

Abstract
Natural products have had an immense influence on science and have directly led to the introduction of many drugs. Organic chemistry, and its unique ability to tailor natural products through synthesis, provides an extraordinary approach to unlock the full potential of natural products. In this Review, an approach based on natural product derived fragments is presented that can successfully address some of the current challenges in drug discovery. These fragments often display significantly reduced molecular weights, reduced structural complexity, a reduced number of synthetic steps, while retaining or even improving key biological parameters such as potency or selectivity. Examples from various stages of the drug development process up to the clinic are presented. In addition, this process can be leveraged by recent developments such as genome mining, antibody–drug conjugates, and computational approaches. All these concepts have the potential to identify the next generation of drug candidates inspired by natural products.
Keywords: drug discovery, medicinal chemistry, natural products, organic chemistry, organic synthesis
1. Introduction
At the advent of research into natural products, organic chemistry was a crucial tool to establish the chemical structure of compounds. As a result, high‐yielding, versatile, and specific synthetic methods were developed, which led, with the help of innovative strategies, to the production of natural products in quantities that otherwise would be inaccessible from natural sources and finally enabled more‐thorough biological evaluation. Once organic chemists could harness the power of synthesis, they were able to modify natural products with an aim to improve the selectivity, potency, stability, and pharmacokinetics. Structure–activity relationship (SAR) properties were investigated by semisynthesis or fragment exchange of natural products, which led to crucial discoveries.
A prime example in this regard has been the development of statins for the treatment of cardiovascular disease, still the leading cause of death worldwide. Although the first generation compounds introduced into the clinic were either identical or closely related to a natural product (lovastatin), the recognition of the crucial 3,5‐dihydroxypentanoate scaffold and optimization by medicinal chemistry led to much more improved derivatives, which had a dramatic impact on medicine and society (Figure 1).1 More stringent criteria of “drug likeness” following Lipinski's “rule of five” and related ADME/pharmacokinetic criteria (ADME=adsorbtion, distribution, metabolism, excretion), along with the introduction of high‐throughput screening (HTS), led to a phasing out of natural products in the lead generation process in drug discovery in the late 1990s. However, in the past 25 years, this paradigm pendent on combinatorial chemistry for medicinal chemistry research has barely provided sufficient numbers of innovative and effective new drugs in Pharma R&D. Interestingly, half of all the new chemical entities (NCEs) introduced over the last 30 years (540 out of 1073) were based on natural products, which underlines their importance for lead generation.2
Figure 1.
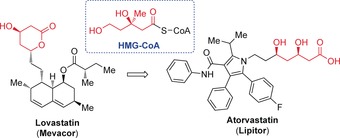
Development of HMG‐CoA reductase inhibitors for the treatment of cardiovascular disease. CoA=coenzyme A, HMG=3‐hydroxy‐3‐methylglutaryl.
Over the last decade, natural products have been experiencing a renaissance in drug discovery; this is based on a number of reasons, in addition to the statistics regarding NCEs presented above. Many studies have revealed striking differences between natural and synthetic compounds, with the former being less hydrophobic, having more stereogenic centers, a larger fraction of sp3‐hybridized carbon atoms, more O and less N, S, and halogen atoms, fewer rotatable bonds, more fused, bridge, or spiro rings, and more solvated hydrogen bond donors and acceptors.3 After all, these compounds produced by nature are a result of millions of years of evolutionary selection and are, hence, biologically prevalidated. All these factors have led to the validation of natural products as prime starting points for drug discovery.
One of the main factors often negatively associated with natural products as lead structures constitutes their limited chemical tractability. The above‐mentioned complex architecture, which sometimes requires lengthy synthetic approaches, often disfavors selection of natural products for further study in a research environment that is heavily under pressure in terms of time and financial constraints. It is the goal of this Review to showcase with several examples from early development to the clinic that the structural complexity of natural products can be reduced by the chemical synthesis of smaller fragments (“less is more”, “reduce to the maximum”), while retaining (or sometimes improving) desired biological parameters such as potency or selectivity. This Review is not aimed at being comprehensive,4 but instead should guide the reader through successful design principles and compounds that are at various stages of the development process, from lead structure identification to the clinic. The ultimate goal is shared or complemented by approaches such as “diverted total synthesis”, “chemical or molecular editing”,5 and “function‐oriented synthesis” (FOS).4a, 6
These approaches all present a unique opportunity of retaining desired effects in minimized, biologically active scaffolds as a consequence of “fragment likeness” with a high degree of three dimensional similarity, yet overcome the limitations of parent natural product structures, such as lack of accessibility or viable synthetic approachability. In addition, these natural product derived fragments could add to the discovery process in the context of fragment‐based drug discovery.7
2. Natural Product Derived Fragments in Drug Discovery
2.1. Anticancer Activity
2.1.1. From Halichondrin B to Eribulin
The polyether macrolide halichondrin B8 was reported in 1986 by Hirata and Uemura (Figure 2).9 It was isolated from the marine sponge Halichondria okadai Kadota, which had been collected off the coast of Japan in the Western Pacific. Later in 1991, Pettit et al. also isolated halichondrin B from an Axinella sp. sponge, collected in Palau.10 This marine macrolide showed potent activity against B16 melanoma cells (IC50=0.093 ng mL−1) and powerful in vivo inhibition of tumor growth (T/C)11 in mice (in up to 375 % of the test group) over the control group and increased the mean survival times for groups with murine B16 melanoma, P388 leukemia, and l1210 leukemia.9b Later studies revealed that halichondrin B acts as a tubulin‐destabilizing agent with a slightly different mechanism of action from other antimitotics, such as the vinca alkaloids.8a, 12 More specifically, it inhibits tubulin polymerization and microtubule assembly, tubulin‐dependent GTP hydrolysis, nucleotide exchange on tubulin, and acts as a noncompetitive inhibitor of the binding of radiolabeled vinblastine to tubulin.8a, 12
Figure 2.
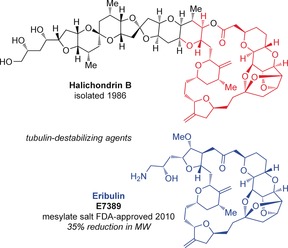
Structural comparison between halichondrin B and eribulin/E7389. FDA=U.S. Food and Drug Administration.
Despite this phenomenal biological activity, the limited supply (less than 2×10−6 wt % yield)9b of the natural product from its marine source and the potential for contamination with other closely related halichondrins or highly toxic metabolites, such as okadaic acid,12 remained serious obstacles that could only seemingly be circumvented by total synthesis. Furthermore, the amount of material needed to support preclinical development and subsequent clinical trials was estimated to be approximately 10 g.8a, 13 If found to be successful, 1–5 kg year−1 would be necessary to meet commercial needs. Although the initial aquaculture with Lissodendoryx sp. seemed promising, the ability of scale‐up to satisfy the commercial demands in a reliable manner could not be achieved.8a, 13
Kishi and co‐workers reported the first total synthesis of halichondrin B, along with norhalichondrin B, in 1992.8a, 14 The route relied on a series of robust Nozaki–Hiyama–Kishi (NHK) reactions, which were used to construct five key bonds in the synthesis.8a, 14a,14d,14e, 15 Further improvements were later developed,16 but the optimized synthetic route contained nearly 120 steps. Despite this, the completion of this landmark total synthesis convinced the U.S. National Cancer Institute (NCI) to move halichondrin B into preclinical development in March 1992.13c The Eisai Research Institute initiated in vitro and in vivo studies of synthetic halichondrin B, along with several analogues provided by the Kishi research group at Harvard University.17 Surprisingly, they found that the C1–C38 macrolide, or the “eastern portion” of the molecule, demonstrated activities within an order of magnitude of the parent compound during an in vivo 3–4 day growth inhibition assay with a DLD‐1 human colon cancer cell line (Figure 2).8a, 18 They found that the macrolactone could be replaced by a nonhydrolyzable ester bioisostere to prevent susceptibility toward nonspecific esterases. Attempts to truncate further while retaining activity were unsuccessful. For example, the 2,6,9‐trioxatricyclo[3.3.2.0]decane was essential for the activity and global conformation of the molecule.9b
Despite the success with total synthesis, the limited supply of halichondrin B was a critical issue in the early 1990s, which was eased, in particular, by the discovery of the potency of simplified analogue eribulin, or E7389 (Figure 2).18a In addition to the synthetic efforts by the Kishi group,19 the synthesis of the C14–C35 fragment (13) of eribulin (Scheme 1) has been optimized for process development on a kilogram scale. This most recent route put forth by Eisai Inc. in 2013 begins with conversion of dihydrofuran (1) into C14–C19 fragment 3, through a tin‐mediated bromoallylation with 2,3‐bromopropene (2; Scheme 2).20 Fragment C20–C26 (5) was synthesized from 1,2‐epoxyhex‐5‐ene (4) in seven steps by a hydrolytic kinetic resolution (HKR) with a Jacobsen catalyst.21 Aldehyde 5 and fragment 3 were then combined in an asymmetric NHK Ni/Cr coupling with chiral ligand 6 a,22 followed by treatment with silica gel to induce formation of the tetrahydrofuran ring. Elaboration of furan 7 through six steps provided the C14–C26 fragment 8. Synthesis of the C27–35 fragment 12 commenced with transformation of readily available d‐glucurono‐3,6‐lactone (9) into C34–C35 diol 10 in eight steps. Reaction of the tetrahydrofuran ring with allyltrimethylsilane, followed by a Horner–Wadsworth–Emmons olefination and a hydroxy‐directed conjugate reduction afforded sulfone 11. Six additional steps provided the desired C27–35 fragment 12 in a final yield of 26 % through an entirely chromatography‐free, 20‐step process.
Scheme 1.
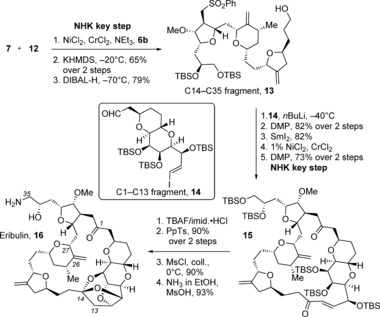
Completion of the synthesis of eribulin (16). Coll.=2,4,6‐collidine or 2,4,6‐trimethylpyridine, DIBAL‐H=diisobutylaluminum hydride, DMP=Dess Martin periodinane, Imid.=imidazole, PpTs=pyridinium para‐toluenesulfonate, TBAF=tetrabutylammonium fluoride, TBS=tert‐butyldimethylsilyl.
Scheme 2.
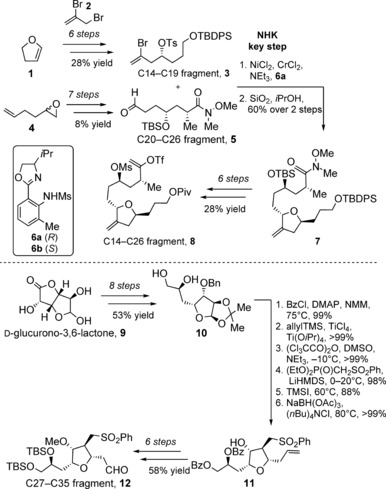
Synthesis of the C14–C26 fragment 8 and C27–C35 fragment 12 of eribulin by Kishi and co‐workers. Bn=benzyl, Bz=benzoyl, DMAP=4‐dimethylaminopyridine, HMDS=hexamethyldisilazide, Ms=mesyl or methanesulfonyl, NMM=N‐methylmorpholine, Piv=pivaloyl, TBDPS=tert‐butyldiphenylsilyl, Tf=trifluloromethanesulfonyl, TMS=trimethylsilyl.
The C14–C26 (8) and C27–C35 (12) subunits were combined in an iterative Nozaki–Hiyama–Kishi reaction and Williamson ether cyclization with ligand 6 b (Scheme 2) in d.r.=20:1 and a 65 % overall yield. A subsequent reductive cleavage afforded the C14–C35 fragment 13 (Scheme 1).23 Sulfone 13 was then combined with the C1–C13 aldehyde 14 from the halichondrin B synthesis, which was assembled in 13 steps from l‐mannonic‐γ‐lactone.16b A SmI2‐mediated desulfonylation, a key Nozaki–Hiyama–Kishi macrocyclization, followed by oxidation were then performed. The trioxatricyclo[3.3.2.0]decane ring system was installed in macrocycle 15 in 90 % yield by treatment with TBAF and then PpTs. A subsequent transformation of the C35 hydroxy group gave the amine of eribulin (16), which was optimized by Kaburagi and Kishi in 2007 to an 84 % overall yield.24 Additionally, the Kishi group reported a special system for the formation of the trioxatricyclo[3.3.2.0]decane ring system, which involved a DOWEX 50WX8‐400 sulfonic acid resin and CaCO3 work‐up of the TBAF‐mediated TBS cleavage reaction, followed by filtration and evaporation.25 Then, passage of the resulting residue through an ion‐exchange resin‐based device completed the formation of the ketal16e in 90 % overall yield.26
In vivo studies revealed that eribulin possesses excellent activities at 0.1–1 mg kg−1 against several chemoresistant human solid tumor xenografts.18a Eribulin displayed superior efficacy over other antimitotics such as even paclitaxel.13c These studies also confirmed that this analogue could induce G2‐M cell‐cycle arrest and disrupt mitotic spindles, and was consistent with the tubulin‐based antimitotic mechanism of the parent halichondrin B. Eisai investigated the safety and effectiveness of eribulin mesylate in a single phase III clinical trial involving 762 women with metastatic breast cancer, during which the median overall survival of patients receiving eribulin mesylate was 13.1 months, whereas this rate was 10.6 months for those receiving only a single agent therapy.27 The U.S. FDA approved eribulin mesylate (Halaven) on November 15, 2010 for patients with metastatic breast cancer, who have received at least two prior chemotherapy treatments for late‐stage disease, including anthracycline‐ and taxane‐based therapies. The European Commission has recently followed suit, issuing their approval on July 3, 2014 under the same conditions as the FDA.28 With this approval in the European Union member states, eribulin mesylate is now approved in more than 50 countries worldwide, including Japan, Singapore, and Switzerland. Eisai is currently investigating eribulin mesylate as a treatment for breast cancer with fewer prior treatments, as well as soft‐tissue sarcoma and non‐small‐cell lung cancer.
Overall, the development of eribulin constitutes a vast improvement over the parent compound as it has a large decrease in complexity, including a 35 % reduction in molecular weight, which requires half the number of synthetic steps to access, and retains the potent antitumor activity, as clearly evident by its use today as a cancer treatment agent worldwide.
2.1.2. From Migrastatin to a Core Analogue
Migrastatin was isolated in 2000 by the Imoto group from a cultured broth of Streptomyces sp. MK929‐43F1 (Figure 3).29 This 14‐membered macrolide with a glutarimide‐terminated side chain inhibited the spontaneous migration of EC17 human esophageal and B16 mouse melanoma cells at a concentration of 10–30 μg mL−1 (IC50=82 μg mL−1).29c
Figure 3.
