

Abstract
BACKGROUND: Clinical testing of new therapeutic interventions requires comprehensive, high-quality preclinical data. Concerns regarding quality of preclinical data have been raised in recent reports. This report examines the data on the interaction of 10 drugs with radiation and provides recommendations for improving the quality, reproducibility, and utility of future studies. The drugs were AZD6244, bortezomib, 17-DMAG, erlotinib, gefitinib, lapatinib, oxaliplatin/Lipoxal, sunitinib (Pfizer, Corporate headquarters, New York, NY), thalidomide, and vorinostat. METHODS: In vitro and in vivo data were tabulated from 125 published papers, including methods, radiation and drug doses, schedules of administration, assays, measures of interaction, presentation and interpretation of data, dosimetry, and conclusions. RESULTS: In many instances, the studies contained inadequate or unclear information that would hamper efforts to replicate or intercompare the studies, and that weakened the evidence for designing and conducting clinical trials. The published reports on these drugs showed mixed results on enhancement of radiation response, except for sunitinib, which was ineffective. CONCLUSIONS: There is a need for improved experimental design, execution, and reporting of preclinical testing of agents that are candidates for clinical use in combination with radiation. A checklist is provided for authors and reviewers to ensure that preclinical studies of drug-radiation combinations meet standards of design, execution, and interpretation, and report necessary information to ensure high quality and reproducibility of studies. Improved design, execution, common measures of enhancement, and consistent interpretation of preclinical studies of drug-radiation interactions will provide rational guidance for prioritizing drugs for clinical radiotherapy trials and for the design of such trials.
Introduction
State-of-the-art cancer treatment includes chemo-/radiotherapy for most tumor sites, but the integration of targeted agents into radio- or chemoradiotherapy will be key to further optimizing treatment responses. The decision to embark on clinical trials of radiotherapy with new anticancer agents should be based on solid preclinical evidence. Because of broadly based concerns about the reproducibility and translation of preclinical studies to the clinic [1], [2], [3], we conducted a critical survey of 125 published preclinical studies testing the interaction with radiation of 10 agents of interest to the Cancer Therapy Evaluation Program of the National Cancer Institute (Figure 1). The rationale was that these agents had demonstrable anticancer activities and had been studied for their potential to enhance radiation response. Supplementary Table 1 shows drug synonyms and properties. The survey covered research reports presenting original data that measured tumor cell killing by radiation plus drug in vitro or tumor response in vivo published before November 13, 2014. Experiments on normal cells and tissues were not evaluated but are important in assessing clinical potential because a therapeutic gain can be achieved only when the effect on the tumor is greater than on the surrounding normal tissues. Mechanistic studies are also important but were not evaluated in this survey. Both would be worthy of separate examination.
Figure 1.
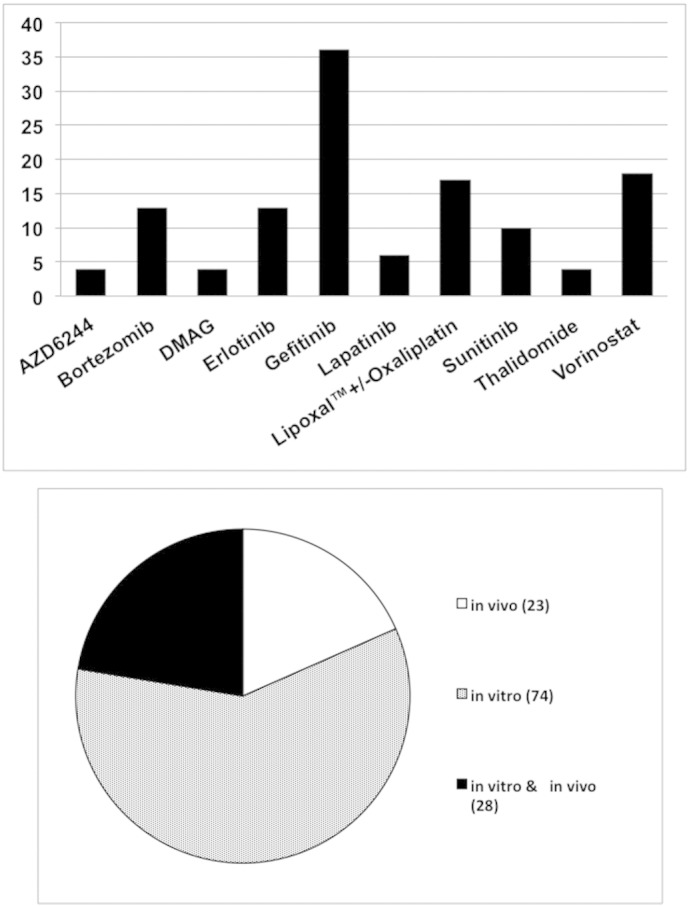
Top: drugs surveyed and numbers of papers reviewed. Bottom: distribution of papers reporting in vitro or in vivo studies or both. Synonyms and properties of drugs are listed in Supplementary Table 1. Papers with both oxaliplatin and LipoxalTM were not counted twice for totals.
Materials and Methods
Searches and Drugs
Searches were conducted using PubMed and the National Library of Medicine, using as search terms the drug name and “radiation,” and limited to English-language papers available through the National Institute of Health Library’s Online Journals. A reference list of papers surveyed is included in Supplementary Table 2.
Analysis Methodology
We examined only data involving single drugs and radiation; these were sometimes control groups in studies of multidrug combinations with radiation. For all studies, we recorded drug, drug concentration or dose, cell line(s) and origin (type of tumor, human or animal), assay and end point, treatment schedule, measure of interaction, radiation parameters (source, energy, dose[s], dose rate, setup, dosimetry), and authors' conclusions and comments. For in vitro studies, we recorded schedule of plating, number of repeats, replicate plates, and plate or flask size. For in vivo studies, we recorded tumor transplant site and size at start of treatment and number of animals/group. To facilitate intercomparison of treatment schedules, day 1 was designated as the day treatment was started.
Results
Tumor lines studied in vitro and/or in vivo are shown in Table 1. They include both human and rodent cells, the majority from long-term cultured cell stocks. Their use in vitro and/or in vivo is indicated by * and # symbols, respectively.
Table 1.
