


Abstract
Transfer RNAs (tRNAs) are essential for encoding the transcribed genetic information from DNA into proteins. Variations in the human tRNAs are involved in diverse clinical phenotypes. Interestingly, all pathogenic variations in tRNAs are located in mitochondrial tRNAs (mt-tRNAs). Therefore, it is crucial to identify pathogenic variations in mt-tRNAs for disease diagnosis and proper treatment. We collected mt-tRNA variations using a classification based on evidence from several sources and used the data to develop a multifactorial probability-based prediction method, PON-mt-tRNA, for classification of mt-tRNA single nucleotide substitutions. We integrated a machine learning-based predictor and an evidence-based likelihood ratio for pathogenicity using evidence of segregation, biochemistry and histochemistry to predict the posterior probability of pathogenicity of variants. The accuracy and Matthews correlation coefficient (MCC) of PON-mt-tRNA are 1.00 and 0.99, respectively. In the absence of evidence from segregation, biochemistry and histochemistry, PON-mt-tRNA classifies variations based on the machine learning method with an accuracy and MCC of 0.69 and 0.39, respectively. We classified all possible single nucleotide substitutions in all human mt-tRNAs using PON-mt-tRNA. The variations in the loops are more often tolerated compared to the variations in stems. The anticodon loop contains comparatively more predicted pathogenic variations than the other loops. PON-mt-tRNA is available at http://structure.bmc.lu.se/PON-mt-tRNA/.
INTRODUCTION
Messenger RNA (mRNA) carries information from DNA and is translated to protein in the ribosome. During translation, transfer RNAs (tRNAs) deliver amino acids to the ribosome for elongation of the synthesized peptide chain. Among the 64 possible codons, 61 encode for 20 amino acids and 3 encode for stop codons that terminate translation. Codons are degenerate, which means that most amino acids are encoded by multiple codons. The codons in mRNA pair during translation with the anticodon of tRNA. Several nuclear genes code for the tRNAs with the same anticodon, on the other hand an anticodon can pair with multiple codons due to wobbling (1). The numbers of tRNA-coding genes vary in organisms. In human, there are 513 nuclear-encoded tRNA genes for 49 isoacceptors and 22 mitochondrial tRNA (mt-tRNA) genes (2).
The human mitochondrial genome (mtDNA) encodes for 13 protein-coding genes, two ribosomal RNAs and 22 tRNAs (3). The major portion (∼93%) of mtDNA codes for genes (4) and the variation rate is several times (10–17x) higher than that of the nuclear genome due to various reasons including inefficient mismatch repair, absence of histones (5,6) and others (7,8). Nuclear and mitochondrial genetic codes have some differences due to different specificities of mt-tRNAs. Each cell contains hundreds or thousands of mitochondria and multiple copies of mtDNA in each of them. Therefore, heteroplasmy is common because both normal and variant mtDNAs can co-exist in the cell. Variations in mtDNA are tolerated to a certain level before they show a biochemical defect (9).
Hundreds of variations in mtDNA have been reported to be associated with diseases and the number of unclassified variants is even larger. MITOMAP (10) stores mitochondrial variations associated with diseases and the Human Mitochondrial Genome Database (mtDB) (11) and the Human Mitochondrial Genome Polymorphism Database (mtSNP) (12) contain likely benign variations. Variations in the mt-tRNAs are even more interesting than those in the nuclear tRNAs because all the reported disease-causing tRNA variations occur in the mt-tRNAs (2,13). Hence, it would be crucial to recognize pathogenic variations in mt-tRNAs to facilitate diagnosis and treatment of mt-tRNA-associated diseases. Based on the canonical criteria for identifying pathogenic variations (14), a method for classifying mt-tRNA variations was developed (15). Subsequently, the authors re-evaluated and adjusted the method and applied it to a larger data set (16). A modification to the classification method has been suggested to penalize the normality in trans-mitochondrial cybrid studies (17). These methods require evidence from several experiments which is expensive and time consuming to obtain. Another method based on sequence conservation and secondary structure base pairs has been published and all possible single nucleotide substitutions in all the 22 mt-tRNAs were classified (18). The method was developed based on a small number of variations and has not been updated or tested with the recent data. The predictions are used by MtSNPScore, a method to analyze the impacts of mitochondrial variations (19). Fast and reliable predictors could be useful to rank variations identified by sequencing and therefore could be part of high throughput analysis pipelines.
Several methods have been developed to predict the effects of variations in human protein-coding genes (20–24). Machine learning (ML)-based tools are powerful for generalizing patterns from the training data and for predicting new cases. ML methods should be trained with sufficiently large data sets, which are not always available. Hence, the predictions of ML methods could be complemented by supporting evidence from various sources. The International Agency for Cancer Research (IARC) has recommended a standard method to categorize variants in cancer into 5 classes based on the posterior probability of pathogenicity (25). This scheme has been widely used to characterize variations in breast cancer susceptibility genes BRCA1 and BRCA2 (26) as well as in mismatch repair genes (27,28). Such methods use pathogenicity scores, based on evolutionary conservation or ML predictors, as prior probability of pathogenicity and combine evidence from additional sources such as segregation, family history and other clinical features. By integrating the evidence from diverse sources, a posterior probability of pathogenicity is computed based on which the variants are finally classified.
