

Abstract
Transforming growth factor-β (TGF-β) and hepatocyte growth factor (HGF) play key roles in regulating the response to renal injury but are thought to mediate divergent effects on cell behavior. However, how TGF-β signaling alters the response to HGF in epithelia, the key site of HGF signaling in the injured kidney, is not well studied. Contrary to our expectation, we showed that deletion of the TGF-β type II receptor in conditionally immortalized proximal tubule (PT) cells impaired HGF-dependent signaling. This reduced signaling was due to decreased transcription of c-Met, the HGF receptor, and the TGF-β-dependent c-Met transcription and increased response to HGF in PT cells were mediated by the Notch pathway. The interactions of TGF-β, HGF, and Notch pathways had biologically significant effects on branching morphogenesis, cell morphology, migration, and proliferation. In conclusion, epithelial TGF-β signaling promotes HGF signaling in a Notch-dependent pathway. These findings suggest that TGF-β modulates PT responses not only by direct effects, but also by affecting other growth factor signaling pathways.
Keywords: transforming growth factor-β type II receptor, growth factors, proximal tubule, Notch signaling
growth factors are critical modulators of the kidney's response to all forms of injury. After renal injury, growth factors are upregulated and coordinate cellular events, such as dedifferentiation, migration, and proliferation, that lead to repair. In the injured kidney, growth factors affect the responses of the injured epithelium, surrounding fibroblasts, endothelium, and infiltrating inflammatory cells, and the balance of these growth factors impacts how the kidney recovers. Two growth factors that play key roles in the response to injury are hepatocyte growth factor (HGF) and transforming growth factor-β (TGF-β).
TGF-β and HGF signaling are increased in the damaged epithelium (15, 21). All three TGF-β ligands (β1, β2, and β3) bind to the ubiquitously expressed TGF-β type II receptor (TβRII), which heterodimerizes with the type I receptor (TβRI) and transduces intracellular signaling through canonical Smads, as well as noncanonical pathways, such as MAPK and GTPases. HGF exerts its effects through the tyrosine kinase receptor c-Met, which becomes autophosphorylated upon ligand binding and then activates intracellular signaling mediators such as ERK and phosphatidylinositol 3-kinase (30). The diverse downstream signaling pathways of both growth factors lead to multiple cellular responses, such as cell growth and differentiation. TGF-β and HGF affect some of these cellular events in a similar manner, while other responses are divergent. Both growth factors promote epithelial dedifferentiation, spreading, and migration after injury: HGF has proproliferative and antiapoptotic effects on the epithelium (41), while TGF-β is cytostatic and increases susceptibility to apoptosis (10). Consistent with these findings, HGF plays a predominantly protective role in renal injury (11, 16, 21, 25), whereas excess TGF-β adversely impacts renal injury (17, 18, 23). Many studies show that TGF-β and HGF negatively regulate each other's production during kidney injury (8, 24). While several investigations have focused on how TGF-β signaling affects HGF production, few have examined how TGF-β alters HGF signaling in renal epithelia, the main target of HGF in the injured kidney and a critical compartment for renal regeneration (41).
The Notch signaling pathway, like TGF-β and HGF, is integral to epithelial cell differentiation and is upregulated in renal injury. In canonical mammalian Notch signaling, the Delta-like and Jagged ligands bind to one of four Notch receptors (13). After ligand binding, γ-secretase cleaves the Notch intracellular domain, which translocates into the nucleus and activates target genes (13). TGF-β stimulates Notch activity in renal tubules, which is important in TGF-β-mediated cell dedifferentiation (22, 37). Although there is little information on how Notch impacts HGF signaling in the kidney, cross talk between these two signaling pathways alters the response to cardiac injury, as well as tumor development (1, 12, 38). TGF-β, Notch, and HGF pathways are upregulated in injured proximal tubules (PTs), but how these signaling pathways interact to alter epithelial responses is not clear.
Our group previously showed that blocking TGF-β signaling in PT epithelium protected against HgCl2-induced acute kidney injury in mice (10). Given the antagonistic relationship between TGF-β and HGF described above, we hypothesized that inhibition of TGF-β may be protective, in part, by augmenting HGF signaling. Contrary to our expectation, we found that TGF-β signaling in PTs augmented responsiveness to HGF through transcriptional upregulation of c-Met. This TGF-β-mediated increase in HGF signaling was dependent on Notch signaling. These findings elucidate novel interactions among TGF-β, Notch, and HGF signaling pathways and suggest that blocking TGF-β signaling also impairs epithelial responses to HGF.
METHODS
Cell culture.
PT cells were generated from the Immortomouse crossed with the Tgfbr2flox/flox (floxed control) mouse as described previously (10). PT cells were grown at 33°C in DMEM/F-12 medium supplemented with 2.5% FBS, hydrocortisone, insulin-transferrin-selenium, triiodothyronine, and penicillin-streptomycin (complete PT medium) with interferon-γ. Prior to experiments, PT cells were moved to a 37°C environment, and interferon-γ was removed to induce differentiation. Cortical fibroblasts were isolated from renal cortices of Tgfbr2flox/flox mice, immortalized by transfection with pSV40 as previously described (7), and grown in DMEM/F-12 medium supplemented with 10% FBS and penicillin-streptomycin. TβRII deletion in PT cells and cortical fibroblasts was achieved by adeno-Cre treatment in vitro as previously verified with immunoblots showing the absence of both TβRII and response to TGF-β1 (10, 27).
All procedures involving mice were approved by the Animal Care and Use Committee of Vanderbilt University.
HGF stimulation.
Cells were serum-starved overnight prior to treatment with HGF (40 ng/ml; Millipore). Unless otherwise noted, cells were subconfluent (<60%) at the time of HGF treatment, which lasted 20 min for all experiments. For phosphatase inhibition, PT cells were treated with 2 mM Na3VO4 30 min prior to and during HGF stimulation. For the low-calcium assay, cells were serum-starved in DMEM containing normal calcium (1 mM CaCl2) or low calcium (5 μM CaCl2) 12 h prior to HGF treatment. To block the TβRI, PT cells were plated with a specific ALK5 inhibitor (10 μM; SB431542, Tocris Biosciences) or an equal volume of DMSO for 5 days prior to HGF stimulation. To inhibit Notch cleavage, cells were incubated with a γ-secretase inhibitor (10 μM; Selleckchem) or an equal volume of DMSO for 2 days prior to HGF stimulation.