Tumor Lines Studied In Vitro and In Vivo
| Drug | Tumor Cell Lines (Human Origin, Unless Otherwise Noted) |
|---|---|
| AZD6244 | Colorectal (HT29#, HCT116#*) |
| NSCLC (Calu-6DC#*, A549#*) | |
| Pancreatic (MiaPaca-2#*) | |
| Prostatic (Du145 wild type# and mutated#) | |
| Bortezomib | Breast (MCF-7#) |
| Cervical (HeLa#, SiHa#*) | |
| Colorectal (WIDR#, LOVO#*, HT29#, KM12L4#) | |
| Esophageal (TE12#) | |
| Glioblastoma (UVW/NAT transfected to express noradrenaline transporter#) | |
| Glioma (TCG3 p53 mutated*, U87 p53 wt*) | |
| Hepatocellular (Huh-7#) | |
| HNSCC (SQ20B with constitutively active EGFR and robust Akt response#, B88*) | |
| Multiple myeloma (MM1#, RPMI 8226#, JJN-3#, ARH-77#) | |
| Neuroblastoma (SK-N-BE[2c]#) | |
| Oral squamous cell carcinoma (B88#, BHY#, HNt#) | |
| Peripheral nerve sheath (NF90.8*) | |
| Prostatic (Du145#, PC-3-Neo (Bcl-2 wild type, deleted PTEN, mutant p53)# and PC3-Bcl-2 (Bcl-2-overexpressing, deleted PTEN, mutant p53)# | |
| 17-DMAG | Breast (NCI/Adr-res#, T47D#) |
| Glioblastoma (U251#) | |
| NSCLC (NCI-H460#, NCI-H460 with scrambled# or KD p53#, A549#) | |
| Pancreatic (PSN1#, MiaPaCa#, AsPC1#) | |
| Prostatic (Du145#*) | |
| Erlotinib | Colon (HT29#, HCT-116#) |
| Glioblastoma (GBM 12*) | |
| HNSCC (SQ20B#*, H226#*, CNE1#, CNE2#*, UT-SCC-5*, SAS*, FaDu*, UT-SCC-14*, CAL33*) | |
| Lung (A973# origin not reported) | |
| Lung large cell (H460 low EGFR variant#) | |
| Lung squamous (H157#) | |
| NSCLC (A549#, Lu99B#, H23#, H3122#, ABC-1#, HCC44#, Calu-6#, H460#, H661#, UM-SCC1#, UM-SCC-6*, 38 lines resistant to Erlotinib ± KRAS mutations#, NCI-H1703, NCI-H1703 with KRAS mutation) | |
| Pancreatic (BxPC-3#*) | |
| Prostate (DU145#*, DU145IRR* subline after 2Gy/d x 5d/w for 6 mo., PC3#, ARCaPE epithelial type#, ARCaPM mesenchymal type#,) | |
| Vulvar squamous carcinoma (A431#) | |
| Gefitinib | Bladder (HT1376#, J82#*, RT112#, RT4#, T24#, UMUC3#, MGH-U1# and its radiosensitive mutant S40b#, 253J B-V#) |
| Breast (MCF-7#, ADR#, MX-1*) | |
| Cholangiocarcinoma (TFK-1# expressing EGFR, HuCCT-1# expressing EGFR) | |
| Colorectal (GEO#*, LoVo#*, LoVo p53 KRAS mutated#) | |
| Ependymoma (IGREP83*) | |
| Esophageal (TE-1#, TE-3#, TE-4#, TE-5#, TE-7#, TE-8#, TE-10#, TE-13#) | |
| Glioblastoma (U251#, U251MG#, U87#, U87MG#) | |
| Glioma (U87MG#, U251#, U-251MG#, SF-767#, BT4C# rat, N0710# stem-like gliomaspheres, P0710# non-stem-like gliomaspheres, IGRG88*) | |
| HNSCC (CAL27#, CAL33#, CAL60#, CAL166#, Hep-2#, Detroit562#, HSC2#*, HSC3#*, HSC4#, SCC-1#*, SCC-6#*) | |
| Lung (H-157#, HCC827#) | |
| Melanoma (M14#, MALME-3M#, SK-MEL 2#, SK-MEL 5#, SK-MEL 28#, UACC 257#) | |
| Mesothelioma (JMN*) | |
| NSCLC (NCI-H460#, VRMC-LCD#, A549#* wt EGFR, H1299#, H596#, Calu-6#, PC9# activating EGFR mutation, SK-LC-16*, HCC827#* activating EGFR mutation, NCI-H1975# EGFR T790M) | |
| Ovarian (OVCAR-3#) | |
| Pancreatic (MiaPaCa#) | |
| Prostate (PC-3#, DU145#, TRAMP-C1*) | |
| Squamous (SCC-VII# mouse, SCCF1# feline, OC-19#, FaDu#) | |
| Thyroid (ARO# anaplastic, WRO# follicular) | |
| Vulvar squamous (A431 expressing EGFR & HER2/neu#*) | |
| Lapatinib | Bladder (RT112 TP53 wt#, RT112 Rad51 KD#, RT112 Ku80 KD#) |
| Breast (SUM102 EGFR+# and subline constitutively expressing Raf#, SUM185# Lapatinib-resistant overexpressing HER2, SUM140#, SUM225#* overexpressing HER2, H16N2-HER2# overexpressing HER2, SUM149* overexpressing EGFR) | |
| NSCLC (A549)* | |
| Pancreatic (T3M4# wt K-ras, MiaPaCa-2#, Capan-2#, Panc-1#) | |
| Oxaliplatin/Lipoxal | Breast (CH3/TIF*) |
| Cervical (HeLa HPV-18+#, SiHa HPV-16+#, CaSki with HPV-16 & -18 genomes#) | |
| Colorectal (HT29#, HCT116 p53 wt#*, S1 from LS-180#, WiDr p53-mutated#, SW 403 p53 wt#) | |
| Glioblastoma (F98#* rat, U87*) | |
| Hepatocellular (SK-Hep1#) | |
| HNSCC (NT8e#, CAL27#, KB#, Hep2#) | |
| NSCLC (A549#) | |
| Pancreatic (BxPC-3#, Panc-1#) | |
| Sunitinib | Breast (4T1 mouse#*, MDA-MB-231#*) |
| Glioblastoma (GL261* mouse, U87*) | |
| Glioma (Implants from primary mouse gliomas* from mice with conditional deletions of PTEN, p53, & stop-floxed luciferase using PDGF-IRES-Cre retrovirus) | |
| Liver (HEP3B#) | |
| Lung (Lewis lung carcinoma* mouse) | |
| Pancreas (MiaPaCa2#, Panc-1#, Panc02*, CAPAN-1*) | |
| Prostate (PC3#*, DU145#, LNCaP# lacks sunitinib target) | |
| Renal (KCI-18/K#*) | |
| Soft tissue sarcoma from genetically engineered mice#* | |
| Thalidomide | Esophageal (TE1#) |
| Fibrosarcoma (FSAII mouse#*) | |
| Glioma (C6/LacZ* rat) | |
| Multiple myeloma (OPM1# mouse, OPM2# mouse) | |
| Squamous cell (SCC-VII# mouse) | |
| Vorinostat | Breast (MCF7#, MDA-MB-231-BR#*, T47D#, 4T1#* mouse) |
| Colorectal (HCT116#*, HT29#, KM20L2#, SW620#*) | |
| HNSCC (SCC-25#) | |
| Liver (HEP3B#) | |
| Medulloblastoma (DAOY#) | |
| Melanoma (A375#, MeWo#) | |
| Multiple myeloma (U266B1#, RPMI8226#, MM1.s#, KMS-11#) | |
| Neuroblastoma (NB1691luc#*, Kelly#, SY5Y#, MYCN inducible Tet21 with MYCN overexpressed# or repressed#) | |
| NSCLC (A549#) | |
| Osteosarcoma (KHOS-24OS#, SAOS2#) | |
| Ovarian (NCI/ADR-RES#) | |
| Pancreatic (MiaPaCa2#, AsPC-1#, Colo357FG#) | |
| Rhabdomyosarcoma (A-204#*, RD#) | |
| Cell line not specified |
#: in vitro studies; *: in vivo studies.
In Vitro (Cell Culture) Studies
Assays and measures of enhancement
In vitro data were reported in 104 papers with 3 to 32 papers for each drug. Seventy-five percent of reports employed clonogenic assays alone to determine drug effects on radiation response, and another 13% employed both clonogenic and short-term assays (≤ 5 days, e.g., dye-based or cell counts). In 12%, only short-term assays were used.
Interactions were quantified by a variety of methods, including: Dose Modification Factor (DMF), Dose Effect Factor or Ratio, or [Sensitizer or Dose] Enhancement Ratios. These are the ratio of radiation doses giving an isoeffect level of cell survival with radiation alone divided by that for radiation plus drug. Other interaction measures used included isobolograms, ratios of SF2 or SF3 (surviving cell fraction at 2 or 3 Gy), derived from clonogenic or limiting dilution assays [4] of survival, Combination Indices [5], ratios of Mean Inactivation Doses [6], Sensitizer Enhancement Ratios (ratios of D0s [a measure of the slope in the linear part of cell survival curves]), or P values of differences in SF with drug alone versus drug plus radiation. In some cases, the method of analysis was not clear, not reported, or apparently by eye. One paper employed spheroid size, morphology, and cell viability in addition to a clonogenic study [7].
Schedules
Treatment schedules for in vitro studies varied widely (Supplementary Table 3). A single schedule was evaluated in 78% of the papers, 2 in 10%, and 3 or more in 12%. One paper did not report the schedule used. When a drug was given after irradiation, the duration of drug exposure was generally reported, but when the drug was given before irradiation, few papers made clear whether the drug was removed immediately before or after irradiation or was present throughout the incubation period.
Drug concentrations and radiation doses
Table 2 shows ranges of radiation doses and drug concentrations used, and Cmax, maximum blood concentration at the maximum tolerable dose, or the recommended dose in patients. Drug concentrations were based on authors’ claims of clinically achievable concentrations or in vitro toxicity studies, but generally a rationale was not given.
Table 2.