We developed a multifactorial probability-based classification method, PON-mt-tRNA, to classify mt-tRNA variations. The method integrates ML prediction together with evidence of segregation, biochemistry and histochemistry to compute the posterior probability of pathogenicity. Variations are classified into 5 classes: pathogenic, likely pathogenic, likely neutral, neutral and variants of uncertain significance (VUS). The method shows high performance with an accuracy and a Matthews correlation coefficient (MCC) of 1.00 and 0.99, respectively. In the absence of the biological evidence, PON-mt-tRNA classifies the variations based on the ML method and the corresponding accuracy and MCC are 0.69 and 0.39, respectively. PON-mt-tRNA has a 2-fold importance for identifying the pathogenicity of mt-tRNA variations. Firstly, the ML method is useful to rank novel variations and prioritize them for experimental evaluation and secondly, the integrated classifier can be used to classify the variations by combining the results of experimental studies together with the ML method.
MATERIALS AND METHODS
Data sets
We obtained classified mt-tRNA variations from the literature (16). In total, there were 55 neutral and 91 pathogenic single nucleotide substitutions. The variations were classified based on evidence from several sources. These variations were used for training and testing in this study. The variation data set is available from VariBench, a database of benchmark variation data sets (http://structure.bmc.lu.se/VariBench/mt-tRNA.php) (29). In addition, there were 46 single nucleotide substitutions that could not be reliably classified as pathogenic or neutral due to lack of sufficient evidence (16).
We collected additional mt-tRNA variations from other sources. From MITOMAP, we obtained 26 variations that were reported more than once in association with disease in the literature and 199 variations reported only once to be associated with disease (10). Benign variations were from mtDB (11) and mtSNP (12) databases. In total, there were 207 unique benign variations in the two databases. The data sets are summarized in Supplementary Table S1 and are available at the PON-mt-tRNA website (http://structure.bmc.lu.se/PON-mt-tRNA/datasets.html/).
Features
We used 12 features to describe variations. The features included evolutionary conservation (3 features), RNA secondary structure (2 features), tertiary interaction (1 feature), sequence context (3 features) and evidence of segregation, biochemistry and histochemistry (3 features). The features are summarized in Supplementary Tables S2 and S3.
Conservation features
We obtained the reference sequences of mt-tRNAs from the mito-tRNAdb (30) and mapped these to the revised Cambridge Reference Sequence (rCRS) of human mtDNA (NC_012920.1). The homologous sequences of the human mt-tRNA sequences from the members of Euarchontoglires superorder were obtained from Mammit-tRNA database (31). We aligned sequences homologous to each mt-tRNA sequence using Clustalw2 (32). From the alignments, we computed Position Specific Scoring Matrices (PSSM) for each mt-tRNA using the AlignInfo module in Biopython (http://biopython.org/DIST/docs/api/Bio.Align.AlignInfo-module.html). The PSSM contains scores for the occurrence of each nucleotide at each position in the human mt-tRNA sequences. We used the PSSM scores for the reference nucleotide and the variant nucleotide as features. In addition, we computed the information content at each position of the mt-tRNA sequences from the alignment with the AlignInfo module and used it as a feature.
Secondary structure
We obtained the secondary structures of the mt-tRNAs from mito-tRNAdb (30). We extracted two features based on the secondary structure of the mt-tRNAs. First, we annotated each position with the secondary structure type, i.e. loop, stem, variable region or other region. Second, we categorized the nucleotides in stem regions into three groups based on the base pairing. Wobble base pairings are common in tRNA secondary structures. We used three categories, (i) pairings that follow Watson–Crick base pair rules; (ii) guanine-uracil base pairs; and (iii) wobble base pairs other than the guanine-uracil base pair.
Tertiary interaction
Suzuki et al. have suggested that each of the human mt-tRNAs have one of the three overall structures (33). We obtained the sites involved in tertiary interaction based on Suzuki et al. (33) and generated a feature based on whether a variation occurs at an interaction site or not.
Sequence context
We extracted one nucleotide before and one after the variation site in the mt-tRNA sequence and formed tri-nucleotide strings. We then classified each nucleotide in the string depending on whether the nucleotide is a purine (A, G) or a pyrimidine (C, T). We grouped the nucleotides as strong (C, G) and weak (A, T) depending on the strength of the hydrogen bonding. We also grouped the nucleotides into keto (G, T) and amino (A, C) based on the presence or absence of a keto-group in the aromatic ring. For example, a string ‘ACT’ where the variation is at ‘C’ was represented as 011, 101 and 110, respectively, as three features. Hence, we extracted three features based on the sequence context.
