- 5.Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988. November 24;336(6197):385–8. [DOI] [PubMed] [Google Scholar]
- 6.Steinhorn BS, Loscalzo J, Michel T. Nitroglycerin and Nitric Oxide—A Rondo of Themes in Cardiovascular Therapeutics. N Engl J Med. 2015. July 16;373(3):277–80. 10.1056/NEJMsr1503311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001. November 27;104(22):2673–8. [DOI] [PubMed] [Google Scholar]
- 8.Simonian NA, Coyle JT. Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 1996;36:83–106. [DOI] [PubMed] [Google Scholar]
- 9.Brandwein HJ, Lewicki JA, Murad F. Reversible inactivation of guanylate cyclase by mixed disulfide formation. J Biol Chem. 1981. March 25;256(6):2958–62. [PubMed] [Google Scholar]
- 10.Fernhoff NB, Derbyshire ER, Marletta MA. A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc Natl Acad Sci U S A. 2009. December 22;106(51):21602–7. 10.1073/pnas.0911083106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braughler JM. Soluble guanylate cyclase activation by nitric oxide and its reversal. Involvement of sulfhydryl group oxidation and reduction. Biochem Pharmacol. 1983. March 1;32(5):811–8. [DOI] [PubMed] [Google Scholar]
- 12.Heckler EJ, Crassous PA, Baskaran P, Beuve A. Protein disulfide-isomerase interacts with soluble guanylyl cyclase via a redox-based mechanism and modulates its activity. Biochem J. 2013. May 15;452(1):161–9. 10.1042/BJ20130298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin E, Lee YC, Murad F. YC-1 activation of human soluble guanylyl cyclase has both heme-dependent and heme-independent components. Proc Natl Acad Sci U S A. 2001. November 6;98(23):12938–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maron BA, Zhang YY, Handy DE, Beuve A, Tang SS, Loscalzo J, et al. Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinyl thiol oxidation in vascular smooth muscle cells. J Biol Chem. 2009. March 20;284(12):7665–72. 10.1074/jbc.M809460200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc Natl Acad Sci U S A. 2007. July 24;104(30):12312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, H SA, Meurer S, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006. September;116(9):2552–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber M, Lauer N, Mulsch A, Kojda G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med. 2001. December 1;31(11):1360–7. [DOI] [PubMed] [Google Scholar]
- 18.Schneidman E, Berry MJ 2nd, Segev R, Bialek W. Weak pairwise correlations imply strongly correlated network states in a neural population. Nature. 2006. April 20;440(7087):1007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garmaroudi FS, Marchant D, Si X, Khalili A, Bashashati A, Wong BW, et al. Pairwise network mechanisms in the host signaling response to coxsackievirus B3 infection. Proc Natl Acad Sci U S A. 2010. September 28;107(39):17053–8. 10.1073/pnas.1006478107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chatterjee MS, Purvis JE, Brass LF, Diamond SL. Pairwise agonist scanning predicts cellular signaling responses to combinatorial stimuli. Nat Biotechnol. 2010. July;28(7):727–32. 10.1038/nbt.1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehar J, Krueger A, Zimmermann G, Borisy A. High-order combination effects and biological robustness. Mol Syst Biol. 2008;4:215 10.1038/msb.2008.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flaherty P, Radhakrishnan ML, Dinh T, Rebres RA, Roach TI, Jordan MI, et al. A dual receptor crosstalk model of G-protein-coupled signal transduction. PLoS Comput Biol. 2008;4(9):e1000185 10.1371/journal.pcbi.1000185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996. May 2;334(18):1150–5. [DOI] [PubMed] [Google Scholar]
- 24.Borisy AA, Elliott PJ, Hurst NW, Lee MS, Lehar J, Price ER, et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci U S A. 2003. June 24;100(13):7977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iyengar R. Complex diseases require complex therapies. EMBO Rep. 2013. December;14(12):1039–42. 10.1038/embor.2013.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keith CT, Borisy AA, Stockwell BR. Multicomponent therapeutics for networked systems. Nat Rev Drug Discov. 2005. January;4(1):71–8. [DOI] [PubMed] [Google Scholar]
- 27.Loscalzo J. Personalized cardiovascular medicine and drug development: time for a new paradigm. Circulation. 2012. January 31;125(4):638–45. 10.1161/CIRCULATIONAHA.111.089243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta EK, Ito MK. Lovastatin and extended-release niacin combination product: the first drug combination for the management of hyperlipidemia. Heart Dis. 2002. Mar-Apr;4(2):124–37. [DOI] [PubMed] [Google Scholar]
- 29.Larder BA, Kemp SD, Harrigan PR. Potential mechanism for sustained antiretroviral efficacy of AZT-3TC combination therapy. Science. 1995. August 4;269(5224):696–9. [DOI] [PubMed] [Google Scholar]
- 30.Arstall MA, Yang J, Stafford I, Betts WH, Horowitz JD. N-acetylcysteine in combination with nitroglycerin and streptokinase for the treatment of evolving acute myocardial infarction. Safety and biochemical effects. Circulation. 1995. November 15;92(10):2855–62. [DOI] [PubMed] [Google Scholar]
- 31.Thompson H. Pipeline for COPD drugs flows with combination candidates. Nat Med. 2013. September;19(9):1079 10.1038/nm0913-1079 [DOI] [PubMed] [Google Scholar]