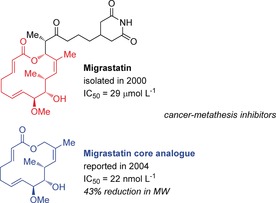
Structural comparison between migrastatin and the migrastatin core analogue. IC50 values are for 4T1 murine breast cancer.
However, the compound demonstrated no appreciable cytotoxicity, inhibition of protein synthesis, or antibiotic/antifungal activities. In a follow‐up study, the ability of migrastatin to inhibit the growth of Ms‐1 human small cell lung carcinoma cells under anchorage‐independent conditions in a dose‐dependent manner (1–100 μg mL−1) implied its influence on integrin signaling, which is also involved in cell migration.29c A year after the absolute configuration was confirmed by X‐ray crystallography,30 the Danishefsky group reported the first synthesis of the macrolide by utilizing a Lewis acid catalyzed diene aldehyde condensation (LACDAC) and ring‐closing metathesis (RCM).31
Follow‐up SAR investigations by the Danishefsky group revealed that, out of eight analogues screened, the migrastatin core surpassed the activity of the parent compound in a Boyden chamber cell migration assay with serum‐induced 4T1 mouse breast tumor cells by three orders of magnitude, with an IC50 value of 22 nmol L−1, versus the 29 μmol L−1 value for migrastatin (Figure 4).32a Analysis of other analogues revealed that reduction of the C2−C3 double bond could be tolerated, while modification of the hydroxy group at C9 or at C13, or at the ketone at C1 could not be tolerated with respect to the potent cell migration suppression activity.31c Eight analogues and migrastatin were also tested for their metabolic stability in mouse plasma and, while the parent compound and compounds containing a glutarimide side chain were inert, the most active compounds including the migrastatin core analogue were hydrolyzed rapidly by esterases. These findings resulted in the macrolactam and macroketone analogues being lead compounds from these studies, with IC50 values of 255 nmol L−1 and 100 nmol L−1, respectively, and with no esterase susceptibility.34a As it was suspected that these analogues influence the angiogenesis process, Danishefsky and co‐workers also investigated endothelial cell migration with human umbilical vein endothelial cells (HUVECs). These results mirrored the previous ones, in that the analogues showed more activity than the parent compound, albeit with some erosion of potency, and they found that the macrolactam, macroketone, and the macrolactone analogues did not have any cytotoxic or antiproliferative effects up to 20 μmol L−1 in an in vitro mouse assay.32a This observation confirmed that inhibition of cell proliferation is not a contributor to the effects observed in the chamber assays. A year later in 2005, Danishefsky and co‐workers reported that several analogues, including the migrastatin macrolactam analogue, inhibit mammary tumor metastasis in mice, which suggests that these compounds interfere with the invasion step by demonstrating that they block Rac activation, lamellipodia formation, and cell migration.34
Figure 4.
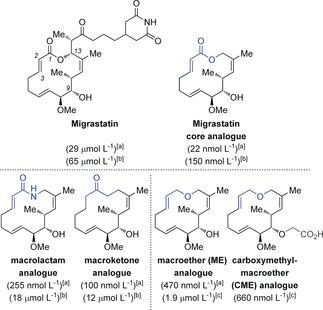
Comparison of the structures and activities of migrastatin analogues from chamber cell migration assays. IC50 values in parantheses are with the following cell lines: [a] 4T1 murine breast cancer,31c, 32 [b] HUVEC human umbilical vein endothelial cells,31c and [c] A549 human lung cancer.33
In a 2010 report, the Danishefsky group reported an even more simple, new macroether (ME) analogue 23 that retains the cancer cell migration inhibition activity of the analogue family (Scheme 3).32b The synthetic sequence towards ME analogue 23 shared the first nine steps of the original route towards the natural product.31a–31c The synthesis began with transformation of tartrate derivative 17 into aldehyde 18 by utilizing a highly diastereoselective divinylzinc addition. The key LACDAC reaction of aldehyde 18 with diene 19 was performed under α‐chelation control in the presence of the Lewis acid TiCl4, and then cyclization with TFA proceeded in 87 % yield to afford dihydropyranone 20. Selective 1,2‐reduction of the ketone and treatment with CSA promoted a Ferrier rearrangement to the corresponding lactol 21, which was then transformed into primary alcohol 22 in three steps. A Williamson etherification reaction and RCM afforded ME analogue 23 in 47 % yield over the five steps. Overall, this analogue had a significant reduction in the complexity of migrastatin, yet retained the activity of the parent compound, and was accessed in 11 fewer synthetic steps.
Scheme 3.
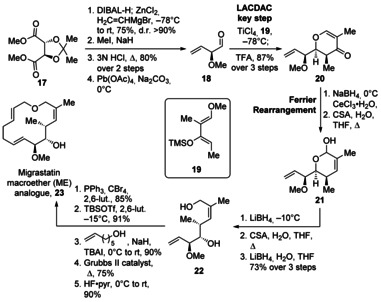
Synthesis of the migrastatin macroether analogue by Danishefsky and co‐workers (ME, 23). CSA=camphorsulfonic acid, DCM=dichlormethane, 2,6‐Lut.=2,6‐lutidine, pyr=pyridine, TBAI=tetrabutylammonium iodide, TFA=trifluoroacetic acid, TBSOTf=tert‐butyldimethylsilyltrifluoromethane sulfonate.
In cell‐migration assays, ME analogue 23 demonstrated migration inhibition against several breast cancer cell lines, including the highly metastatic human LM2, without affecting the viability of the cells.32b This analogue also inhibited tumor invasion and metathesis, and prolonged the overall survival in NOD/SCID mice injected with human breast cancer MDA‐MD‐231 cells in the abdominal mammary fat pad (human breast cancer xenograft model).33 In the course of these studies, the authors also proposed that 23 interferes with fascin‐1‐dependent migratory behavior. This interaction was confirmed by Chen et al. in 2010 with the X‐ray crystal structure of the macroketone analogue in actin‐binding sites of fascin.35 However, there was some debate over this result, as the X‐ray structure of the macroketone analogue contained an E‐ instead of the Z‐configured double bond between C4 and C5, and the C6 stereocenter was inverted to the S configuration.36
In 2011, Danishefsky and co‐workers reported that 23 was considered to be the most promising analogue on the basis of its in vitro and in vivo activities, physical properties, ease of preparation, and biological stability (Figure 4).33 This publication details the promising in vitro and in vivo migration inhibition activity of ME against metastatic small‐cell lung carcinoma (SCLC), and also introduced yet another promising candidate, carboxymethyl‐ME (CME), which was synthesized with an aim to enhance the bioavailability and pharmacostability, and achieved a higher potency with lower toxicity than ME. This CME analogue was also used as evidence against the X‐ray crystallography studies of Chen et al.: since the carboxylic acid functionality reduced cell membrane permeability, and this compound retained its potent metastasis inhibition, the findings implied that the primary target is not an intracellular protein such as Fascin, but a protein target on the cell surface. In 2013, Majchrzak et al. added to this debate by reporting that the migrastatin core analogue showed antibranching activity and interfered with the mechanisms of filopdia assembly, thereby preventing cross‐linking of fascin‐1‐dependent actin filaments in vitro.37 Overexpression of fascin1 in cancer cells has been linked with clinically aggressive tumors, poor prognosis, and shortened disease‐free survival. Further studies on these migrastatin analogues and additional structures are ongoing.
2.1.3. From Anguinomycin C/D to an Anguinomycin Analogue
In 1995, Hayakawa et al. reported the structures of anguinomycins C and D, which were isolated from cultures of Streptomyces sp. (Figure 5).38 These antitumor antibiotics induced cell death in pRB‐inactivated glia cells at picomolar concentrations and, surprisingly, cell‐cycle arrest at only the G1 phase was observed for normal rat glia cells. Retinoblastoma tumor suppressor protein (pRB) is often inactivated during the development of a variety of cancers.
Figure 5.
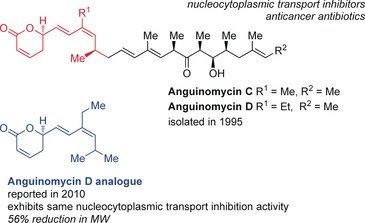
Structural comparison between anguinomycin C/D and the anguinomycin D analogue.
The absolute configuration of anguinomycin C was confirmed by the first total synthesis in 2007 by Gademann and co‐workers. This route had a total of 29 steps, with 18 steps in the longest linear sequence, and an overall yield of 6.7 %.39 Biological studies demonstrated that this natural product is a potent inhibitor of CRM1‐dependent protein export from the cell nucleus, an activity shared by a close structural relative, leptomycin B. It is known that leptomycin B covalently modifies chromosomal region maintenance 1 (CRM1; exportin 1) at the nucleophillic sulphydryl group of Cys 528 by utilizing its α,β‐unsaturated δ‐lactone, thus preventing export of protein cargoes that rely on this cleft by preventing formation of the ternary CRM1/cargo substrate/Ran complex, or the binary complex CRM1/cargo substrate in the absence of Ran.40 More recent computational studies of inhibitors, such as leptomycin B, to CRM1 revealed that the mechanism of inhibition goes beyond simple Michael addition, and is followed by a CRM1‐mediated hydrolysis of the lactone.41 Mutagenesis revealed that at least one of the residues Arg 543, Lys 548, or Cys 579 needs to be present to stabilize the resulting anionic tetrahedral intermediate and lower the energy barrier to drive the reaction. Furthermore, evidence not only supported that hydrolysis must follow conjugate addition, as the corresponding hydroxy acids do not inhibit CRM1, but also that hydrolysis decreases the reversibility of the Michael addition and enables persistent binding of the inhibitors to CRM1. This argument is supported by the fact that reversibility of the conjugate addition should be kinetically controlled and the α‐proton of the hydrolyzed (carboxylate) intermediate is much less acidic than the corresponding proton of the lactone.41
Although dose‐limiting toxicity prevented further development of leptomycin B as an antitumor agent,42 Gademann and co‐workers saw the potential of the anguinomycin core and went on to synthesize anguinoymcin D and analogues for further studies with the aim of investigating if the polyketide side chain mimics the hydrophobic leucine‐rich nuclear export signal of the cargo protein and is necessary for activity.43 The route employed was similar to the synthesis of the anguinomycin C derivative, and began with an asymmetric hetero‐Diels–Alder reaction between diene 24 and aldehyde 25 with catalyst 26 according to Jacobsen et al.,44 which afforded pyran 27 in 86 % yield and with 96 % ee (Scheme 4). The acetal was inverted to the more thermodynamically stable compound and a hydrozirconation of the alkyne with the Schwartz reagent, followed by transmetalation with Zn for a Negishi cross‐coupling utilizing [PdCl2(DPEphos)] with dibromoolefin 28 45 to afford diene 29, but with inversion to the undesired isomer. Inversion to the desired Z olefin was accomplished with another Negishi cross‐coupling reaction to install the ethyl group in 84 % yield and with good selectivity. Silyl ether 30 was then converted into boronate 31. A classic, Evans syn‐aldol strategy was employed to assemble vinyl iodide 33 with the DIOZ (4‐isopropyl‐5,5‐diphenyloxazolidin‐2‐one) auxiliary 32 developed by Hintermann and Seebach.46 The 13 step route, which was executed in 24 % overall yield, employed the DIOZ auxiliary three times and two boron‐mediated aldol reactions. Boronate 31 and vinyl iodide 33 were combined under Suzuki coupling conditions developed by Marshall et al.47 With a particular batch of the Suzuki reaction, there was a minor by‐product, which was taken with desired product 34 through the last three steps and purified by column chromatography to afford not only anguinomycin D (35), but also aldehyde analogue 36 and truncated analogue 37, likely a degradation product of the boronate intermediate from the previous Suzuki coupling.
Scheme 4.
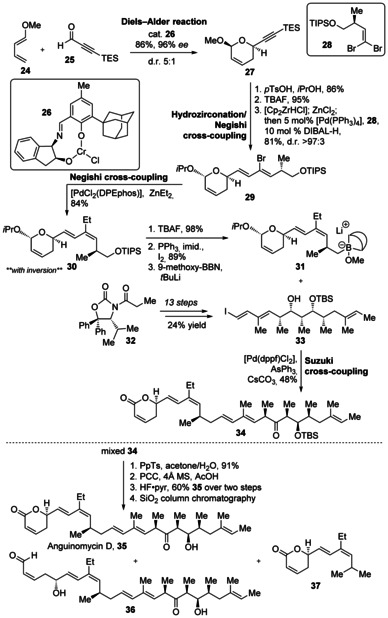
Synthesis of anguinomycin D (35) and analogues by Gademann and co‐workers. Cp=C5H5, DPEphos=bis[(2‐diphenylphosphino)phenyl]ether, dppf=1,1′‐bis(diphenylphosphino)ferrocene, 9‐methoxy‐BBN=9‐methoxy‐9‐borabicyclo[3.3.1]nonane, PCC=pyridinium chlorochromate, TES=triethylsilyl, TIPS=triisopropylsilyl, TsOH=p‐toluenesulfonic acid.
These compounds were then screened for their ability to inhibit CRM1‐dependent protein export by measuring the accumulation of Rio2 protein in the HeLa cell nucleus by indirect immunofluorescence analysis. Anguinomycin C and D (Figure 5) both displayed partial inhibition at 5 nmol L−1 and full inhibition at 10 nmol L−1 while, surprisingly, aldehyde analogue 36 showed full inhibition at 50 nmol L−1 and truncated analogue 37 exhibited an unexpected strong inhibition at 25 nmol L−1. This study demonstrated that the activity could be retained even when the anguinomycin polyketide chain was replaced by a hydrophobic moiety or when completely removed, and reinforced the importance of the conjugate acceptor ability of the α,β‐unsaturated δ‐lactone.41 This concept that the anguinomycin core could be reduced by nearly 60 % in molecular weight and still retain its activity was supported by molecular modeling studies of the (R)‐α,β‐unsaturated δ‐lactone in the NES (nuclear export signals) binding pocket of CRM140c–40e with all‐atom energy minimization (Figure 6).49 Further investigations into this more synthetically accessible, truncated analogue 37 of anguinomycin could provide more powerful or selective nucleocytoplasmic transport inhibitors for cancer treatment.50
Figure 6.