Other evidence
We used the evidence of segregation of the variation with disease, involvement in biochemical defects in complexes I, III and IV, and histochemical evidence of mitochondrial disease for developing a multifactorial likelihood model. These features were obtained together with the training data set from Yarham et al. (16). A score greater than zero was used as the presence of evidence and a score equal to zero for the lack of evidence.
Classifier development
We developed a two-step method for classification of mt-tRNA variants. In the first step, an ML method was developed to predict the probability of pathogenicity of variations. In the second step, the probability of pathogenicity predicted by the ML method was used as prior probability and was integrated with evidence of segregation, biochemistry and histochemistry to compute the posterior probability of pathogenicity.
ML method
We used a random forest (RF) algorithm to train an ML method (34). The randomForest package (version 4.6–12) in the R statistical software (version 3.0.2) (https://www.r-project.org/) was used to implement the RF algorithm. We used default parameters for training the RF algorithm where the number of trees grown (ntree) was 500 and the number of features used at each split (mtry) was 3. We trained the predictor using nine features. Features representing segregation, biochemistry and histochemistry were not used.
Posterior probability of pathogenicity
In the second step, we computed the likelihood ratio (LR) of the evidence of segregation, biochemistry and histochemistry in pathogenic and neutral data sets as described by Lindor et al. (26). LRs for the evidence of segregation, biochemical and histochemical tests were computed as follows:
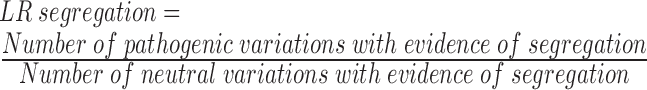 |
Then, the combined LR based on the evidence of all three sources is given by the product of the LRs for each source.
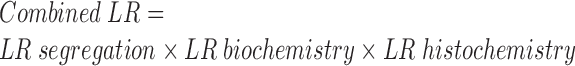 |
The combined LR was integrated together with the ML-based probability of pathogenicity to compute the posterior probability of pathogenicity (26). The ML method-based probability of pathogenicity was used as prior probability for the second step.
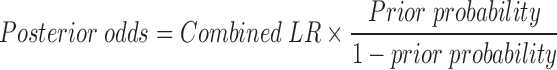 |
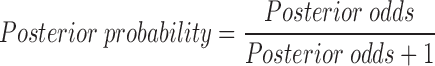 |
Training and testing
We used balanced training data sets to train the ML method and to estimate LRs. As the training and test data sets were small, we trained and tested the method 2000 times by introducing variability in the training and test data sets. First, we randomly sampled without replacement 15 pathogenic and 15 neutral variants for testing the method. From among the remaining variants, we randomly sampled 40 pathogenic and 40 neutral variants and trained the ML predictor and calculated LRs 100 times. The performance of the trained methods was evaluated using the same test data. This approach introduced variability to the training data and the performance of methods trained on different training data could be evaluated on the same test data. To introduce variability to the test data, the same procedure of randomly choosing test data and then training 100 predictors was repeated 20 times. The variability was introduced to reduce bias in the training and testing. Thus, 2000 predictors were trained and their performance was evaluated. PON-mt-tRNA uses all the 2000 predictors for predicting the pathogenicity of variants.
Classification
Two schemes were designed to classify variations depending on the input data for PON-mt-tRNA. When evidence from any one of the sources, i.e. segregation, biochemistry or histochemistry, is available, the method classifies variations based on the posterior probability of pathogenicity obtained by integrating the ML method and evidence information. The variations were classified into five groups – pathogenic, likely pathogenic, likely neutral, neutral and VUS. The cut-offs for classifying variations were adopted from Lindor et al. and presented in Supplementary Table S4.
In the absence of evidence from the three sources, PON-mt-tRNA classifies the variants based on the ML method. In this case, the variants are classified into four groups – pathogenic, likely pathogenic, likely neutral and neutral. The cut-offs for each group are presented in the Supplementary Table S4. If at least 90% (1800) of the predictors in the ML method predict the probability of pathogenicity greater than or equal to 0.5, the variant is classified as pathogenic and if at least 90% (1800) of the predictors predict the probability of pathogenicity to be smaller than 0.5, the variant is classified as neutral. The remaining variants are classified as likely pathogenic if their average predicted probability of pathogenicity is greater than or equal to 0.5 and likely neutral if their average predicted probability of pathogenicity is smaller than 0.5.