Ranges of Radiation Doses and Drug Concentrations Tested In Vitro and Drug Concentrations Achievable in Patients
| Drug | Range of Radiation Doses, Gy | Range of Drug Concentrations | Cmax at MTD* or Recommended Dose** |
|---|---|---|---|
| AZD6244 | 2-10 | 100-250 nM (46–114 ng/ml) |
486-718 ng/ml** (1060-1600 nM) [58] |
| Bortezomib | 1-16 | 0.68 nM-50 μM (0.4-19,000 ng/ml) |
20 (s.c.)-160 (i.v.) ng/ml** (50-420 nM) [59] |
| 17-DMAG | 2-8 | 10-100 nM (6.2-62,000 ng/ml) |
500 ng/ml* (800 nM) [60] |
| Erlotinib | 0.5-10 | 0.1-10 μM (0.04-4 μg/ml) 150mM (probable error); dose not given |
1340-1450 ng/ml** (3.4-3.7 μM) [61] |
| Gefitinib | 0.5-10 | 0.005-200 μM (2.2-900 ng/ml) |
159 ng/ml @250 mg dose (oral)** (0.36 μM) [62], [63] |
| Lapatinib | 1-10 | 0.6-2.5 μM (0.35-1.45 μg/ml) |
2.43 μg/ml @ 1250 mg dose** (4.2 μM)) [64], [65] |
| Oxaliplatin | 0.5-15 | 0.5-1000 μM (200–400,000 ng/ml) |
3.5-3.8 ng/ml** (0.009-0.010 μM) [66] |
| Lipoxal | 2.2-2.3 | 1.25-20 μM (0.5-8 μg/ml) |
9.2-12 μg/ml (23-30 μM) [67] |
| Sunitinib | 2-10 | 0.01-1 μM (40ng-4 μg/ml) |
73 ng/ml* (0.2 μM) [68] |
| Thalidomide | 0.4-12 | 2-150 μM (0.5-39 μg/ml) |
1.81-2.82 μg/ml @ 400 mg dose** (7-11 μM) [69], [70], [71] |
| Vorinostat | 0.5-40 | 0.2-2.5 μM (528 – 1320 ng/ml) |
1.3 ng/ml* (0.005 μM) [72], [73] |
MTD, maximum tolerable dose. The single asterix (*) indicates dose at the MTD the double asterix (**) indicates recommended dose for use in patients.
Drug preparation
Information on preparation and storage of the drug is important because some drugs are unstable. Twenty-three percent of the papers reported preparing the drug in DMSO, a known radioprotectant, without reporting the final DMSO concentration used or whether it was protective at that concentration.
Statistical considerations: equality of colony counts, repeats, replicates
Of the 77 in vitro papers that tested cell survival using a clonogenic assay at more than one radiation dose level, only 32% mentioned adjusting plate size and/or number of cells plated to achieve statistically comparable numbers of colonies counted in controls and irradiated groups at all radiation doses [8]. None indicated selecting an incubation time on the plateau of the increase in colony number over time or adjusting the termination date of experiments to achieve comparable colony sizes to account for drug-induced growth delay [8]. Sixty-eight percent of papers reported repeating the experiments; 32% did not provide information on the number of repeats; 42% did not report having replicate plates.
Irradiation
Radiation sources used for both in vitro and in vivo studies are shown in Supplementary Table 4. X-ray, 137Cs, and 60Co sources were the most commonly used. Linear accelerators and neutrons were less frequently used. Fourteen percent of reports were unclear or did not report the radiation source.
Problems
Eighty-three percent of the in vitro studies had one or more problems that would make interpretation or replication of the study difficult, or could lead to erroneous conclusions (Table 3). The most common problem was incomplete or unclear description of methods. The next most common error was a prolonged delay between plating of cells and irradiation, which raises questions as to the multiplicity of cells (single or clusters of two or more cells) at the time of radiation exposure; some papers did not report this information. Only one paper with in vitro studies reported blinded data collection. Many publications did not report (or were unclear about) necessary radiation parameters: source (14%), energy (19%), dose rate (38%), setup (76%), and equipment calibration (92%).
Table 3.
In Vitro Papers with Problems, of 104 Papers
| Number of Papers | Problem |
|---|---|
| 42 | Incomplete or unclear methods |
| 45 | Possible multiplicity (clumps) of cells at time of irradiation: > 8 h between plating and irradiation or time between plating and irradiation not reported |
| 24 | DMSO (a radioprotectant) used as vehicle, concentration not given |
| 24 | Statistical problems (including error bars not identified, too many significant figures) |
| 16 | Conclusions not supported by data (e.g., claim of radiosensitization despite large overlap of data, “therapeutic gain” without studies in normal tissue cells |
| 16 | Textual discrepancies or proofing errors |
| 17 | Data plotted improperly: radiation dose not on linear scale, surviving fraction not on log scale, radiation dose in μM units, ratio to drug dose or IC50 |
| 14 | Data not shown, or only “typical” or “representative” data shown |
| 12 | Short-term assay only (dye-based or cell counts), with radiation |
| 4 | Vehicle not reported |
| 3 | Incorrect or missing reference in methods |
| 3 | Controls missing or not shown |
| 1 | Plating efficiency determined in separate experiment |
| 1 | Data not discussed |
In Vivo (Animal) Studies
Assays and schedules of treatment
In vivo studies were reported in 51 papers, with 1 to 11 papers for each drug. Most (90%) employed a growth delay assay in mice with transplanted tumors. Tumors were treated and their growth followed either for the number of days to reach a given multiple of tumor volume at the start of treatment or to the relative volume at a given number of days after treatment. Tumors implanted orthotopically in the brain or kidney were measured using magnetic resonance imaging, bioluminescent imaging, or weight after sacrifice. Other assays used were animal survival duration (mean or median survival time, Kaplan-Meier plots [20%]), tumor-free survival (2%), TCD50 (radiation dose for local tumor control in 50% of subjects, 2%), size of lytic bone lesions (2%), and in vivo treatment followed by an in vitro assay (2%). In some papers, multiple assays were used. The schedules used in in vivo studies are shown in Supplementary Table 5.
Group sizes and repeats
Seventy-eight percent of the papers had at least five 5 subjects per group in all groups. Eighteen percent had one or more groups with less than five 5 subjects, and 4% did not report group size. Only 12% of papers with in vivo studies reported that they repeated their experiments; the rest presumably ran their studies only once.
Problems
Problems identified in the in vivo studies are shown in Table 4. The exact timing of drug and radiation was rarely reported for the days when both were given together in multifraction treatment. Only two papers with in vivo studies reported data collection by an observer blinded to the treatments received. Randomization of animals to treatment groups was reported in 76% of the papers, but none reported the method of randomization. Only one paper reported using power calculations to determine appropriate sample sizes, and none of the others justified their choice of group sizes. As with in vitro studies, necessary radiation parameters were not reported or unclear: source (20%), energy (37%), dose rate (43%), setup (45%), and equipment calibration (88%).
Table 4.
In Vivo Papers with Problems, of 51 Papers
| Number of Papers | Problem |
|---|---|
| 25 | Exact timing between drug and irradiation not given |
| 17 | Tumor size at start of treatment: Tumors ≤ 5 mm diameter, unequal size among groups, or not reported |
| 12 | Unexplained discrepancies in text, figures, or figure legends |
| 13 | Statistical problems: error bars not identified or not shown, mixed use of SE and SD, mean and median, large differences in tumor size at start of treatment, growth delay data censored to time and not to size |
| 7 | Drug dissolved in DMSO, a radioprotectant, with final concentration not given, or vehicle not identified |
| 5 | Methods unclear |
| 6 | Conclusions not supported by data: claim for differing growth rates with growth curves congruent or parallel after lag, growth delay curves continued after loss of animals from group, therapeutic benefit claimed with no normal tissue studies |
| 4 | Data not shown, or only "typical" or "representative" data shown |
| 3 | Radiation or drug dose not reported, or duration of treatment not given |
| 3 | Interaction terms undefined (synergism, etc.) |
| 3 | Group size small (N ≤ 5) |
| 2 | Control groups not done or not shown |
| 2 | DMF/Dose Effect Factor calculated with single radiation dose level |
| 1 | Whole-body irradiation for localized tumor |
| 1 | Curves from control and experimental groups plotted separately |
| 3 | Inconsistent/incomplete reporting of data |
| 1 | Data pooled from several schedules |
| 1 | Unusual responses not discussed or explained (disappearance of 6- to 8-mm–diameter tumors after 5 Gy) |
Enhancement of Radiation Response
After compiling the above data, we asked whether the preclinical data for any of these drugs would show sufficient promise to warrant clinical trials. Among the 125 papers reviewed, 282 drug-tumor line combinations were studied with radiation, with a total of 517 experiments. We analyzed the results in two steps (Supplementary Tables 6 and 7).
