


Abstract
RNase P is the enzyme that removes 5′ extensions from tRNA precursors. With its diversity of enzyme forms—either protein- or RNA-based, ranging from single polypeptides to multi-subunit ribonucleoproteins—the RNase P enzyme family represents a unique model system to compare the evolution of enzymatic mechanisms. Here we present a comprehensive study of substrate recognition and cleavage-site selection by the nuclear single-subunit proteinaceous RNase P PRORP3 from Arabidopsis thaliana. Compared to bacterial RNase P, the best-characterized RNA-based enzyme form, PRORP3 requires a larger part of intact tRNA structure, but little to no determinants at the cleavage site or interactions with the 5′ or 3′ extensions of the tRNA. The cleavage site depends on the combined dimensions of acceptor stem and T domain, but also requires the leader to be single-stranded. Overall, the single-subunit PRORP appears mechanistically more similar to the complex nuclear ribonucleoprotein enzymes than to the simpler bacterial RNase P. Mechanistic similarity or dissimilarity among different forms of RNase P thus apparently do not necessarily reflect molecular composition or evolutionary relationship.
INTRODUCTION
RNase P is the endonuclease responsible for the removal of transcriptional 5′-leader sequences from tRNA precursors (pre-tRNAs) (1,2). The (nearly) ubiquitous enzyme is found in two fundamentally different forms: (i) as a complex of an ancient, structurally conserved RNA molecule with a variable number of proteins (1 in Bacteria, 4–5 in Archaea, up to 10 in Eukarya), where the RNA is the actual catalyst and active alone under specific in vitro conditions (3–7); (ii) as a ∼60-kDa protein called PRORP (proteinaceous or protein-only RNase P), which does not contain a nucleic acid as enzyme subunit, although it requires additional proteins in some cases (8–12); PRORP is found in various Eukarya, but not in Bacteria or Archaea. The ribonucleoprotein (RNP) and protein-only forms of RNase P are apparently the result of convergent evolution, and even though they lack any structural homology, they are functionally largely exchangeable in genetic swap experiments (9,11,13). The fundamental difference in molecular composition and their independent evolutionary origin raise the question of whether the different types of RNase P use similar or different mechanisms to perform their enzymatic function. Their basic catalytic strategy for phosphodiester hydrolysis appears similar despite differences in the way metal ions are coordinated at the pre-tRNA's scissile bond (14–16), yet little is known on how the two enzyme types compare with respect to substrate recognition and cleavage-site selection.
Substrate recognition by RNA-based RNase P has been well characterized in the bacterial system through genetic and biochemical studies (see ref. 17 and refs. therein) and, finally, by the crystal structure of a holoenzyme in complex with a tRNA (18). The RNA subunit (P RNA) is primarily responsible for tRNA recognition on the basis of three major interactions: (i) stacking between bases in the specificity domain of the P RNA and bases in the tRNA's TΨC and D loops; (ii) a conserved adenosine in the specificity domain entering the minor groove of the tRNA acceptor stem; (iii) intermolecular base pairing between nucleotides in the catalytic domain of the P RNA and the DCCA motif at the tRNA's 3′ end. The protein subunit does not contact any region of the mature tRNA, but interacts with the 5′ leader of the tRNA precursor (18–22). The bacterial enzyme efficiently cleaves hairpin-like substrates composed of a 5′ leader, a helix resembling the stacked aminoacyl acceptor and T stems, and a 3′-CCA motif (23), and even substrates further minimized to short helices that maintain only the determinants at the cleavage site, were shown to be processed at the canonical site (24–26).
Fewer studies are available on substrate recognition by eukaryal (nuclear) RNP enzymes, but they nevertheless revealed some peculiarities. Rather than recognizing determinants at the cleavage site like the bacterial P RNA, the eukaryal nuclear RNase P RNP appears to select the cleavage site primarily by ‘measuring’ the combined lengths of the stacked aminoacyl acceptor and T domains (27,28). Artificial stem-loop substrates mimicking these two stacked pre-tRNA domains, which are efficiently cleaved by bacterial RNase P (23), were reported to require a bulge interrupting the helix at the 5′ junction of acceptor and T stem for substantial cleavage by the eukaryal RNP enzymes to occur (28,29). However, overall, substrate recognition and the contribution of the multiple components of the more complex nuclear RNPs are only poorly understood.
Protein-only RNase P (PRORP) consists of an N-terminal pentatricopeptide-repeat (PPR) domain and a C-terminal metallonuclease domain, connected by a central structural zinc-binding domain (12,30); the PPR is an RNA-binding motif proposed to mediate a base-specific interaction (31). Whereas in animal mitochondria, this PRORP protein needs to be complemented by a two-protein methyltransferase complex to function as an RNase P (8,32), it acts as an RNase P on its own in plants and protists (9–11). Despite the availability of the crystal structure of a plant PRORP protein (30), and initial studies addressing the interaction of this ‘single-subunit’ PRORP with tRNAs and the role of the PPR domain (33,34), the concepts of substrate recognition and cleavage-site selection by PRORP are currently mostly based on modeling rather than on experimental evidence. Here, we have investigated substrate recognition and cleavage-site selection by a prototypical nuclear single-subunit PRORP from Arabidopsis thaliana (PRORP3). Starting from a well-studied and conformationally stable pre-tRNA, we deleted or varied conserved structural elements and nucleotide identities. Processing of the tRNA variants was analyzed by thorough enzyme kinetics. This allowed us to pinpoint specific determinants for substrate recognition and positioning of the cleavage site, and to define minimal substrate(s) for single-subunit PRORPs.
MATERIALS AND METHODS
Expression and purification of recombinant PRORP3
The coding sequence of A. thaliana PRORP3 [locus tag At4g21900; 517 amino acids corresponding to the presumably mature form (9)] was cloned into the NcoI/XhoI sites of pET-28b(+) (Novagen) using the primers listed in Supplementary Table S1. The recombinant protein carries a C-terminal 6×His tag attached via a spacer of two amino acids. PRORP3 was expressed in Escherichia coli BL21(DE3).
Bacteria were lysed by sonication and recombinant PRORP3 was purified on a HisTrap HP column using an ÄKTApurifier chromatography system (GE Healthcare); buffer A (150 mM NaCl, 50 mM Tris·Cl pH 7.4, 10% glycerol, 1 mM DTT), buffer A’ (buffer A, but with 1 M NaCl), buffer B (buffer A plus 500 mM imidazole). The lysate was loaded and washed with 5 and 10% buffer B, then washed with buffer A, with buffer A’, and again with buffer A, and finally eluted with 50% buffer B. The purity of the recombinant protein was assessed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie brilliant blue staining. The concentration of purified PRORP3 was calculated from the absorbance at 280 nm, its molar extinction coefficient (ϵ280 = 81360 M−1 cm−1) and molecular weight (58.84 kDa); the measurement was confirmed by a Bradford protein assay (BioRad) using BSA standards. According to an A260/A280 ratio of 0.6, as well as gel electrophoresis after phenol-chloroform extraction, purified PRORP3 was judged to be free of nucleic acids.
Mutations were introduced into the PRORP3 expression plasmid by one or two rounds of site-directed mutagenesis using the QuikChange protocol (Agilent Technologies) and the primers listed in Supplementary Table S1. Mutant proteins were expressed and purified as described for the wild-type protein, but using His SpinTrap columns (GE Healthcare). The concentration of the mutant proteins relative to the wild-type protein was estimated by SDS-PAGE and Coomassie brilliant blue staining.
Preparation of Bacillus subtilis RNase P
Bacillus subtilis P RNA was in vitro transcribed from pDW66 (35); B. subtilis P protein was expressed, purified, and the RNase P holoenzyme reconstituted as previously described (36).
Preparation of RNA substrates
The Thermus thermophilus pre-tRNAGly was transcribed from plasmid pSBpt3’hh (37). Plasmids for the in vitro transcription of variants with U1–A72, U−1, G−1, A−1 and A73, of variants with substitutions C56→U56, G18→A18, A57→C57 and G19→A19, and of variants Aa−2bp, Aa+2bp and Aab1T, were produced from pSBpt3’hh (or a derivative with U65→C65) by one or two rounds of site-directed mutagenesis using the QuikChange protocol (Agilent Technologies). Plasmids for the transcription of Aab4T and Aab9T were produced by polymerase chain reaction (PCR) of two overlapping fragments, overlap extension and amplification, restriction digestion and cloning into the EcoRI/HindIII sites of pSP64 (Promega). A plasmid for the transcription of AaT was produced by annealing of two complementary oligonucleotides and cloning into the EcoRI/BamHI sites of pSP64. Oligonucleotides (and templates) for site-directed mutagenesis and (PCR) cloning are listed in Supplementary Table S2. The plasmids were digested with BamHI or NheI prior to in vitro transcription with T7 RNA polymerase (transcripts carried self-cleaving cis-hammerhead ribozymes generating identical 3′ ends).
Plasmids for the transcription of leader-length variants were produced by ‘inside-out’ PCR mutagenesis (38) using pSBpt3’hh as a template, followed by circularization of the products by T4 DNA ligase. They contain an additional hammerhead ribozyme inserted between the T7 promoter and the 5′-leader sequence. Oligonucleotides used are listed in Supplementary Table S3. The plasmids were digested with XbaI prior to in vitro transcription with T7 RNA polymerase.
DNA for the in vitro transcription of the three pre-tRNAGly-trailer variants, of the previously described ΔD and ΔAc variants (39), of Aab9T variants with substitutions U54→C54, U55→C55, C56→U56, A57→G57, A57→C57, A57→U57, A58→G58 and Aab9TCCUUUUA, of Aab9T variants Aa+2bp, T+2bp, Aa−2bp T+2bp and T4loop, and of pre-tRNAGly variants Aa+4bp, Aa+m3GC, Aa+m3AU, G−1–C73, U−1–A73 and Aa+3AU, were produced by PCR amplification using either pSBpt3’hh (or its derivatives with G−1 or U−1) as a template, or a DNA oligonucleotide, or no template in case of two overlapping PCR primers. Primers and oligonucleotide templates are listed in Supplementary Tables S4 and S5. All the PCR products start with a T7 promoter and end with the pre-tRNA's 3′-trailer sequence.