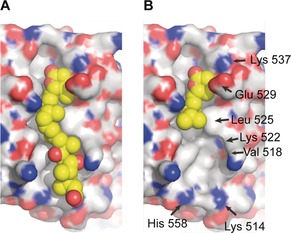
Computational model of A) leptomycin B and B) truncated analogue 37 binding to CRM1 (C: gray, O: red, N: blue; C in inhibitor molecule: yellow).48
2.1.4. From Duocarmycin SA to N‐Boc‐DSA
Duocarmycin SA was isolated from Streptomyces sp. DO113 collected from a soil sample at the Rokkakudo temple in Kyoto, Japan in 1990 by Takahashi and co‐workers, and given its name because it was more stable and demonstrated more potent Gram‐positive antibacterial (minimum inhibition concentrations (MICs): 2.7 nmol L−1 for Staphylococcus aureus and 1.4 nmol L−1 for Bacillus subtilis) and cytotoxic activities (against murine lymphocytic leukemia P388 and murine sarcoma 180) than close relative duocarmycin A (Figure 7).51 The duocarmycins act as sequence‐selective DNA alkylating agents with 3′→5′ binding directionality, specific for AT‐rich regions in the minor groove, where stabilizing van der Waals contacts are maximized.52 Binding of the agent to DNA induces a conformational change, which disrupts the stabilization of the vinylogous amide and activates the cyclopropane ring of the cyclopropapyrroloindole in the left‐hand side of the core for the reversible, stereoelectronically controlled adenine‐N3 addition in an SN2 fashion (shape‐dependent catalysis).52b The bound agent spans 3.5 base pairs and complements the topological curvature and pitch of the minor groove, with the hydrophobic face of the molecule implanted deeply within.53 SAR studies of another close relative, (+)‐CC‐1065, identified the required, but not necessarily optimal, pharmacophore for this family of molecules, thus rendering further studies on the more active and stable duocarmycin SA necessary.
Figure 7.
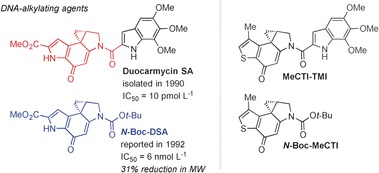
Structural comparison of duocarmycin SA and analogues. IC50 values are for L1210 mouse lymphocytic leukemia. Boc=tert‐butoxycarbonyl, MeCTI=7‐methyl‐1,2,8,8a‐tetrahydrocyclopropa[c]thieno[3,2‐e]indol‐4‐one, TMI=5,6,7‐trimethoxyindole‐2‐carboxylate.
The first total synthesis of duocarmycin SA (46), as well as the synthesis of analogue 44 was reported in 1992 by Boger and Machiya (Scheme 5).54 Two successive regioselective alkylations of substituted diimide quinone 39 (synthesized in 5 steps and 38 % yield)55 were performed at C5 with dimethyl malonate (38), and then at C6 with pyrrolidine enamine of pyruvaldehyde dimethyl acetal 40 56 to afford aniline 41. Acidic conditions removed the acetal group and promoted indole formation, which was followed by cyclization of the resulting diol to complete the dihydropyrroloindole core 42. Eight additional steps, including chiral resolution of the bis‐(R)‐O‐acetylmandelate esters, provided (+)‐dihydropyrroloindole 43. This intermediate could then be directly transformed into (+)‐N‐Boc‐DSA analogue 44 in 85 % yield, or to the natural antipode of (+)‐duocarmycin SA (46) by coupling with the 5,6,7‐trimethoxyindole‐2‐carboxyclic acid salt 45 (made in 3 steps and 74 % yield from 3,4,5‐trimethoxybenzaldehyde)53 in 53 % yield over the two steps. The unnatural (−)‐N‐Boc‐DSA and (−)‐duocarmycin SA enantiomers were also synthesized from (−)‐ent‐43.
Scheme 5.
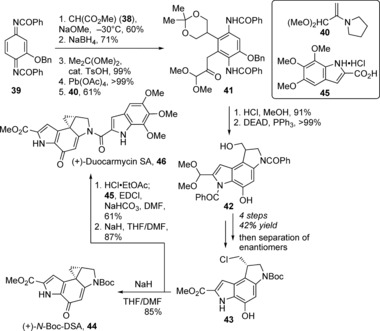
Total synthesis of (+)‐duocarmycin SA (46) and (+)‐N‐Boc‐DSA (44) by Machiya and Boger. DEAD=diethyl azodicarboxylate, EDCI=1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide, DSA=duocarmycin SA.
The natural (+)‐enantiomers were found to have 10 times more potent in vitro cytotoxicity than the corresponding unnatural (−) enantiomers, with IC50 values (L1210 mouse lymphocytic leukemia) of 10 pmol L−1 for (+)‐duocarmycin SA and 6 nmol L−1 for (+)‐N‐Boc‐DSA.54b N‐Boc‐DSA was found to be a substantially less efficient (ca. 104 times) DNA alkylator, with a less strict selectivity profile than the parent natural product (SA), and with both enantiomers showing the same site reactivity profile.52a The latter property was also unusual, but a natural consequence of the diastereomeric relationship of the adducts. N‐Boc‐DSA was found to be the most stable of the duocarmycin analogues, as measured by chemical solvolysis, with complete stability at pH 7 and a half‐life of 177 h at pH 3.54b Gas‐phase heats of reaction for the alkylation of N‐methyladenine with N‐acetylduocarmycin A and N‐acetylduocarmycin SA were calculated by using semiempirical methods (AM1, MNDO). The results reflected the experimental observations that the alkylation of DNA is reversible (unlike with (+)‐CC‐1065), as the model calculations with methyladenine gave a near thermally neutral reaction.54b This underlines the importance of noncovalent interactions in the minor groove (hydrophobic binding/van der Waals contacts), as DNA is nonetheless effectively alkylated by duocarmycins (binding‐driven bonding).
A fundamental parabolic relationship for all the duocarmycins was established between the reactivity of the electrophilic cyclopropane ring and the cytotoxic activity spanning a 104–106 range, where an increased solvolytic stability or decreased reactivity correlates to an increased cytotoxic potency.54b, 57 Highly stable analogues do not efficiently alkylate the DNA, while highly reactive analogues do not reach the biological target. For proof of concept, Boger and co‐workers introduced the rationally designed thiophene analogue N‐Boc‐MeCTI in 2007, which reflected the optimal point of balanced stability and reactivity in the parabolic relationship, being slightly more stable, but 5–6 times less potent than N‐Boc‐DSA (Figure 7).57, 58 In this study, they also reported analogue MeCTI‐TMI, which has an even higher cytotoxic potency than duocarmycin SA (IC50=5 pmol L−1 versus 10 pmol L−1). The retained single‐digit nanomolar potency of N‐Boc‐DSA despite a significant reduction in structural complexity rendered it an excellent example of capturing biological activity with a natural product fragment.
2.1.5. From Dynemicin A to a Dynemicin Analogue
The violet dynemicin A was discovered in 1989 in the fermentation broth of Micromonospora chersina sp. (M956‐1), isolated from a soil sample from Gujarat State, India (Figure 8).59 It demonstrated potent Gram‐positive antibacterial activity with low toxicity, especially against Staphylococcus aureus Smith infection (mouse i.p., PD50=0.13 mg kg−1), and potent in vivo antitumor activity against B16 melanoma, Moser human carcinoma, HCT‐116 human carcinoma, and normal/vincristine‐resistant P388 leukemia (IC50=7–9 nmol L−1), even prolonging the life span of mice with P388 leukemia or B16 melanoma. Structurally, it contains a unique hybrid of an anthraquinone and a 1,5‐diyn‐3‐ene system embedded within a strained 10‐membered ring. The enediyne unit makes it a relative of the esperamicin/calicheamicin family. The mechanism of action involves intercalation of the anthraquinone core of into the minor groove of the DNA helix, followed by attack of the phenyl diradical resulting from the enediyne core, thereby leading to DNA strand scission of the sugar–phosphate backbone three base‐pairs apart.60 The phenyl diradical is proposed to form by two sequential one‐electron reductions of the hydroquinone to the hydroquinonediol, epoxide opening (key to activation), nucleophilic attack (i.e. water) on the p‐quinone methide intermediate or simple tautomerization, and then Bergman cyclization of the strained (Z)‐enediyne to the diaryl radical. Dynemicin A preferentially cuts the 3′ side of purine bases with a preference for guanines, thus differing from its esperamicin/calicheamicin relatives, and demonstrates a preference for double‐stranded (B‐form) and stem regions of single‐stranded DNA.
Figure 8.
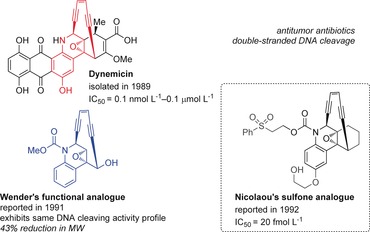
Structural comparison of dynemicin A and analogues. IC50 values are for Molt‐4 T‐cell leukemia.
Along with several pioneering synthetic studies,61 the first total synthesis and absolute configuration of (+)‐dynemicin A was reported by Myers et al. in 1995.62 In total, this route was completed in 26 steps in 0.3 % yield and employed an exo‐selective Diels–Alder reaction as the key step. Several analogues were also made by using the same approach. In the following years, copious synthetic and SAR studies of enediyne analogues of the dynemicin core were performed by the groups of Schreiber,61a,61d Wender,63 Nicolaou,61b, 64 Isobe,65 Myers,62a, 66 Danishefsky,61g, 67 Maier,68 Magnus,69 and others.64a,64d, 70 These studies probed the effects of triggering groups or initiators on the nitrogen atom or aryl ring, which could be activated under basic or photochemical conditions that could be mimicked by intracellular processes. Tethering devices were also investigated to aid target delivery, as were deactivating groups that would modulate enediyne activity, as well as detection devices that would facilitate mechanistic studies.64a One of the most potent dynemicin analogues was the sulfone analogue of Nicolaou et al. with an IC50 value of 20 fm against Molt‐4 T‐cell leukemia, as compared to an IC50 range of 0.1 nmol L−1‐0.1 μmol L−1 for dynemicin A (Figure 8)64a, 64b,64d, 71 However, one of the most simplified analogues was accessed by Wender et al. in just 7 steps (Scheme 6).63a The route began with the reduction of commercially available quinoline carboxyaldehyde (47) and then stepwise incorporation of the enediyne bridge, first by ethynyl Grignard addition to the imine to give alkyne 48, followed by formation of the key epoxide and Sonogashira coupling with enyne 49. Oxidation of alcohol 50 to the aldehyde and treatment with cesium fluoride permitted closure of the strained 10‐membered ring, thereby producing analogue 51. As compared to the parent compound, this greatly simplified analogue exhibited the same activity profile for cleaving plasmid DNA.63b,63d
Scheme 6.
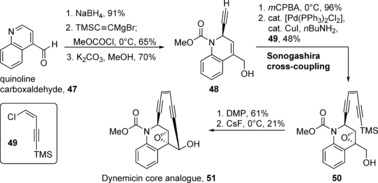
Synthesis of the dynemicin core analogue (51) by Wender and co‐workers. mCPBA=meta‐chloroperbenzoic acid.
2.1.6. From Bryostatin 1 to Picolog
The bryostatin family consists of 20 complex natural products that were isolated from the marine bryozoan Bulgula neritina, starting in the late 1960s.72 They all share a 20‐membered macrolide core with three densely substituted tetrahydropyran rings and a signature exocyclic enoate appended to at least one of these rings. The actual source of these molecules has been narrowed down to a bacterial symbiont of B. neritina, Endobugula sertula, which uses them to protect its larvae from predators.73 The most well‐studied member, bryostatin 1, has a diverse and extremely important range of biological activities stemming from its ability to modulate protein kinase C (PKC; Figure 9).74 Bryostatin 1 exhibits potent anticancer activity by reversing drug resistance, stimulating the immune system, or restoring apopotic function.75 It either has been or is currently being evaluated in 37 clinical trials for various types of cancer.76 It has also been shown to enhance memory and learning in animals, reduce Aβ peptide build‐up in mice, induce synaptic contact formation, and reduce postischemic/hypoxic damage resulting from stroke.77 As such, it has been evaluated in two clinical trials for Alzheimer's disease, with another trial currently ongoing. In addition, bryostatin 1 is also currently being evaluated for HIV/AIDS treatment in yet another clinical trial. A recent study confirmed its ability to activate latent viral reservoirs and downregulate the CD4 cell surface receptor synthesis/expression necessary for viral entry into uninfected cells.78 Other potential therapeutic applications include treatments for cardiovascular disease, stroke, pain, or cognitive dysfunction.
Figure 9.
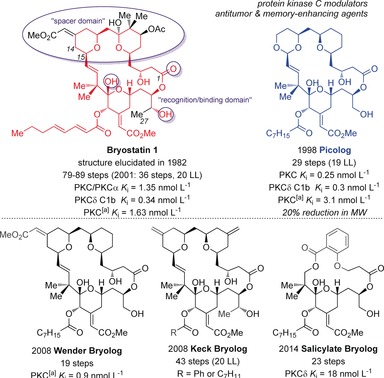
Structural comparison of bryostatin 1 to bryologs (LL=longest linear sequence). [a] Protein kinase C rat brain mix.
Despite the remarkable biological activity of the bryostatins, the clinical supply by good manufacturing practice (GMP) is dwindling because of the natural scarcity (isolation yields of 1×10−3 to 1×10−8 %), the lack of sustainable success with aquaculture or biosynthesis, and the lack of facile or scalable routes to these molecules, notwithstanding the synthetic advances of the last 30 years. Currently, seven total syntheses of bryostatins 7, 2, 3, 16, 1, and 9 have been reported by the groups of Masamune,79 Evans,80 Yamamura,81 Trost,82 Keck,83 Wender,84 and Krische.85 The initial routes were completed in 79–89 steps, with the most recent route towards bryostatin 7 by Krische and co‐workers in 2011 being the most efficient with a total step count of 36 (longest linear sequence: 20 steps).
In an effort to avoid stepwise limitations of total synthesis, and the dose‐limiting toxicities of the parent compound, extensive studies on the synthesis and evaluation of bryostatin analogues (bryologs) have been performed, most comprehensively by the Wender research group since the 1980s. To date, over 100 analogues have been synthesized and screened for various activities, and nearly one‐third exhibit single‐digit nanomolar or even picomolar potencies for PKC binding.6d These studies were primarily driven by the guiding principle that the bryostatin core structure could be divided into the upper “binding” or “recognition domain” (C1–C14) and lower “spacer domain” (C15–C27) according to its interactions with PKC (Figure 9).86 Studies on bryologs have examined the significance of the A ring,87 the B ring,88 the C2089 and C7 side chains,90 as well as de novo structures,91 among others.92 Notable examples of analogues include picolog synthesized by Wender et al. (reported in 2002 with a PKC binding affinity of 0.25 nmol L−1), which was synthesized in 29 steps with a longest linear sequence of 19 steps.93 This represented one of the most potent and promising simplified bryologs, as this was realized in 50 fewer synthetic steps than the syntheses towards the parent structure at that time, and surpassed its activity (bryostatin 1, PKC K i=1.35 nmol L−1).