- 32.Bellamy TC, Wood J, Goodwin DA, Garthwaite J. Rapid desensitization of the nitric oxide receptor, soluble guanylyl cyclase, underlies diversity of cellular cGMP responses. Proc Natl Acad Sci U S A. 2000. March 14;97(6):2928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halvey EJ, Vernon J, Roy B, Garthwaite J. Mechanisms of activity-dependent plasticity in cellular nitric oxide-cGMP signaling. J Biol Chem. 2009. September 18;284(38):25630–41. 10.1074/jbc.M109.030338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J, Clark JW, Bryan RM, Robertson CS. Mathematical modeling of the nitric oxide/cGMP pathway in the vascular smooth muscle cell. Am J Physiol Heart Circ Physiol. 2005. August;289(2):H886–97. [DOI] [PubMed] [Google Scholar]
- 35.Batchelor AM, Bartus K, Reynell C, Constantinou S, Halvey EJ, Held KF, et al. Exquisite sensitivity to subsecond, picomolar nitric oxide transients conferred on cells by guanylyl cyclase-coupled receptors. Proc Natl Acad Sci U S A. 2010. December 21;107(51):22060–5. 10.1073/pnas.1013147107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Condorelli P, George SC. In vivo control of soluble guanylate cyclase activation by nitric oxide: a kinetic analysis. Biophys J. 2001. May;80(5):2110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garthwaite J. Dynamics of cellular NO-cGMP signaling. Front Biosci. 2005;10:1868–80. [DOI] [PubMed] [Google Scholar]
- 38.Held KF, Dostmann WR. Sub-nanomolar sensitivity of nitric oxide mediated regulation of cGMP and vasomotor reactivity in vascular smooth muscle. Front Pharmacol. 2012;3:130 10.3389/fphar.2012.00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy B, Garthwaite J. Nitric oxide activation of guanylyl cyclase in cells revisited. Proc Natl Acad Sci U S A. 2006. August 8;103(32):12185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitrovic V, Hernandez AF, Meyer M, Gheorghiade M. Role of guanylate cyclase modulators in decompensated heart failure. Heart Fail Rev. 2009. December;14(4):309–19. 10.1007/s10741-009-9149-7 [DOI] [PubMed] [Google Scholar]
- 41.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004. June 16;43(12 Suppl S):13S–24S. [DOI] [PubMed] [Google Scholar]
- 42.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006. September;2(9):458–66. [DOI] [PubMed] [Google Scholar]
- 43.Araujo RP, Petricoin EF, Liotta LA. A mathematical model of combination therapy using the EGFR signaling network. Biosystems. 2005. April;80(1):57–69. [DOI] [PubMed] [Google Scholar]
- 44.Russo I, Del Mese P, Doronzo G, Mattiello L, Viretto M, Bosia A, et al. Resistance to the nitric oxide/cyclic guanosine 5'-monophosphate/protein kinase G pathway in vascular smooth muscle cells from the obese Zucker rat, a classical animal model of insulin resistance: role of oxidative stress. Endocrinology. 2008. April;149(4):1480–9. [DOI] [PubMed] [Google Scholar]
- 45.Stone JR, Marletta MA. Heme stoichiometry of heterodimeric soluble guanylate cyclase. Biochemistry. 1995. November 14;34(45):14668–74. [DOI] [PubMed] [Google Scholar]
- 46.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991. June;43(2):109–42. [PubMed] [Google Scholar]
- 47.Conti M, Nemoz G, Sette C, Vicini E. Recent progress in understanding the hormonal regulation of phosphodiesterases. Endocr Rev. 1995. June;16(3):370–89. [DOI] [PubMed] [Google Scholar]
- 48.Marczin N, Ryan US, Catravas JD. Effects of oxidant stress on endothelium-derived relaxing factor-induced and nitrovasodilator-induced cGMP accumulation in vascular cells in culture. Circ Res. 1992. February;70(2):326–40. [DOI] [PubMed] [Google Scholar]
- 49.Burke-Wolin TM, Wolin MS. Inhibition of cGMP-associated pulmonary arterial relaxation to H2O2 and O2 by ethanol. Am J Physiol. 1990. May;258(5 Pt 2):H1267–73. [DOI] [PubMed] [Google Scholar]
- 50.Bliss CI. The toxicity of poisons applied jointly. Arzneimittelforschung. 1939;26:585–615. [Google Scholar]
- 51.Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung. 1953. June;3(6):285–90. [PubMed] [Google Scholar]
- 52.Berenbaum MC. Synergy, additivism and antagonism in immunosuppression. A critical review. Clin Exp Immunol. 1977. April;28(1):1–18. [PMC free article] [PubMed] [Google Scholar]
- 53.Berenbaum MC. The expected effect of a combination of agents: the general solution. J Theor Biol. 1985. June 7;114(3):413–31. [DOI] [PubMed] [Google Scholar]
- 54.Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995. June;47(2):331–85. [PubMed] [Google Scholar]
- 55.Chou TC, Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. J Biol Chem. 1977. September 25;252(18):6438–42. [PubMed] [Google Scholar]
- 56.Tallarida RJ. Drug synergism: its detection and applications. J Pharmacol Exp Ther. 2001. September;298(3):865–72. [PubMed] [Google Scholar]
- 57.Agoston V, Csermely P, Pongor S. Multiple weak hits confuse complex systems: a transcriptional regulatory network as an example. Phys Rev E Stat Nonlin Soft Matter Phys. 2005;71:051909 [DOI] [PubMed] [Google Scholar]
- 58.Dworzanski W, Jaskolska A, Szumilo M, Switalska A, Burdan F. [Cardiovascular complications of phosphodiesterase-5 inhibitors]. Pol Merkur Lekarski. 2011. February;30(176):139–42. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(TIF)
Data Availability Statement
All relevant data are within the paper and supporting information.