The template for E. coli pre-tRNAHis was a kind gift of Michael E. Harris (40).
In vitro transcription, 5′-end labeling with 32P and gel purification were carried out as previously described (15,41,42).
RNA processing assays
Single-turnover experiments were performed in 50 mM Tris·Cl pH 7.1, 20 mM NaCl, 4.5 mM MgCl2, 20 μg/ml BSA, 5 mM DTT, 0.4 units/μl Ribolock RNase Inhibitor (Fermentas), at 20°C. PRORP3 was preincubated in processing buffer (without RNase Inhibitor) for 5 min at 20°C. RNA substrates were preincubated in processing buffer (without DTT) for 5 min at 55°C and for 25 min at 20°C. The reactions were started by combining enzyme and substrate solutions (final enzyme concentration varying from 1 nM to 10 μM; final substrate concentration <0.5 nM). Aliquots of the reactions were taken at different time points and stopped by addition of an equal volume of stop/loading buffer [7 M urea, 200 mM sucrose, 10 mM EDTA, 0.02% bromophenol blue; or composed as described previously (15)]. RNAs were separated by 10, 12, 15 or 20% denaturing (7 M urea) PAGE. After phosphorimaging, substrates and 5′-cleavage products were quantified with either ImageQuant TL (GE Healthcare) or AIDA (raytest) image analysis software. First-order rate constants of cleavage (kobs) were calculated by nonlinear regression analysis fitting the data to the equation for a single exponential: fcleaved = fendpoint × (1 − e−(kobs) × t), where fcleaved = fraction of substrate cleaved, t = time, fendpoint = maximum cleavable substrate (Prism, GraphPad Software; or Grafit, Erithacus Software). To determine the maximal rate constant (kreact) and the enzyme concentration at which the half-maximal rate constant is achieved (KM(sto)), kobs values for at least 5 different enzyme concentrations (distributed below and above KM(sto)) from at least three replicate experiments each were plotted against the enzyme concentration, and kreact and KM(sto) calculated by nonlinear regression analysis, fitting the data to a ‘Michaelis-Menten-like’ enzyme kinetics model: kobs = kreact × [PRORP3]/(KM(sto) + [PRORP3]) (see also Supplementary Figure S1); result tables list the best-fit values ± curve-fit standard error (Prism, GraphPad Software).
Processing reactions with pre-tRNAGly variants carrying a dephosphorylated 1- or 2-nt leader were performed with 10 nM of unlabeled substrate and 80 nM PRORP3. Aliquots of the reactions were taken and immediately frozen in liquid nitrogen. The RNA was extracted, subjected to 5′-end labeling with 32P, and analyzed by 20% denaturing PAGE (Supplementary Figure S2A). The labeled, uncleaved substrate was quantified and first-order rate constants of cleavage (kobs) were derived as described above (Supplementary Figure S2B).
Bioinformatics
Chloroplastida/Viridiplantae-PRORP sequences were retrieved from UniProtKB (www.uniprot.org) by BLAST and aligned using Clustal Omega (43); incomplete sequences missing one of the three core domains of PRORP (12,30) were excluded. The nucleotide-specifying residues 1, 4 and 34 (44,45; numbering according to the Pfam PPR model) of the PPR motifs of the three A. thaliana PRORPs were determined and a sequence logo was created from the alignment using WebLogo (46).
The 598 nuclear tRNA genes of A. thaliana were retrieved from the PlantRNA database (47) and analyzed with Microsoft Excel. Sequence logos were generated with WebLogo (46).
RESULTS
To investigate substrate recognition and cleavage-site selection by PRORP we studied the processing of substrate variants derived from the precursor of T. thermophilus tRNAGly, a class I tRNA of canonical sequence and structure (Figure 1A). The helical arms of this tRNA are mostly composed of G–C base pairs and its structure therefore appears particularly stable and predictable when parts of the pre-tRNA molecule are altered. The pre-tRNAGly model substrate has been used in numerous studies with a wide variety of RNase P enzymes (11,15,38,48–52).
Figure 1.

Structure of Thermus thermophilus pre-tRNAGly, derived deletion variants and minimal substrates. (A) Classical cloverleaf representation of pre-tRNAGly. The structural domains are color-coded: magenta, aminoacyl acceptor stem; blue, D domain; red, anticodon domain; gold, variable domain; green, TΨC domain. The positions of selected nucleotides are numbered according to convention (70). The canonical RNase P cleavage site is between nucleotides −1 and 1. (B) Predicted secondary structures of pre-tRNAGly variants without anticodon (Ac) or D domain, or composed of the aminoacyl acceptor stem (Aa) and TΨC domain (T) only, some with a bulge (b) of variable length inserted; the sequence of all 5′ leaders (not shown) is identical to wild-type pre-tRNAGly shown in (A).
The three PRORPs of A. thaliana are the currently best-characterized single-subunit protein-only RNase P enzymes (9,10,13,15,30,33,53); PRORP1 functions in mitochondria and chloroplasts, PRORP2 and PRORP3 redundantly serve as nuclear RNase P. As the purification of PRORP3 turned out to be most straightforward and resulting preparations consistently superior in terms of specific activity, purity and yield, we decided to use PRORP3 throughout this study. We employed recombinant PRORP3 purified to apparent homogeneity.
Enzymatic reactions were carried out in a low-salt buffer with 4.5 mM Mg2+. A pH of 7.1 and a temperature of 20°C, close to the optimal growth temperature of A. thaliana (23°C), were chosen to slow down catalysis for better handling of manual kinetics. Processing of the pre-tRNA substrate variants was analyzed under single-turnover conditions (trace amounts of substrate, excess of enzyme; see Supplementary Figure S1 for representative plots), thus product release could be excluded as the rate-limiting step in the reaction kinetics. The determined kinetic parameters kreact (the maximal rate constant) and KM(sto) (the enzyme concentration at which the rate constant is half-maximal) are equivalent to kcat and KM in classical Michaelis-Menten kinetics where substrate is in excess of enzyme; likewise, the two kinetic parameters are informative with respect to the efficiency of the chemical/cleavage step and enzyme-substrate affinity.
The study was conducted in parallel in two laboratories, and extensive efforts were made to standardize all experimental procedures (including the use of the same enzyme preparations). Almost identical kreact of 1.67 ± 0.03 and 1.72 ± 0.04 min−1 were determined for the wild-type pre-tRNAGly substrate by both groups (Supplementary Figure S1A). However, for the KM(sto) we obtained slightly divergent measurements of 4.8 ± 0.4 and 1.5 ± 0.2 nM, an issue that we could not resolve. As related substrate variants were anyway analyzed in parallel, the kinetic parameters are always given in comparison to the wild-type substrate analyzed by the same laboratory.
5′ and 3′ extensions: the pre-tRNA leader and trailer
Interactions with the extensions of the tRNA structure are crucial for substrate recognition and cleavage-site selection by bacterial RNase P: while the protein subunit contacts the pre-tRNA leader, the RNA subunit interacts with the conserved DCCA motif at the tRNA's 3′-end (18–22,54,55). The 3′-terminal CCA is not naturally found in eukaryal pre-tRNAs, and it was in fact proposed to act as an antideterminant for A. thaliana PRORP1 (33). To examine the role of 5′ and 3′ extensions in pre-tRNA processing by PRORP3, we produced variants of the tRNAGly model substrate with a leader sequentially shortened from 14 nt to 1, or with a trailer either modified to consist of the (mature) CCA sequence only, entirely deleted (discriminator U73 only), or extended to 40 nt. Shortening of the leader down to 2 nt or deletion/extension of the trailer had no effect, and all these variants were processed with the same efficiency as the wild-type pre-tRNA substrate (Table 1). The processing kinetics of a precursor with a 1-nt leader showed no significant change in KM(sto), but a 10-fold reduction in kreact. To clarify whether this reduction in cleavage efficiency was due to the additional negative charge of the 5′-terminal phosphate moved into the proximity of the cleavage site, we determined the cleavage rate (kobs) of substrates with a 5′-hydroxyl end. However, shortening the leader length of pre-tRNAGly variants without a 5′ phosphate from 2 nt to 1 still resulted in a drop of the cleavage rate (Supplementary Figure S2; note: the specific experimental approach required for this type of substrate only allowed an approximation of kobs under presumably enzyme-saturated conditions). Taken together our results demonstrate that neither leader nor trailer sequences, nor a 3′-terminal CCA contribute to or interfere with pre-tRNA binding by PRORP3. A minimum leader length of 2 nt nevertheless appears to be required for efficient catalysis.
Table 1. The role of 5′ and 3′ extensions: pre-tRNA leader and trailer length variations.
| kreact (min−1) | KM(sto) (nM) | |
|---|---|---|
| wild-typea | 1.67 ± 0.03 | 4.8 ± 0.4 |
| 7-nt leader | 1.7 ± 0.1 | 3.1 ± 0.7 |
| 4-nt leader | 1.7 ± 0.1 | 3.4 ± 0.7 |
| 2-nt leaderb | 1.6 ± 0.1 | 3.4 ± 0.8 |
| 1-nt leader | 0.17 ± 0.02 | 5.4 ± 2.2 |
| (mature) CCA | 1.6 ± 0.1 | 4.9 ± 1.0 |
| no trailerc | 1.5 ± 0.1 | 4.6 ± 0.7 |
| 40-nt trailer | 1.5 ± 0.1 | 5.3 ± 1.1 |
Single-turnover kinetic constants of PRORP3 for the processing of pre-tRNAGly with leader and trailer sequences of different length (best-fit values ± standard error from the fitting of at least three replicate experiments each).
aWild-type pre-tRNAGly has a leader of 14 nt and a trailer of 6 nt (including the CCA sequence; see Figure 1A).
bThe sequence of the leader is CC and thereby differs from the wild-type leader at position −2.
cThe aminoacyl acceptor stem of this pre-tRNAGly variant is extended by the discriminator nucleotide only at its 3′ end.