The synthesis of picolog (61) began with the conversion of diol 52 into aldehyde 53 in four steps so that an asymmetric Keck allylation could set the stereocenter at C23 and an acid‐catalyzed cyclization/dehydration could form lactone 54 (Scheme 7).93b,93c An additional, straightforward seven steps elaborated the pyran ring, including installation of the acyl octanoic acid side chain at C20, to give aldehyde 55. The enal was installed at C15 through a zinc‐mediated addition of (Z)‐bromo‐2‐ethoxyethene, followed by acid‐induced elimination and then Sharpless dihydroxylation, cleavage of the pyran ketal, and protection with a silyl group gave intermediate 56, precursor to the recognition domain. The spacer domain 60 was synthesized in 10 steps from 4‐benzyloxy‐2‐butanone (57) and methyl 5‐chloro‐5‐oxovalerate (58) by employing two asymmetric Noyori hydrogenations and a dienolate addition of ethyl acetoacetate (59) in between. The two domains were combined by utilizing a PyBrop‐mediated esterification and a deprotection/macro‐transacetalization step to afford picolog (61) in a 63 % yield over the two steps.
Scheme 7.
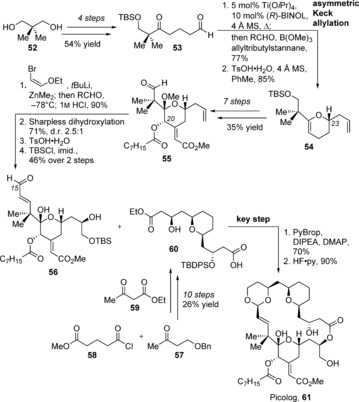
Synthesis of picolog (61) by Wender and co‐workers. DIPEA=diisopropylethylamine, PyBrop=bromotripyrrolidinophosphonium hexafluorophosphate.
As the original synthetic routes towards the parent bryostatin compounds were limited by a Julia olefination/lactonization sequence, the groups of both Wender and Keck sought a new approach and developed a Prins macrocyclization94 as a key step in back‐to‐back reports in 2008 (Figure 9). This novel tactic allowed access to more complex bryologs in fewer steps than the parent compounds and with even higher potency than picolog.95 This same powerful strategy was used by Wender and co‐workers in 2012 to access bryologs that demonstrated 1000‐fold higher potency than prostratin, the leading clinical candidate at the time.78b More recent progress has culminated in the Merle series of bryologs from Keck and co‐workers,96 and the most simplified bryologs to‐date from Wender et al.—the salicylate‐derived class (Figure 9).97 This bryolog shown can be accessed in only 23 steps and displays nanomolar PKC binding affinities. The evolution of the bryolog structures and their broad therapeutic applications has been advancing now for more than 40 years and continues to provide a source of inspiration for discovery.
2.2. Antibiotic Activity
2.2.1. From Caprazamycin B to an Oxazolidine analogue
Caprazamycin B (CPZ‐B) was isolated in 2003 by Igarashi et al. from the culture broth of the actinomycete, Streptomyces sp. MK730‐62F2 (Figure 10).98 This antimicrobial agent demonstrated excellent activity against drug‐susceptible and multidrug‐resistant M. tuberculosis (MDR‐TB or XDR‐TB), without significant toxicity in mice. The caprazamycin family (CPZs) are part of the 6′‐N‐alkyl‐5′‐β‐O‐aminoribosoyl‐glycluridine class of antibiotics, and include liposidomycins (LPMs), which exhibit excellent antimicrobial activities against Gram‐positive bacteria.99 The mode of action of these liponucleosides is inhibition of peptidoglycan synthesis by inhibition of phospho‐MurNAc‐pentapeptide translocase (MraY translocase I). MraY is essential for bacterial cell growth, thus making it a target of interest for the treatment of tuberculosis (TB),100 and even vancomycin‐ and methicillin‐resistant Staphylococcus aureus (VRSA, MRSA).101
Figure 10.
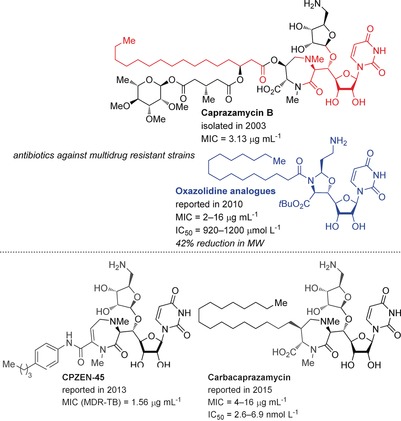
Structural comparison of caprazamycin B and analogues. MIC values are given against MDR‐TB, XDR‐TB, MRSA, and/or VRE strains. IC50 values are given for MraY inhibition.
Although a very promising antibacterial treatment, CPZ‐B has remained synthetically challenging, especially in regard to attaching the unstable fatty acid side chain. This fact, combined with an onerous HPLC separation and a poor water‐solubility profile, has prevented it from becoming a therapeutically viable candidate.102 Only recently has a total synthesis been reported for the less‐active caprazamycin A103 by Takemoto and co‐workers,104 while Watanabe and co‐workers have reported a partial synthesis of the western portion of CPZ‐B.105 Thus, the primary focus over the years has been on analogues of CPZ, developed primarily by Ichikawa, Matsuda, and co‐workers.106 During their studies, the potency of the diketopiperazine and acyclic analogues revealed that the diazepanone moiety was not essential, but contributory to antibacterial activity.106b, 107 Replacing the diazepanone and aminoribose units with an oxazolidine moiety restricted the flexibility sufficiently to lead to an increase in potency of MIC=2–16 μg mL−1 against MRSA and VRE (vancomycin‐resistent enterococci) strains (oxazolidine analogue; Figure 10).107 The most recent studies include CPZ analogues such as CPZEN‐45 with particularly high potency against MDR‐TB strains.102 As the oxazolidine analogues were revealed to be weak MraY inhibitors (IC50=920–1200 μm), Ichikawa et al. recently reported the carbacaprazamycin analogues, with MIC values of 4–16 μg mL−1 and MraY inhibition with IC50 values of 2.6–6.9 nmol L−1.108 However, the morphological changes observed in S. aureus indicated a mode of action that may be completely different from existing peptidoglycan inhibitors.
The most potent oxazolidine analogue 65 can be accessed in just 12 steps, starting with an oxidation, two‐carbon elongation, and an aminohydroxylation109 of uridine 62 to afford amino alcohol 63 (Scheme 8).107 Protecting group manipulations and installation of the oxaziridine ring by condensation with azidoacetaldehyde, followed by acylation afforded N‐palmitoyloxazolidine 64.107 Finally, conversion of the methyl ester into the tert‐butyl ester by using an isourea derivative followed by reduction of the azide to the amine afforded oxazolidine analogue 65. The overall reduction in the complexity of this analogue compared to the parent compound is striking, all the while retaining its potent antibiotic activity against multidrug‐resistant strains.
Scheme 8.
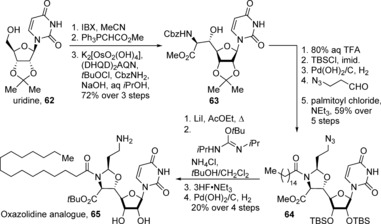
Synthesis of the oxazolidine analogue (65) by Ichikawa, Matsuda, and co‐workers. Cbz=benzyloxycarbonyl, (DHQD)2AQN=1,4‐bis(dihydroquinidinyl)anthraquinone, IBX=2‐iodoxybenzoic acid.
2.3. Neuritogenic Activity
2.3.1. From Militarinone D/Farinosone A to Pyridone Analogues
A plethora of biologically active pyridone alkaloids have been isolated from entomopathogenic Deuteromycetes fungi. Militarinone D was isolated in 2002110 and later identified together with several related molecules in 2003111 by Hamburger and co‐workers through bioassay‐guided fractionation of a mycelial extract of Paecilomyces militaris (RCEF 0095), which displayed pronounced neuritogenic activity in PC‐12 cells (Figure 11). Isolated from a mycelial extract of a related fungus, farinosones A and B structurally resemble the militarinones, retain similar neurite outgrowth activity, and exhibit no appreciable cytotoxicity.112 A number of other pyridone alkaloids have been synthesized and evaluated in terms of their biological activity.111, 113
Figure 11.
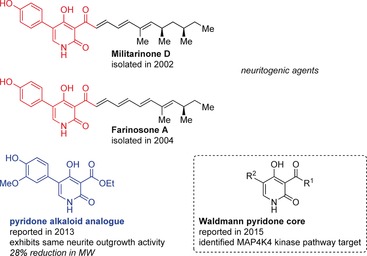
Structural comparison of militarinone D and farinosone A with pyridone alkaloid analogues.
In 2011, Gademann and co‐workers developed a unified approach toward several related pyridone alkaloid natural products.114 They found that these natural products, their enantiomers, and some additional isomers with a Z‐configured double bond in the side chain demonstrated neuritogenic activity in a standardized PC‐12 assay, thereby revealing that neither the length of the side chain nor the absolute configuration were essential to the pharmacophore of this molecule family. In 2013, the Gademann group followed up with further SAR studies and found truncated analogue 69 to have a comparable neurite outgrowth capacity as farinosone A at 20 μm.115 Easily synthesized by utilizing the same general route as for the pyridone natural products, pyridone analogue 69 was accessed in just seven steps from ethyl cyanoacetate (66) and 12 % overall yield by utilizing a Suzuki–Miyaura coupling between the brominated pyridone 67 and pinacol borane 68 (Scheme 9). The neuritogenic effects of pyridone analogue 69 could even be observed down to concentrations of 1 μm. Further investigations mirrored prior biological studies116 and revealed that this potent analogue, as well as militarinone D, influence the MAP kinase pathway, as the neuritogenic effects could be blocked by co‐incubation with the ERK1/2 inhibitor PD98059 (at 5 μm). Recently, Waldmann and co‐workers reported a militarinone‐inspired collection of pyridones, and identified the stress pathway kinase MAP4K4 as the target (Figure 11).117 Both examples show that by reducing the number of carbon atoms by nearly a half, much more simplified, but still potent natural product fragments could be successfully produced.
Scheme 9.
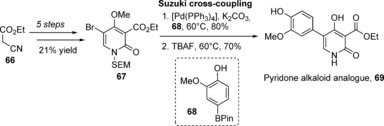
Synthesis of pyridone analogue 69 by Gademann and co‐workers. Pin=Pinacol, SEM=trimethylsilylethoxymethyl.
2.4. Miscellaneous Biological Activity
In addition to those presented within this Review, there are many other notable examples of biologically active, natural product derived fragments (Figure 12). Myers and Herzon demonstrated that avrainvillamide had the same nanomicromolar antiproliferative activity (IC50=50–100 nmol L−1) against several cancer cell lines as its structural dimer, stephacidin B.118 These investigations, including SAR studies of several analogues, established the absolute stereochemical configuration, found that this reversible dimerization occurs through the 3‐alkylidene‐3H‐indole 1‐oxide functional group, attributed the activity of stephacidin B to arise from dissociation to avrainvillamide (as hypothesized), and identified the nucleolar phosphoprotein nucleophosmin as a biological target. The elegant studies of the well‐known antibiotic vancomycin and its aglycon by Crowley and Boger found the simplified structure to have more potent activity against a vancomycin‐resistant strain than the parent compound (μg mL−1 versus mg mL−1 activity).119 These investigations reaffirmed the dimeric binding model120 and addressed the growing vancomycin‐resistance issue. Another example is the truncation of the 14‐mer cyclic peptide hormone somatostatin (SRIF14) to the 8‐mer, octreotide.6c, 121 Now marketed by Novartis as the acetate salt, this drug is 70 times more potent than its parent peptide in vivo, and is used in the treatment of acromegaly as well as symptoms associated with metastatic carcinoid tumors or vasoactive intestinal peptide‐secreting adenomas. This example showed that it is possible to mimic the activity of natural hormones with a truncated, synthetic analogue, and resulted in a clinically successful drug that improves thousands of lives annually. Another clinically relevant example is that of rostafuroxin, which is a simplified version of the toxic cardiac glycoside ouabain.122 This antihypertensive agent is currently in phase IIb clinical trials and has been proven to be especially effective for those with genetic abnormalities of adducin and EO (endogenous ouabain) blood pressure regulation. Gademann and co‐workers demonstrated that the known siderophore aachelin H could be truncated to its chromophore and reapplied as an anchor to create antifouling and antimicrobial surface coatings for potential use in biocompatible medical devices.123 In 1994, Nicolaou et al. designed and synthesized truncated brevetoxin analogue [AFGHIJK], which was able to bind to the voltage‐gated sodium channel and exhibit the same electrophysiological activities as the parent, “red‐tide” ladder polyether toxin with an additional four rings.119b, 124 More recently, Overman and co‐workers reported the tBu‐MacE analogue to have the same Golgi‐modifying properties as the parent spongian diterpene macfarlandin E, but with even less cytotoxicity.125
Figure 12.
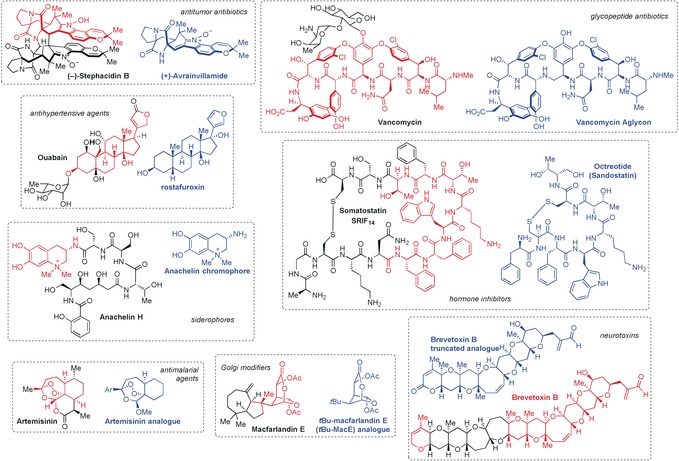
Structural comparison of additional natural product derived, biologically active fragments and their parent compounds.
Our last example is from the studies by Posner and O'Neill on analogues of the sesquiterpene trioxane artemisinin. They have reported many simplified aryl derivatives with comparable in vitro antimalarial activities in the nanomolar range, which also function by the iron‐induced triggering mechanism and were furthermore effective in rodent models.126
3. Additional Sources of Natural Product Fragment Lead Structures
This Review has focused on the identification of suitable fragments through chemical synthesis. Looking toward the future, however, recent advancements in structural biology, computational chemistry, and genome mining show the potential to identify suitable natural product fragments that might be further refined by chemical synthesis.
A different approach to access novel, natural product fragments based on genome data is fueled by advancements in fields such as molecular biology, bioinformatics, genetic mapping, and structural biology. Within the realm of nature, DNA‐encoding fragments of compounds are exchanged between organisms and incorporated in new structures, which can sometimes be found in different kingdoms.127 The natural product fragments are exchanged within organisms by horizontal gene or fragment gene transfer. A representative example is found in the pederin, theopederin A, and onnamide A family of natural products, which share common fragments.128 The revolution in genome mining and advances in metabolomics will continue to unearth even more natural product structures, at an ever‐increasing speed.