Real-time PCR analysis.
Total RNA was extracted from PT cells according to the instructions provided with the Qiagen RNeasy kit, and reverse transcription was performed using the iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR (qPCR) was performed with 100 ng of cDNA, primers at 400 nM, and SYBR Green supermix (Bio-Rad) using a thermocycler (model CFX96, Bio-Rad). Relative mRNA expressions were determined by the comparative cycle threshold (ΔΔCT) method, and, after validation with a panel of housekeeping genes, GAPDH was used as a reference gene. Primers are as follows: 5′-AGGTCGGTGTGAACGGATTTG (forward) and 5′-TGTAGACCATGTAGTTGAGGTCA (reverse) for GAPDH, 5′-CACCACTTGGGAGTATTGTGC (forward) and 5′-GGGACATCAGTCTCATTCACAG (reverse) for HGF, 5′-CCCTGCTGAGAAACTCTTCC (forward) and 5′-TTGATGAAGGTGGAGATGGA (reverse) for c-Met, 5′-TGCAAAGACAGTGAGGGAAG (forward) and 5′-AGTGTGCTCGGAGATGTGAG (reverse) for Sp1, 5′-TTGGCTTCTGCACAGTTAGG (forward) and 5′-GGAAGGACATACTGCCCACT (reverse) for Sp3, 5′-CGGTCTACACCAGCAACAGT (forward) and 5′-CACATGGAGTCCGAAGTGAG (reverse) for Hes1, 5′-GCAGATGACTGTGGATCACC (forward) and 5′-CCCAAACTCCGATAGTCCAT (reverse) for Hey1, and 5′-GGATTGCCCACTTCGAGTAT (forward) and 5′-TATTGCAGCCAAAGCCATAG (reverse) for Jagged1.
Immunoblots.
Cells were lysed in ice-cold RIPA buffer plus protease and phosphatase inhibitors (Sigma cocktail), homogenized by shearing through a syringe, clarified by centrifugation, and quantified using bicinchoninic acid protein assay (Thermo Scientific). For tissue lysates, the γGT-Cre;Tgfbr2flox/flox mice, in which recombination was previously confirmed, were injured with a single injection of HgCl2 (30 μmol/kg in saline), and cortical lysates were generated as described previously (10). For cell and tissue lysates, proteins were separated by SDS-polyacrylamide gel, transferred onto polyvinylidene difluoride or nitrocellulose membranes, blocked in 5% milk or BSA, and incubated with the following primary antibodies: c-Met, phosphorylated c-Met, phosphorylated ERK, phosphorylated Akt, total Akt, and cleaved Notch1 (Cell Signaling), total ERK (Santa Cruz Biotechnology), and E-cadherin (BD Biosciences). GAPDH, focal adhesion kinase (Santa Cruz), and α-tubulin (Cell Signaling) were validated as loading controls (data not shown), chosen on the basis of the size of the target protein, and used to control for protein loading. Bands on autoradiography film were quantified using Java-based image-processing software (ImageJ).
Three-dimensional culture assay.
PT cells (n = 20,000) were plated in gels containing collagen I and Matrigel as described previously (2, 3) and, once gels solidified, 100 μl of complete PT medium (see above) with or without HGF was added. After 5 days, gels were washed, fixed with 4% paraformaldehyde, and either stained with rhodamine-phalloidin (after permeabilization with 0.025% saponin and quenching with 75 mM NH4Cl and 20 mM glycine in PBS with CaCl2 and MgCl2) for confocal imaging or photographed with an inverted microscope and camera, and 10 random tubules were imaged per sample, with branches measured by ImageJ.
Cell migration assay.
PT cells (n = 20,000) in serum-free medium were plated on Transwell inserts (8 μm) precoated with Matrigel and incubated for 6 h. Cells on top of the membrane (i.e., cells that did not migrate) were removed with a cotton swab, and the bottom was fixed in 4% paraformaldehyde for 45 min. The membrane was stained with 2% crystal violet overnight, images were obtained at ×200 magnification with a Nikon Eclipse TE300 inverted microscope (10 randomly chosen fields per sample), and the number of migrated cells was counted and quantified in a blinded fashion. HGF-treated samples were exposed to 40 ng/ml HGF for 24 h before and throughout migration. Cells treated with the γ-secretase inhibitor (10 μM) were pretreated for 3 days (controls received equivalent volumes of DMSO).
Cell morphology.
PT cells were plated on Matrigel (BD Biosciences)-coated chamber-well slides in serum-free medium with or without HGF (40 ng/ml) for 24 h and then stained with rhodamine-phalloidin. For γ-secretase studies, PT cells were incubated with the inhibitor or equal amounts of DMSO for 2 days before they were plated on chamber-well slides and stimulated with HGF as described above. Images were obtained using a fluorescence microscope (model BX51, Olympus).
MTS cell proliferation assay.
PT cells were plated in 12-well plates, serum-starved overnight, and then treated with HGF for 24 h. To ensure equal numbers of cells, the number of cells was quantified using the CellTiter 96 Aqueous One Solution (Promega) at the time of HGF stimulation and again after 24 h in the presence and absence of HGF.
Isolation of membrane proteins.
Subconfluent, serum-starved (overnight) PT cells were placed on ice, washed with PBS (pH 8.0) plus CaCl2 and MgCl2 (PBS-CM), and incubated with 1 mM EZ-Link Sulfo-NSS-SS-Biotin (Thermo Scientific) in DMEM/F-12 medium supplemented with protease and phosphatase inhibitors (Sigma) for 1 h at 4°C. After PT cells were washed, unbound biotin was quenched by incubation with 0.1% BSA in PBS-CM at 4°C, and cells were washed in PBS-CM, lysed in basic lysis buffer (20 mM Tris·HCl, pH 8, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and protease and phosphatase inhibitors), scraped, and centrifuged for 15 min at 13,000 rpm at 4°C. Then 50–60 μg of protein per sample were incubated for 16 h with streptavidin-agarose beads (Thermo Scientific) at 4°C, washed, and centrifuged, and the pellet was saved.
Isolation of cytosolic and nuclear proteins.
Cytosolic and nuclear fractions were isolated from subconfluent, serum-starved PT cells using a protocol described elsewhere (33).