Nucleotide identities at the cleavage site
Nucleotides at the cleavage site play an important role for pre-tRNA binding, cleavage-site recognition and catalysis by bacterial RNase P (see ref. 17 and refs. therein). A guanosine is the most frequent nucleotide immediately downstream of an RNase P cleavage site, at the 5′ end of tRNAs, irrespective of their organismal origin (56). Changing the G1–C72 base pair to the rare U1–A72 negatively affects binding and catalysis by the bacterial RNA enzyme (57,58). The same change introduced into our model substrate, however, had no substantial effect on cleavage by PRORP3 (Table 2).
Table 2. The effect of varying base identity at the cleavage site.
| kreact (min−1) | KM(sto) (nM) | kreact/KM(sto) (min−1 nM−1) | |
|---|---|---|---|
| wild-typea | 1.67 ± 0.03 | 4.8 ± 0.4 | 0.35 |
| U1–A72 | 2.2 ± 0.1 | 5.3 ± 0.9 | 0.41 |
| U−1 | 2.9 ± 0.1 | 8.1 ± 1.4 | 0.36 |
| G−1, A73b | 2.3 ± 0.1 | 6.5 ± 1.0 | 0.35 |
| A−1, A73b | 5.1 ± 0.2 | 7.8 ± 1.4 | 0.66 |
| A73 | 1.67 ± 0.04 | 4.5 ± 0.6 | 0.37 |
Single-turnover kinetic constants of PRORP3 for the processing of pre-tRNAGly variants with different identity of nucleotides −1 and 1 (best-fit values ± standard error from the fitting of at least three replicate experiments each).
aIn the wild-type pre-tRNAGly the following nucleotides are found at the cleavage site: C−1, a G1–C72 base pair and U73.
bThe identity of U73 was changed to A73 to prevent base pairing with nucleotide −1.
The nucleotide at −1 (N−1), immediately upstream of the cleavage site, interacts with bacterial RNase P RNA and a U−1 appears preferred and conserved in bacteria and archaea (59). We systematically tested a possible role of N−1 by substituting the C−1 of pre-tRNAGly with U, G or A (Table 2); for the variants with a purine at −1, we changed the discriminator identity from U73 to A73, to hinder base pairing with N−1. The substitution of C−1 with U−1 or G−1 had a slightly positive effect on catalysis (kreact) and a minor negative effect on KM(sto). Only A−1 resulted in an overall twofold increased processing efficiency (kreact/KM(sto)). Altering the identity of the discriminator base alone did not affect the kinetics of cleavage. None of these base changes next to the cleavage site resulted in any form of miscleavage (partial or complete shift to adjacent phosphodiester bonds). We conclude that nucleotide identities at the cleavage site do not per se affect cleavage-site selection or enzyme-substrate interaction, but an A upstream of the cleavage site accelerates catalysis 3-fold.
Structural domains of the tRNA, minimal substrates
Bacterial, archaeal and the eukaryal nuclear RNP form of RNase P appear to primarily contact the arm of the L-shaped tRNA structure that corresponds to the T domain stacked on the aminoacyl acceptor stem, and model substrates without D and anticodon domains are efficiently cleaved by these enzymes (23,28,29,39,60). We analyzed the processing of a set of pre-tRNAGly variants with deleted anticodon and/or D domains (Figure 1B) to determine the minimal structural requirements of a PRORP substrate. Deletion of the anticodon domain (ΔAc) had no substantial effect on processing efficiency, deletion of the D domain (ΔD), however, negatively affected binding and cleavage (Table 3). A minimized substrate, composed of the acceptor stem and T stem-loop in uninterrupted continuity (AaT), was also cleaved at the canonical site, although with extremely low efficiency. Introducing a bulge at the 5′ junction of the acceptor and T stems into this substrate (Aab1T, Aab4T, Aab9T) profoundly improved cleavage (kreact), regardless whether 1, 4 or 9 nt were inserted. However, while the short bulges (Aab1T, Aab4T) did not substantially improve KM(sto), a bulge of 9 nt (Aab9T) resulted in a substrate that was even slightly more efficiently processed than the pre-tRNAGly variant lacking the D domain (ΔD). Acceptor stem and T domain thus appear to constitute the minimal structural elements of a PRORP3 substrate, with a little ‘kink’ between acceptor and T stems required for efficient catalysis, and a bigger RNA bulge for efficient binding.
Table 3. The effect of tRNA-domain deletions and cleavage of minimal substrates.
| kreact (min−1) | KM(sto) (nM) | |
|---|---|---|
| wild-type | 1.72 ± 0.04 | 1.5 ± 0.2 |
| ΔAc | 1.48 ± 0.04 | 1.7 ± 0.3 |
| ΔD | 0.36 ± 0.02 | 86 ± 16 |
| AaT | 0.066 ± 0.002 | 1839 ± 168 |
| Aab1T | 0.33 ± 0.01 | 1685 ± 218 |
| Aab4T | 0.26 ± 0.01 | 1151 ± 125 |
| Aab9T | 0.42 ± 0.01 | 40 ± 6 |
Single-turnover kinetic constants of PRORP3 for the processing of pre-tRNAGly variants without anticodon (Ac) or D domain, or composed of the aminoacyl acceptor stem (Aa) and TΨC domain (T) only (see Figure 1B for secondary structures; best-fit values ± standard error from the fitting of at least three replicate experiments each).
Conserved sequences of the D and TΨC loops, and PRORP's PPR domain
The N-terminal domain of PRORP comprises five in-tandem PPR motifs (30); PPRs are RNA-binding modules proposed to recognize a nucleobase via two or three amino acid residues that determine their specificity according to a code (44,45). This suggests some sequence specificity to be involved in PRORP's interaction with pre-tRNAs. Although tRNAs show little overall primary sequence conservation, the D and particularly the TΨC loop contain a number of conserved and semiconserved nucleotides (61). Based on the cleavage of mitochondrial pre-tRNACys model-substrate variants and nuclease footprinting, A. thaliana PRORP1 was proposed to contact nucleotides C56, R57 and G18 (33). However, as conserved nucleotides in the D and TΨC loops are involved in the tertiary interactions that stabilize the L-shaped tRNA structure (Figure 2A), processing defects of mutant substrates could be due to misfolding rather than to the disruption of local interactions with the enzyme. Therefore, we first altered the TΨC-loop sequence in the context of the minimized substrate Aab9T (Figure 2B) to exclude effects resulting from global structural changes rather than local base-specific interactions with PRORP. With the exception of the conservative A57→G57, all base substitutions had a negative effect on processing (Table 4). C56→U56, A57→C57 and A57→U57 most strongly affected KM(sto) (more than 3-fold increased), whereas the cleavage rate (kreact) was decreased 7- to 47-fold for A58→G58, A57→C57, U54→C54 and A57→U57. Purine to pyrimidine substitutions at position 57 thus most severely affected the processing of the model substrate and U55→C55 the least. However, at least U54→C54 and A58→G58 also change the intra-loop geometry by disrupting the conserved U54·A58 reverse Hoogsteen base pair that closes the T loop structure (62). Moreover, a substrate with the TΨC loop sequence CCUUUUA that combined the different base exchanges did not show a cumulative processing deficiency, suggesting that in fact alterations of the conserved T loop structure rather than disruptions of single, base-specific interactions underlie most (if not all) mutations’ negative effect on processing.
Figure 2.

Tertiary-structure position of conserved nucleotides whose identity was altered. Two-dimensional representations of the (L-shaped) tertiary structures (domains color-coded as in Figure 1) with tertiary interactions indicated by broken lines; solid gray lines indicate phosphodiester bonds of adjacent nucleotides that are displayed distant in the two-dimensional representation of the tertiary structure. (A) Structure of pre-tRNAGly and position of conserved nucleotides in the TΨC or D loop that were altered in the substrate variants; base substitutions with nucleotide number indicated. (B) Predicted structure of the minimal substrate Aab9T and position of conserved nucleotides in the TΨC loop that were altered in the substrate variants.
Table 4. The effect of altering the sequence of the D and TΨC loops.
| Aab9T | pre-tRNAGly | |||
|---|---|---|---|---|
| kreact (min−1) | KM(sto) (nM) | kreact (min−1) | KM(sto) (nM) | |
| wild-type | 0.42 ± 0.01 | 40 ± 6 | 1.67 ± 0.03 | 4.8 ± 0.4 |
| G18→A18 | 1.87 ± 0.07 | 22 ± 3 | ||
| G19→A19, C56→U56a | 1.78 ± 0.06 | 7.7 ± 1.2 | ||
| U54→C54 | 0.017 ± 0.001 | 88 ± 12 | ||
| U55→C55 | 0.159 ± 0.005 | 80 ± 9 | ||
| C56→U56 | 0.13 ± 0.01 | 208 ± 27 | 1.81 ± 0.05 | 6.4 ± 0.9 |
| A57→G57 | 0.47 ± 0.02 | 34 ± 6 | ||
| A57→C57 | 0.018 ± 0.001 | 462 ± 63 | 1.56 ± 0.05 | 6.7 ± 0.9 |
| A57→U57 | 0.009 ± 0.001 | 144 ± 52 | ||
| A58→G58 | 0.060 ± 0.001 | 108 ± 7 | ||
| TCCUUUUAb | 0.017 ± 0.001 | 72 ± 18 | ||
Single-turnover kinetic constants of PRORP3 for the processing of minimal-substrate (Aab9T) and pre-tRNAGly variants with different identity of conserved nucleotides in the D and/or TΨC loops (see Figure 2A and B for position within the secondary structures; best-fit values ± standard error from the fitting of at least three replicate experiments each).
aThe identity of C56 was varied in order to maintain tertiary Watson–Crick base pairing with nucleotide 19.
bThe sequence of the wild-type TΨC loop of Aab9T and pre-tRNAGly is UUCAAGU.