Performance evaluation
We tested the performance of PON-mt-tRNA by using the predictions of the method for the test data in the 2000 iterations. The variations in the training and test data were disjoint in each iteration. The overall performance of the methods was calculated by averaging the performance scores. We used six standard performance measures (35) following the guidelines (36). The performance measures were computed using the following equations:
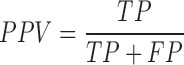 |
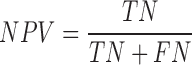 |
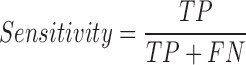 |
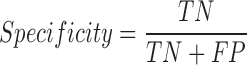 |
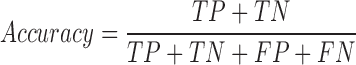 |
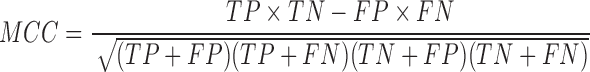 |
where, TP is correctly predicted pathogenic variations, TN is correctly predicted neutral variations, FP is incorrectly predicted neutral variations, FN is incorrectly predicted pathogenic variations, PPV is Positive Predictive Value, NPV is Negative Predictive Value and MCC is Matthews Correlation Coefficient. Receiver Operating Characteristics (ROC) curves were also used to assess the performance of methods and they were obtained using the ROCR package in the R statistical software (37).
Classification of unknown cases
Among the 200 variations studied by Yarham et al. (16), 46 single nucleotide substitutions could not be classified as ‘definitely pathogenic’ or ‘definitely neutral’ due to lack of sufficient evidence. We classified these variations using PON-mt-tRNA. First, we predicted the probabilities of pathogenicity using the ML method. Then, we combined the average of the predicted probabilities of pathogenicity with the LRs to compute the posterior probabilities of pathogenicity. The variations were classified into five classes based on the posterior probability of pathogenicity (Supplementary Table S4).
Prediction for all possible substitutions
We used PON-mt-tRNA to predict the pathogenicity for all possible single nucleotide substitutions in all 22 human mt-tRNAs. Only the ML method was used for classification. We mapped the variations to the mt-tRNA secondary structures obtained from mito-tRNAdb (30) and visualized the numbers of predicted pathogenic variations at each position in the 22 mt-tRNAs on a three-dimensional structure of yeast phenylalanine tRNA (pdb id: 1EHZ) using UCSF Chimera (38). The yeast tRNA structure was used for visualization due to lack of structure for any human tRNA.
RESULTS
PON-mt-tRNA
We developed a multifactorial probability-based classification method, PON-mt-tRNA, to predict the pathogenicity of single nucleotide substitutions in human mt-tRNAs. The method integrates an ML method and evidence of segregation, biochemistry and histochemistry. In the first step, the ML method is used to predict the probability of pathogenicity of variations. Then, the predicted probability is used as prior probability and integrated with the evidence information to predict the posterior probability of pathogenicity (Figure 1). The ML method was trained on a balanced set of known pathogenic and neutral variations. We trained altogether 2000 ML predictors using different sets of pathogenic and neutral variations chosen by random sampling without replacement. The accuracy and MCC for the ML method are 0.69 and 0.39, respectively (Table 1). The performance scores for PON-mt-tRNA are much higher when evidence of segregation, biochemistry and histochemistry is known. The accuracy and MCC are in this case 1.00 and 0.99, respectively (Table 1). We compared the performance of PON-mt-tRNA with previously published method which was based on sequence conservation and secondary structure (referred as Kondrashov) (18). Both versions of PON-mt-tRNA showed superior performance compared to the Kondrashov method (Table 1 and Supplementary Figure S1). The sensitivity of the Kondrashov method is higher than for the ML predictor but with a very low specificity (0.47) indicating that the method is severely unbalanced.
Figure 1.

Schematic outline of PON-mt-tRNA. The method predicts the probability of pathogenicity using 2000 ML predictors and integrates with evidence of segregation, biochemistry and histochemistry. If the evidence is not known, PON-mt-tRNA predicts the pathogenicity based on the ML predictors only.
Table 1. Performance comparison of Kondrashov method and PON-mt-tRNA (ML method and integrated predictor).
| TP | TN | FP | FN | PPVa | NPVa | Sensitivitya | Specificitya | Accuracya | MCCa | |
|---|---|---|---|---|---|---|---|---|---|---|
| Kondrashovb | 13.10 | 7.10 | 7.90 | 1.90 | 0.63 (0.07) | 0.79 (0.14) | 0.87 (0.08) | 0.47 (0.14) | 0.67 (0.09) | 0.38 (0.18) |
| PON-mtRNA (ML probc) | 10.35 | 10.45 | 4.55 | 4.65 | 0.70 (0.08) | 0.70 (0.06) | 0.69 (0.09) | 0.70 (0.12) | 0.69) (0.06) | 0.39 (0.13) |
| PON-mtRNA (Posterior probc, d) | 11.80 | 11.35 | 0 | 0.10 | 1.00 (0.00) | 0.99 (0.02) | 0.99 (0.02) | 1.00 (0.00) | 1.00 (0.01) | 0.99 (0.02) |
aPerformance scores are averages of 2000 iterations. The standard deviations are indicated in parentheses.
bMethod developed by Kondrashov (18). The performance of the method may be over-estimated as some of the variations were used to train the method.
cProb, Probability.
dPredicted pathogenic and likely pathogenic are considered as pathogenic and neutral and likely neutral are together considered as neutral. The variants of uncertain significance are not included.