The in vitro studies that could be used to evaluate the drug-radiation combinations (designated “useable”) were those conducted at clinically achievable drug concentrations, reported drug and radiation doses and schedules, used clonogenic assays and reported a DMF, or had survival curves from which DMFs could be estimated. “Positive” studies had a DMF > 1.1. Experiments using other measures of effectiveness, including subjective assessments, were then added. All in vitro studies with oxaliplatin and vorinostat were tested at drug concentrations above those clinically achievable, and in vivo results were mixed. For most of the other drugs, results varied with tumor line, drug and radiation dose, and schedule.
The in vivo studies that could be used for similar intercomparisons (designated “useable”) were those that reported drug and radiation doses and schedules and reported data from growth delay, tumor volume, or survival studies, or those from which these end points could be estimated from the published figures. Only one paper reported DMFs, using TCD50 assays. We did not attempt to compare these drug doses to those in patients; tolerance levels and pharmacodynamics can differ significantly between species. In only five cases were replicate studies reported, with the same tumor line, drug and radiation doses, and schedules. In all cases, the replicates were from the same laboratory but not always with identical results. Thus, despite the many experiments performed, there were no assessments by different laboratories of any unique combination of variables including drug, tumor line, drug and radiation dose, and schedule.
We then considered the effects of drug doses and schedules (Supplementary Table 7) and attempted to determine if there was a consistent message about the interactions of these drugs with radiation. It was helpful when a series of tests was done in which only a single parameter was varied: tumor line, drug concentration or dose, radiation dose or dose range, or treatment schedule. Higher drug doses or durations, higher radiation doses, or fractionation of radiation treatment were not always more effective. Different schedules did not always result in different responses. Of the 10 drugs surveyed, only 6 had 1 or more tumor lines with “useable” data both in vitro and in vivo.
Sunitinib failed to enhance radiation response either in vitro or in vivo. AZD6244, bortezomib, 17-DMAG, and gefitinib showed positive results in vitro, but had few studies and/or mixed results in vivo. Erlotinib showed mixed results in vitro but was mostly ineffective in vivo. Lapatinib, LipoxalTM (Regulon, Inc, Athens, Greece), and thalidomide had mixed results but insufficient in vivo data to determine which variables might account for the differences. Oxaliplatin showed mixed results in vivo; one study indicated that higher drug doses were effective and lower doses ineffective, but other studies were positive at those lower doses. Vorinostat showed no clear patterns or consistent results in vivo even when nearly identical schedules and drug doses were used; radiation doses and fractionation varied widely, so comparisons among studies were not possible.
Discussion
In Vitro Studies
Assays
In patients, it is the survival of the few cancer cells, not the death of the many, that leads to recurrences and new metastases months to years after therapy. Clonogenic assays are therefore considered the gold standard for assessing cell survival in vitro following radiation exposure [9] because they measure cell survival and are independent of modes of cell death (mitotic catastrophe, apoptosis, senescence, necrosis, or autophagy [10]) and the time intervals over which they occur. Lethally irradiated cells usually have prolonged cell cycles and die after one or several cell divisions [11]. Clonogenic assays allow time for elimination of cells not capable of sustained proliferation. The incubation period used should be on the plateau of the plot of colony number versus time, which can be different for controls and drug/radiation groups. Cell lines that do not attach well or that tend to migrate can be used for colony-forming assays if measures are taken to immobilize the cells during colony formation, such as growth in agar. Alternatively, survival can be determined through Poisson statistical analysis of limiting dilution clonogenicity, as adapted from Lefkovits [4].
Short-term assays such as cell counts or the MTT or other dye-based assays tend to overestimate cell survival because they count as survivors metabolically active cells that will ultimately die. Whereas short-term dye-based assays are commonly used to assess drug toxicity or cell growth rates, they are unreliable for determining radiation cell survival. Although short-term clonogenic assays have been proposed for high-throughput screening of drugs and radiation [7], [12], [13], [14], they should not be used with slowly proliferating cell lines, for lines that do not form discrete colonies [14], or for drugs that delay or slow cell proliferation. They must be validated for both cell lines and drugs, and leads verified with more detailed studies using standard clonogenic assays.
Drugs that slow or halt cell division or kill cells affect both plating efficiency and optimal incubation time for colony formation. Therefore, colonies from drug-treated cultures should be counted when the colony size of the unirradiated cultures treated with drug is equivalent to that of untreated controls. This may require incubating drug-treated cultures several days longer than controls. None of the papers reported adjusting incubation time for drug effects on colony growth rate. This is especially problematic when drug exposure is continued during colony development after irradiation. Drug-induced growth inhibition can also delay tumor growth and must be reported and considered in interpretation of the data.
Survival curves from drug-treated groups that are normalized for toxicity of the drug in the absence of radiation can be more easily compared with the radiation-only curve, but plating efficiencies of all control and test groups should be reported as an indication of drug toxicity, which contributes to the total response. Colonies with ≥ 50 cells represent > 5 cell divisions, if all dividing cells survive.
Choice of tumor lines
The choice of cell/tumor lines and models for study is important, although the best model for a given assessment is still a matter of debate. Our suggestion is that for targeted agents, lines having the target should be used and compared with effects in isogenic lines and others lacking the target to verify that the agent is affecting radiation response through the predicted mechanism [15]. Voskoglou-Nomikos et al. [16] examined the predictive value of in vitro, mouse allograft, and human xenograft models for Phase II clinical response to 26 cytotoxic cancer drugs in breast, non–small cell lung, ovarian, and colon cancers. The mouse allograft model was not predictive, but results with in vitro and xenograft models varied with tumor type and model. These studies did not, however, involve radiation. The use of a single cell line or tumor model is clearly insufficient as a guide for eventual clinical testing. Extensively cultured cell lines may result in tumors that no longer reflect the characteristics of the tumors from which they originated [17]. A panel of low-passage, genetically annotated cells might provide more accurate assessment of clinical potential but should be validated. The identity of all cell lines should be validated. Testing for pathogens that can impact radiosensitivity or tumor growth (e.g., mycoplasmas and murine viruses) should be carried out before the studies and reported.
Survival curve interpretation
Reduction in the shoulder of the radiation survival curve may result from cell cycle-selective sensitization [18] by a drug or from a reduction in the capacity to repair radiation damage [19]. Studies carried out at a single radiation dose level are insufficient to reveal whether the drug shows merely additive toxicity or affects the slope or shoulder of the radiation cell survival curve, with implications for multifraction irradiation. Therefore, full radiation survival curves should be obtained to determine whether the drug changes the slope of the survival curve (radiosensitization) or reduces the shoulder. In the latter case, multifraction studies would be informative. Only two papers reported in vitro data using more than one fraction of radiation, but both used only a single radiation dose level, and one used only a short-term dye-based assay. None reported studies with synchronous cells.
Microenvironmental effects
Microenvironmental influences such as hypoxia and cell-to-cell and cell-matrix interactions can have a major impact on survival [20], [21], [22], [23]. Cell culture conditions do not usually mimic the heterogeneity in solid tumors growing in a patient or orthotopically implanted in immunosuppressed animals, where oxygen, nutrient, and drug concentrations vary in space and time [24], [25], [26], [27]. Vascular and supportive tissues are present in vivo, cell densities are generally much higher than those in vitro, and tumor size can affect response. Most in vitro studies employed exponentially growing cultures or those treated soon after plating, with cells in lag phase. Slowly or nonproliferating stem cells may predominate in patients and may govern response to therapy. If they are recruited to active proliferation during the course of fractionated treatments, models that more closely mimic their biology and radiobiology might be more predictive of clinical responses [28]. Use of confluent cultures replated for clonogenic survival immediately after treatment may partly overcome this deficiency. Hypoxic conditions and 3D cultures that allow cellular interactions with matrix or stromal elements, etc., can reveal how these factors influence tumor response in vivo. Cells that do not form colonies easily or have low plating efficiencies may be assessed in 3D cultures, conditioned medium, or sterilized feeder layers from appropriate cells [29]. These methodologies are not currently used for routine screening.