Statistics.
Student's t-test with unequal variance was used to compare two sets of data; P < 0.05 was considered statistically significant. Each experiment was repeated three times, and data are shown as means ± SE.
RESULTS
Blocking TGF-β signaling in PT cells impairs the response to HGF.
We used PT cells, the target of acute kidney injury, to determine how TGF-β signaling affects epithelial responsiveness to HGF. PT cells, with and without TβRII (10), were exposed to HGF for 20 min, 2 h, and 6 h. Activation (i.e., phosphorylation) of the HGF receptor c-Met was reduced in TβRII−/− compared with TβRIIflox/flox PT cells (Fig. 1, A and B). Also, total expression of the c-Met receptor was reduced in TβRII−/− PT cells (Fig. 1C), which was not significantly different from the difference in c-Met activation (Fig. 1B). Although TGF-β signaling has been shown to alter HGF expression in fibroblasts (5), there was no significant difference in HGF transcript levels between TβRIIflox/flox and TβRII−/− PT cells (Fig. 1D), and minimal HGF protein expression was detected in conditioned medium of PT cells as measured by ELISA (data not shown). Consistent with decreased c-Met phosphorylation, activation of the downstream signaling proteins AKT and ERK in response to HGF was impaired in TβRII−/− PT cells (Fig. 1, E–H). We stimulated renal cortical fibroblasts in the presence and absence of TβRII (27) with HGF and observed augmented c-Met activation in TβRII−/− fibroblasts (Fig. 1, I and J), suggesting that TGF-β signaling alters the response to HGF in a cell-specific manner.
Fig. 1.

Transforming growth factor-β (TGF-β) receptor type II (TβRII)-deficient (TβRII−/−) proximal tubule (PT) cells have impaired hepatocyte growth factor (HGF) signaling. A–C: TβRIIflox/flox (floxed control) and TβRII−/− PT cells were treated with HGF (40 ng/ml) for 20 min, 2 h, and 6 h and immunoblotted for c-Met expression and c-Met phosphorylation (pc-Met), which were quantified using α-tubulin as a loading control. D: quantitative PCR (qPCR) analysis of HGF transcription by TβRIIflox/flox and TβRII−/− PT cells normalized to GAPDH. E–H: TβRIIflox/flox and TβRII−/− PT cells were treated with HGF (40 ng/ml) for 20 min and immunoblotted for AKT and ERK phosphorylation (pAKT and pERK), which was quantified. FAK, focal adhesion kinase. I and J: lysates of TβRIIflox/flox and TβRII−/− cortical fibroblasts were treated with HGF (40 ng/ml) for 20 min, 2 h, and 6 h and immunoblotted for c-Met phosphorylation, which was quantified using α-tubulin as a loading control. Values are means ± SE of results from 3 separate experiments. **P < 0.01; ***P < 0.0001.
TβRII−/− PT cells have reduced c-Met membrane expression and transcript levels.
We then examined whether these TβRII-dependent changes in c-Met expression and phosphorylation were present in PTs in vivo. Expression and phosphorylation of c-Met were significantly reduced in mice lacking TβRII in PTs (γGT-Cre;Tgfbr2flox/flox) (10) compared with floxed controls after HgCl2-induced acute kidney injury (Fig. 2, A–C), an injury model previously shown to increase HGF transcript levels and activity (14). As TβRII-dependent changes in c-Met expression and activation were present in vivo and in vitro, we further investigated how TβRII alters c-Met expression in vitro. c-Met expression and membrane localization were reduced in TβRII−/− compared with TβRIIflox/flox PT cells (Fig. 2, D–F). c-Met expression in TβRII−/− PT cells was decreased to a similar extent in whole cell lysates and membrane preparations (47 ± 6% and 45 ± 6%, respectively; Fig. 2, E and F), suggesting that decreased c-Met expression in TβRII−/− PT cells was not due to increased endocytosis of the receptor. c-Met transcript levels as measured by qPCR were decreased in TβRII−/− PT cells (Fig. 2G), indicating that the reduced c-Met protein expression is due to transcriptional changes.
Fig. 2.

PTs lacking TβRII in vitro have reduced c-Met membrane localization and transcript levels. A–C: cortical tissue lysates of γGT-Cre;Tgfbr2flox/flox (conditional deletion of TβRII in the PT) and Tgfbr2flox/flox (floxed control) mice were subjected to HgCl2-induced acute kidney injury and immunoblotted for c-Met expression and phosphorylation with α-tubulin as a loading control, and protein expression was quantified. D: lysates of whole cell and membrane preparations (see methods) were immunoblotted for c-Met expression with α-tubulin, which was used to show the purity of membrane preparations, and β1-integrin, which was used as a loading control for membrane expression. E and F: quantification of c-Met expression in whole cell lysates and membrane preparations, with PT cell expression of TβRIIflox/flox in each experiment (n = 3) adjusted to 1. G: qPCR analysis of c-Met mRNA in TβRIIflox/flox and TβRII−/− PT cells normalized to GAPDH expression (n = 3). H and I: PT cells pretreated for 4 days with an ALK5 inhibitor (SB431542) or vehicle control (DMSO) were stimulated with HGF and immunoblotted for c-Met phosphorylation, and protein expression was quantified using α-tubulin as loading control (n = 3). J: PT cells were pretreated for 4 days with an ALK5 inhibitor (SB431542) or vehicle control (DMSO), RNA was isolated and converted to cDNA, and c-Met transcription was quantified by qPCR. Values are means ± SE. *P < 0.05.
To determine whether decreased c-Met expression and activation of TβRII−/− PT cells are due to impaired TGF-β signaling, we used a well-characterized inhibitor of ALK5, the TβRI. ALK5 inhibition significantly reduced HGF activation of c-Met in both cell populations (Fig. 2, H and I) but reduced c-Met transcript levels only in TβRIIflox/flox PT cells (Fig. 2J). These results imply that the reduced c-Met expression of TβRII−/− PT cells is due to impaired signaling through the TGF-β receptors.