PON-mt-tRNA classifies the variations into 5 classes: pathogenic, likely pathogenic, likely neutral, neutral and VUS (Supplementary Table S4). The classification of variations in the four classes (excluding VUS) is highly reliable (Table 1). On average 20.6% (3.10/15) of pathogenic and 24.3% (3.65/15) of neutral variations are classified as VUS in our test data set (Supplementary Table S5). In the absence of evidence from segregation, biochemistry and histochemistry, PON-mt-tRNA classifies the variations into four classes pathogenic, likely pathogenic, likely neutral and neutral based on the ML method (Supplementary Table S4).
The ML method was trained using nine features representing evolutionary conservation, sequence context, RNA secondary structure and tertiary interactions (Supplementary Table S2). We evaluated the importance of the features based on the importance scores obtained from the RF algorithm. The sequence context and evolutionary conservation are the most important features while the secondary structure and tertiary interaction are the least important features (Supplementary Table S2). Evidence of segregation, biochemical test and histochemical test were used for the multifactorial probability-based PON-mt-tRNA. We compared the importance of the sources of evidence for pathogenicity. All three sources provide evidence in favor of pathogenicity (Supplementary Table S3). The evidence of the biochemical test showed the highest LR (8.68) and the evidence of segregation the lowest LR (2.17). The LR for evidence of the histochemical test is 6.68. The contributions of individual features in the overall performance of the method could not be tested because of the small size of the training data.
Classification of unknown cases
We classified 46 variants with unknown outcome (16) into 5 classes using PON-mt-tRNA. Most of these variants are present in the MITOMAP database which includes variations reported to be associated with diseases. However, Yarham et al. could not classify them as definitely pathogenic due to lack of sufficient evidence. Of the 23 classified variations, 11 are classified as pathogenic, 10 likely pathogenic, 1 likely neutral and 1 neutral (Table 2 and Supplementary Table S6). The remaining 23 variations are considered to be VUS due to lack of sufficient evidence.
Table 2. Classification of unknown cases using PON-mt-tRNA. The variations were classified as ‘likely pathogenic’ by Yarham et al. due to lack of sufficient evidence.
| Pathogenic | Likely pathogenic | Unknown | Likely neutral | Neutral | Total | |
|---|---|---|---|---|---|---|
| RF probabilitya | 13 | 9 | NA | 14 | 10 | 46 |
| Posterior probability | 11 | 10 | 23 | 1 | 1 | 46 |
aVariations are classified into 4 classes. If more than 90% of RF predictors predict the prior probability to be greater or smaller than 0.5, the variations are classified as pathogenic or neutral, respectively. Otherwise, the variations are classified as likely pathogenic or likely neutral based on the average of the predicted probabilities.
Prediction of reported disease-associated and benign variations
To further test the method, we predicted the pathogenicity of additional variations obtained from MITOMAP, mtDB and mtSNP. From each data set, we eliminated variants present in the training data set. As additional evidence is not available for these cases, we used the ML method for classification. The mtDB and mtSNP databases contain likely benign variations. PON-mt-tRNA predicted 85.5% (177/207) of the cases in these databases as being neutral or likely neutral (Table 3). MITOMAP contains variations reported to be associated with diseases, however, only about half of them are predicted to be pathogenic or likely pathogenic. When we investigated the overlap between MITOMAP and the benign variation data sets, 23 variations overlapped in the two data sets. After eliminating the overlapping variations, 58.2% (46/79) of the remaining MITOMAP variations (called as MITOMAP filtered) were predicted to be pathogenic or likely pathogenic. Among the 79 MITOMAP filtered variations, 4 had the ‘confirmed’ status in the database which means that at least two independent laboratories have reported the pathogenicity of the variations. Among the ‘confirmed’ variations, 1 was predicted as pathogenic, 2 as likely pathogenic and 1 as neutral (Table 3).
Table 3. Prediction of mt-tRNA variations from mtDB, mtSNP and MITOMAP using PON-mt-tRNA. The variants present in PON-mt-tRNA training and test data sets were excluded.
| Data set | Data description | Pathogenic | Likely pathogenic | Likely neutral | Neutral | Total |
|---|---|---|---|---|---|---|
| mtDB + mtSNPa | Non-disease-associated | 12 (5.79%) | 18 (8.70%) | 46 (22.22%) | 131 (63.29%) | 207 |
| MITOMAPb | Disease-associated | 28 (27.45%) | 21 (20.59%) | 20 (19.61%) | 33 (32.35%) | 102 |
| MITOMAP (filtered)a | Disease-associated | 27 (34.18%) | 19 (24.05%) | 16 (20.25%) | 17 (21.52%) | 79 |
| MITOMAP (filtered and confirmed)b | Disease-associated | 1 (25%) | 2 (50%) | 0 (0%) | 1 (25%) | 4 |
aVariations in MITOMAP not present in mtDB and mtSNP.
bVariations in MITOMAP with status ‘confirmed’.