Measurement of enhancement and terminology of interactions
As described by Steel and Peckham [30], there are four ways in which combinations of drug and radiation can improve therapeutic efficacy: spatial cooperation, where both modalities interact in a defined and limited anatomical site such as the tumor; nonoverlapping toxicity; protection of normal tissue; and enhanced tumor response. Here we focus on the last of these possible interactions. Studies at a single radiation dose level using the Combination Index of Chou and Talalay [5] or ratios of SF2Gy are difficult to compare. Small uncertainties in these estimates will be magnified if extrapolated to 30 fractions. Nonlinear regression analysis should be used to compare radiation survival curves [31]. To determine DMFs at the standard 10% SF, the highest radiation doses must reduce SFs to ≤ 10% in all groups, with normalization of drug data to 100% SF. However, it is important to keep in mind that DMFs depend on the shapes of the normalized survival curves and the surviving fraction at which they were measured. In addition, the cumulative effects of any toxicity to, and radiosensitization of normal tissues and cells should be less than that observed for tumors. Enhancement of treatment effects assessed by a standard method would allow straightforward comparisons among drugs, laboratories, and tumor lines. Possibilities for spatial cooperation and nonoverlapping toxicities in sparing normal tissues should also be considered in evaluating the activity of radiation-drug combinations. In considering the ultimate clinical application of a combination, the volume of normal tissue and the extent of its damage in relation to total organ volume may allow for use of a therapy that produces major damage but spares an adequate functional volume.
Statistics
Statistical concerns in published papers have been presented [2], [32]. Similar problems were observed in the papers we reviewed, including mis- or nonidentification of error bars, masking of variability by presentation of "representative" data, inappropriate calculation of P values and error bars, and use of inappropriate statistical tests. These will not be enumerated here. The most commonly reported statistic was the P value, which measures the probability of obtaining the given test value when control and test groups are in fact not different (the null hypothesis). However, as stated by Gertrude Stein, "A difference, to be a difference, must make a difference." Because an important goal in preclinical studies of drugs and radiation is to identify and prioritize combinations for clinical trials, a more useful and robust statistic is a measure of the magnitude of the enhancement, such as a DMF with a 95% confidence interval. This requires testing at multiple radiation dose levels. The minimum DMF that should be required before proceeding with clinical radiotherapy trials has not been settled, but the 95% confidence interval for the DMF should at a minimum exclude 1. In addition, any drug giving a DMF of less than 1.2 is not likely to be successful in the clinic.
Drug concentrations
Drugs should be tested at clinically relevant exposure times and concentrations: at or below the Cmax from Phase I trial data, if known. Lacking this information, a range of drug concentrations should be tested and combined with tests of the effects on target proteins and/or pathways. A reasonable approximation of the duration of drug exposure in patients should be made. When in vivo tolerability information is obtained, a decision can then be made on feasibility and design of animal studies.
In Vivo Studies
As with in vitro studies, cell lines used for in vivo evaluation of drug-radiation interactions should be representative of human tumors and have the relevant targets of the drug being tested. Subcutaneous tumors can be measured directly with calipers. Orthotopic tumors are more like tumors in patients in temperature, metabolism, stromal composition, vascularization, and chemosensitivity [33], [34] but are more difficult to measure, usually requiring either advanced imaging technology or sacrifice of the animal.
Tumor lines that have been passaged exhibit genetic and physiologic drift from their primary tumors and may not respond in the same manner as the primary tumor in situ. Camphausen et al. [35] have found differences in gene expression in untreated and irradiated cells from brain tumors grown in vitro or as subcutaneous or orthotopic xenografts, but the effects of these differences on radiation response have not been tested in clonogenic, growth delay, or TCD50 assays. Recent adoption of genetically engineered mouse models and patient-derived xenografts may mitigate the drawbacks of established cell line xenografts but are costly. Both genetically engineered mouse models and patient-derived xenograft models appear to have good predictive value regarding tumor responses to therapy [36] but need further verification.
Assays
The growth delay assay is the most frequently used assay for in vivo studies, but it can overestimate the effectiveness of drugs that temporarily inhibit growth but do not kill cells [37], [38]. The time for tumor regrowth, particularly for doses greater than 12 to 15 Gy, can be influenced by the tumor bed effect (slower growth of a tumor from vascular/stromal damage in the irradiated site) [39]. This effect can be minimized if the size of the tumor at the conclusion of the assay is ≤ 2 × treatment volume. The TCD50 assay for local tumor control is a measure of complete eradication of tumor cells but is more difficult, takes longer to carry out, requires more animals, and is more costly.
The value of single-fraction radiation studies is controversial. As indicated above, they can be useful for determining the effect of a drug on the survival curve of proliferating cells and the effects of scheduling of drug and irradiation on interactions in in vitro studies. Two-fraction experiments can then indicate whether the interaction is enhanced or reduced by fractionation. Similar comparisons with in vivo data can also indicate whether enhancement decreases with fractionation. Multifraction experiments can be prohibitively costly in time and animal health, especially if repeated anesthesia is required, but are more “clinically mimetic” than a single dose. Whether 30-fraction experiments that model the typical clinical fractionation regimens are necessary or whether fewer fractions would suffice has not been established. Drug-radiation interactions may be influenced more by the radiation dose per fraction than the number of fractions.
Timing of start of treatment
Tumors were treated starting either at a fixed interval after implantation, when tumor sizes vary considerably, or on the day each tumor reached a given size. Some of the papers reviewed did not report this information. When growth curves were normalized, group differences in size at the start of treatment could not be discerned, but if tumor characteristics changed with size, this could have affected treatment response and conclusions. Although an ideal starting tumor size cannot be prescribed, the tumors should be fully established and their size distribution at the start of treatment reported. Tumor growth rates in patients are generally slower than spontaneous or transplanted human or rodent tumors in rodents. Serial transplantation selects the fastest-growing subpopulations [40]. In some studies, treatment was initiated when tumors were ≤ 25 to 50 mm3. It was not clear how or how accurately such small tumors were measured, or whether they had the response characteristics that might be relevant to tumors in patients.
Statistics
A minimum of five animals per group has been recommended for growth delay experiments, but when tumor growth rates in untreated animals vary considerably, more are needed to distinguish treatment effects from underlying variability in tumor growth rates. Larger group sizes are also needed when tumor size is measured less precisely, as with fluorescence imaging with orthotopically implanted tumors. More precise measurements (magnetic resonance imaging, computed tomography, ultrasound) are preferable, but the additional radiation dose to the tumor from computed tomography must be considered and reported.
Physics and Dosimetry
With deficiencies in reporting dosimetry and irradiation setup, it was not possible to know whether researchers had accounted for such factors as dose from backscatter, uniformity of dose across the radiation field or through the depth of the tumor, absorption by overlying culture medium, culture vessel or tissues, and whether dosimetry had been carried out under the same conditions as the experimental setup or was traceable to equipment calibrated by the National Institute of Standards and Technology. Lack of this information makes it impossible to replicate the studies and to compare results within and among laboratories [41], [42].
Rationale
The rationale for the design of the studies was rarely given in the papers we reviewed, and few reported systematic studies to determine optimal combinations of drug and radiation doses and schedules that, with information on pharmacokinetics and pharmacodynamics in rodents and humans, could be helpful for translating preclinical data into clinical trials. Smith and Houghton have pointed out the importance of clinically relevant drug concentrations for studies in vitro[43]. If this is not known at the time of study, a range of drug concentrations/doses should be studied so that when human pharmacokinetic studies have been performed, the effectiveness of the relevant concentration can be inferred from data already available. A variety of schedules should be studied to determine whether interactions vary with schedule. For a molecularly targeted drug, it is important to determine if the target is affected by that drug concentration or dose. The lowest drug concentration that affects the target should be used for radiation studies. If radiosensitization occurs only at higher concentrations, “off-target” effects may be occurring. Carefully designed in vivo studies should be more informative about a drug's likely performance in more complex environments in patients. Future preclinical and clinical studies will be based on tumor biology rather than histological subtype, using biomarkers to assess response [44].
The Next Steps
Methodologies for assessing tumor cell and tumor radioresponse are continually evolving, and new assays and methods may enhance the speed, sensitivity, selectivity, and predictive power of preclinical studies. In addition to the high-throughput clonogenic assays mentioned above, other innovations include methods and reagents that allow for control and modifications to culture substrata and oxygenation [22], [26], [45], [46], [47]. Genetically engineered mouse models are being developed that mimic conditions in spontaneous tumors [48]. Patient-derived xenografts also offer the potential for conducting “co-clinical trials” with tumors that retain characteristics of the parent tumor for several passages [49]. Although adoption of these new technologies may provide significant advantages for preclinical testing involving radiation, the results must be validated with patient clinical trial data.