We also investigated whether mechanisms other than alteration of c-Met transcription, reduced HGF signaling in TβRII−/− PT cells. We investigated the role of E-cadherin, since this adherens junction protein can inhibit tyrosine kinase growth factor signaling (29) and is suppressed by TGF-β. Consistent with this, E-cadherin expression and membrane localization were augmented in TβRII−/− PT cells (Fig. 3, A–C). Furthermore, the difference in HGF signaling was present only in subconfluent conditions, during which time E-cadherin is suppressed (Fig. 3, D and E). We disrupted E-cadherin using low-calcium (5 μM) medium and confirmed that this augmented c-Met activation in confluent TβRIIflox/flox PT cells (Fig. 3, F and G). However, low-calcium medium did not alter differences in HGF signaling between TβRII−/− and TβRIIflox/flox PT cells in subconfluent conditions (Fig. 3, H and I). Thus, augmented E-cadherin expression does not play a role in reduced HGF signaling in TβRII−/− PT cells.
Fig. 3.

E-cadherin expression and phosphatase activity do not account for reduced HGF responsiveness in TβRII−/− PT cells. A–C: total cell lysates and membrane preparations of TβRIIflox/flox and TβRII−/− PT cells were immunoblotted for E-cadherin expression with α-tubulin and β1-integrin as loading controls, and E-cadherin expression in whole cell and membrane preparations was quantified. D and E: TβRIIflox/flox and TβRII−/− PT cells were stimulated with HGF in confluent (>90%) or subconfluent (<60%) conditions, c-Met phosphorylation was measured by immunoblotting, and differences between confluent and subconfluent conditions were quantified. F and G: confluent TβRIIflox/flox PT cells were incubated in low-calcium (5 μM) or normal-calcium (1 mM) medium (see methods), stimulated with HGF, immunoblotted for c-Met phosphorylation expression, and quantified using densitometry. H and I: subconfluent TβRIIflox/flox and TβRII−/− PT cells incubated in low- or normal-calcium medium were treated with HGF and immunoblotted for c-Met phosphorylation, and expression was quantified. J and K: PT cells were pretreated with a tyrosine phosphatase inhibitor (Na3VO4) 20 min prior to HGF stimulation, and c-Met phosphorylation was measured by immunoblotting and quantified with focal adhesion kinase as a loading control. Values are means ± SE; n = 3. *P < 0.05; **P < 0.01; ***P < 0.0001. White lines indicate where lanes within the same blot have been moved.
We also investigated whether altered phosphatase activity might account for differences in HGF signaling, as TGF-β alters other tyrosine kinase growth factor signaling pathways through phosphatases (35). As expected, pretreatment of PT cells with Na3VO4, a tyrosine phosphatase inhibitor, augmented c-Met phosphorylation in response to HGF in TβRII−/− and TβRIIflox/flox PT cells (Fig. 3, J and K). However, the decreased responsiveness of TβRII−/− PT cells to HGF persisted even with the phosphatase inhibitor (Fig. 3, J and K). Taken together, our data show that inhibition of TGF-β signaling either genetically or pharmacologically reduces HGF signaling through transcriptional changes in c-Met and not by alteration of E-cadherin expression or phosphatase activity.
Notch signaling mediates reduced HGF signaling of TβRII−/− PT cells.
Our data indicated that the decreased c-Met expression of TβRII−/− PT cells was transcriptionally mediated, so we investigated putative transcription factors that link TGF-β signaling and c-Met expression. The transcription factors Sp1 and Sp3 have been shown to regulate c-Met expression (39), but we found no significant differences in their transcript levels between TβRIIflox/flox and TβRII−/− PT cells by qPCR (Fig. 4, A and B). We then investigated whether the Notch signaling pathway may be involved, since this pathway is a downstream target of TGF-β signaling (22) and interacts with the HGF/c-Met pathway in other organs (1, 12). TGF-β has been shown to induce expression of the ligand Jagged1 (22, 28), and, consistent with this finding, Jagged1 mRNA expression was suppressed in TβRII−/− PT cells (Fig. 4C). Notch transcriptional targets, Hes1 and Hey1, were also diminished in PT cells lacking TβRII (Fig. 4, D and E). Furthermore, cleaved Notch1, an indicator of Notch signaling, was significantly decreased in nuclear isolates of TβRII−/− PT cells (Fig. 4, F and G). Thus our data indicate that Notch signaling is reduced in TβRII−/− compared with TβRIIflox/flox PT cells.
Fig. 4.

Notch signaling is impaired in TβRII−/− PT cells. A–E: qPCR was used to quantify transcription of Sp1, Sp3, Jagged1, Hes1, and Hey1. F: TβRIIflox/flox and TβRII−/− PT cells were fractionated into cytosolic and nuclear components (see methods), and cleaved Notch1 (cNotch1) was measured with GAPDH and poly(ADP-ribose)polymerase 1 (PARP1) as loading controls for cytosolic and nuclear compartments, respectively. G: nuclear expression of cleaved Notch1 in TβRIIflox/flox and TβRII−/− PT cells (n = 3). Values are means ± SE. *P < 0.05.
To determine whether the suppressed Notch signaling in TβRII−/− PT cells accounts for its reduced responsiveness to HGF, we inhibited Notch activity using a γ-secretase inhibitor. As expected, the γ-secretase inhibitor blocked Notch1 cleavage and Hes1 transcription but also reduced c-Met protein expression and transcription in TβRIIflox/flox PT cells (Fig. 5, A–E). Furthermore, addition of the γ-secretase inhibitor reduced the HGF-induced c-Met activation to levels similar to those of TβRII−/− PT cells (Fig. 5, F and G), implying that the difference in c-Met activation between TβRIIflox/flox and TβRII−/− PT cells is Notch-dependent.
Fig. 5.

Blocking Notch with γ-secretase reduces c-Met activation in TβRIIflox/flox PT cells. A: PT cells in the presence and absence of γ-secretase inhibitor (γSI) were immunoblotted for cleaved Notch1 and c-Met expression, with α-tubulin used as a loading control. B and C: effects of γ-secretase inhibitor on c-Met protein expression and Notch1 cleavage. D and E: qPCR was used to determine Hes1 and c-Met transcripts in PT cells pretreated with γ-secretase inhibitor or vehicle control (DMSO) (n = 3). F and G: PT cells pretreated with γ-secretase inhibitor or DMSO were stimulated with HGF and immunoblotted for c-Met phosphorylation and quantified, with α-tubulin used as a loading control (n = 3). All experiments were repeated 3 times. Values are means ± SE. *P < 0.05.
TβRII−/− PT cells have reduced biological responses to HGF.