Two conserved guanosines in the D loop are further potential candidates for a specific interaction with PRORP enzymes. G18 is involved in a tertiary interaction with Ψ55 and its substitution was reported to strongly impair pre-tRNA processing by A. thaliana PRORP1; in contrast, substitution of the neighboring G19 that base pairs with C56, did not substantially affect processing (33). We tested substitutions of the two, as well as TΨC loop substitutions C56→U56 and A57→C57, in the context of the full-length tRNA structure (Figure 2A). The nucleotide substitutions in the TΨC loop and G19→A19 in the D loop had no significant effect on processing in this structural context (Table 4); only G18→A18 resulted in a moderately increased KM(sto), possibly the result of a mild tertiary structural disturbance. This suggests either the presence of sufficient compensatory recognition elements in pre-tRNAGly compared with the minimized substrate Aab9T, which result in a masking of the structural alterations of the T loop, or that single mutations in pre-tRNAGly do not disrupt the T loop structure, embedded and redundantly stabilized in the genuine tRNA tertiary fold, to the same extent as in the context of Aab9T.
To reveal potential base-specific interaction(s) between the tRNA and the PPR domain of PRORP3 from the enzyme rather than the substrate side, we identified the conjectural nucleotide-specifying amino acids 1, 4 and 34 (44,45) of the five PPRs and analyzed their evolutionary conservation among plant PRORPs (Figure 3; the PPR domain was not found to be sufficiently conserved at the primary sequence level to allow an automated alignment of plant to non-plant PRORP sequences). PPR1 and 5 show no appreciable conservation of their nucleotide-specifying candidate residues. In PPR2 and even more so in PPR4, the crucial residue at PPR-position 4 is not sufficiently conserved to be responsible for an important specific interaction with a conserved tRNA nucleotide. For PPR3, however, the invariable threonine as a nucleotide-specifying residue in position 4 combined with the conserved basic amino acid at 34 would be predicted to confer purine specificity to this PPR (44,45). The purine at tRNA position 57 is highly conserved, is not involved in an intra-T loop interaction, and its substitution by a pyrimidine most severely affected processing of our minimal substrate Aab9T (Table 4). To analyze whether PPR3 ‘recognizes’ R57 (in accordance with the proposed rules), we ‘re-programmed’ this PPR for the specific recognition of all four base identities at position 57. The different PRORP3 variants carrying mutations of residues T113 and/or R145 were analyzed with position-57 variants of pre-tRNAGly and the minimized substrate Aab9T. The single-amino acid substitutions had either no or only a minor effect on the processing of wild-type pre-tRNAGly, but with the exception of T113S, they were all less active on the (wild-type) minimal substrate Aab9T, even the supposedly A-specifying variant R145N (Table 5); the double mutants were either severely impaired or not active at all. The activity of none of the PRORP3 variants could be rescued by employing a substrate with the supposedly matching base identity at position 57; all active enzyme variants cleaved substrates with A57 or G57 more efficiently than those with C57 or U57. Thus, while both, PPR3 and R57, appear to play an important role in the interaction of PRORP with pre-tRNA model substrates, the two either do not directly interact with each other, or their interaction is not governed by the rules proposed to generally specify PPR-RNA interactions.
Figure 3.
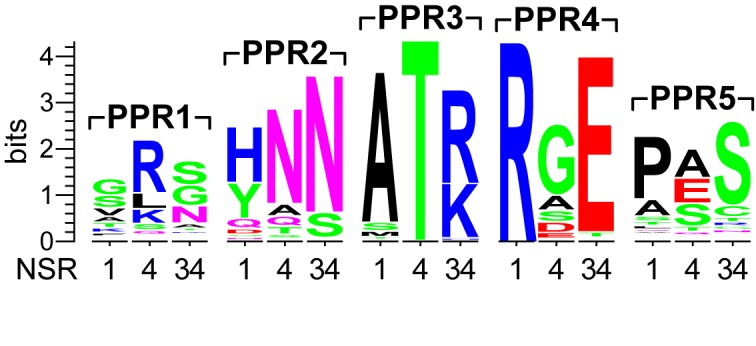
Nucleotide-specifying residues of plant-PRORP PPR motifs. Conjectural nucleotide-specifying residues of the five PPR motifs found in PRORPs of Chloroplastida/Viridiplantae. From the structure of Arabidopsis thaliana PRORP1 (30) the nucleotide-specifying residues (NSR) 1, 4 and 34 (44,45; numbering according to the Pfam PPR model) of the PPR motifs of the three A. thaliana PRORPs were derived and a sequence logo was generated from the alignment of 175 PRORP sequences.
Table 5. Substrate cleavage by PRORP3 variants with a ‘re-programmed’ PPR3.
| Predicted target nucleotide(s) | pre-tRNAGly | Aab9T | |||||
|---|---|---|---|---|---|---|---|
| wild-type | A57→C57 | wild-type | A57→G57 | A57→U57 | A57→C57 | ||
| kobs (min−1)a | kobs (min−1)b | ||||||
| wild-type | A, G | 1.8 ± 0.1 | 1.4 ± 0.1 | 0.40 ± 0.02c | 0.41 ± 0.02c | 0.0071 ± 0.0002c | 0.011 ± 0.001 |
| T113S | A, G, U | 2.0 ± 0.1 | 1.6 ± 0.1 | 0.34 ± 0.01 | 0.37 ± 0.02 | 0.004 ± 0.001 | 0.005 ± 0.001 |
| R145N | A | 2.0 ± 0.1 | 1.33 ± 0.05 | 0.06 ± 0.01 | 0.09 ± 0.03 | n.d. | n.d. |
| R145D | G | 1.15 ± 0.02 | 0.25 ± 0.02 | n.d. | n.d. | n.d. | n.d. |
| T113N | C, U | 1.56 ± 0.04 | 1.1 ± 0.1 | 0.017 ± 0.002 | 0.015 ± 0.001 | n.d. | n.d. |
| T113N-R145N | C | 0.38 ± 0.02 | 0.104 ± 0.004 | n.d. | n.d. | n.d. | n.d. |
| T113N-R145D | U | 0.047 ± 0.002 | 0.010 ± 0.002 | n.d. | n.d. | n.d. | n.d. |
Single-turnover rate constants (kobs) of wild-type PRORP3 and its ‘re-programmed’ variants for the processing of pre-tRNAGly and minimal-substrate (Aab9T) variants with different identity of position 57 (best-fit values ± standard error from the fitting of at least three replicate experiments each; n.d., not determinable because of no or too slow product formation). PRORP3 variants (first column) are identified by the substitution of the presumptive nucleotide-specifying amino acid residues T113 and R145 of PPR3. Target nucleotide(s) (second column) were predicted using the general recognition rules proposed for PPRs (44,45).
Enzyme concentrations were chosen to be saturating based on the KM(sto) for the wild-type enzyme-substrate combination (compare Table 3).
akobs of pre-tRNAGly variants determined at 500 nM PRORP3.
bkobs of Aab9T variants determined at 1 μM PRORP3, unless otherwise specified (c).
ckobs determined at 800 nM PRORP3.
Cleavage-site selection by PRORP
Neither sequence variations at the cleavage site, nor the length of the 5′ and 3′ extensions, nor deletion of the D and/or anticodon domain affected the position of cleavage by PRORP3; all the pre-tRNA and minimal substrate variants were precisely and exclusively processed at the canonical RNase P cleavage site. The conserved structure of the aminoacyl acceptor and T domain thus appear to contain all the information for cleavage-site selection/positioning. To investigate whether simply the combined lengths of the two stacked helices and the T loop determine the cleavage site, we first varied the length of the acceptor stem of pre-tRNAGly. Deletion (Aa−2bp) or insertion (Aa+2bp) of 2 bp into the aminoacyl acceptor stem shifted the main cleavage site from the end of the acceptor helix by 2 nt in both cases, either into the single-stranded leader or into the double-stranded stem, resulting in the release of a shorter and longer leader, respectively (Figure 4A and B); the cleavage site's distance from the tRNA body thereby remained constant, although we observed some additional minor cleavage 1 nt closer to the end of the acceptor helix in both cases. Alteration of the acceptor stem length was also associated with a drop in cleavage efficiency, primarily manifesting as an increase in KM(sto) (Table 6). Varying the length of the stems and the size of the TΨC loop of the Aab9T minimal substrate was detrimental to cleavage efficiency (Table 6), but nevertheless largely confirmed the importance of the overall dimensions of acceptor and T domain (Figure 4C and D): (i) extending the length of the acceptor stem (Aa+2bp) again partially shifted the cleavage site by 2 nt to within the acceptor stem; (ii) extending the T stem in a similar way rendered the substrate (T+2bp) nearly uncleavable, but cleavage could be rescued by proportionally shortening the acceptor stem (Aa−2bp T+2bp), which also redirected cleavage to the end of the now only 5 bp long acceptor stem; (iii) reducing the size of the conserved 7-nt TΨC loop to 4 nt (T4loop) finally resulted in a partial relocation of the cleavage site to 1 or 2 nt upstream. Taken together these results indicate a pre-tRNA-binding mode that positions the site to be cleaved indeed primarily as a consequence of its distance relative to the T-loop, i.e. the combined length of the aminoacyl acceptor stem and the T domain is an important determinant for docking the correct phosphodiester bond into the active site of PRORP.