Anticancer antibody–drug conjugates (ADCs) are currently the biopharmaceutical standard for innovative cancer treatments, with over 30 entities in clinical development.129 As the name suggests, an antibody is linked to a cytotoxic, natural product derived “payload” to maximize targeting and efficacy of the anticancer agent toward the diseased tissue. Lead optimization in this field involves the development of chemical conjugation methods to couple the natural product to the antibody, the design of the linker, and the type of antibody used.130 Gemtuzumab ozogamicin (Mylotarg) was the first clinically approved ADC. It was launched in 2000 in the USA for the treatment of refractory acute myeloid leukemia (AML), specifically for cells bearing the CD33 antigen.129a, 131 It contains an N‐acetyl‐γ‐calicheamicin dimethyl hydrazide derivative as its active agent, linked by a pH‐labile hydrazone fragment to a recombinant, humanized IgG4 κ antibody, developed by Wyeth and UCB Pharma. This success has led to the evaluation of other cytotoxins for this type of cancer chemotherapy, including taxanes, ansamycins, and duocarmycins.130
More recently, natural product derived fragments are leading the next generation of ADCs for clinical development.129a There are currently 33 ADCs with natural product derived warheads, of which 10 are in phase II trials and 23 in phase I trials. They are derivatives of calicheamicin γ1, dolastatin 10 (monomethylaurisatin E and F, MMAE and MMAF), maytansine (DM1 and DM4), doxorubicin, SN‐38 (camptothecin, irinotecan derivative), and anthramycin (pyrrolobenzodiazepine, or PBD, dimer; SGD‐1882). It is an exciting time in this relatively new field, as the technologies continue to develop for the efficient production of these natural product derived ADCs. Successful lead optimization will likely launch this class of anticancer chemotherapies into predominant clinical use, and eventually application toward treatments of other diseases.
Although fragment‐based drug design (FBDD) or screening libraries from diversity‐oriented synthesis (DOS) seemed to be the contending methods to approach drug development in recent years, in the past 2–3 years we have seen a novel tactic that combines these two methods and involves computational analysis and data mining to create or repurpose natural product derived fragments (NPDFs).132 This allows for an elegant and systematic identification of biological targets for “orphan” natural products and/or NPDFs, which can then be validated through chemical synthesis and subsequent biological assessment. In the pioneering report by Waldmann and co‐workers in 2013, they disclosed an innovative algorithm in which 2000 clusters of natural NPDFs were generated from 180 000 natural products, rich in sp3‐hybridized centers.133 The overall concept was to identify unexplored ligand classes and areas of chemical space for established drug targets, especially those that have been difficult to address. This approach combined the advantages of the chemical space offered by fragments with the rich chemistry and geometry that natural products bring, meanwhile leaving a possibility for optimization for biological recognition and modulation.132 As a proof‐of‐concept, they screened and identified novel scaffolds as inhibitors of p38α MAP kinase and tyrosine/dual‐specificity phosphatases by using protein X‐ray crystallography. The term “biology‐oriented synthesis” (BIOS) has been coined by Waldmann and co‐workers for this method.134
In another example from 2014, Schneider and co‐workers developed a ligand‐based model to predict biological targets for NPDFs that does not require a three‐dimensional target model, but instead relies on the hypothetical polypharmacological character of natural products.135 To validate their method, they analyzed the antitumor agent archazolid A, and identified eight new biochemical targets for this molecule, all of which have been associated with antitumor effects. Additional work from the Schneider group has focused on repurposing of de novo created compounds from computational models to predict bioactivity,136 or on chemography by creating a generative topographic map (GTM) to visualize chemical space populated by natural products and synthetic drugs.137
In 2015, Quinn and co‐workers demonstrated that their self‐generated NPDF library captured more than half of the small pharmacophore triplet diversity than other natural product datasets,138 and Lanz and Riedl used NPDFs as seeds for the de novo discovery of lead structures for therapeutically relevant targets.139
Overall, these examples delineate a future for this field by proving that the combination of bioinformatic/cheminformatic tools with organic synthesis will enable scientists to expand the boundaries of biologically relevant chemical space.
4. Summary and Outlook
In this Review, we aimed to highlight how a desired property of a natural product, such as its powerful biological activity, can be retained by a smaller, truncated compound (“reduce to the max”)—a refined fragment of the parent natural product. This approach addresses some of the shortcomings of the use of natural products in drug discovery. The chemical tractability can be greatly improved by structurally more simple analogues, with fewer stereogenic centers and often an overall drastically shorter synthetic route. In fact, some of the resulting fragments resemble classical medicinal chemistry targets with regard to size and complexity. In addition, although considerable advances have been made in synthetic methods over the last decades, chemical synthesis alone cannot likely keep up with the de novo creation of interesting natural product like scaffolds. As a result, a number of potential avenues have opened up the future of this field, including the use of natural product derived fragments in antibody–drug conjugates, stereochemically complex modules for fragment‐based drug discovery, or scaffold repurposing, as the next generation of natural product‐inspired therapies.
These, among other advancements in such fields as structural biology, genetic mapping, protein crystallography, computational chemistry, nanotechnology, and genome mining show how natural product fragments can be used to address some of the foreseeable challenges of innovative drug development. All these combined approaches clearly present a promising future role for natural products in drug development. Innovation in these trajectories is key to the future of natural product diversity having an impact on and expanding the breadth of the drug discovery field.
Biographical Information
Karl Gademann (born 1972) was educated at the ETH Zürich and Harvard University (PhD with Prof. Dr. Dieter Seebach, postdoctoral studies with Prof. Dr. Eric N. Jacobsen, and habilitation with Prof. Dr. Erick M. Carreira). He started his independent research at the EPFL Lausanne, before moving to the University of Basel in 2010 as a full professor. He has received several awards, including the Novartis Early Career Award, the National Latsis Prize, the Ruzicka Medal, and the European Young Investigator Award. In August 2015 he moved to the University of Zürich.

Biographical Information
Erika Crane was born in Kettering, Ohio (USA) in 1985. Her research career began as an undergraduate with Prof. Robert S. Coleman at The Ohio State University. She received her BS in Biochemistry from Ohio Northern University in 2007, and then her PhD in Organic Chemistry with Prof. Karl A. Scheidt from Northwestern University in 2012. She is currently a Marie Curie Postdoctoral Fellow in the group of Prof. Dr. Karl Gademann at the University of Basel (Switzerland), directing projects which explore the isolation, total synthesis, and biological evaluation of neuritogenic natural products.

Acknowledgements
Financial support was provided in part by Novartis (NIBR Postgraduate Fellowship to E.C). E.C. is currently supported by a Marie Curie International Incoming Fellowship within the 7th European Community Framework Program. We gratefully acknowledge Dr. Christof Sparr for helpful discussions and Dr. Regina Berg for critically proofreading the manuscript during translation into the German version.
E. A. Crane, K. Gademann, Angew. Chem. Int. Ed. 2016, 55, 3882.
References
- 1.
- 1a. Istvan E. S., Deisenhofer J., Science 2001, 292, 1160–1164; [DOI] [PubMed] [Google Scholar]
- 1b. Jain K. S., Kathiravan M. K., Somani R. S., Shishoo C. J., Bioorg. Med. Chem. 2007, 15, 4674–4699. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Szychowski J., Truchon J. F., Bennani Y. L., J. Med. Chem. 2014, 57, 9292–9308; [DOI] [PubMed] [Google Scholar]
- 2b. Newman D. J., Cragg G. M., J. Nat. Prod. 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Feher M., Schmidt J. M., J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [DOI] [PubMed] [Google Scholar]
- 4.For additional reviews, see
- 4a. Szpilman A. M., Carreira E. M., Angew. Chem. Int. Ed. 2010, 49, 9592–9628; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9786–9823; [Google Scholar]
- 4b. Wender P. A., Donnelly A. C., Loy B. A., Near K. E., Staveness D. in Natural Products in Medicinal Chemistry, 1st ed. (Ed.: S. Hanessian), Wiley-VCH, Weinheim, 2014, pp. 475–543. [Google Scholar]
- 5. Wilson R. M., Danishefsky S. J., J. Org. Chem. 2006, 71, 8329–8351. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wender P. A., Verma V. A., Paxton T. J., Pillow T. H., Acc. Chem. Res. 2008, 41, 40–49; [DOI] [PubMed] [Google Scholar]
- 6b. Wach J. Y., Gademann K., Synlett 2012, 163–170; [Google Scholar]
- 6c. Wender P. A., Tetrahedron 2013, 69, 7529–7550; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Wender P. A., Quiroz R. V., Stevens M. C., Acc. Chem. Res. 2015, 48, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Erlanson D. A., McDowell R. S., O'Brien T., J. Med. Chem. 2004, 47, 3463–3482; [DOI] [PubMed] [Google Scholar]
- 7b. Murray C. W., Rees D. C., Nat. Chem. 2009, 1, 187–192; [DOI] [PubMed] [Google Scholar]
- 7c. Scott D. E., Coyne A. G., Hudson S. A., Abell C., Biochemistry 2012, 51, 4990–5003; [DOI] [PubMed] [Google Scholar]
- 7d. Bathula S. R., Akondi S. M., Mainkar P. S., Chandrasekhar S., Org. Biomol. Chem. 2015, 13, 6432–6448. [DOI] [PubMed] [Google Scholar]
- 8.For extensive reviews, see
- 8a. Jackson K. L., Henderson J. A., Phillips A. J., Chem. Rev. 2009, 109, 3044–3079; [DOI] [PubMed] [Google Scholar]
- 8b. Yu M. J., Kishi Y., Littlefield B. A. in Anticancer Agents from Natural Products (Eds.: G. M. Cragg, D. G. I. Kingston, D. J. Newman), CRC, Taylor & Francis, Boca Raton, 2012, pp. 317–345. [Google Scholar]
- 9.
- 9a. Uemura D., Takahashi K., Yamamoto T., Katayama C., Tanaka J., Okumura Y., Hirata Y., J. Am. Chem. Soc. 1985, 107, 4796–4798; [Google Scholar]
- 9b. Hirata Y., Uemura D., Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar]
- 10. Pettit G. R., Herald C. L., Boyd M. R., Leet J. E., Dufresne C., Doubek D. L., Schmidt J. M., Cerny R. L., Hooper J. N., Rutzler K. C., J. Med. Chem. 1991, 34, 3339–3340. [DOI] [PubMed] [Google Scholar]
- 11.T/C=size of treated tumor/size of control tumor.
- 12. Bai R. L., Paull K. D., Herald C. L., Malspeis L., Pettit G. R., Hamel E., J. Biol. Chem. 1991, 266, 15882–15889. [PubMed] [Google Scholar]
- 13.
- 13a. Munro M. H., Blunt J. W., Dumdei E. J., Hickford S. J., Lill R. E., Li S., Battershill C. N., Duckworth A. R., J. Biotechnol. 1999, 70, 15–25; [DOI] [PubMed] [Google Scholar]
- 13b. Hart J. B., Lill R. E., Hickford S. J. H., Blunt J. W., Munro M. H. G. in Drugs from the Sea (Ed.: N. Fusetani), Karger, Basel, 2000, pp. 134–153; [Google Scholar]
- 13c. Ref. [8b].
- 14.
- 14a. Brimacombe J. S., Mofti A. M., Tucker L. C. N., J. Chem. Soc. C 1971, 2911–2915; [DOI] [PubMed] [Google Scholar]
- 14b. Wong M. Y. H., Gray G. R., J. Am. Chem. Soc. 1978, 100, 3548–3553; [Google Scholar]
- 14c. Vekemans J. A. J. M., Boerekamp J., Godefroi E. F., Chittenden G. J. F., Recl. Trav. Chim. Pays-Bas 1985, 104, 266–272; [Google Scholar]
- 14d. Aicher T. D., Kishi Y., Tetrahedron Lett. 1987, 28, 3463–3466; [Google Scholar]
- 14e. Aicher T. D., Buszek K. R., Fang F. G., Forsyth C. J., Jung S. H., Kishi Y., Matelich M. C., Scola P. M., Spero D. M., Yoon S. K., J. Am. Chem. Soc. 1992, 114, 3162–3164; [Google Scholar]
- 14f. Aicher T. D., Buszek K. R., Fang F. G., Forsyth C. J., Jung S. H., Kishi Y., Scola P. M., Tetrahedron Lett. 1992, 33, 1549–1552; [Google Scholar]
- 14g. Buszek K. R., Fang F. G., Forsyth C. J., Jung S. H., Kishi Y., Scola P. M., Yoon S. K., Tetrahedron Lett. 1992, 33, 1553–1556. [Google Scholar]
- 15.
- 15a. Takai K., Kimura K., Kuroda T., Hiyama T., Nozaki H., Tetrahedron Lett. 1983, 24, 5281–5284; [Google Scholar]
- 15b. Hughes N. A., Carbohydr. Res. 1968, 7, 474–479; [Google Scholar]
- 15c. Lohray B. B., Kalantar T. H., Kim B. M., Park C. Y., Shibata T., Wai J. S. M., Sharpless K. B., Tetrahedron Lett. 1989, 30, 2041–2044; [Google Scholar]
- 15d. Tomooka K., Okinaga T., Suzuki K., Tsuchihashi G., Tetrahedron Lett. 1989, 30, 1563–1566; [Google Scholar]
- 15e. Colvin E. W., Hamill B. J., J. Chem. Soc. Chem. Commun. 1973, 151–152; [Google Scholar]
- 15f. Colvin E. W., Hamill B. J., J. Chem. Soc. Perkin Trans. 1 1977, 869–874; [Google Scholar]
- 15g. Gilbert J. C., Weerasooriya U., J. Org. Chem. 1979, 44, 4997–4998; [Google Scholar]
- 15h. Inanaga J., Hirata K., Saeki H., Katsuki T., Yamaguchi M., Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- 16.