The percentages are indicated in the parentheses.
Classification of all possible variations
We classified all the possible single nucleotide substitutions in the human mt-tRNAs using PON-mt-tRNA. Among the 4521 possible variations, 51.0% were predicted as pathogenic. Among them, 73.5% (1695) variations occur in stem regions and 18.2% (419) in the loops. We mapped the numbers of predicted pathogenic variations at each position in the 22 human mt-tRNAs to the corresponding positions in the three-dimensional structure of yeast nuclear phenylalanine tRNA (pdb id: 1EHZ). The proportion of predicted pathogenic variations is higher in stems (61.5%) than in loops (34.1%) (Figure 2A). Among the stems, the D-stem is the shortest but had the highest proportion of pathogenic variations (82.7%) and among the loops, the anticodon loop has the highest proportion of pathogenic variations (57.1%) (Figures 2 and 3 and Supplementary Table S7). The variations in the anticodon loop are likely to affect the anticodon and mRNA recognition and therefore are more often pathogenic than in other loops.
Figure 2.

Distribution of predicted pathogenic variations in mt-tRNA structure. All possible single nucleotide substitutions at each position of the 22 mt-tRNAs are mapped to the three-dimensional structure of the yeast phenylalanine tRNA (pdb id: 1EHZ). (A) The numbers of predicted pathogenic mt-tRNA variations at each position. There are 66 possible variations per site except for sites which are missing from some mt-tRNAs. (B) The numbers of mt-tRNAs containing at least one predicted pathogenic variation at each position. Ac-stem, Anticodon stem; Ac-loop, Anticodon loop.
Figure 3.

The distribution of predicted pathogenic variations in mt-tRNAs. All possible single nucleotide substitutions at each position of the 22 mt-tRNAs are classified using PON-mt-tRNA. There are three possible substitutions at each position. The secondary structures were obtained from mito-tRNAdb. Shading indicates the numbers of predicted pathogenic variants per site. Acc-stem, Acceptor stem; Ac-stem, Anticodon stem; Ac-loop, Anticodon loop; V-region, Variable region.
We compared the predictions of PON-mt-tRNA with those of the Kondrashov method. Both methods agreed in the predictions for 71.1% (3216/4521) of the possible single nucleotide substitutions. Of the remaining 1301 variations, PON-mt-tRNA predicted 96 as pathogenic and 1205 as neutral. PON-mt-tRNA and the Kondrashov method agree the most for variations in the D-stems and T-loops. Over 80% of the possible variations in D-stem are predicted as pathogenic by both methods and over 70% of the variations in the T-loop are predicted as neutral (Supplementary Table S7).
Web application
The PON-mt-tRNA web interface is available at http://structure.bmc.lu.se/PON-mt-tRNA/. The method requires the reference position in mtDNA, the reference (original) nucleotide and the altered nucleotide for each variation as inputs. In addition, the user can submit evidence for segregation, biochemical and histochemical features. The evidence field is optional as the data are not always available. If the evidence is provided, PON-mt-tRNA integrates the evidence with predictions of ML method to classify the variations, otherwise the predictions of the ML method are used for classification. The predictions for all possible nucleotide substitutions in mt-tRNA genes are available for download at http://structure.bmc.lu.se/PON-mt-tRNA/datasets.html/.
DISCUSSION
Large numbers of tRNA variations have been identified and all of those associated with disease appear in mt-tRNAs (2,13). Due to the degeneracy of the genetic code and the presence of isoacceptors for nuclear tRNAs, the phenotype may not necessarily appear even for harmful variants. The variations identified in patients are often filtered based on the common variants in the haplogroups and other benign variations reported in databases. Several databases including MITOMAP (10), mammit-tRNA (31), mtDB (11), mtSNP (12) and others collect and store variations reported in literature. These databases are useful for interpreting previously reported variations. We found that these resources contain contradictory reports for a number of cases. This may be partly because of technical errors or shortcomings in interpreting the variation effects (39). Other possible reasons could be phenotypic heterogeneity and variable penetrance. Several factors including threshold of heteroplasmy, mitotic segregation, clonal expansion and genetic bottleneck may affect the clinical outcome of mitochondrial variations (6). Therefore, it is essential to accurately classify the variations in order to correctly diagnose and provide genetic advice to patients.