Figure 4.

Cleavage-site selection by PRORP3. Variants of pre-tRNAGly and the minimal-substrate Aab9T with varying length and structure of the aminoacyl acceptor stem and/or T domain, or pre-tRNAGly variants with base-paired nucleotides −1 and 73 were subjected to cleavage by PRORP3, and the cleavage site determined by mapping the length of the released 5′ leader. Processing assays with the different substrates were incubated with different concentrations of PRORP3 (for pre-tRNAGly wild-type, Aa−2bp, Aa+2bp and wild-type Aab9T: 200 nM; for Aab9T Aa+2bp, T+2bp, T+2bp Aa−2bp and T4loop: 1 μM; for pre-tRNAGly Aa+4bp, Aa+m3GC and Aa+m3AU: 500 nM; for pre-tRNAHis, pre-tRNAGly G−1–C73, U−1–A73 and Aa+3AU: 200 nM) or with 10 nM Bacillus subtilis RNase P (pre-tRNAHis) until sufficient product had formed. RNAs were separated by 12% (B, D and F) or 15% (G and I) denaturing PAGE (only the part showing the 5′-cleavage products is shown). Alkaline hydrolysis ladders were generated from wild-type pre-tRNAGly (due to 2′,3′-cyclic-phosphate ends their migration is slightly offset relative to the RNase P cleavage products with 3′-hydroxyl ends). (A) Pre-tRNAGly variants with an acceptor stem extended or shortened by inserting or deleting 2 bp; the other tRNA domains (not shown) are identical to the wild-type (see Figure 1A). The indicated reference positions 1 and −1 are for the purpose of this study defined as the seventh and eighth nucleotide in the 5′ strand of the aminoacyl acceptor stem counting (in 3′-to-5′ direction) from the base of the stem. Arrows of different size indicate major and minor cleavage sites. (B) Cleavage-site determination of the variants shown in (A). (C) Acceptor stem and T domain variants of the minimal substrate Aab9T. (D) Cleavage-site determination of the variants shown in (C). (E) Pre-tRNAGly variants with an acceptor stem extended by 4, by a mismatch and 3 G–C, or by a mismatch and 3 A–U bp. (F) Cleavage-site determination of the variants shown in (E). (G) Cleavage of E. coli pre-tRNAHis by B. subtilis RNase P and PRORP3. (H) Pre-tRNAGly variants with base-paired nucleotides −1 and 73, or with an acceptor stem extended by 3 A–U bp. (I) Cleavage-site determination of the variants shown in (H).
Table 6. The effects of varying the length and structure of the aminoacyl acceptor stem and/or T domain, and of introducing base pairing upstream of the cleavage site.
| pre-tRNAGly | Aab9T | |||||
|---|---|---|---|---|---|---|
| cleavage site(s)a | kreact (min−1) | KM(sto) (nM) | cleavage site(s)a | kreact (min−1) | KM(sto) (nM) | |
| wild-type | 1(100%) | 1.72 ± 0.04 | 1.5 ± 0.2 | 1(100%) | 0.42 ± 0.01 | 40 ± 6 |
| Aa+2bp | 1(81%), −1(19%) | 0.32 ± 0.03 | 36 ± 12 | 1(51%), −2(35%), −3(14%) | 0.0015 ± 0.0005b | n.d. |
| Aa−2bp | 1(76%), 2(24%) | 0.8 ± 0.1 | 33 ± 8 | |||
| T+2bp | 1(52%), −1(30%), 3(18%) | n.d. | n.d. | |||
| Aa−2bp T+2bp | 3(85%), 2(15%) | 0.0126 ± 0.0004 | 134 ± 21 | |||
| T4loop | 1(43%),−1(43%), −2(14%) | 0.019 ± 0.001b | n.d. | |||
| Aa+4bp | −1, −2(33%), −3, −4, −5, −6 | 0.023 ± 0.002c | n.d. | |||
| Aa+m3GC | 1(76%), −1(16%), −2(8%) | 0.022 ± 0.001 | 11 ± 3 | |||
| Aa+m3AU | 1(87%), −1(13%) | 0.44 ± 0.02 | 1.5 ± 0.9 | |||
| G−1–C73 | −1(100%) | 1.46 ± 0.02 | 2.0 ± 0.1 | |||
| U−1–A73 | 1(80%), −1(20%) | 3.6 ± 0.1 | 2.1 ± 0.3 | |||
| Aa+3AU | 1(88%), −1(12%) | 1.5 ± 0.1 | 2.3 ± 0.5 | |||
Major cleavage site(s), their relative cleavage rate and inclusive single-turnover kinetic constants of PRORP3 for the processing of pre-tRNAGly and minimal-substrate (Aab9T) variants with varied lengths and structure of the acceptor stem and/or T domain, or pre-tRNAGly variants with base-paired nucleotides −1 and 73 (see Figure 4 for the details of the structural changes and cleavage-site mapping; reference positions 1 and −1 are for the purpose of this study defined as the seventh and eighth nucleotide in the 5′ strand of the aminoacyl acceptor stem counting (in 3′-to-5′ direction) from the base of the stem; kinetic constants (best-fit values ± standard error from the fitting of at least three replicate experiments each) were derived from rate constants determined for cleavage at all sites; n.d., not determined.
aThe position of the nucleotide downstream of the cleaved phosphodiester bond is specified, i.e. cleavage occurred 5′ of the indicated nucleotide, and the relative product quantity is indicated in superscript parenthesis.
bRate constant (kobs) determined with 1 μM PRORP3 (best-fit values ± standard error from the fitting of at least three replicate experiments each).
cRate constant (kobs) determined with 500 nM PRORP3 (best-fit value ± standard error from the fitting of at least three replicate experiments).
The partial ‘out of register’ cleavage of pre-tRNAGly variants with shortened or extended acceptor stem (Aa−2bp and Aa+2bp) 1 nt closer to the end of the acceptor helix, together with the reduced affinity of PRORP3 for such substrates, suggests an additional preference of the enzyme to cleave at the end of a helix, implying that the nucleotide immediately upstream of the cleavage site must be ‘flexible’ and, if base-paired, has to be melted to fit into the active site. Consistently, extension of the acceptor stem by 4 bp (Aa+4bp) strongly impaired cleavage (Table 6) and led to weak cleavages all along its distal extension (Figure 4E and F). Introducing a mismatch into the extended stem (Aa+m3GC) largely restored cleavage-site positioning and KM(sto), but not the cleavage rate (kreact). However, replacing the 3 G–C bp upstream of the mismatch with A–U (Aa+m3AU) bp largely restored the kinetic parameters, indicating that melting of any upstream base pairing is required for efficient substrate docking, site selection and cleavage.
‘Miscleavage’ of pre-tRNAGly variants also suggests some flexibility in the ‘measuring mechanism’ of PRORP, i.e. a substrate-binding mode capable of productively positioning also slightly longer substrates. This is reminiscent of a distinct feature of bacterial RNase P that selectively cleaves pre-tRNAHis 1 nt upstream of the canonical site, i.e. between −2 and−1, to release a tRNAHis with an 8-bp acceptor stem. Among the features redirecting the cleavage, a G−1 base-paired to the discriminator C73 of bacterial tRNAHis is the most critical. Whereas a similar extension of the acceptor stem, or a T domain of non-canonical size, are not found in any nucleus-encoded tRNA species in plants, their mitochondria and chloroplasts have bacterial-type tRNAHis genes with a G−1–C73 base pair (47). PRORP1, the mitochondrial/chloroplastic RNase P of A. thaliana, was in fact reported to cleave a potato mitochondrial pre-tRNAHis at both, the canonical site (−1/1), as well as upstream (−2/−1) like the bacterial enzyme (63). When we tested E. coli pre-tRNAHis with PRORP3, however, cleavage occurred exclusively at the ‘bacterial cleavage site’ (Figure 4G). To further investigate this observation, we modified our model substrate T. thermophilus pre-tRNAGly to introduce G–C or U–A base pairing between position −1 and the discriminator at 73 (Figure 4H; the latter found in 16% of the nuclear tRNA genes in A. thaliana). Whereas the U−1–A73 variant was efficiently cleaved at the canonical site (with only minor miscleavage 1 nt upstream), the pre-tRNAHis look-alike (G−1–C73) was exclusively cleaved at the upstream position, resulting in a tRNAGly with an 8-bp acceptor stem (Figure 4H and I; Table 6). Surprisingly, the G−1–C73 substrate was cleaved with wild-type-like efficiency too, revealing an unexpected flexibility in the binding and active-site docking of substrates with 12- or 13-bp acceptor-T stem modules.
The sequences flanking tRNA structures in primary transcripts are not random. The 5′ leaders of A. thaliana pre-tRNAs are rich in adenosines, particularly close to the cleavage site, and the 3′ end is generally followed either directly or after a few nucleotides by a run of uridines derived from the polymerase III terminator (Supplementary Figure S3). This bias in the leader and trailer sequences of A. thaliana pre-tRNAs results in a high frequency of short A/U-rich acceptor stem extensions (Supplementary Table S6). To clarify whether PRORP3 can cope with such extensions, we finally tested a pre-tRNAGly with an acceptor stem extension of 3 A–U bp (Aa+3AU; extensions of this kind are found in 3.5% of A. thaliana tRNA genes). In line with its role as a nuclear RNase P in A. thaliana, PRORP3 efficiently cleaved this pre-tRNA variant, with only little miscleavage at the upstream phosphodiester bond (Figure 4H and I; Table 6).