- 16a. Duan J. J. W., Kishi Y., Tetrahedron Lett. 1993, 34, 7541–7544; [Google Scholar]
- 16b. Stamos D. P., Kishi Y., Tetrahedron Lett. 1996, 37, 8643–8646; [Google Scholar]
- 16c. Liu S. B., Kim J. T., Dong C. G., Kishi Y., Org. Lett. 2009, 11, 4520–4523; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Stamos D. P., Chen S. S., Kishi Y., J. Org. Chem. 1997, 62, 7552–7553; [Google Scholar]
- 16e. Namba K., Jun H. S., Kishi Y., J. Am. Chem. Soc. 2004, 126, 7770–7771. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Wang Y., Habgood G. J., Christ W. J., Kishi Y., Littlefield B. A., Yu M. J., Bioorg. Med. Chem. Lett. 2000, 10, 1029–1032; [DOI] [PubMed] [Google Scholar]
- 17b. Seletsky B. M., Wang Y., Hawkins L. D., Palme M. H., Habgood G. J., DiPietro L. V., Towle M. J., Salvato K. A., Wels B. F., Aalfs K. K., Kishi Y., Littlefield B. A., Yu M. J., Bioorg. Med. Chem. Lett. 2004, 14, 5547–5550. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Towle M. J., Salvato K. A., Budrow J., Wels B. F., Kuznetsov G., Aalfs K. K., Welsh S., Zheng W., Seletsky B. M., Palme M. H., Habgood G. J., Singer L. A., Dipietro L. V., Wang Y., Chen J. J., Quincy D. A., Davis A., Yoshimatsu K., Kishi Y., Yu M. J., Littlefield B. A., Cancer Res. 2001, 61, 1013–1021; [PubMed] [Google Scholar]
- 18b. Zheng W., Seletsky B. M., Palme M. H., Lydon P. J., Singer L. A., Chase C. E., Lemelin C. A., Shen Y., Davis H., Tremblay L., Towle M. J., Salvato K. A., Wels B. F., Aalfs K. K., Kishi Y., Littlefield B. A., Yu M. J., Bioorg. Med. Chem. Lett. 2004, 14, 5551–5554. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Kim D. S., Dong C. G., Kim J. T., Guo H. B., Huang J., Tiseni P. S., Kishi Y., J. Am. Chem. Soc. 2009, 131, 15636–15641; [DOI] [PubMed] [Google Scholar]
- 19b. Yang Y. R., Kim D. S., Kishi Y., Org. Lett. 2009, 11, 4516–4519; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Shan M. D., Kishi Y., Org. Lett. 2012, 14, 660–663. [DOI] [PubMed] [Google Scholar]
- 20. Austad B. C., Benayoud F., Calkins T. L., Campagna S., Chase C. E., Choi H. W., Christ W., Costanzo R., Cutter J., Endo A., Fang F. G., Hu Y. B., Lewis B. M., Lewis M. D., McKenna S., Noland T. A., Orr J. D., Pesant M., Schnaderbeck M. J., Wilkie G. D., Abe T., Asai N., Asai Y., Kayano A., Kimoto Y., Komatsu Y., Kubota M., Kuroda H., Mizuno M., Nakamura T., Omae T., Ozeki N., Suzuki T., Takigawa T., Watanabe T., Yoshizawa K., Synlett 2013, 24, 327–332. [Google Scholar]
- 21. Tokunaga M., Larrow J. F., Kakiuchi F., Jacobsen E. N., Science 1997, 277, 936–938. [DOI] [PubMed] [Google Scholar]
- 22. Kurosu M., Lin M. H., Kishi Y., J. Am. Chem. Soc. 2004, 126, 12248–12249. [DOI] [PubMed] [Google Scholar]
- 23.See Ref. 18b.
- 24. Kaburagi Y., Kishi Y., Tetrahedron Lett. 2007, 48, 8967–8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaburagi Y., Kishi Y., Org. Lett. 2007, 9, 723–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dong C. G., Henderson J. A., Kaburagi Y., Sasaki T., Kim D. S., Kim J. T., Urabe D., Guo H., Kishi Y., J. Am. Chem. Soc. 2009, 131, 15642–15646. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. FDA approves new treatment option for late-stage breast cancer, press release, U.S. Food and Drug Administration, 2010; available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm233863.htm, last accessed: October 12, 2015;
- 27b. E7389 Versus Treatment of Physician's Choice in Patients With Locally Recurrent or Metastatic Breast Cancer, U.S. National Institutes of Health, 2006; available from: https://clinicaltrials.gov/show/NCT00388726NLM identifier: NCT00388726, last accessed: January 27, 2015.
- 28.Eisai Receives European Commission Approval of Indication Expansion for Anticancer Agent Halaven for Advanced Breast cancer After Only One Prior Chemotherapy, press release, Eisai, 2014; available from: http://www.eisai.com/news/news201437.html, last accessed: October 12, 2015.
- 29.
- 29a. Nakae K., Yoshimoto Y., Sawa T., Homma Y., Hamada M., Takeuchi T., Imoto M., J. Antibiot. 2000, 53, 1130–1136; [DOI] [PubMed] [Google Scholar]
- 29b. Nakae K., Yoshimoto Y., Ueda M., Sawa T., Takahashi Y., Naganawa H., Takeuchi T., Imoto M., J. Antibiot. 2000, 53, 1228–1230; [DOI] [PubMed] [Google Scholar]
- 29c. Takemoto Y., Nakae K., Kawatani M., Takahashi Y., Naganawa H., Imoto M., J. Antibiot. 2001, 54, 1104–1107. [DOI] [PubMed] [Google Scholar]
- 30. Nakamura H., Takahashi Y., Naganawa H., Nakae K., Imoto M., Shiro M., Matsumura K., Watanabe H., Kitahara T., J. Antibiot. 2002, 55, 442–444. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Gaul C., Danishefsky S. J., Tetrahedron Lett. 2002, 43, 9039–9042; [Google Scholar]
- 31b. Gaul C., Njardarson J. T., Danishefsky S. J., J. Am. Chem. Soc. 2003, 125, 6042–6043; [DOI] [PubMed] [Google Scholar]
- 31c. Gaul C., Njardarson J. T., Shan D., Dorn D. C., Wu K. D., Tong W. P., Huang X. Y., Moore M. A. S., Danishefsky S. J., J. Am. Chem. Soc. 2004, 126, 11326–11337; [DOI] [PubMed] [Google Scholar]
- 31d.for additional synthetic studies towards migrastatin, see
- 31e. Reymond S., Cossy J., Eur. J. Org. Chem. 2006, 4800–4804; [Google Scholar]
- 31f. Reymond S., Cossy J., Tetrahedron 2007, 63, 5918–5929; [Google Scholar]
- 31g. Anquetin G., Horgan G., Rawe S., Murray D., Madden A., MacMathuna P., Doran P., Murphy P. V., Eur. J. Org. Chem. 2008, 1953–1958; [Google Scholar]
- 31h. Yadav J. S., Lakshmi P. N., Synlett 2010, 1033–1036; [Google Scholar]
- 31i. Dias L. C., Finelli F. G., Conegero L. S., Krogh R., Andricopulo A. D., Eur. J. Org. Chem. 2009, 6748–6759; [Google Scholar]
- 31j. Gade N. R., Iqbal J., Tetrahedron Lett. 2013, 54, 4225–4227; [Google Scholar]
- 31k. Gade N. R., Iqbal J., Eur. J. Org. Chem. 2014, 6558–6564. [Google Scholar]
- 32.
- 32a. Njardarson J. T., Gaul C., Shan D., Huang X. Y., Danishefsky S. J., J. Am. Chem. Soc. 2004, 126, 1038–1040; [DOI] [PubMed] [Google Scholar]
- 32b. Oskarsson T., Nagorny P., Krauss I. J., Perez L., Mandal M., Yang G. L., Ouerfelli O., Xiao D. H., Moore M. A. S., Massague J., Danishefsky S. J., J. Am. Chem. Soc. 2010, 132, 3224–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lecomte N., Njardarson J. T., Nagorny P., Yang G. L., Downey R., Ouerfelli O., Moore M. A. S., Danishefsky S. J., Proc. Natl. Acad. Sci. USA 2011, 108, 15074–15078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Shan D. D., Chen L., Njardarson J. T., Gaul C., Ma X. J., Danishefsky S. J., Huang X. Y., Proc. Natl. Acad. Sci. USA 2005, 102, 3772–3776; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34b.additional studies also found that the migrastatin macroketone analogue also significantly increase the immobile fraction E-cadherin in vivo by FRAP (fluorescence recovery after photobleaching) in 2014 by Murphy and co-workers; see Lo Re D., Zhou Y., Nobis M., Anderson K. I., Murphy P. V., ChemBioChem 2014, 15, 1459–1464. [DOI] [PubMed] [Google Scholar]
- 35. Chen L., Yang S., Jakoncic J., Zhang J. J., Huang X. Y., Nature 2010, 464, 1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nagorny P., Krauss I., Njardarson J. T., Perez L., Gaul C., Yang G. L., Ouerfelli O., Danishefsky S. J., Tetrahedron Lett. 2010, 51, 3873–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Majchrzak K., Lo Re D., Gajewska M., Bulkowska M., Homa A., Pawlowski K., Motyl T., Murphy P. V., Krol M., PLoS One 2013, 8, e76789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hayakawa Y., Sohda K., Shinya K., Hidaka T., Seto H., J. Antibiot. 1995, 48, 954–961. [DOI] [PubMed] [Google Scholar]
- 39. Bonazzi S., Guttinger S., Zemp I., Kutay U., Gademann K., Angew. Chem. Int. Ed. 2007, 46, 8707–8710; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8862–8865. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. Kudo N., Wolff B., Sekimoto T., Schreiner E. P., Yoneda Y., Yanagida M., Horinouchi S., Yoshida M., Exp. Cell Res. 1998, 242, 540–547; [DOI] [PubMed] [Google Scholar]
- 40b. Kudo N., Matsumori N., Taoka H., Fujiwara D., Schreiner E. P., Wolff B., Yoshida M., Horinouchi S., Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40c. Dong X. H., Biswas A., Chook Y. M., Nat. Struct. Mol. Biol. 2009, 16, 558–560; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40d. Dong X. H., Biswas A., Suel K. E., Jackson L. K., Martinez R., Gu H. M., Chook Y. M., Nature 2009, 458, 1136-1141; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40e. Monecke T., Guttler T., Neumann P., Dickmanns A., Gorlich D., Ficner R., Science 2009, 324, 1087–1091. [DOI] [PubMed] [Google Scholar]
- 41. Sun Q. X., Carrasco Y. P., Hu Y. C., Guo X. F., Mirzaei H., MacMillan J., Chook Y. M., Proc. Natl. Acad. Sci. USA 2013, 110, 1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Newlands E. S., Rustin G. J. S., Brampton M. H., Br. J. Cancer 1996, 74, 648–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bonazzi S., Eidam O., Guttinger S., Wach J. Y., Zemp I., Kutay U., Gademann K., J. Am. Chem. Soc. 2010, 132, 1432–1442. [DOI] [PubMed] [Google Scholar]
- 44. Plater M. J., Aiken S., Bourhill G., Tetrahedron 2002, 58, 2415–2422. [Google Scholar]
- 45. Komatsu K., Tanino K., Miyashita M., Angew. Chem. Int. Ed. 2004, 43, 4341–4345; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4441–4445. [Google Scholar]
- 46. Hintermann T., Seebach D., Helv. Chim. Acta 1998, 81, 2093–2126. [Google Scholar]
- 47.
- 47a. Marshall J. A., Bourbeau M. P., J. Org. Chem. 2002, 67, 2751–2754; [DOI] [PubMed] [Google Scholar]
- 47b. Marshall J. A., Mikowski A. M., Bourbeau M. P., Schaaf G. M., Valeriote F., Bioorg. Med. Chem. Lett. 2006, 16, 320–323. [DOI] [PubMed] [Google Scholar]
- 48.Adapted from Ref. 43 with permission.
- 49. Kalyanaraman C., Bernacki K., Jacobson M. P., Biochemistry 2005, 44, 2059–2071. [DOI] [PubMed] [Google Scholar]
- 50. Gademann K., Curr. Drug Targets 2011, 12, 1574–1580. [DOI] [PubMed] [Google Scholar]
- 51. Ichimura M., Ogawa T., Takahashi K., Kobayashi E., Kawamoto I., Yasuzawa T., Takahashi I., Nakano H., J. Antibiot. 1990, 43, 1037–1038. [DOI] [PubMed] [Google Scholar]
- 52.
- 52a. Boger D. L., Johnson D. S., Proc. Natl. Acad. Sci. USA 1995, 92, 3642–3649; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52b. MacMillan K. S., Boger D. L., J. Med. Chem. 2009, 52, 5771–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Boger D. L., Ishizaki T., Zarrinmayeh H., Munk S. A., Kitos P. A., Suntornwat O., J. Am. Chem. Soc. 1990, 112, 8961–8971. [Google Scholar]
- 54.
- 54a. Boger D. L., Machiya K., J. Am. Chem. Soc. 1992, 114, 10056–10058; [Google Scholar]
- 54b. Boger D. L., Machiya K., Hertzog D. L., Kitos P. A., Holmes D., J. Am. Chem. Soc. 1993, 115, 9025–9036. [Google Scholar]
- 55. Boger D. L., Coleman R. S., J. Am. Chem. Soc. 1988, 110, 4796–4807. [Google Scholar]
- 56. Taylor E. C., Dumas D. J., J. Org. Chem. 1981, 46, 1394–1402. [Google Scholar]
- 57. Tichenor M. S., MacMillan K. S., Stover J. S., Wolkenberg S. E., Pavani M. G., Zanella L., Zaid A. N., Spalluto G., Rayl T. J., Hwang I., Baraldi P. G., Boger D. L., J. Am. Chem. Soc. 2007, 129, 14092–14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. MacMillan K. S., Lajiness J. P., Cara C. L., Romagnoli R., Robertson W. M., Hwang I., Baraldi P. G., Boger D. L., Bioorg. Med. Chem. Lett. 2009, 19, 6962–6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.
- 59a. Konishi M., Ohkuma H., Matsumoto K., Tsuno T., Kamei H., Miyaki T., Oki T., Kawaguchi H., Vanduyne G. D., Clardy J., J. Antibiot. 1989, 42, 1449–1452; [DOI] [PubMed] [Google Scholar]
- 59b. Konishi M., Ohkuma H., Tsuno T., Oki T., Vanduyne G. D., Clardy J., J. Am. Chem. Soc. 1990, 112, 3715–3716. [Google Scholar]
- 60.
- 60a. Sugiura Y., Shiraki T., Konishi M., Oki T., Proc. Natl. Acad. Sci. USA 1990, 87, 3831–3835; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60b. Wender P. A., Kelly R. C., Beckham S., Miller B. L., Proc. Natl. Acad. Sci. USA 1991, 88, 8835–8839; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60c. Semmelhack M. F., Gallagher J., Cohen D., Tetrahedron Lett. 1990, 31, 1521–1522. [Google Scholar]
- 61.