We defined the biological significance of reduced HGF signaling in TβRII−/− PT cells by investigating responses to cellular events known to be modulated by HGF signaling. Initially, we explored how TGF-β and HGF signaling interact to regulate branching morphogenesis. TβRII−/− PT cells grown in three-dimensional gels (collagen I-Matrigel) had significantly more branching than TβRIIflox/flox PT cells a finding not due to increased tubule length (Fig. 6, A–C). However, HGF stimulation significantly increased branching in TβRIIflox/flox PT cells (from 5.8 to 10.3 branches per tubule) compared with a minimal change in TβRII−/− PT cells (from 10.1 to 11.8 branches per tubule; Fig. 6, A–C). HGF is known to induce morphological changes, whereby the cell dedifferentiates and has a more fibroblast-like shape (19, 32). We observed that TβRII−/− PT cells spread less at baseline than TβRIIflox/flox PT cells (Fig. 6D). After HGF treatment, TβRIIflox/flox PT cells had a more fibroblast-like appearance, consistent with HGF-induced dedifferentiation, whereas there was little change in the TβRII−/− PT cells (Fig. 6D). We next defined how HGF altered epithelial migration of PT cells with and without TβRII on Matrigel-coated Transwell inserts (Fig. 6, E and F). HGF treatment dramatically increased migration of TβRIIflox/flox, but not TβRII−/−, PT cells (Fig. 6, E and F). Similarly, HGF promoted more proliferation, and there was a greater increase in the number of HGF-treated TβRIIflox/flox (>60%) than HGF-treated TβRII−/− (20%) PT cells (Fig. 6G). Thus, consistent with the decreased signaling in response to HGF, TβRII−/− PT cells had reduced functional responses as well.
Fig. 6.

TβRII−/− PT cells have diminished biological responses to HGF. TβRIIflox/flox and TβRII−/− PT cells were grown in 3-dimensional gels containing Matrigel and collagen I for branching morphogenesis studies (see methods). A–C: representative confocal images of PT cells in the presence and absence of HGF (40 ng/ml), and the number of branches and tubule length (μm):branch number were quantified using ImageJ on 10 tubules per experiment (n = 3). The tubule length (in μm) was determined by the longest tubule and divided by the number of branches. D: HGF-induced effects on cell morphology were assessed by rhodamine-phalloidin staining of TβRIIflox/flox and TβRII−/− PT cells plated on Matrigel-coated chamber slides in the presence and absence of HGF. Magnification ×400; scale bar = 50 μm. E: representative images of migration assessed by plating TβRIIflox/flox and TβRII−/− PT cells on Matrigel-coated Transwell inserts in the presence and absence of 40 ng/ml HGF and staining with crystal violet. Magnification ×200; scale bars = 50 μm. F: number of migrated cells was counted in a blinded manner in 10 ×200-magnified images per sample. Values are means ± SE; n = 3. G: HGF-induced increase in proliferation was measured by the MTS assay (see methods) and reported as percent increase in HGF-treated cells compared with untreated cells after 24 h. *P < 0.05; **P < 0.01; ***P < 0.001.
γ-Secretase inhibitor abrogated HGF signaling in TβRIIflox/flox PT cells.
We then investigated whether inhibiting Notch signaling using the γ-secretase inhibitor could block the biological responses to HGF in TβRIIflox/flox PT cells. The HGF-induced increase in cell dedifferentiation and lamellopodia formation in TβRIIflox/flox PT cells was reduced by treatment with the γ-secretase inhibitor, whereas there was little change in the actin cytoskeleton of TβRII−/− PT cells (Fig. 7A). Similarly, pretreatment with a γ-secretase inhibitor dramatically reduced TβRIIflox/flox, but not TβRII−/−, PT cell migration (Fig. 7, B and C). These data suggest that the reduced responsiveness of TβRII−/− PT cells to HGF is mediated through suppressed Notch signaling.
Fig. 7.

γ-Secretase inhibitor reduces the response of TβRIIflox/flox, but not TβRII−/−, PT cells to HGF. A: representative images of PT cells pretreated with γ-secretase or DMSO and stimulated with HGF (see methods), plated on Matrigel-coated chamber slides, and stained with rhodamine-phalloidin. Magnification ×400; scale bar = 50 μm. B: representative images showing migration on Matrigel-coated Transwell inserts repeated with all cells stimulated by HGF + γ-secretase or DMSO. Magnification ×200, scale bars = 50 μm. C: migrated cells were counted in a blinded manner in 10 fields per sample (n = 3). Values are means ± SE. *P < 0.05.
DISCUSSION
TGF-β directly alters many epithelial responses to injury but may also modulate cellular events through effects on other key growth factor pathways. We showed that deletion of TβRII in PTs significantly suppressed HGF signaling and biological responses due to transcriptional reduction of the c-Met receptor. Our data suggest that Notch signaling plays an important role in regulation of the TGF-β-dependent increase in HGF signaling. Thus our data indicate that the interaction between TGF-β and downstream Notch signaling pathways in PT cells is necessary for a full biological response to HGF.
Our finding that TGF-β signaling promotes epithelial HGF signaling was somewhat surprising, as others have shown that these growth factors mediate divergent responses to renal injury (20, 24). These differences may be explained by our data showing that the TGF-β-dependent changes in HGF signaling were cell type-specific. TGF-β signaling inhibited HGF signaling in cortical fibroblasts, which is consistent with other reports showing that blocking TβRII in mesenchymal cells augments HGF signaling (5). However, our finding that TGF-β does not alter HGF production in epithelial cells contrasts with the suppressive effect of TGF-β on HGF synthesis in fibroblasts reported by others (26), further showing the cell-specific interactions of TGF-β and HGF signaling. Although mesenchymal cells are important sources of HGF, injured renal epithelia are considered the most important sites of HGF signaling (41). Perhaps our finding that epithelial TGF-β promotes HGF-dependent signaling is not too surprising, given that both HGF and TGF-β promote cell dedifferentiation, spreading, and migration. Thus it is possible that these two growth factors cooperatively interact to regulate cellular responses critical to the injured epithelia.