DISCUSSION
At first glance, different forms of RNase P appear to recognize and bind their pre-tRNA substrates in a similar way, by docking to the stacked aminoacyl acceptor and T domains of the tRNA structure, and modeling of PRORP–tRNA complexes has been used to emphasize this similarity to the RNA-based, bacterial RNase P (33,64,65). Our close-up, however, shows that the elements that make an RNA a substrate for RNase P and direct cleavage to the correct site differ distinctly between the bacterial RNP and the eukaryal protein-only enzyme. In short, A. thaliana PRORP3, in comparison with bacterial RNase P, requires more tRNA structure, particularly a largely intact ‘acceptor-T domain’, but little to no determinants at the cleavage site, nor interactions with the extensions of the tRNA (5′ leader, 3′ trailer or CCA). Cleavage-site positioning by PRORP3 also mostly depends on the dimensions of the ‘acceptor-T domain’ combined with the constraint that the leader has to be unpaired; again, determinants at the cleavage site used by the bacterial enzyme are not involved. Intriguingly, PRORP3 nevertheless displays the same ‘flexibility’ to shift cleavage to 1 nt upstream on specific substrates. Overall, in substrate recognition and cleavage-site selection PRORP appears more similar to the complex eukaryal nuclear RNP enzymes, although those have not been studied in sufficient detail to finally assess the full extent of this mechanistic convergence.
The lack of any apparent interaction with the 5′ or 3′ extension of the tRNA is a major distinction between PRORP and bacterial RNase P. Pre-tRNA interaction with PRORP3, as reflected by the KM(sto), remained unchanged down to a leader of 1 nt, or upon removal or extension of the trailer. Active-site contacts involving nucleotide −2 nevertheless appear to be important for catalysis, as its removal resulted in a sudden 10-fold drop of the cleavage rate. In contrast, the protein subunit of the bacterial RNP contacts the leader up to −7, enhancing the enzyme's affinity for the pre-tRNA and assisting in positioning the cleavage site (19–22). Naturally, the crucial DCCA interaction of the bacterial enzyme (18,54) is not found in a eukaryal RNase P, and we could not confirm the previously suggested adverse effect of a 3′-terminal CCA on PRORP (33). The pre-tRNA 3′ extension simply appears irrelevant for a productive interaction with PRORP3.
The role of the pre-tRNA 5′ leader and 3′ trailer was previously studied for the yeast nuclear RNase P RNP (66). A pre-tRNA with a leader shortened to 2 nt showed no decrease in cleavage efficiency (a variant with a 1-nt leader was not investigated). Intriguingly however, competition experiments indicated that a single-stranded 3′ trailer supported binding to the RNP enzyme, and the authors concluded that yeast nuclear RNase P exposes two binding sites involved in substrate recognition, one that interacts with the tRNA body and one that binds the 3′ trailer. Thus, while the nuclear RNP and PRORP appear similar in their lack of contacts to the pre-tRNA 5′ extension, they apparently differ with respect to interactions with 3′-extensions.
In contrast to bacterial RNase P (57,58), PRORP3 is not affected in its cleavage activity by the identity of the 5′-terminal nucleotide/base pair of the tRNA domain (at least in canonically sized tRNAs), despite the same strong bias in favor of a terminal G1–C72 base pair in the tRNAs of A. thaliana (Supplementary Figure S3 and Table S6). An equally obvious bias toward an A immediately upstream of the cleavage site (−1; Supplementary Figure S3) is reflected by stimulated catalysis of A−1 substrates compared to substrates with more rare U−1, G−1 or C−1 identities. In bacteria, however, the base at −1 does not affect catalysis, but interaction with the enzyme, and a U−1 is preferred and most frequent (59). The role of both presumptive determinants at the cleavage site has not been investigated for the eukaryal nuclear RNP enzymes.
Not unexpectedly, RNase P enzymes that make little use of determinants close to the cleavage site, require more ‘information’ from the tRNA structure itself to reliably recognize their substrates. The ‘acceptor-T domain’ presented as an uninterrupted continuous stem-loop structure (AaT), a minimal substrate efficiently cleaved by the bacterial enzyme (23), is a poor substrate for PRORP. As in the case of the RNA-based nuclear RNase P (28,29), insertion of a bulge of at least one, but preferably more nucleotides, at the site where the two helices are normally interrupted by their connections to the remaining domains, was required to convert the ‘acceptor-T domain’ mimic into a decent substrate for PRORP. While a 1-nt bulge was sufficient to rescue catalysis, a longer inset was required to lower the KM(sto), suggesting that a small kink or flexibility is required for positioning the minimal substrate in the active site, yet more conformational flexibility mediates additional interactions for efficient substrate docking. Still such substrates perform worse than a largely intact pre-tRNA and only the anticodon domain seems entirely dispensable.
While PRORP does not recognize specific bases at the cleavage site, the conserved presence of a domain comprised of consecutive PPRs suggests the involvement of some sequence specificity in substrate recognition. PPR motifs are proposed to recognize nucleobases according to a code specified by two or three of their amino acid residues, with the tandem arrangement of several motifs resulting in the reading of a stretch of RNA sequence (44,45). In tRNAs, the only consecutive stretch of conserved primary sequence is found in the TΨC loop, a prototypic T-loop structure and anchor for tertiary interactions with the D loop (61,62). A recent study indeed proposed PPR2 and PPR3 to be involved in a specific interaction with the TΨC loop's C56 and R57, respectively (34). Our data, however, do not confirm the predicted type of base-specific interaction with the PPR motifs of PRORP. (i) The nucleotide-specifying residues of four of the five PPRs of PRORP are not sufficiently conserved to be consistent with the predicted ‘code’ when more plant sequences are taken into account. (ii) Any of our attempts to ‘re-program’ the single conserved PPR motif (PPR3), predicted to specify purines by the rules of the code and proposed to recognize R57 within the TΨC loop (34), failed; R57 variants, particularly in the context of the minimal substrate Aab9T, were strictly preferred over Y57 substrates by all the PPR3 mutants, even by those that had been ‘re-programmed’ to recognize pyrimidines. Mutagenesis of PRORP3's PPR3 nevertheless demonstrated its importance for substrate recognition, all the variants, except for T113S, had impaired cleavage activity, at least when acting on the minimal substrate. Also R57 and the other T-loop bases appear to play a role in substrate recognition, again particularly in the context of the minimal substrate lacking D domain determinants. However, rather than the primary sequence only, it appears to also be the conserved structure of the loop that is relevant for a productive interaction with PRORP. Finally, testing T-loop variants in the context of the complete tRNA structure also showed that, similar to bacterial RNase P, not all the different substrate-recognition determinants are strictly required, but deficiencies in one can be fully compensated by the (optimal) manifestation of others.
Substrate recognition has to tie in with cleavage-site selection, and the two processes are naturally not separable. For bacterial RNase P, the features ‘recognized’ at the cleavage site also largely determine the position of cleavage (17). Again, the eukaryal enzymes, both RNA- and protein-based, are different and display some mechanistic convergence. Here, the overall dimensions (length) of the ‘acceptor-T domain’ determines primarily where the cleavage is positioned; metaphorically called ‘measuring’ (27,28), this obviously reflects a defined distance between the T loop interaction site and the active site of the enzyme. Another important feature to allow cleavage at the site determined in this way is the single-strandedness or flexibility of the 5′ leader, particularly of the nucleotide immediately upstream of the cleavage site (−1). Any base pairing beyond the cleavage site needs to be melted to allow recognition/binding and cleavage. This requirement appears to be shared not only between PRORP and the nuclear RNP enzyme (66,67), but also with bacterial RNase P, although in this latter case single-strandedness of the leader is required to enable the crucial interactions with the RNA and protein subunit of this enzyme (17).
Another similarity to bacterial RNase P, yet again with a different mechanistic basis, is the ability of PRORP3 to efficiently redirect its cleavage site to 1 nt upstream on substrates where a G−1 is base-paired to a discriminator C73, a characteristic feature of bacterial tRNAHis. G−1–C73 pairing makes N−1 and N73 inaccessible for specific contacts within the active site of bacterial RNase P RNA, which are required to expose the canonical cleavage site and to properly position the Mg2+ ions involved in the catalytic process (17). In the case of PRORP3, this phenomenon appears to indicate the flexibility of the enzyme's substrate-binding pocket to efficiently accommodate substrates of slightly differing length. Substrates with a stable G−1–C73 base pair are thereby cleaved upstream of −1 resulting in an 8-bp acceptor stem, whereas for pre-tRNAs with a weak, e.g. U−1–A73, melting of this extra base pair and canonical cleavage appears to be the preferred pathway. The biological significance of this property of PRORP3 is elusive, as a G−1–C73 is not found in eukaryal nuclear tRNAsHis, where the discriminator generally is A73 (56). At least in A. thaliana, strong base pairing to the discriminator appears to be under negative selection and is extremely rare, and two consecutive G–C base pairs upstream of the canonical cleavage site are not found at all in nuclear pre-tRNAs (Supplementary Table S6). Weak base pairing between −1 and the discriminator 73, as well as further potential weak acceptor stem extensions, are nevertheless relatively frequent, yet they do not represent a major obstacle to canonical cleavage. Miscleaved pre-tRNAs, extended by an extra nucleotide at their 5′ end, could theoretically undergo another cycle of processing to finally remove the short extension, though this would occur at a 10-fold lower rate only (see Table 1).
Remarkably, the ‘naked’ human nuclear RNase P RNA was reported to cleave a bacterial pre-tRNAHis at the −1/−2 upstream site too (68), yet no eukaryal RNP holoenzyme form has been tested on such a substrate so far. The Xenopus RNP enzyme, however, was reported to remove the 5′-terminal base-paired G of an acceptor stem-extended yeast tRNAPhe (27). So in the end it remains unclear whether or not the eukaryal nuclear RNP enzymes also shift their cleavage site on G−1–C73 acceptor stem-extended substrates.