- 61a. Porco J. A., Schoenen F. J., Stout T. J., Clardy J., Schreiber S. L., J. Am. Chem. Soc. 1990, 112, 7410–7411; [Google Scholar]
- 61b. Nicolaou K. C., Hwang C. K., Smith A. L., Wendeborn S. V., J. Am. Chem. Soc. 1990, 112, 7416–7418; [Google Scholar]
- 61c. Nishikawa T., Isobe M., Goto T., Synlett 1991, 393–395; [Google Scholar]
- 61d. Taunton J., Wood J. L., Schreiber S. L., J. Am. Chem. Soc. 1993, 115, 10378–10379; [Google Scholar]
- 61e. Yoon T. Y., Shair M. D., Danishefsky S. J., Shulte G. K., J. Org. Chem. 1994, 59, 3752–3754; [Google Scholar]
- 61f. Shair M. D., Yoon T. Y., Danishefsky S. J., Angew. Chem. Int. Ed. Engl. 1995, 34, 1721–1723; [Google Scholar]; Angew. Chem. 1995, 107, 1883–1885; [Google Scholar]
- 61g. Shair M. D., Yoon T. Y., Mosny K. K., Chou T. C., Danishefsky S. J., J. Am. Chem. Soc. 1996, 118, 9509–9525. [Google Scholar]
- 62.
- 62a. Myers A. G., Fraley M. E., Tom N. J., Cohen S. B., Madar D. J., Chem. Biol. 1995, 2, 33–43; [DOI] [PubMed] [Google Scholar]
- 62b. Myers A. G., Fraley M. E., Tom N. J., J. Am. Chem. Soc. 1994, 116, 11556–11557. [Google Scholar]
- 63.
- 63a. Wender P. A., Zercher C. K., J. Am. Chem. Soc. 1991, 113, 2311–2313; [Google Scholar]
- 63b. Wender P. A., Zercher C. K., Beckham S., Haubold E. M., J. Org. Chem. 1993, 58, 5867–5869; [Google Scholar]
- 63c. Wender P. A., Beckham S., Oleary J. G., Synthesis 1994, 1278–1282; [Google Scholar]
- 63d. Wender P. A., Beckham S., Mohler D. L., Tetrahedron Lett. 1995, 36, 209–212. [Google Scholar]
- 64.
- 64a. Nicolaou K. C., Dai W. M., Tsay S. C., Estevez V. A., Wrasidlo W., Science 1992, 256, 1172–1178; [DOI] [PubMed] [Google Scholar]
- 64b. Nicolaou K. C., Smith A. L., Acc. Chem. Res. 1992, 25, 497–503; [Google Scholar]
- 64c. Nicolaou K. C., Dai W. M., J. Am. Chem. Soc. 1992, 114, 8908–8921; [Google Scholar]
- 64d. Nicolaou K. C., Smith A. L., Yue E. W., Proc. Natl. Acad. Sci. USA 1993, 90, 5881–5888; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64e. Nicolaou K. C., Dai W. M., Hong Y. P., Tsay S. C., Baldridge K. K., Siegel J. S., J. Am. Chem. Soc. 1993, 115, 7944–7953. [Google Scholar]
- 65.
- 65a. Unno R., Michishita H., Inagaki H., Suzuki Y., Baba Y., Jomori T., Nishikawa T., Isobe M., Bioorg. Med. Chem. 1997, 5, 883–901; [DOI] [PubMed] [Google Scholar]
- 65b. Unno R., Michishita H., Inagaki H., Suzuki Y., Baba Y., Jomori T., Moku M., Nishikawa T., Isobe M., Bioorg. Med. Chem. 1997, 5, 903–919; [DOI] [PubMed] [Google Scholar]
- 65c. Unno R., Michishita H., Inagaki H., Suzuki Y., Baba Y., Jomori T., Nishikawa T., Isobe M., Bioorg. Med. Chem. 1997, 5, 987–999. [DOI] [PubMed] [Google Scholar]
- 66.
- 66a. Myers A. G., Cohen S. B., Tom N. J., Madar D. J., Fraley M. E., J. Am. Chem. Soc. 1995, 117, 7574–7575; [Google Scholar]
- 66b. Myers A. G., Tom N. J., Fraley M. E., Cohen S. B., Madar D. J., J. Am. Chem. Soc. 1997, 119, 6072–6094. [Google Scholar]
- 67.
- 67a. Shair M. D., Yoon T., Chou T. C., Danishefsky S. J., Angew. Chem. Int. Ed. Engl. 1994, 33, 2477–2479; [Google Scholar]; Angew. Chem. 1994, 106, 2578–2580; [Google Scholar]
- 67b. Danishefsky S. J., Shair M. D., J. Org. Chem. 1996, 61, 16–44. [Google Scholar]
- 68. Bosse F., Tunoori A. R., Maier M. E., Tetrahedron 1997, 53, 9159–9168. [Google Scholar]
- 69.
- 69a. Magnus P., Eisenbeis S. A., Rose W. C., Zein N., Solomon W., J. Am. Chem. Soc. 1993, 115, 12627–12628; [Google Scholar]
- 69b. Magnus P., Eisenbeis S. A., Fairhurst R. A., Iliadis T., Magnus N. A., Parry D., J. Am. Chem. Soc. 1997, 119, 5591–5605. [Google Scholar]
- 70.
- 70a. Maier M. E., Bosse F., Niestroj A. J., Eur. J. Org. Chem. 1999, 1–13; [Google Scholar]
- 70b. Sinha S. C., Li L. S., Miller G. P., Dutta S., Rader C., Lerner R. A., Proc. Natl. Acad. Sci. USA 2004, 101, 3095–3099; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70c. Riva R., Banfi L., Basso A., Gandolfo V., Guanti G., Eur. J. Org. Chem. 2010, 2768–2787. [Google Scholar]
- 71. Nicolaou K. C., Maligres P., Suzuki T., Wendeborn S. V., Dai W. M., Chadha R. K., J. Am. Chem. Soc. 1992, 114, 8890–8907. [Google Scholar]
- 72.
- 72a. Pettit G. R., Herald C. L., Doubek D. L., Herald D. L., Arnold E., Clardy J., J. Am. Chem. Soc. 1982, 104, 6846–6848; [Google Scholar]
- 72b.for an extensive review on the byrostatin studies by the Wender group, see Wender P. A., Donnelly A. C., Loy B. A., Near K. E., Staveness D. in Natural Products in Medicinal Chemistry, 1st ed. (Ed.: S. Hanessian), Wiley-VCH, Weinheim, 2014, pp. 475–543. [Google Scholar]
- 73.
- 73a. Hildebrand M., Waggoner L. E., Liu H. B., Sudek S., Allen S., Anderson C., Sherman D. H., Haygood M., Chem. Biol. 2004, 11, 1543–1552; [DOI] [PubMed] [Google Scholar]
- 73b. Sudek S., Lopanik N. B., Waggoner L. E., Hildebrand M., Anderson C., Liu H. B., Patel A., Sherman D. H., Haygood M. G., J. Nat. Prod. 2007, 70, 67–74. [DOI] [PubMed] [Google Scholar]
- 74. Szallasi Z., Smith C. B., Pettit G. R., Blumberg P. M., J. Biol. Chem. 1994, 269, 2118–2124. [PubMed] [Google Scholar]
- 75.
- 75a. Elgie A. W., Sargent J. M., Alton P., Peters G. J., Noordhuis P., Williamson C. J., Taylor C. G., Leuk. Res. 1998, 22, 373–378; [DOI] [PubMed] [Google Scholar]
- 75b. Wall N. R., Mohammad R. M., Al-Katib A. M., Leuk. Res. 1999, 23, 881–888; [DOI] [PubMed] [Google Scholar]
- 75c. Oz H. S., Hughes W. T., Rehg J. E., Thomas E. K., Microb. Pathog. 2000, 29, 187–190. [DOI] [PubMed] [Google Scholar]
- 76.
- 76a. Kortmansky J., Schwartz G. K., Cancer Invest. 2003, 21, 924–936; [DOI] [PubMed] [Google Scholar]
- 76b. Shaha S. P., Tomic J., Shi Y., Pham T., Mero P., White D., He L., Baryza J. L., Wender P. A., Booth J. W., Spaner D. E., Clin. Exp. Immunol. 2009, 158, 186–198; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76c.for current clinical information, see http://clinicaltrials.gov [last accessed: May 2015].
- 77.
- 77a. Etcheberrigaray R., Tan M., Dewachter I., Kuiperi C., Van der Auwera I., Wera S., Qiao L. X., Bank B., Nelson T. J., Kozikowski A. P., Van Leuven F., Alkon D. L., Proc. Natl. Acad. Sci. USA 2004, 101, 11141–11146; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77b. Hongpaisan J., Alkon D. L., Proc. Natl. Acad. Sci. USA 2007, 104, 19571–19576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.
- 78a. Boto W. M. O., Brown L., Chrest J., Adler W. H., Cell Regul. 1991, 2, 95–103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78b. DeChristopher B. A., Loy B. A., Marsden M. D., Schrier A. J., Zack J. A., Wender P. A., Nat. Chem. 2012, 4, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kageyama M., Tamura T., Nantz M. H., Roberts J. C., Somfai P., Whritenour D. C., Masamune S., J. Am. Chem. Soc. 1990, 112, 7407–7408. [Google Scholar]
- 80.
- 80a. Evans D. A., Carreira E. M., Tetrahedron Lett. 1990, 31, 4703–4706; [Google Scholar]
- 80b. Evans D. A., Carter P. H., Carreira E. M., Prunet J. A., Charette A. B., Lautens M., Angew. Chem. Int. Ed. 1998, 37, 2354–2359; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2526–2530; [Google Scholar]
- 80c. Evans D. A., Carter P. H., Carreira E. M., Charette A. B., Prunet J. A., Lautens M., J. Am. Chem. Soc. 1999, 121, 7540–7552. [Google Scholar]
- 81. Ohmori K., Ogawa Y., Obitsu T., Ishikawa Y., Nishiyama S., Yamamura S., Angew. Chem. Int. Ed. 2000, 39, 2290–2294; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2376–2379. [Google Scholar]
- 82. Trost B. M., Dong G. B., Nature 2008, 456, 485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Keck G. E., Poudel Y. B., Cummins T. J., Rudra A., Covel J. A., J. Am. Chem. Soc. 2011, 133, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wender P. A., Schrier A. J., J. Am. Chem. Soc. 2011, 133, 9228–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lu Y., Woo S. K., Krische M. J., J. Am. Chem. Soc. 2011, 133, 13876–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.
- 86a. Wender P. A., Koehler K. F., Sharkey N. A., Dellaquila M. L., Blumberg P. M., Proc. Natl. Acad. Sci. USA 1986, 83, 4214–4218; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86b. Wender P. A., Cribbs C. M., Koehler K. F., Sharkey N. A., Herald C. L., Kamano Y., Pettit G. R., Blumberg P. M., Proc. Natl. Acad. Sci. USA 1988, 85, 7197–7201; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86c. Wender P. A., DeBrabander J., Harran P. G., Jimenez J. M., Koehler M. F. T., Lippa B., Park C. M., Siedenbiedel C., Pettit G. R., Proc. Natl. Acad. Sci. USA 1998, 95, 6624–6629; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86d. Kimura K., Mizutani M. Y., Tomioka N., Endo Y., Shudo K., Itai A., Chem. Pharm. Bull. 1999, 47, 1134–1137. [Google Scholar]
- 87.
- 87a. Wender P. A., Lippa B., Tetrahedron Lett. 2000, 41, 1007–1011; [Google Scholar]
- 87b. Wender P. A., Clarke M. O., Horan J. C., Org. Lett. 2005, 7, 1995–1998; [DOI] [PubMed] [Google Scholar]
- 87c. Wender P. A., Horan J. C., Org. Lett. 2006, 8, 4581–4584; [DOI] [PubMed] [Google Scholar]
- 87d. Wender P. A., Reuber J., Tetrahedron 2011, 67, 9998–10005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87e. Wender P. A., Baryza J. L., Brenner S. E., DeChristopher B. A., Loy B. A., Schrier A. J., Verma V. A., Proc. Natl. Acad. Sci. USA 2011, 108, 6721–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wender P. A., Horan J. C., Verma V. A., Org. Lett. 2006, 8, 5299–5302. [DOI] [PubMed] [Google Scholar]
- 89.
- 89a. Wender P. A., Hinkle K. W., Tetrahedron Lett. 2000, 41, 6725–6729; [Google Scholar]
- 89b. Wender P. A., Baryza J. L., Org. Lett. 2005, 7, 1177–1180. [DOI] [PubMed] [Google Scholar]
- 90. Wender P. A., Verma V. A., Org. Lett. 2008, 10, 3331–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Trost B. M., Yang H. B., Thiel O. R., Frontier A. J., Brindle C. S., J. Am. Chem. Soc. 2007, 129, 2206–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.
- 92a. Hale K. J., Frigerio M., Manaviazar S., Hummersone M. G., Fillingham I. J., Barsukov I. G., Damblon C. F., Gescher A., Roberts G. C. K., Org. Lett. 2003, 5, 499–502; [DOI] [PubMed] [Google Scholar]
- 92b. Keck G. E., Poudel Y. B., Welch D. S., Kraft M. B., Truong A. P., Stephens J. C., Kedei N., Lewin N. E., Blumberg P. M., Org. Lett. 2009, 11, 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.
- 93a. Wender P. A., De Brabander J., Harran P. G., Jimenez J. M., Koehler M. F. T., Lippa B., Park C. M., Shiozaki M., J. Am. Chem. Soc. 1998, 120, 4534–4535; [Google Scholar]
- 93b. Wender P. A., Baryza J. L., Bennett C. E., Bi C., Brenner S. E., Clarke M. O., Horan J. C., Kan C., Lacote E., Lippa B., Nell P. G., Turner T. M., J. Am. Chem. Soc. 2002, 124, 13648–13649; [DOI] [PubMed] [Google Scholar]
- 93c. Wender P. A., Mayweg A. V. W., VanDeusen C. L., Org. Lett. 2003, 5, 277–279; [DOI] [PubMed] [Google Scholar]
- 93d. DeChristopher B. A., Fan A. C., Felsher D. W., Wender P. A., Oncotarget 2012, 3, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.For a minireview on Prins macrocyclization approaches in polyketide natural product synthesis, see Crane E. A., Scheidt K. A., Angew. Chem. Int. Ed. 2010, 49, 8316–8326; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8494–8505. [Google Scholar]
- 95.
- 95a. Wender P. A., DeChristopher B. A., Schrier A. J., J. Am. Chem. Soc. 2008, 130, 6658–6659; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95b. Keck G. E., Kraft M. B., Truong A. P., Li W., Sanchez C. C., Kedei N., Lewin N. E., Blumberg P. M., J. Am. Chem. Soc. 2008, 130, 6660–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.
- 96a. Keck G. E., Poudel Y. B., Rudra A., Stephens J. C., Kedei N., Lewin N. E., Peach M. L., Blumberg P. M., Angew. Chem. Int. Ed. 2010, 49, 4580–4584; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4684–4688; [Google Scholar]
- 96b. Kraft M. B., Poudel Y. B., Kedei N., Lewin N. E., Peach M. L., Blumberg P. M., Keck G. E., J. Am. Chem. Soc. 2014, 136, 13202–13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.