The Kondrashov method does not require experimental evidence to rank and prioritize variations. The method has not been updated for several years and it showed poor performance in our evaluation (Table 1). We developed a novel multifactorial probability-based method, PON-mt-tRNA, to accurately classify variations in human mt-tRNAs. The method integrates an ML method and evidence from three sources for classification of variations. As far as we know, this is the first method that implements an ML algorithm for predicting the pathogenicity of mt-tRNA variants. The ML method does not require any experimental evidence and can rank all novel variations. PON-mt-tRNA showed higher performance scores than the Kondrashov method (Table 1 and Supplementary Figure S1). The integrated predictor has a sensitivity of 0.99 and a specificity of 1.00. Although we cannot compare the performance of PON-mt-tRNA to that of Yarham et al. because that would introduce circularity, we were able to classify half of the variations (23/46) that Yarham et al. could not classify reliably. The evidence-based experimental method requires evidence from several sources and therefore takes a long time to classify a novel variation. PON-mt-tRNA can rank novel variations using the ML method and prioritize likely harmful variations for further experiments. When the results from the three sources (segregation, biochemistry and histochemistry) are available, the method can classify variations with an almost perfect accuracy (Table 1).
We classified all possible single nucleotide substitutions in the mt-tRNA genes using PON-mt-tRNA. The results show that the pathogenic variations are most frequent in the stems (Figures 2 and 3) where the variations are likely to disrupt the hydrogen bonding and affect the mt-tRNA structure and consequently translation. Among the loops, the anticodon loops are the most vulnerable for variations because of the effects on codon recognition. These results are in line with those from an independent previous study (15). The predicted pathogenic variations are likely harmful but may not be pathogenic in all individuals due to the complexity of the mitochondrial genetics. The pathogenicity of variations in patients can be revealed by integrating the predictions and additional sources of evidence.
Single-fiber and trans-mitochondrial cybrid studies are considered as the gold standard for the evaluation of pathogenicity of mtDNA variations (15,16) but such experiments are time-consuming. To confirm the highly accurate predictions of PON-mt-tRNA, we advise to perform such experiments, when possible. Heteroplasmy is an important aspect of mitochondrial genetics and plays a role in mtDNA variations’ pathogenicity. However, PON-mt-tRNA does not incorporate heteroplasmy for prediction. PON-mt-tRNA facilitates the classification of mt-tRNA variations in two steps. First, the method can be used to rank novel variations based on the prediction of the ML method when no other evidence is known. Based on the ranking, the variants can be prioritized for experiments to collect further evidence of pathogenicity. This version of PON-mt-tRNA can be used immediately after identifying the variations by genome sequencing. Thus, it can be integrated into a high-throughput analysis pipeline to rank mt-tRNA variations. Second, the method can be used to classify variations when evidence of segregation, and results of biochemical and histochemical tests are available.
AVAILABILITY
PON-mt-tRNA is available at http://structure.bmc.lu.se/PON-mt-tRNA/.
Supplementary Material
Acknowledgments
Faculty of Medicine, Lund University; Barncancerfonden; and Vetenskapsrådet. We thank Gerard Schaafsma for proofreading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Lund University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kutter C., Brown G.D., Goncalves A., Wilson M.D., Watt S., Brazma A., White R.J., Odom D.T. Pol III binding in six mammals shows conservation among amino acid isotypes despite divergence among tRNA genes. Nat. Genet. 2011;43:948–955. doi: 10.1038/ng.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirchner S., Ignatova Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2015;16:98–112. doi: 10.1038/nrg3861. [DOI] [PubMed] [Google Scholar]
- 3.Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H.L., Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P., Roe B.A., Sanger F., et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 4.Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khrapko K., Coller H.A., André P.C., Li X.C., Hanekamp J.S., Thilly W.G. Mitochondrial mutational spectra in human cells and tissues. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13798–13803. doi: 10.1073/pnas.94.25.13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuppen H.A.L., Blakely E.L., Turnbull D.M., Taylor R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta. 2010;1797:113–128. doi: 10.1016/j.bbabio.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Wallace D.C., Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013;5:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 10.Lott M.T., Leipzig J.N., Derbeneva O., Xie H.M., Chalkia D., Sarmady M., Procaccio V., Wallace D.C. mtDNA variation and analysis using MITOMAP and MITOMASTER. Curr. Protoc. Bioinformatics. 2013;1:1.23.21–21.23.26. doi: 10.1002/0471250953.bi0123s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ingman M., Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34:D749–D751. doi: 10.1093/nar/gkj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka M., Takeyasu T., Fuku N., Li-Jun G., Kurata M. Mitochondrial genome single nucleotide polymorphisms and their phenotypes in the Japanese. Ann. N. Y. Acad. Sci. 2004;1011:7–20. doi: 10.1007/978-3-662-41088-2_2. [DOI] [PubMed] [Google Scholar]