Given that in most organisms a single RNase P enzyme per tRNA-expressing compartment is responsible for the processing of all the encoded tRNAs, it seems reasonable to assume that most if not all single-subunit PRORPs employ mechanisms of substrate recognition and cleavage-site selection that are very similar to those identified here for A. thaliana PRORP3. This implies that the enzymatic mechanisms established here for (variants of) the pre-tRNAGly model substrate in principle also apply to all other pre-tRNAs (of canonical structure). Yet in part different mechanisms likely govern the action of human/animal mitochondrial PRORPs (69). Not only do they make use of two additional protein subunits to cleave mitochondrial pre-tRNAs (8,32), but they also act on a much wider structural diversity of pre-tRNAs (61).
In conclusion, the general mechanisms employed by different forms of RNase P either differ substantially (PRORP versus bacterial RNase P), or display substantial commonalities (PRORP versus nuclear RNP RNase P). Thus with respect to substrate recognition, mechanistic similarity does not simply reflect molecular composition or evolutionary relationship. And although the extent of mechanistic convergence between the highly complex eukaryal nuclear RNP and the seemingly simple single-subunit PRORPs cannot yet be fully assessed, some of the functional implications of the herewith described mechanistic differences and similarities, respectively, have already been indicated by genetic transplantation/exchange experiments. Whereas in yeast an RNase P RNP-for-PRORP swap remained without any apparent functional or fitness consequences (13), complementation of a bacterial RNase P deficiency by PRORP revealed clear-cut limitations, particularly in the processing of certain non-tRNA substrates by the latter (M. Gößringer et al., in preparation). Thus, in the end, the RNase P family remains a unique model system for studies of enzyme evolution or enzyme-substrate coevolution.
Supplementary Material
Acknowledgments
We acknowledge the excellent technical assistance of Aurélie Buzet and Dominik Helmecke, as well as the help of Lukas Kerul and Julia Roka with some of the experiments. We thank Elisa Vilardo for discussions and critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Austrian Science Fund (FWF) [I299, W1207 to W.R.]; German Research Foundation (DFG) [HA 1672/17-1 to R.K.H.]. Funding for open access charge: FWF [I299].
Conflict of interest statement. None declared.
REFERENCES
- 1.Lai L.B., Vioque A., Kirsebom L.A., Gopalan V. Unexpected diversity of RNase P, an ancient tRNA processing enzyme: challenges and prospects. FEBS Lett. 2010;584:287–296. doi: 10.1016/j.febslet.2009.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu F., Altman S., editors. Ribonuclease P. NY: Springer; 2010. [Google Scholar]
- 3.Hartmann R.K., Gößringer M., Späth B., Fischer S., Marchfelder A. The making of tRNAs and more - RNase P and tRNase Z. Prog. Nucleic Acid Res. Mol. Biol. 2009;85:319–368. doi: 10.1016/S0079-6603(08)00808-8. [DOI] [PubMed] [Google Scholar]
- 4.Ellis J.C., Brown J.W. The evolution of RNase P and its RNA. In: Liu F, Altman S, editors. Ribonuclease P. NY: Springer; 2010. pp. 17–40. [Google Scholar]
- 5.Esakova O., Krasilnikov A.S. Of proteins and RNA: the RNase P/MRP family. RNA. 2010;16:1725–1747. doi: 10.1261/rna.2214510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lai L.B., Cho I.-M., Chen W.-Y., Gopalan V. Archaeal RNase P: a mosaic of its bacterial and eukaryal relatives. In: Liu F, Altman S, editors. Ribonuclease P. NY: Springer; 2010. pp. 153–172. [Google Scholar]
- 7.Walker S.C., Marvin M.C., Engelke D. Eukaryote RNase P and RNase MRP. In: Liu F, Altman S, editors. Ribonuclease P. NY: Springer; 2010. pp. 173–202. [Google Scholar]
- 8.Holzmann J., Frank P., Löffler E., Bennett K.L., Gerner C., Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008;135:462–474. doi: 10.1016/j.cell.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Gobert A., Gutmann B., Taschner A., Gößringer M., Holzmann J., Hartmann R.K., Rossmanith W., Giegé P. A single Arabidopsis organellar protein has RNase P activity. Nat. Struct. Mol. Biol. 2010;17:740–744. doi: 10.1038/nsmb.1812. [DOI] [PubMed] [Google Scholar]
- 10.Gutmann B., Gobert A., Giegé P. PRORP proteins support RNase P activity in both organelles and the nucleus in Arabidopsis. Genes Dev. 2012;26:1022–1027. doi: 10.1101/gad.189514.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taschner A., Weber C., Buzet A., Hartmann R.K., Hartig A., Rossmanith W. Nuclear RNase P of Trypanosoma brucei: a single protein in place of the multi-component RNA-protein complex. Cell Rep. 2012;2:19–25. doi: 10.1016/j.celrep.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lechner M., Rossmanith W., Hartmann R.K., Thölken C., Gutmann B., Giegé P., Gobert A. Distribution of ribonucleoprotein and protein-only RNase P in eukarya. Mol. Biol. Evol. 2015;32:3186–3193. doi: 10.1093/molbev/msv187. [DOI] [PubMed] [Google Scholar]
- 13.Weber C., Hartig A., Hartmann R.K., Rossmanith W. Playing RNase P evolution: swapping the RNA catalyst for a protein reveals functional uniformity of highly divergent enzyme forms. PLoS Genet. 2014;10:e1004506. doi: 10.1371/journal.pgen.1004506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas B.C., Li X., Gegenheimer P. Chloroplast ribonuclease P does not utilize the ribozyme-type pre-tRNA cleavage mechanism. RNA. 2000;6:545–553. doi: 10.1017/s1355838200991465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pavlova L.V., Gößringer M., Weber C., Buzet A., Rossmanith W., Hartmann R.K. tRNA processing by protein-only versus RNA-based RNase P: kinetic analysis reveals mechanistic differences. Chembiochem. 2012;13:2270–2276. doi: 10.1002/cbic.201200434. [DOI] [PubMed] [Google Scholar]
- 16.Howard M.J., Klemm B.P., Fierke C.A. Mechanistic studies reveal similar catalytic strategies for phosphodiester bond hydrolysis by protein-only and RNA-dependent ribonuclease P. J. Biol. Chem. 2015;290:13454–13464. doi: 10.1074/jbc.M115.644831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirsebom L.A. RNase P RNA mediated cleavage: substrate recognition and catalysis. Biochimie. 2007;89:1183–1194. doi: 10.1016/j.biochi.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Reiter N.J., Osterman A., Torres-Larios A., Swinger K.K., Pan T., Mondragon A. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature. 2010;468:784–789. doi: 10.1038/nature09516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niranjanakumari S., Stams T., Crary S.M., Christianson D.W., Fierke C.A. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc. Natl. Acad. Sci. U.S.A. 1998;95:15212–15217. doi: 10.1073/pnas.95.26.15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurz J.C., Niranjanakumari S., Fierke C.A. Protein component of Bacillus subtilis RNase P specifically enhances the affinity for precursor-tRNAAsp. Biochemistry. 1998;37:2393–2400. doi: 10.1021/bi972530m. [DOI] [PubMed] [Google Scholar]
- 21.Rueda D., Hsieh J., Day-Storms J.J., Fierke C.A., Walter N.G. The 5′ leader of precursor tRNAAsp bound to the Bacillus subtilis RNase P holoenzyme has an extended conformation. Biochemistry. 2005;44:16130–16139. doi: 10.1021/bi0519093. [DOI] [PubMed] [Google Scholar]
- 22.Koutmou K.S., Zahler N.H., Kurz J.C., Campbell F.E., Harris M.E., Fierke C.A. Protein-precursor tRNA contact leads to sequence-specific recognition of 5′ leaders by bacterial ribonuclease P. J. Mol. Biol. 2010;396:195–208. doi: 10.1016/j.jmb.2009.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McClain W.H., Guerrier-Takada C., Altman S. Model substrates for an RNA enzyme. Science. 1987;238:527–530. doi: 10.1126/science.2443980. [DOI] [PubMed] [Google Scholar]
- 24.Forster A.C., Altman S. External guide sequences for an RNA enzyme. Science. 1990;249:783–786. doi: 10.1126/science.1697102. [DOI] [PubMed] [Google Scholar]
- 25.Hansen A., Pfeiffer T., Zuleeg T., Limmer S., Ciesiolka J., Feltens R., Hartmann R.K. Exploring the minimal substrate requirements for trans-cleavage by RNase P holoenzymes from Escherichia coli and Bacillus subtilis. Mol. Microbiol. 2001;41:131–143. doi: 10.1046/j.1365-2958.2001.02467.x. [DOI] [PubMed] [Google Scholar]
- 26.Brännvall M., Kikovska E., Wu S., Kirsebom L.A. Evidence for induced fit in bacterial RNase P RNA-mediated cleavage. J. Mol. Biol. 2007;372:1149–1164. doi: 10.1016/j.jmb.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 27.Carrara G., Calandra P., Fruscoloni P., Doria M., Tocchini-Valentini G.P. Site selection by Xenopus laevis RNAase P. Cell. 1989;58:37–45. doi: 10.1016/0092-8674(89)90400-5. [DOI] [PubMed] [Google Scholar]