In the clinic, radiotherapy is rarely used as the sole treatment modality. New drugs are tested in clinical trials as additions to “standard of care” regimens comprised of surgery and other drugs. Although we examined only data for single agents tested with radiation, several of the papers in this survey reported studies of combinations of two or more drugs with radiation, and relevant combinations of agents should be studied before initiating clinical trials [50].
It is important for researchers to understand both the limitations of their models and the complexity of the disease and its treatment in patients. Intratumor heterogeneity of the target can reduce the effectiveness of molecularly targeted agents in patients. Many differences between cell culture, rodent models, and human patients have been pointed out [16] and must be considered when designing, conducting, and interpreting results of preclinical studies and, in particular, in recommending and designing subsequent clinical trials.
Checklist of Information Required for Publications on Drug-Radiation Effectiveness
A proposed checklist for researchers and reviewers of preclinical studies evaluating the effectiveness of drugs with ionizing radiation is given in Table 5.
Table 5.
Checklist
| To Be Reported | For All Studies | In Vitro Studies | In Vivo Studies |
|---|---|---|---|
| Drug(s) used | • Names (if more than one) • References to structure and characteristics • Preparation, storage, vehicle, dilution, final concentration |
||
| Radiation | • Details on radiation source, dosimetry, dose rate, beam characteristics and filtration for x-ray/linac sources and radiation setup including buildup or backscatter characteristics • Statement of irradiator constancy measurements and output traceability to National Standards. • References to written irradiation standards and/or protocols used. Examples of relevant radiation factors and their concise description for publication are given in the appendix of [41], [42] http://dx.doi.org/10.6028/jres.118.021. |
• Setup used for the study | • Setup, shielding and shielding efficacy • Use of restraint and/or anesthesia. |
| Cell/tumor line(s) and culture conditions | • Name, tumor type, tissue and species of origin and relevant molecular profile, if known • Test results for validation of identity and pathogen status. |
• Proliferation status and multiplicity at the start of treatment • Medium and serum concentrations, use of other additives such as antibiotics, incubator oxygen and CO2 concentrations and any non-standard incubation conditions, including hypoxia, variations in temperatures used, plating in suspension (e.g. methylcellulose, agar, Matrigel, etc.) or on other matrix or substrate |
• Tumor size at start of treatment, metastatic potential, tumor doubling time, and whether treatment was started when tumors reached a given size or at a given time after implantation. • Tumors should be sufficiently large at the start of treatment to have biological properties of established tumors and to facilitate accurate measurement. • Tumor transplantation procedure, site, method and frequency of measurement. |
| Assay(s) used | • Assay used | • Clonogenic assay: minimum cells/colony, plate size, adjustments in number of cells plated and/or plate size, duration of incubation time to achieve equal colony sizes and counts among drug-treated, irradiated, and combined-treatment groups • Justification for duration of posttreatment incubation time (i.e., on plateau of curve of colony counts vs incubation time). |
• Growth delay: measurements made and cutoff criteria: tumor size, number of days after treatment, euthanasia criteria |
| • Dye-based short-term assays (e.g., MTT and similar assays) or those focused on a single mode of death (e.g., apoptosis) are not appropriate for use with radiation without verification by comparison with standard clonogenic assays in the cell lines used for the study. • Promising results from pilot studies at a single radiation dose level or short-term high throughput clonogenic assays should be followed up with standard clonogenic assays using ≥ 3 radiation dose levels plus unirradiated controls. |
• Survival time: euthanasia criteria • Animal husbandry practices (e.g., conventional or defined flora colony) • Possible cage effects, if animals in a given treatment group were caged together or distributed among different cages |
||
| • For other in vitro assays (e.g. spheroids): criteria and end points used | • TCD50 assays: maximum follow-up time, euthanasia criteria |
||
| Controls | • Appropriate concurrent drug-only, radiation-only, vehicle-only controls | ||
| Schedule(s) | • Drug administration and irradiation schedules and their rationale. | • Drug duration and removal | • Route of drug administration and exact timing, especially when treatments are given over several days. |
| Plotting of data | • Clonogenic assay with multiple radiation dose levels: Log of surviving fraction vs linear radiation dose | ||
| Statistics and software | • Statistics and software used • Error bars used (conf. interval, SD, or SE, and appropriate use of the latter) • Number of independent experiments performed • Inclusion of full data set in supplement if "representative data" are shown • Detailed methodology for measuring effects (e.g. enhancement) • Consult with a statistician both during planning of experiments and analysis of data to determine appropriate statistical tests and group sizes |
• Number of replicate plates used for each group • Curve-fitting model and DMF when full survival curves are obtained, not just P value. |
• Number of animals/group • Power calculations to justify group size • Number of independent experiments performed • Method for handling of censored data. • Initial variance within groups |
| Blinding | • Perform blinded counting of colonies or other end point measurements | • Method of randomization to treatment groups and whether experiment was carried out double-blinded | |
| Interpretation of data | • Verify appropriateness of interpretation of data | ||
| Final check | • Proofread to ensure clarity, consistency and accuracy of methods in text, figures, and figure legends. • Verify references to previously published methods and report any differences. |
Conclusions
Several of the drugs surveyed may warrant further study to define their optimal use: bortezomib, 17-DMAG, gefitinib, lapatinib, Lipoxal, and vorinostat. AZD6244, erlotinib, oxaliplatin, and thalidomide do not appear promising, although they have not been extensively studied. Sunitinib, despite few in vitro studies that could be evaluated, showed positive results in only 1 of 16 in vivo studies and is not recommended for clinical radiotherapy trials.
Many of the problems identified in these preclinical studies are not unique to the field of drug-radiation studies. Multiple recommendations have been made to improve the quality of preclinical scientific research and publications (e.g. [2], [51], [52], [53], [54]). Guidance specific to the study of drug-radiation combinations has been published (e.g. [55], [56], [57]) but has not identified the problems reported here and has not focused on the need to be able to compare results from multiple drugs and multiple laboratories. The number and diversity of variables among the studies surveyed here, including tumor sizes, types and transplant sites, drug and radiation doses, schedules of drug and radiation administration, assays and end points, and criteria for interpretation of interactions, made it difficult to collate this information into robust guidance for clinical trials. In preclinical research, there is a need to balance the freedom to explore many tumor lines, drug doses, radiation doses, mechanisms, and schedules with the larger mission of improving tumor response in cancer patients treated with radiotherapy. This requires sufficient, relevant, robust, accurately reported data. Effects should be quantified with common measures so that data from different laboratories can be compared, and to provide a sound basis for decisions and designs for clinical trials [3]. It is important for researchers to fully justify the rationale for experimental design. Although no preclinical model or study will perfectly predict clinical response, it should provide clinically informative data. When clinical trials are completed, it will be important to compare the results with the preclinical data so that the models used and predictive value of preclinical studies can be improved. Investigators, grant and journal reviewers, and editors have a critical role in ensuring that studies are responsibly conducted and reported, and reviewed by persons with a relevant background in radiation biology.
Acknowledgement
This study was funded by the National Cancer Institute (NCI) which employs co-authors E. J. B., C. N. C., J. D., J. C., and J. B. M. J. M. B is funded in part by R01CA149318. The work represents the opinion of all the authors and does not reflect official NCI positions or policies.
Footnotes
This study was funded by the National Cancer Institute (NCI) which employs co-authors E. J. B., C. N. C., J. D., J. C., and J. B. M. J. M. B is funded in part by R01CA149318.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.tranon.2016.01.002.
Appendix A. Supplementary data
Supplementary materials.