Deletion of TβRII in PTs reduced c-Met expression in cells in vitro and in HgCl2-injured kidneys. It is unclear if c-Met expression was reduced because TGF-β signaling was abrogated or because the kidneys sustained less injury as we previously reported (10). Since epithelial injury upregulates c-Met, reduced injury could also reduce c-Met expression; however, our in vitro data suggest that impaired TGF-β signaling contributes to decreased c-Met expression. Our finding that TGF-β augmented HGF signaling through a transcriptional increase in the c-Met receptor is consistent with the finding of Zhang et al. (40) that c-Met transcription is upregulated by exogenous TGF-β in human PT cells (40). However, this earlier study showed that the transcription factor Sp1 was critical to c-Met transcription induced by exogenous TGF-β1 (40), while we showed no difference in levels of Sp1 expression between TβRIIflox/flox and TβRII−/− PT cells. Differences in the cells used (conditionally immortalized murine PT cells vs. immortalized human PT cells) may account for discrepant mechanisms, but the relationship between renal epithelial TGF-β and HGF appears conserved between species.
To confirm that the reduced HGF signaling in TβRII−/− PT cells was due to suppressed TGF-β signaling, we pharmacologically inhibited the TβRI using the well-established ALK5 inhibitor. This inhibitor reduced responsiveness to HGF and c-Met expression in the TβRIIflox/flox PT cells, but also reduced responsiveness to HGF in TβRII−/− PT cells without significantly changing c-Met expression. TGF-β-independent ligands (e.g., activins) may activate ALK5 in a TβRII-independent manner. Our data suggest that these TGF-β ligand-independent signaling pathways also alter HGF signaling, but not through c-Met transcription.
Our studies also illustrate how deletion of TβRII from PTs fundamentally changes the cells: reducing cell spreading, altering actin cytoskeleton, and augmenting E-cadherin expression. Some of these changes are consistent with our previous findings that deletion of TβRII in collecting duct epithelia altered stress fiber formation and augmented Rho GTPase activity (9). Although many studies have focused on how excessive TGF-β signaling dramatically alters the epithelial cytoskeleton as part of an epithelial-to-mesenchymal transition, our results suggest that even basal TGF-β signaling is integral to epithelial cell structure and organization.
Our data convincingly show that TGF-β signaling in PT cells increases Notch activity, which is consistent with previous reports showing that exogenous TGF-β1 augments epithelial Notch signaling (22, 34, 36, 37). The mechanism whereby TGF-β signaling upregulates Notch activity was beyond the scope of this study, but our results showing decreased Jagged1 transcription in TβRII−/− PT cells are consistent with other studies indicating that TGF-β increases Notch signaling through transcriptional upregulation of Jagged1 (22, 28, 37). Others have shown that Smad3 is critical to TGF-β-induced Jagged1/Notch signaling in renal epithelia (37). Taken together with earlier studies showing that TGF-β induced c-Met expression through Smads (40), it is possible that Smad-dependent induction of Jagged1 augments Notch activity, which is key to TGF-β-dependent c-Met expression and augmented HGF signaling (Fig. 8). While little is known about Notch interactions with c-Met in the kidney, there is evidence of cross talk between c-Met and Notch signaling pathways in cancer biology (38). However, there are conflicting results regarding how Notch affects c-Met transcription: suppression of Notch1 reduced c-Met expression in one study, but constitutively active Notch repressed c-Met transcription in another (1, 31). To block the effects of Notch, we used a γ-secretase inhibitor, which prevented Notch cleavage in our studies and has been widely used by others to inhibit Notch signaling (4, 6, 34). However, γ-secretase inhibitors are not specific for Notch signaling, and it is possible that another γ-secretase target mediates TGF-β-dependent c-Met transcription.
Fig. 8.
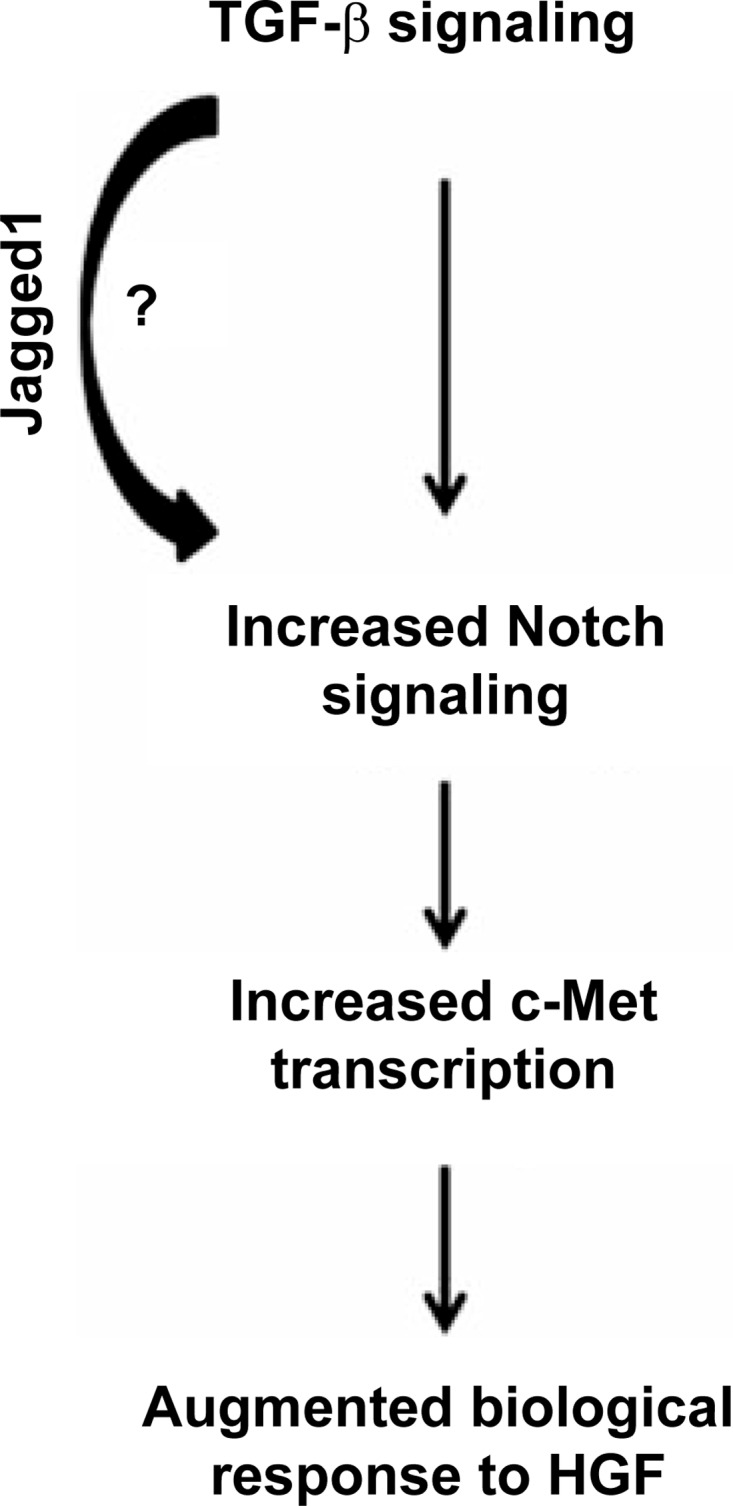
Schematic showing how TGF-β signaling increases epithelial responsiveness to HGF through the Notch signaling pathway.