- 97a. Wender P. A., Nakagawa Y., Near K. E., Staveness D., Org. Lett. 2014, 16, 5136–5139; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97b. Wender P. A., Staveness D., Org. Lett. 2014, 16, 5140–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Igarashi M., Nakagawa N., Doi N., Hattori S., Naganawa H., Hamada M., J. Antibiot. 2003, 56, 580–583. [DOI] [PubMed] [Google Scholar]
- 99. Kimura K., Miyata N., Kawanishi G., Kamio Y., Izaki K., Isono K., Agric. Biol. Chem. 1989, 53, 1811–1815. [Google Scholar]
- 100. Dini C., Curr. Top. Med. Chem. 2005, 5, 1221–1236. [DOI] [PubMed] [Google Scholar]
- 101. Kimura K., Bugg T. D. H., Nat. Prod. Rep. 2003, 20, 252–273. [DOI] [PubMed] [Google Scholar]
- 102. Takahashi Y., Igarashi M., Miyake T., Soutome H., Ishikawa K., Komatsuki Y., Koyama Y., Nakagawa N., Hattori S., Inoue K., Doi N., Akamatsu Y., J. Antibiot. 2013, 66, 171–178. [DOI] [PubMed] [Google Scholar]
- 103. Igarashi M., Takahashi Y., Shitara T., Nakamura H., Naganawa H., Miyake T., Akamatsu Y., J. Antibiot. 2005, 58, 327–337. [DOI] [PubMed] [Google Scholar]
- 104. Nakamura H., Tsukano C., Yasui M., Yokouchi S., Igarashi M., Takemoto Y., Angew. Chem. Int. Ed. 2015, 54, 3136–3139; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3179–3182. [Google Scholar]
- 105.
- 105a. Gopinath P., Watanabe T., Shibasaki M., J. Org. Chem. 2012, 77, 9260–9267; [DOI] [PubMed] [Google Scholar]
- 105b.in 2014, another synthesis of (+)-caprazol was published: Gopinath P., Wang L., Abe H., Ravi G., Masuda T., Watanabe T., Shibasaki M., Org. Lett. 2014, 16, 3364–3367. [DOI] [PubMed] [Google Scholar]
- 106.
- 106a. Hirano S., Ichikawa S., Matsuda A., Angew. Chem. Int. Ed. 2005, 44, 1854–1856; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 1888–1890; [Google Scholar]
- 106b. Hirano S., Ichikawa S., Matsuda A., J. Org. Chem. 2008, 73, 569–577; [DOI] [PubMed] [Google Scholar]
- 106c. Yamaguchi M., Matsuda A., Ichikawa S., Org. Biomol. Chem. 2015, 13, 1187–1197. [DOI] [PubMed] [Google Scholar]
- 107. Ii K., Ichikawa S., Al-Dabbagh B., Bouhss A., Matsuda A., J. Med. Chem. 2010, 53, 3793–3813. [DOI] [PubMed] [Google Scholar]
- 108. Ichikawa S., Yamaguchi M., Hsuan L. S., Matsuda A., ACS Infect. Dis. 2015, 1, 151–156. [DOI] [PubMed] [Google Scholar]
- 109.
- 109a. Rudolph J., Sennhenn P. C., Vlaar C. P., Sharpless K. B., Angew. Chem. Int. Ed. Engl. 1996, 35, 2810–2813; [Google Scholar]; Angew. Chem. 1996, 108, 2991–2995; [Google Scholar]
- 109b. Tao B., Schlingloff G., Sharpless K. B., Tetrahedron Lett. 1998, 39, 2507–2510; [Google Scholar]
- 109c. Morgan A. J., Masse C. E., Panek J. S., Org. Lett. 1999, 1, 1949–1952. [DOI] [PubMed] [Google Scholar]
- 110. Schmidt K., Gunther W., Stoyanova S., Schubert B., Li Z. Z., Hamburger M., Org. Lett. 2002, 4, 197–199. [DOI] [PubMed] [Google Scholar]
- 111. Schmidt K., Riese U., Li Z. Z., Hamburger M., J. Nat. Prod. 2003, 66, 378–383. [DOI] [PubMed] [Google Scholar]
- 112. Cheng Y. X., Schneider B., Riese U., Schubert B., Li Z. Z., Hamburger M., J. Nat. Prod. 2004, 67, 1854–1858. [DOI] [PubMed] [Google Scholar]
- 113.
- 113a. Isaka M., Chinthanom P., Supothina S., Tobwor P., Hywel-Jones N. L., J. Nat. Prod. 2010, 73, 2057–2060; [DOI] [PubMed] [Google Scholar]
- 113b. Jessen H. J., Barbaras D., Hamburger M., Gademann K., Org. Lett. 2009, 11, 3446–3449; [DOI] [PubMed] [Google Scholar]
- 113c. Burch P., Chicca A., Gertsch J., Gademann K., ACS Med. Chem. Lett. 2014, 5, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.
- 114a. Jessen H. J., Schumacher A., Shaw T., Pfaltz A., Gademann K., Angew. Chem. Int. Ed. 2011, 50, 4222–4226; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4308–4312; [Google Scholar]
- 114b.for a similar synthetic approach towards (+)-torrubiellone C, see Jessen H. J., Schumacher A., Schmid F., Pfaltz A., Gademann K., Org. Lett. 2011, 13, 4368–4370. [DOI] [PubMed] [Google Scholar]
- 115. Schmid F., Jessen H. J., Burch P., Gademann K., Med. Chem. Commun. 2013, 4, 135–139. [Google Scholar]
- 116. Riese U., Ziegler E., Hamburger M., FEBS Lett. 2004, 577, 455–459. [DOI] [PubMed] [Google Scholar]
- 117. Schröder P., Förster T., Kleine S., Becker C., Richters A., Ziegler S., Rauh D., Kumar K., Waldmann H., Angew. Chem. Int. Ed. 2015, 54, 12398–12403. [DOI] [PubMed] [Google Scholar]
- 118.
- 118a. Myers A. G., Herzon S. B., J. Am. Chem. Soc. 2003, 125, 12080–12081; [DOI] [PubMed] [Google Scholar]
- 118b. Herzon S. B., Myers A. G., J. Am. Chem. Soc. 2005, 127, 5342–5344; [DOI] [PubMed] [Google Scholar]
- 118c. Wulff J. E., Herzon S. B., Siegrist R., Myers A. G., J. Am. Chem. Soc. 2007, 129, 4898–4899; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118d. Wulff J. E., Siegrist R., Myers A. G., J. Am. Chem. Soc. 2007, 129, 14444–14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.
- 119a. Crowley B. M., Boger D. L., J. Am. Chem. Soc. 2006, 128, 2885–2892; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119b.for a more thorough review of this topic, see Ref. [4a].
- 120.
- 120a. Sundram U. N., Griffin J. H., Nicas T. I., J. Am. Chem. Soc. 1996, 118, 13107–13108; [Google Scholar]
- 120b. Malabarba A., Nicas T. I., Thompson R. C., Med. Res. Rev. 1997, 17, 69–137; [DOI] [PubMed] [Google Scholar]
- 120c. Mackay J. P., Gerhard U., Beauregard D. A., Maplestone R. A., Williams D. H., J. Am. Chem. Soc. 1994, 116, 4573–4580; [Google Scholar]
- 120d. Mackay J. P., Gerhard U., Beauregard D. A., Westwell M. S., Searle M. S., Williams D. H., J. Am. Chem. Soc. 1994, 116, 4581–4590. [Google Scholar]
- 121.
- 121a. Bauer W., Briner U., Doepfner W., Haller R., Huguenin R., Marbach P., Petcher T. J., Pless J., Life Sci. 1982, 31, 1133–1140; [DOI] [PubMed] [Google Scholar]
- 121b. Battershill P. E., Clissold S. P., Drugs 1989, 38, 658–702. [DOI] [PubMed] [Google Scholar]
- 122.
- 122a. Quadri L., Bianchi G., Cerri A., Fedrizzi G., Ferrari P., Gobbini M., Melloni P., Sputore S., Torri M., J. Med. Chem. 1997, 40, 1561–1564; [DOI] [PubMed] [Google Scholar]
- 122b. Staessen J. A., Kuznetsova T., Acceto R., Bacchieri A., Brand E., Burnier M., Celis H., Citterio L., de Leeuw P. W., Filipovsky J., Fournier A., Kawecka-Jaszcz K., Manunta P., Nikitin Y., O'Brien E. T., Redon J., Thijs L., Ferrari P., Valentini G., Bianchi G., Pharmacogenomics 2005, 6, 755–775; [DOI] [PubMed] [Google Scholar]
- 122c. Schoner W., Scheiner-Bobis G., Am. J. Physiol. Cell Physiol. 2007, 293, C509–C536; [DOI] [PubMed] [Google Scholar]
- 122d. Ferrari P., Ferrandi M., Valentini G., Manunta P., Bianchi G., Med. Hypotheses 2007, 68, 1307–1314; [DOI] [PubMed] [Google Scholar]
- 122e. Ferrari P., Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 1254–1258; [DOI] [PubMed] [Google Scholar]
- 122f. First Patient Enrolled in Phase IIb Clinical Trial of Rostafuroxin in Italy, press release, 2013, available from: http://www.leespharm.com/file/news/FOR%20IMMEDIATE%20RELEASE_Rostafuroxin%20first%20patient.pdf, last accessed: October 12, 2015.
- 123.
- 123a. Zürcher S., Wäckerlin D., Bethuel Y., Malisova B., Textor M., Tosatti S., Gademann K., J. Am. Chem. Soc. 2006, 128, 1064–1065; [DOI] [PubMed] [Google Scholar]
- 123b. Wach J. Y., Bonazzi S., Gademann K., Angew. Chem. Int. Ed. 2008, 47, 7123–7126; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 7232–7235; [Google Scholar]
- 123c.highlighted in: Wender P. A., Donnelly A. C., Loy B. A., Near K. E., Staveness D. in Natural Products in Medicinal Chemistry, 1st ed. (Ed.: S. Hanessian), Wiley-VCH, Weinheim, 2014, pp. 475–543; and [Google Scholar]; Wach J. Y., Gademann K., Synlett 2012, 163–170. [Google Scholar]
- 124.
- 124a. Nicolaou K. C., Tiebes J., Theodorakis E. A., Rutjes F. P. J. T., Koide K., Sato M., Untersteller E., J. Am. Chem. Soc. 1994, 116, 9371–9372; [Google Scholar]
- 124b. Gawley R. E., Rein K. S., Jeglitsch G., Adams D. J., Theodorakis E. A., Tiebes J., Nicolaou K. C., Baden D. G., Chem. Biol. 1995, 2, 533–541. [DOI] [PubMed] [Google Scholar]
- 125.
- 125a. Molinski T. F., Faulkner D. J., He C. H., Vanduyne G. D., Clardy J., J. Org. Chem. 1986, 51, 4564–4567; [Google Scholar]
- 125b. Schnermann M. J., Beaudry C. M., Egorova A. V., Polishchuk R. S., Sutterlin C., Overman L. E., Proc. Natl. Acad. Sci. USA 2010, 107, 6158–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Posner G. H., O'Neill P. M., Acc. Chem. Res. 2004, 37, 397–404. [DOI] [PubMed] [Google Scholar]
- 127. Helfrich E. J., Reiter S., Piel J., Curr. Opin. Biotechnol. 2014, 29, 107–115. [DOI] [PubMed] [Google Scholar]
- 128. Piel J., Hui D., Wen G., Butzke D., Platzer M., Fusetani N., Matsunaga S., Proc. Natl. Acad. Sci. USA 2004, 101, 16222–16227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.
- 129a. Butler M. S., Robertson A. A. B., Cooper M. A., Nat. Prod. Rep. 2014, 31, 1612–1661; [DOI] [PubMed] [Google Scholar]
- 129b.for more thorough and recent reviews on natural product derived antibody–drug conjugates, see
- 129c. Newman D. J., Cragg G. M., Mar. Drugs 2014, 12, 255–278; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129d. Chari R. V. J., Miller M. L., Widdison W. C., Angew. Chem. Int. Ed. 2014, 53, 3796–3827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3872–3904; [Google Scholar]
- 129e. Gerber H. P., Koehn F. E., Abraham R. T., Nat. Prod. Rep. 2013, 30, 625–639; [DOI] [PubMed] [Google Scholar]
- 129f. Flygare J. A., Pillow T. H., Aristoff P., Chem. Biol. Drug Des. 2013, 81, 113–121; [DOI] [PubMed] [Google Scholar]
- 129g. Mullard A., Nat. Rev. Drug Discovery 2013, 12, 329–332; [DOI] [PubMed] [Google Scholar]
- 129h. Sievers E. L., Senter P. D., Annu. Rev. Med. 2013, 64, 15–29. [DOI] [PubMed] [Google Scholar]
- 130. Carter G. T., Nat. Prod. Rep. 2011, 28, 1783–1789. [DOI] [PubMed] [Google Scholar]
- 131. Hamann P. R., Hinman L. M., Hollander I., Beyer C. F., Lindh D., Holcomb R., Hallett W., Tsou H. R., Upeslacis J., Shochat D., Mountain A., Flowers D. A., Bernstein I., Bioconjugate Chem. 2002, 13, 47–58. [DOI] [PubMed] [Google Scholar]
- 132. Shoichet B. K., Nat. Chem. 2013, 5, 9–10. [DOI] [PubMed] [Google Scholar]
- 133. Over B., Wetzel S., Grutter C., Nakai Y., Renner S., Rauh D., Waldmann H., Nat. Chem. 2013, 5, 21–28. [DOI] [PubMed] [Google Scholar]
- 134.
- 134a. Wetzel S., Bon R. S., Kumar K., Waldmann H., Angew. Chem. Int. Ed. 2011, 50, 10800–10826; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10990–11018; [Google Scholar]
- 134b. van Hattum H., Waldmann H., J. Am. Chem. Soc. 2014, 136, 11853–11859. [DOI] [PubMed] [Google Scholar]
- 135. Reker D., Perna A. M., Rodrigues T., Schneider P., Reutlinger M., Monch B., Koeberle A., Lamers C., Gabler M., Steinmetz H., Muller R., Schubert-Zsilavecz M., Werz O., Schneider G., Nat. Chem. 2014, 6, 1072–1078. [DOI] [PubMed] [Google Scholar]
- 136. Rodrigues T., Lin Y. C., Hartenfeller M., Renner S., Lima Y. F., Schneider G., Chem. Commun. 2015, 51, 7478–7481. [DOI] [PubMed] [Google Scholar]
- 137. Miyao T., Reker D., Schneider P., Funatsu K., Schneider G., Planta Med. 2015, 81, 429–435. [DOI] [PubMed] [Google Scholar]
- 138. Pascolutti M., Campitelli M., Nguyen B., Pham N., Gorse A. D., Quinn R. J., PLoS One 2015, 10, e0120942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lanz J., Riedl R., ChemMedChem 2015, 10, 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]