References
- 1.Hackam DG, Redelmeier DA. Translation of research evidence from animals to humans. JAMA. 2006;296:1731–1732. doi: 10.1001/jama.296.14.1731. [DOI] [PubMed] [Google Scholar]
- 2.Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, Crystal RG, Darnell RB, Ferrante RJ, Fillit H. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–191. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu FF, Participants ftw, Okunieff P, Bernhard EJ, Stone HB, Yoo S, Coleman CN, Vikram B, Brown M, Buatti J. Lessons learned from radiation oncology clinical trials. Clin Cancer Res. 2013;19:6089–6100. doi: 10.1158/1078-0432.CCR-13-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefkovits I. Limiting dilution analysis. Immunol Methods. 1979;1:355–370. [Google Scholar]
- 5.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzym Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 6.Fertil B, Dertinger H, Courdi A, Malaise EP. Mean inactivation dose: a useful concept for intercomparison of human cell survival curves. 1984. Radiat Res. 2012;178:AV237–AV243. doi: 10.1667/rrav20.1. [DOI] [PubMed] [Google Scholar]
- 7.Wang M, Kern AM, Hulskotter M, Greninger P, Singh A, Pan Y, Chowdhury D, Krause M, Baumann M, Benes CH. EGFR-mediated chromatin condensation protects KRAS-mutant cancer cells against ionizing radiation. Cancer Res. 2014;74:2825–2834. doi: 10.1158/0008-5472.CAN-13-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dewey WC. In vitro systems: standardization of endpoints. Int J Radiat Oncol Biol Phys. 1979;5:1165–1174. doi: 10.1016/0360-3016(79)90636-9. [DOI] [PubMed] [Google Scholar]
- 9.Kahn J, Tofilon PJ, Camphausen K. Preclinical models in radiation oncology. Radiat Oncol. 2012;7:223. doi: 10.1186/1748-717X-7-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12(Suppl 2):1463–1467. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- 11.Chu K, Leonhardt EA, Trinh M, Prieur-Carrillo G, Lindqvist J, Albright N, Ling CC, Dewey WC. Computerized video time-lapse (CVTL) analysis of cell death kinetics in human bladder carcinoma cells (EJ30) X-irradiated in different phases of the cell cycle. Radiat Res. 2002;158:667–677. doi: 10.1667/0033-7587(2002)158[0667:cvtlca]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 12.Wang M, Morsbach F, Sander D, Gheorghiu L, Nanda A, Benes C, Kriegs M, Krause M, Dikomey E, Baumann M. EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Res. 2011;71:6261–6269. doi: 10.1158/0008-5472.CAN-11-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin SH, Zhang J, Giri U, Stephan C, Sobieski M, Zhong L, Mason KA, Molkentine J, Thames HD, Yoo SS. A high content clonogenic survival drug screen identifies mek inhibitors as potent radiation sensitizers for KRAS mutant non-small-cell lung cancer. J Thorac Oncol. 2014;9:965–973. doi: 10.1097/JTO.0000000000000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiwana GS, Prevo R, Buffa FM, Yu S, Ebner DV, Howarth A, Folkes LK, Budwal B, Chu K-Y, Durrant L. Identification of vitamin B1 metabolism as a tumor-specific radiosensitizing pathway using a high-throughput colony formation screen. Oncotarget. 2015:1–12. doi: 10.18632/oncotarget.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res. 2003;9:4227–4239. [PubMed] [Google Scholar]
- 17.Gillet JP, Varma S, Gottesman MM. The clinical relevance of cancer cell lines. J Natl Cancer Inst. 2013;105:452–458. doi: 10.1093/jnci/djt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denekamp J. Cell kinetics and radiation biology. Int J Radiat Biol Relat Stud Phys Chem Med. 1986;49:357–380. doi: 10.1080/09553008514552591. [DOI] [PubMed] [Google Scholar]
- 19.Alper T, Cramp WA. The role of repair in radiobiology. Experientia. 1989;45:21–33. doi: 10.1007/BF01990449. [DOI] [PubMed] [Google Scholar]
- 20.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 21.Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat. 2012;15:39–49. doi: 10.1016/j.drup.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eke I, Cordes N. Radiobiology goes 3D: how ECM and cell morphology impact on cell survival after irradiation. Radiother Oncol. 2011;99:271–278. doi: 10.1016/j.radonc.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Hammond EM, Asselin MC, Forster D, O'Connor JP, Senra JM, Williams KJ. The meaning, measurement and modification of hypoxia in the laboratory and the clinic. Clin Oncol. 2014;26:277–288. doi: 10.1016/j.clon.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9:539–549. doi: 10.1038/bjc.1955.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tannock IF. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br J Cancer. 1968;22:258–273. doi: 10.1038/bjc.1968.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koch CJ, Jenkins WT, Jenkins KW, Yang XY, Shuman AL, Pickup S, Riehl CR, Paudyal R, Poptani H, Evans SM. Mechanisms of blood flow and hypoxia production in rat 9L-epigastric tumors. Tumor Microenviron Ther. 2013;1:1–13. doi: 10.2478/tumor-2012-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaupel P, Mayer A. Hypoxia in tumors: pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv Exp Med Biol. 2014;812:19–24. doi: 10.1007/978-1-4939-0620-8_3. [DOI] [PubMed] [Google Scholar]
- 28.Krause M, Prager J, Zhou X, Yaromina A, Dorfler A, Eicheler W, Baumann M. EGFR-TK inhibition before radiotherapy reduces tumour volume but does not improve local control: differential response of cancer stem cells and nontumourigenic cells? Radiother Oncol. 2007;83:316–325. doi: 10.1016/j.radonc.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 29.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 30.Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int J Radiat Oncol Biol Phys. 1979;5:85–91. doi: 10.1016/0360-3016(79)90044-0. [DOI] [PubMed] [Google Scholar]
- 31.Ashton JC. Drug combination studies and their synergy quantification using the Chou-Talalay method—letter. Cancer Res. 2015;75:2400. doi: 10.1158/0008-5472.CAN-14-3763. [DOI] [PubMed] [Google Scholar]
- 32.Vaux DL. Research methods: know when your numbers are significant. Nature. 2012;492:180–181. doi: 10.1038/492180a. [DOI] [PubMed] [Google Scholar]
- 33.Rofstad EK. Orthotopic human melanoma xenograft model systems for studies of tumour angiogenesis, pathophysiology, treatment sensitivity and metastatic pattern. Br J Cancer. 1994;70:804–812. doi: 10.1038/bjc.1994.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo TH, Kubota T, Watanabe M, Furukawa T, Kase S, Tanino H, Saikawa Y, Ishibiki K, Kitajima M, Hoffman RM. Site-specific chemosensitivity of human small-cell llung carcinoma growing orthotopically compared to subcutaneously in SCID mice: the importance of orthotopic models to obtain relevant drug evaluation data. Anticancer Res. 1993;13:627–630. [PubMed] [Google Scholar]
- 35.Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF, Tofilon PJ. Orthotopic growth of human glioma cells quantitatively and qualitatively influences radiation-induced changes in gene expression. Cancer Res. 2005;65:10389–10393. doi: 10.1158/0008-5472.CAN-05-1904. [DOI] [PubMed] [Google Scholar]
- 36.Anderson JC, Duarte CW, Welaya K, Rohrbach TD, Bredel M, Yang ES, Choradia NV, Thottassery JV, Yancey Gillespie G, Bonner JA. Kinomic exploration of temozolomide and radiation resistance in glioblastoma multiforme xenolines. Radiother Oncol. 2014;111:468–474. doi: 10.1016/j.radonc.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krause M, Baumann M, Thames HD. In regard to Solomon et al.: EGFR blockade with ZD1839 ("Iressa") potentiates the antitumor effects of single and multiple fractions of ionizing radiation in human A431 squamous cell carcinoma. IJROBP 2003;55:713-723. Int J Radiat Oncol Biol Phys. 2003;57:300–301. doi: 10.1016/s0360-3016(03)00512-1. [author reply 301] [DOI] [PubMed] [Google Scholar]
- 38.Baumann M, Krause M, Zips D, Eicheler W, Dörfler A, Ahrens J, Petersen C, Brüchner K, Hilberg F. Selective inhibition of the epidermal growth factor receptor tyrosine kinase by BIBX1382BS and the improvement of growth delay, but not local control, after fractionated irradiation in human FaDu squamous cell carcinoma in the nude mouse. Int J Radiat Biol. 2003;79:547–559. doi: 10.1080/0955300031000112839. [DOI] [PubMed] [Google Scholar]