The interactions between TGF-β and HGF in our studies were shown to be biologically relevant, as TβRII−/− PT cells had impaired HGF-mediated cellular responses. Furthermore, we showed that TGF-β enhances the response to HGF through Notch signaling, as both HGF-dependent morphological changes and migration in TβRIIflox/flox PT cells were blocked with the γ-secretase inhibitor. The signaling interactions among TGF-β, HGF, and Notch pathways in epithelial cells have not been well studied, but each of these pathways has been shown to be upregulated in tubular injury and to promote epithelial dedifferentiation. In the injured renal epithelium, coordinated signaling among growth factor pathways that control differentiation may be important to allow regeneration after renal injury.
In conclusion, this study demonstrates biologically significant interactions between TGF-β and HGF signaling pathways in PT epithelial cells. In addition, we show that Notch signaling plays an important role in regulation of these interactions. These findings suggest that intact epithelial TGF-β signaling is necessary for a full biological response to HGF. Furthermore, TGF-β signaling alters epithelial cell behavior not only by direct effects, but also by changing how these cells respond to other growth factors.
GRANTS
This work was supported by a Career Development Award from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development (L. Gewin); Veterans Affairs Merit Awards 2I01BX000320 (R. C. Harris) and 1I01BX002196 (R. Zent); and National Institute of Diabetes and Digestive and Kidney Diseases Grants RO1-DK-51265 (R. C. Harris), RO1-DK-95785 (R. C. Harris), RO1-DK-083187 (R. Zent), RO1-DK-075594 (R. Zent), and RO1-DK-069221 (R. Zent). This material is based on work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Biomedical Laboratory Research and Development. This material is the result of work supported with resources and the use of facilities at the VA Tennessee Valley Healthcare System.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.N.K., S.N., L.W., E.G., and L.G. performed the experiments; S.N.K., S.N., L.W., and L.G. analyzed the data; S.N.K., S.N., and L.G. interpreted the results of the experiments; S.N.K. and L.G. prepared the figures; S.N.K. and L.G. drafted the manuscript; R.C.H., R.Z., and L.G. developed the concept and designed the research; R.C.H., R.Z., and L.G. edited and revised the manuscript; R.Z. and L.G. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Scott Baldwin and his laboratory for assistance with and use of their Nikon Eclipse TE300 inverted microscope.
REFERENCES
- 1.Apostolou P, Toloudi M, Ioannou E, Kourtidou E, Chatziioannou M, Kopic A, Komiotis D, Kiritsis C, Manta S, Papasotiriou I. Study of the interaction among Notch pathway receptors, correlation with stemness, as well as their interaction with CD44, dipeptidyl peptidase-IV, hepatocyte growth factor receptor and the SETMAR transferase, in colon cancer stem cells. J Recept Signal Transduct Res 33: 353–358, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Cantley LG, Barros EJ, Gandhi M, Rauchman M, Nigam SK. Regulation of mitogenesis, motogenesis, and tubulogenesis by hepatocyte growth factor in renal collecting duct cells. Am J Physiol Renal Fluid Electrolyte Physiol 267: F271–F280, 1994. [DOI] [PubMed] [Google Scholar]
- 3.Chen D, Roberts R, Pohl M, Nigam S, Kreidberg J, Wang Z, Heino J, Ivaska J, Coffa S, Harris RC, Pozzi A, Zent R. Differential expression of collagen- and laminin-binding integrins mediates ureteric bud and inner medullary collecting duct cell tubulogenesis. Am J Physiol Renal Physiol 287: F602–F611, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Chen JK, Conway EM, Harris RC. Survivin mediates renal proximal tubule recovery from AKI. J Am Soc Nephrol 24: 2023–2033, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. Enhanced hepatocyte growth factor signaling by type II transforming growth factor-β receptor knockout fibroblasts promotes mammary tumorigenesis. Cancer Res 67: 4869–4877, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Curry CL, Reed LL, Golde TE, Miele L, Nickoloff BJ, Foreman KE. γ-Secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi's sarcoma tumor cells. Oncogene 24: 6333–6344, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Elias BC, Mathew S, Srichai MB, Palamuttam R, Bulus N, Mernaugh G, Singh AB, Sanders CR, Harris RC, Pozzi A, Zent R. The integrin β1-subunit regulates paracellular permeability of kidney proximal tubule cells. J Biol Chem 289: 8532–8544, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esposito C, Parrilla B, De Mauri A, Cornacchia F, Fasoli G, Foschi A, Mazzullo T, Plati A, Scudellaro R, Dal Canton A. Hepatocyte growth factor (HGF) modulates matrix turnover in human glomeruli. Kidney Int 67: 2143–2150, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Gewin L, Bulus N, Mernaugh G, Moeckel G, Harris RC, Moses HL, Pozzi A, Zent R. TGF-β receptor deletion in the renal collecting system exacerbates fibrosis. J Am Soc Nephrol 21: 1334–1343, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gewin L, Vadivelu S, Neelisetty S, Srichai MB, Paueksakon P, Pozzi A, Harris RC, Zent R. Deleting the TGF-β receptor attenuates acute proximal tubule injury. J Am Soc Nephrol 23: 2001–2011, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gong R, Rifai A, Dworkin LD. Anti-inflammatory effect of hepatocyte growth factor in chronic kidney disease: targeting the inflamed vascular endothelium. J Am Soc Nephrol 17: 2464–2473, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Gude NA, Emmanuel G, Wu W, Cottage CT, Fischer K, Quijada P, Muraski JA, Alvarez R, Rubio M, Schaefer E, Sussman MA. Activation of Notch-mediated protective signaling in the myocardium. Circ Res 102: 1025–1035, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet 13: 654–666, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Igawa T, Matsumoto K, Kanda S, Saito Y, Nakamura T. Hepatocyte growth factor may function as a renotropic factor for regeneration in rats with acute renal injury. Am J Physiol Renal Fluid Electrolyte Physiol 265: F61–F69, 1993. [DOI] [PubMed] [Google Scholar]