- 13.Abbott J.A., Francklyn C.S., Robey-Bond S.M. Transfer RNA and human disease. Front. Genet. 2014;5:158. doi: 10.3389/fgene.2014.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiMauro S., Schon E.A. Mitochondrial DNA mutations in human disease. Am. J. Med. Genet. 2001;106:18–26. doi: 10.1002/ajmg.1392. [DOI] [PubMed] [Google Scholar]
- 15.McFarland R., Elson J.L., Taylor R.W., Howell N., Turnbull D.M. Assigning pathogenicity to mitochondrial tRNA mutations: when ‘definitely maybe’ is not good enough. Trends Genet. 2004;20:591–596. doi: 10.1016/j.tig.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Yarham J.W., Al-Dosary M., Blakely E.L., Alston C.L., Taylor R.W., Elson J.L., McFarland R. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum. Mutat. 2011;32:1319–1325. doi: 10.1002/humu.21575. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Vioque E., Bornstein B., Gallardo M.E., Fernandez-Moreno M.A., Garesse R. The pathogenicity scoring system for mitochondrial tRNA mutations revisited. Mol. Genet. Genomic Med. 2014;2:107–114. doi: 10.1002/mgg3.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondrashov F.A. Prediction of pathogenic mutations in mitochondrially encoded human tRNAs. Hum. Mol. Genet. 2005;14:2415–2419. doi: 10.1093/hmg/ddi243. [DOI] [PubMed] [Google Scholar]
- 19.Bhardwaj A., Mukerji M., Sharma S., Paul J., Gokhale C.S., Srivastava A.K., Tiwari S. MtSNPscore: a combined evidence approach for assessing cumulative impact of mitochondrial variations in disease. BMC Bioinformatics. 2009;10(Suppl. 8):S7. doi: 10.1186/1471-2105-10-S8-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niroula A., Urolagin S., Vihinen M. PON-P2: prediction method for fast and reliable identification of harmful variants. PLoS One. 2015;10:e0117380. doi: 10.1371/journal.pone.0117380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niroula A., Vihinen M. Classification of amino acid substitutions in mismatch repair proteins using PON-MMR2. Hum. Mutat. 2015;36:1128–1134. doi: 10.1002/humu.22900. [DOI] [PubMed] [Google Scholar]
- 22.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calabrese R., Capriotti E., Fariselli P., Martelli P.L., Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 24.Olatubosun A., Valiaho J., Harkonen J., Thusberg J., Vihinen M. PON-P: integrated predictor for pathogenicity of missense variants. Hum. Mutat. 2012;33:1166–1174. doi: 10.1002/humu.22102. [DOI] [PubMed] [Google Scholar]
- 25.Plon S.E., Eccles D.M., Easton D., Foulkes W.D., Genuardi M., Greenblatt M.S., Hogervorst F.B., Hoogerbrugge N., Spurdle A.B., Tavtigian S.V. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindor N.M., Guidugli L., Wang X., Vallee M.P., Monteiro A.N., Tavtigian S., Goldgar D.E., Couch F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS) Hum. Mutat. 2012;33:8–21. doi: 10.1002/humu.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson B.A., Spurdle A.B., Plazzer J.P., Greenblatt M.S., Akagi K., Al-Mulla F., Bapat B., Bernstein I., Capella G., den Dunnen J.T., et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014;46:107–115. doi: 10.1038/ng.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson B.A., Goldgar D.E., Paterson C., Clendenning M., Walters R., Arnold S., Parsons M.T., Michael D.W., Gallinger S., Haile R.W., et al. A multifactorial likelihood model for MMR gene variant classification incorporating probabilities based on sequence bioinformatics and tumor characteristics: a report from the Colon Cancer Family Registry. Hum. Mutat. 2013;34:200–209. doi: 10.1002/humu.22213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nair P.S., Vihinen M. VariBench: a benchmark database for variations. Hum. Mutat. 2013;34:42–49. doi: 10.1002/humu.22204. [DOI] [PubMed] [Google Scholar]
- 30.Juhling F., Morl M., Hartmann R.K., Sprinzl M., Stadler P.F., Putz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Putz J., Dupuis B., Sissler M., Florentz C. Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA. 2007;13:1184–1190. doi: 10.1261/rna.588407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki T., Nagao A., Suzuki T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011;45:299–329. doi: 10.1146/annurev-genet-110410-132531. [DOI] [PubMed] [Google Scholar]
- 34.Breiman L. Random Forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 35.Vihinen M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genomics. 2012;13(Suppl. 4):S2. doi: 10.1186/1471-2164-13-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vihinen M. Guidelines for reporting and using prediction tools for genetic variation analysis. Hum. Mutat. 2013;34:275–282. doi: 10.1002/humu.22253. [DOI] [PubMed] [Google Scholar]
- 37.Sing T., Sander O., Beerenwinkel N., Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 38.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera - a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 39.Bandelt H.J., Yao Y.G., Salas A., Kivisild T., Bravi C.M. High penetrance of sequencing errors and interpretative shortcomings in mtDNA sequence analysis of LHON patients. Biochem. Biophys. Res. Commun. 2007;352:283–291. doi: 10.1016/j.bbrc.2006.10.131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.