- 28.Yuan Y., Altman S. Substrate recognition by human RNase P: identification of small, model substrates for the enzyme. EMBO J. 1995;14:159–168. doi: 10.1002/j.1460-2075.1995.tb06986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrara G., Calandra P., Fruscoloni P., Tocchini-Valentini G.P. Two helices plus a linker: a small model substrate for eukaryotic RNase P. Proc. Natl. Acad. Sci. U.S.A. 1995;92:2627–2631. doi: 10.1073/pnas.92.7.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howard M.J., Lim W.H., Fierke C.A., Koutmos M. Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc. Natl. Acad. Sci. U.S.A. 2012;109:16149–16154. doi: 10.1073/pnas.1209062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filipovska A., Rackham O. Pentatricopeptide repeats: modular blocks for building RNA-binding proteins. RNA Biol. 2013;10:1426–1432. doi: 10.4161/rna.24769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vilardo E., Nachbagauer C., Buzet A., Taschner A., Holzmann J., Rossmanith W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012;40:11583–11593. doi: 10.1093/nar/gks910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gobert A., Pinker F., Fuchsbauer O., Gutmann B., Boutin R., Roblin P., Sauter C., Giegé P. Structural insights into protein-only RNase P complexed with tRNA. Nat. Commun. 2013;4:1353. doi: 10.1038/ncomms2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imai T., Nakamura T., Maeda T., Nakayama K., Gao X., Nakashima T., Kakuta Y., Kimura M. Pentatricopeptide repeat motifs in the processing enzyme PRORP1 in Arabidopsis thaliana play a crucial role in recognition of nucleotide bases at TΨC loop in precursor tRNAs. Biochem. Biophys. Res. Commun. 2014;450:1541–1546. doi: 10.1016/j.bbrc.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 35.Smith D., Burgin A.B., Haas E.S., Pace N.R. Influence of metal ions on the ribonuclease P reaction. Distinguishing substrate binding from catalysis. J. Biol. Chem. 1992;267:2429–2436. [PubMed] [Google Scholar]
- 36.Niranjanakumari S., Kurz J.C., Fierke C.A. Expression, purification and characterization of the recombinant ribonuclease P protein component from Bacillus subtilis. Nucleic Acids Res. 1998;26:3090–3096. doi: 10.1093/nar/26.13.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busch S., Kirsebom L.A., Notbohm H., Hartmann R.K. Differential role of the intermolecular base-pairs G292-C75 and G293-C74 in the reaction catalyzed by Escherichia coli RNase P RNA. J. Mol. Biol. 2000;299:941–951. doi: 10.1006/jmbi.2000.3789. [DOI] [PubMed] [Google Scholar]
- 38.Li D., Willkomm D.K., Schön A., Hartmann R.K. RNase P of the Cyanophora paradoxa cyanelle: a plastid ribozyme. Biochimie. 2007;89:1528–1538. doi: 10.1016/j.biochi.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 39.Hardt W.-D., Schlegl J., Erdmann V.A., Hartmann R.K. Role of the D arm and the anticodon arm in tRNA recognition by eubacterial and eukaryotic RNase P enzymes. Biochemistry. 1993;32:13046–13053. doi: 10.1021/bi00211a014. [DOI] [PubMed] [Google Scholar]
- 40.Sun L., Campbell F.E., Zahler N.H., Harris M.E. Evidence that substrate-specific effects of C5 protein lead to uniformity in binding and catalysis by RNase P. EMBO J. 2006;25:3998–4007. doi: 10.1038/sj.emboj.7601290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rossmanith W., Bettinger E., Cerni C., Karwan R.M. Expression of mouse RNase MRP RNA in human embryonic kidney 293 cells. Mol. Biol. Rep. 1997;24:221–230. doi: 10.1023/a:1006882704481. [DOI] [PubMed] [Google Scholar]
- 42.Rossmanith W., Tullo A., Potuschak T., Karwan R., Sbisà E. Human mitochondrial tRNA processing. J. Biol. Chem. 1995;270:12885–12891. doi: 10.1074/jbc.270.21.12885. [DOI] [PubMed] [Google Scholar]
- 43.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barkan A., Rojas M., Fujii S., Yap A., Chong Y.S., Bond C.S., Small I. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 2012;8:e1002910. doi: 10.1371/journal.pgen.1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yagi Y., Hayashi S., Kobayashi K., Hirayama T., Nakamura T. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One. 2013;8:e57286. doi: 10.1371/journal.pone.0057286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crooks G.E., Hon G., Chandonia J.-M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cognat V., Pawlak G., Duchêne A.M., Daujat M., Gigant A., Salinas T., Michaud M., Gutmann B., Giegé P., Gobert A., et al. PlantRNA, a database for tRNAs of photosynthetic eukaryotes. Nucleic Acids Res. 2013;41:D273–D279. doi: 10.1093/nar/gks935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schlegl J., Fürste J.P., Bald R., Erdmann V.A., Hartmann R.K. Cleavage efficiencies of model substrates for ribonuclease P from Escherichia coli and Thermus thermophilus. Nucleic Acids Res. 1992;20:5963–5970. doi: 10.1093/nar/20.22.5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warnecke J.M., Held R., Busch S., Hartmann R.K. Role of metal ions in the hydrolysis reaction catalyzed by RNase P RNA from Bacillus subtilis. J. Mol. Biol. 1999;290:433–445. doi: 10.1006/jmbi.1999.2890. [DOI] [PubMed] [Google Scholar]
- 50.Pfeiffer T., Tekos A., Warnecke J.M., Drainas D., Engelke D.R., Séraphin B., Hartmann R.K. Effects of phosphorothioate modifications on precursor tRNA processing by eukaryotic RNase P enzymes. J. Mol. Biol. 2000;298:559–565. doi: 10.1006/jmbi.2000.3655. [DOI] [PubMed] [Google Scholar]
- 51.Marszalkowski M., Willkomm D.K., Hartmann R.K. 5′-end maturation of tRNA in Aquifex aeolicus. Biol. Chem. 2008;389:395–403. doi: 10.1515/BC.2008.042. [DOI] [PubMed] [Google Scholar]
- 52.Li D., Willkomm D.K., Hartmann R.K. Minor changes largely restore catalytic activity of archaeal RNase P RNA from Methanothermobacter thermoautotrophicus. Nucleic Acids Res. 2009;37:231–242. doi: 10.1093/nar/gkn915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou W., Karcher D., Fischer A., Maximova E., Walther D., Bock R. Multiple RNA processing defects and impaired chloroplast function in plants deficient in the organellar protein-only RNase P enzyme. PLoS One. 2015;10:e0120533. doi: 10.1371/journal.pone.0120533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirsebom L.A., Svärd S.G. Base pairing between Escherichia coli RNase P RNA and its substrate. EMBO J. 1994;13:4870–4876. doi: 10.1002/j.1460-2075.1994.tb06814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hardt W.-D., Schlegl J., Erdmann V.A., Hartmann R.K. Kinetics and thermodynamics of the RNase P RNA cleavage reaction: analysis of tRNA 3′-end variants. J. Mol. Biol. 1995;247:161–172. doi: 10.1006/jmbi.1994.0130. [DOI] [PubMed] [Google Scholar]
- 56.Jühling F., Mörl M., Hartmann R.K., Sprinzl M., Stadler P.F., Pütz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kikovska E., Brännvall M., Kirsebom L.A. The exocyclic amine at the RNase P cleavage site contributes to substrate binding and catalysis. J. Mol. Biol. 2006;359:572–584. doi: 10.1016/j.jmb.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 58.Kikovska E., Brännvall M., Kufel J., Kirsebom L.A. Substrate discrimination in RNase P RNA-mediated cleavage: importance of the structural environment of the RNase P cleavage site. Nucleic Acids Res. 2005;33:2012–2021. doi: 10.1093/nar/gki344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zahler N.H., Christian E.L., Harris M.E. Recognition of the 5′ leader of pre-tRNA substrates by the active site of ribonuclease P. RNA. 2003;9:734–745. doi: 10.1261/rna.5220703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sinapah S., Wu S., Chen Y., Pettersson B.M.F., Gopalan V., Kirsebom L.A. Cleavage of model substrates by archaeal RNase P: role of protein cofactors in cleavage-site selection. Nucleic Acids Res. 2011;39:1105–1116. doi: 10.1093/nar/gkq732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giegé R., Jühling F., Pütz J., Stadler P., Sauter C., Florentz C. Structure of transfer RNAs: similarity and variability. WIREs RNA. 2012;3:37–61. doi: 10.1002/wrna.103. [DOI] [PubMed] [Google Scholar]
- 62.Chan C.W., Chetnani B., Mondragón A. Structure and function of the T-loop structural motif in noncoding RNAs. WIREs RNA. 2013;4:507–522. doi: 10.1002/wrna.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Placido A., Sieber F., Gobert A., Gallerani R., Giegé P., Maréchal-Drouard L. Plant mitochondria use two pathways for the biogenesis of tRNAHis. Nucleic Acids Res. 2010;38:7711–7717. doi: 10.1093/nar/gkq646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howard M.J., Liu X., Lim W.H., Klemm B.P., Koutmos M., Engelke D.R., Fierke C.A. RNase P enzymes: divergent scaffolds for a conserved biological reaction. RNA Biol. 2013;10:909–914. doi: 10.4161/rna.24513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pinker F., Bonnard G., Gobert A., Gutmann B., Hammani K., Sauter C., Gegenheimer P.A., Giegé P. PPR proteins shed a new light on RNase P biology. RNA Biol. 2013;10:1457–1468. doi: 10.4161/rna.25273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ziehler W.A., Day J.J., Fierke C.A., Engelke D.R. Effects of 5′ leader and 3′ trailer structures on pre-tRNA processing by nuclear RNase P. Biochemistry. 2000;39:9909–9916. doi: 10.1021/bi000603n. [DOI] [PubMed] [Google Scholar]
- 67.Lee Y., Kindelberger D.W., Lee J.Y., McClennen S., Chamberlain J., Engelke D.R. Nuclear pre-tRNA terminal structure and RNase P recognition. RNA. 1997;3:175–185. [PMC free article] [PubMed] [Google Scholar]
- 68.Kikovska E., Svärd S.G., Kirsebom L.A. Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2062–2067. doi: 10.1073/pnas.0607326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holzmann J., Rossmanith W. tRNA recognition, processing, and disease: hypotheses around an unorthodox type of RNase P in human mitochondria. Mitochondrion. 2009;9:284–288. doi: 10.1016/j.mito.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 70.Sprinzl M., Horn C., Brown M., Ioudovitch A., Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26:148–153. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.