- 15.Inoue T, Okada H, Kobayashi T, Watanabe Y, Kanno Y, Kopp JB, Nishida T, Takigawa M, Ueno M, Nakamura T, Suzuki H. Hepatocyte growth factor counteracts transforming growth factor-β1, through attenuation of connective tissue growth factor induction, and prevents renal fibrogenesis in 5/6 nephrectomized mice. FASEB J 17: 268–270, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Kawaida K, Matsumoto K, Shimazu H, Nakamura T. Hepatocyte growth factor prevents acute renal failure and accelerates renal regeneration in mice. Proc Natl Acad Sci USA 91: 4357–4361, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R, Glick AB, Hahnel B, Hosser H, Grone HJ, Kriz W. Tubular overexpression of transforming growth factor-β1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol 177: 632–643, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Bottinger EP, Klotman PE, Thorgeirsson SS. Transgenic mice with increased plasma levels of TGF-β1 develop progressive renal disease. Lab Invest 74: 991–1003, 1996. [PubMed] [Google Scholar]
- 19.Lai JK, Wu HC, Shen YC, Hsieh HY, Yang SY, Chang CC. Kruppel-like factor 4 is involved in cell scattering induced by hepatocyte growth factor. J Cell Sci 125: 4853–4864, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol 287: F7–F16, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Rajur K, Tolbert E, Dworkin LD. Endogenous hepatocyte growth factor ameliorates chronic renal injury by activating matrix degradation pathways. Kidney Int 58: 2028–2043, 2000. [DOI] [PubMed] [Google Scholar]
- 22.Matsuno Y, Coelho AL, Jarai G, Westwick J, Hogaboam CM. Notch signaling mediates TGF-β1-induced epithelial-mesenchymal transition through the induction of Snai1. Int J Biochem Cell Biol 44: 776–789, 2012. [DOI] [PubMed] [Google Scholar]
- 23.Miyajima A, Chen J, Lawrence C, Ledbetter S, Soslow RA, Stern J, Jha S, Pigato J, Lemer ML, Poppas DP, Vaughan ED, Felsen D. Antibody to transforming growth factor-β ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int 58: 2301–2313, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Mizuno S, Matsumoto K, Kurosawa T, Mizuno-Horikawa Y, Nakamura T. Reciprocal balance of hepatocyte growth factor and transforming growth factor-β1 in renal fibrosis in mice. Kidney Int 57: 937–948, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Mizuno S, Matsumoto K, Nakamura T. Hepatocyte growth factor suppresses interstitial fibrosis in a mouse model of obstructive nephropathy. Kidney Int 59: 1304–1314, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Mungunsukh O, Day RM. Transforming growth factor-β1 selectively inhibits hepatocyte growth factor expression via a micro-RNA-199-dependent posttranscriptional mechanism. Mol Biol Cell 24: 2088–2097, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neelisetty S, Alford C, Reynolds K, Woodbury L, Nlandu-Khodo S, Yang H, Fogo AB, Hao CM, Harris RC, Zent R, Gewin L. Renal fibrosis is not reduced by blocking transforming growth factor-β signaling in matrix-producing interstitial cells. Kidney Int 88: 503–514, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nyhan KC, Faherty N, Murray G, Cooey LB, Godson C, Crean JK, Brazil DP. Jagged/Notch signalling is required for a subset of TGFβ1 responses in human kidney epithelial cells. Biochim Biophys Acta 1803: 1386–1395, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J 23: 1739–1748, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodrigues GA, Park M. Autophosphorylation modulates the kinase activity and oncogenic potential of the Met receptor tyrosine kinase. Oncogene 9: 2019–2027, 1994. [PubMed] [Google Scholar]
- 31.Stella MC, Trusolino L, Pennacchietti S, Comoglio PM. Negative feedback regulation of Met-dependent invasive growth by Notch. Mol Cell Biol 25: 3982–3996, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442–454, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson AG, Kim K, Melancon B, Ghidu VP, Sulikowski GA, LaFleur B, Salic A, Lee LA, Miller DM 3rd, Lee E. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat Chem Biol 6: 829–836, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueno T, Kobayashi N, Nakayama M, Takashima Y, Ohse T, Pastan I, Pippin JW, Shankland SJ, Uesugi N, Matsusaka T, Nagata M. Aberrant Notch1-dependent effects on glomerular parietal epithelial cells promotes collapsing focal segmental glomerulosclerosis with progressive podocyte loss. Kidney Int 83: 1065–1075, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu Y, Baker D, Quan T, Baldassare JJ, Voorhees JJ, Fisher GJ. Receptor type protein tyrosine phosphatase-κ mediates cross-talk between transforming growth factor-β and epidermal growth factor receptor signaling pathways in human keratinocytes. Mol Biol Cell 21: 29–35, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc Natl Acad Sci USA 98: 6686–6691, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J 23: 1155–1165, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang S, Chung WC, Miele L, Xu K. Targeting Met and Notch in the Lfng-deficient, Met-amplified triple-negative breast cancer. Cancer Biol Ther 15: 633–642, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Li Y, Dai C, Yang J, Mundel P, Liu Y. Sp1 and Sp3 transcription factors synergistically regulate HGF receptor gene expression in kidney. Am J Physiol Renal Physiol 284: F82–F94, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Yang J, Li Y, Liu Y. Both Sp1 and Smad participate in mediating TGF-β1-induced HGF receptor expression in renal epithelial cells. Am J Physiol Renal Physiol 288: F16–F26, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Zhou D, Tan RJ, Lin L, Zhou L, Liu Y. Activation of hepatocyte growth factor receptor, c-Met, in renal tubules is required for renoprotection after acute kidney injury. Kidney Int 84: 509–520, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]