- 39.Urano M, Suit HD. Experimental evaluation of tumor bed effect for C3H mouse mammary carcinoma and for C3H mouse fibrosarcoma. Radiat Res. 1971;45:41–49. [PubMed] [Google Scholar]
- 40.Steel GG, Hodgett J, Janik P. Cell population kinetics of a spontaneous rat tumour during serial transplantation. Br J Cancer. 1971;25:802–811. doi: 10.1038/bjc.1971.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desrosiers MF, DeWerd LA, Deye J, Lindsay P, Murphy MK, Mitch MG, Macchiarini F, Stojadinovic S, Stone HB. The importance of dosimetry standardization in radiobiology. NIST J Res. 2013;118:1–15. doi: 10.6028/jres.118.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zoetelief J, Broerse JJ, Davies RW, Octave-Prignot M, Rezvani M, Vergara JC, Toni MP. Protocol for X-ray dosimetry in radiobiology. Int J Radiat Biol. 2001;77:817–835. doi: 10.1080/09553000110050605. [DOI] [PubMed] [Google Scholar]
- 43.Smith MA, Houghton P. A proposal regarding reporting of in vitro testing results. Clin Cancer Res. 2013;19:2828–2833. doi: 10.1158/1078-0432.CCR-13-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Redig AJ, Janne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol. 2015;33:975–977. doi: 10.1200/JCO.2014.59.8433. [DOI] [PubMed] [Google Scholar]
- 45.Nam JM, Chung Y, Hsu HC, Park CC. Beta1 integrin targeting to enhance radiation therapy. Int J Radiat Biol. 2009;85:923–928. doi: 10.3109/09553000903232876. [DOI] [PubMed] [Google Scholar]
- 46.Pires IM, Bencokova Z, Milani M, Folkes LK, Li JL, Stratford MR, Harris AL, Hammond EM. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010;70:925–935. doi: 10.1158/0008-5472.CAN-09-2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Potiron VA, Abderrahmani R, Giang E, Chiavassa S, Di Tomaso E, Maira SM, Paris F, Supiot S. Radiosensitization of prostate cancer cells by the dual PI3K/mTOR inhibitor BEZ235 under normoxic and hypoxic conditions. Radiother Oncol. 2013;106:138–146. doi: 10.1016/j.radonc.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 48.Perez BA, Ghafoori AP, Lee CL, Johnston SM, Li Y, Moroshek JG, Ma Y, Mukherjee S, Kim Y, Badea CT. Assessing the radiation response of lung cancer with different gene mutations using genetically engineered mice. Front Oncol. 2013;3 doi: 10.3389/fonc.2013.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flatmark K, Ree AH. Radiosensitizing drugs: lessons to be learned from the oxaliplatin story. J Clin Oncol. 2010;28:e577–e578. doi: 10.1200/JCO.2010.30.0921. [author reply e581-573] [DOI] [PubMed] [Google Scholar]
- 51.Begley CG, Ellis LM. Drug development: raise standards for preclinical cancer research. Nature. 2012;483:531–533. doi: 10.1038/483531a. [DOI] [PubMed] [Google Scholar]
- 52.Bohannon J. Who's afraid of peer review? Science. 2013;342:60–65. doi: 10.1126/science.2013.342.6154.342_60. [DOI] [PubMed] [Google Scholar]
- 53.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- 55.Coleman CN, Lawrence TS, Kirsch DG. Enhancing the efficacy of radiation therapy: premises, promises, and practicality. J Clin Oncol. 2014;32:2832–2835. doi: 10.1200/JCO.2014.57.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colevas AD, Brown JM, Hahn S, Mitchell J, Camphausen K, Coleman CN, Radiation Modifier Working Group of the National Cancer I Development of investigational radiation modifiers. J Natl Cancer Inst. 2003;95:646–651. doi: 10.1093/jnci/95.9.646. [DOI] [PubMed] [Google Scholar]
- 57.Lawrence YR, Vikram B, Dignam JJ, Chakravarti A, Machtay M, Freidlin B, Takebe N, Curran WJ, Jr., Bentzen SM, Okunieff P. NCI-RTOG translational program strategic guidelines for the early-stage development of radiosensitizers. J Natl Cancer Inst. 2013;105:11–24. doi: 10.1093/jnci/djs472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–2146. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moreau P, Karamanesht II, Domnikova N, Kysellyova MY, Vilchevska KV, Doronin VA, Cakana A, van de Velde H, Deraedt W, Facon T. Pharmacokinetic, pharmacodynamic and covariate analysis of subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma. Clin Pharmacokinet. 2012;51:823–829. doi: 10.1007/s40262-012-0010-0. [DOI] [PubMed] [Google Scholar]
- 60.Kummar S, Gutierrez ME, Gardner ER, Chen X, Figg WD, Zajac-Kaye M, Chen M, Steinberg SM, Muir CA, Yancey MA. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur J Cancer. 2010;46:340–347. doi: 10.1016/j.ejca.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frohna P, Lu J, Eppler S, Hamilton M, Wolf J, Rakhit A, Ling J, Kenkare-Mitra SR, Lum BL. Evaluation of the absolute oral bioavailability and bioequivalence of erlotinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in a randomized, crossover study in healthy subjects. J Clin Pharmacol. 2006;46:282–299. doi: 10.1177/0091270005284193. [DOI] [PubMed] [Google Scholar]
- 62.Swaisland HC, Smith RP, Laight A, Kerr DJ, Ranson M, Wilder-Smith CH, Duvauchelle T. Single-dose clinical pharmacokinetic studies of gefitinib. Clin Pharmacokinet. 2005;44:1165–1177. doi: 10.2165/00003088-200544110-00004. [DOI] [PubMed] [Google Scholar]
- 63.van Zandwijk N. Tolerability of gefitinib in patients receiving treatment in everyday clinical practice. Br J Cancer. 2003;89:S9–S14. doi: 10.1038/sj.bjc.6601477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chu QS, Cianfrocca ME, Goldstein LJ, Gale M, Murray N, Loftiss J, Arya N, Koch KM, Pandite L, Fleming RA. A Phase I and pharmacokinetic study of lapatinib in combination with letrazole in patients with advanced cancer. Clin Cancer Res. 2008;14:4484–4490. doi: 10.1158/1078-0432.CCR-07-4417. [DOI] [PubMed] [Google Scholar]
- 65.Brain E, Isambert N, Dalenc F, Diéras V, Bonneterre J, Rezai K, Jimenez M, Mefti-Lacheraf F, Cottura E, Tresca P. Phase I study of lapatinib plus vinorelbine in patients with locally advanced or metastatic breast cancer overexpressing HER2. Br J Cancer. 2012;106:673–677. doi: 10.1038/bjc.2011.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fizazi K, Ducreux M, Ruffié P, Bonnay M, Daniel C, Soria JC, Hill C, Fandi A, Poterre M, Smith M. Phase I, dose-finding, and pharmacokinetic study of ralitrexed combined with oxaliplatin in patients with advanced cancer. J Clin Oncol. 2000;18:2293–2300. doi: 10.1200/JCO.2000.18.11.2293. [DOI] [PubMed] [Google Scholar]
- 67.Stathopoulos GP, Boulikas T, Kourvetaris A, Stathopoulos J. Liposomal oxaliplatin in the treatment of advanced cancer: a phase I study. Anticancer Res. 2006;26:1489–1494. [PubMed] [Google Scholar]
- 68.Sweeney CJ, Chiorean EG, Verschraegen CF, Lee FC, Jones S, Royce M, Tye L, Liau KF, Bello A, Chao R. A phase I study of sunitinib plus capecitabine in patients with advanced solid tumors. J Clin Oncol. 2010;28:4513–4520. doi: 10.1200/JCO.2009.26.9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Teo SK, Colburn WA, Thomas SD. Single-dose oral pharmacokinetics of three formulations of thalidomide in healthy male volunteers. J Clin Pharmacol. 1999;39:1162–1168. [PubMed] [Google Scholar]
- 70.Teo SK, Scheffler MR, Kook KA, Tracewell WG, Colburn WA, Stirling DI, Thomas SD. Thalidomide dose proportionality assessment following single doses to healthy subjects. J Clin Pharmacol. 2001;41:662–667. doi: 10.1177/00912700122010555. [DOI] [PubMed] [Google Scholar]
- 71.Shia H-S, Chao Y, Chen L-T, Yao T-J, Huang J-D, Chang J-Y, Chen P-J, Chuang T-R, Cihin Y-H, Whang-Peng J. Phase I and pharmacokinetic study of oral thalidomide in patients with advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2006;58:654–664. doi: 10.1007/s00280-006-0203-z. [DOI] [PubMed] [Google Scholar]
- 72.Deming DA, Ninan J, Bailey HH, Kolesar JM, Eickhoff J, Reid JM, Ames MM, McGovern RM, Alberti D, Marnocha R. A Phase I study of intermittently dosed vorinostat in combination with bortezomib in patients with advanced solid tumors. Invest New Drugs. 2014;32:323–329. doi: 10.1007/s10637-013-0035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, Harris C, Zweibel J, Wright JJ, Espinoza-Delgado I. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res. 2009;15:5250–5257. doi: 10.1158/1078-0432.CCR-08-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials.