

Abstract
In the 2008 WHO classification, chronic myeloid malignancies that share both myelodysplastic and myeloproliferative features define the myelodysplastic/myeloproliferative group, which includes chronic myelomonocytic leukemia, juvenile myelomonocytic leukemia, atypical chronic myeloid leukemia, refractory anemia with ring sideroblasts and thrombocytosis, and myelodysplastic/myeloproliferative unclassified. With the notable exception of refractory anemia with ring sideroblasts and thrombocytosis, there is much overlap among the various subtypes at the molecular and clinical levels, and a better definition of these entities, an understanding of their biology and an identification of subtype-specific molecular or cellular markers are needed. To address some of these challenges, a panel comprised of laboratory and clinical experts in myelodysplastic/myeloproliferative was established, and four independent academic MDS/MPN workshops were held on: 9th March 2013, in Miami, Florida, USA; 6th December 2013, in New Orleans, Louisiana, USA; 13th June 2014 in Milan, Italy; and 5th December 2014 in San Francisco, USA. During these meetings, the current understanding of these malignancies and matters of biology, diagnosis and management were discussed. This perspective and the recommendations on molecular pathogenesis, diagnosis and clinical characterization for adult onset myelodysplastic/myeloproliferative is the result of a collaborative project endorsed and supported by the MDS Foundation.
Introduction
The chronic myeloproliferative neoplasms are made up of diverse disorders, some with proliferative features and others with dysplastic hematopoiesis. They arise from a pluripotent lymphoid-myeloid stem cell or in some cases a more committed myeloid progenitor.1 In an attempt to improve their classification, the World Health Organization (WHO) divided them into three distinct categories: myeloproliferative neoplasms (MPNs), myelodysplastic syndromes (MDS) and a category with overlapping characteristics of both MDS and MPNs, referred to as myelodysplastic/myeloproliferative neoplasms (MDS/MPN) or ‘overlap MDS/MPN’.2 The MDS/MPN group is made up of chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia (JMML), atypical chronic myeloid leukemia (aCML), a ‘provisional entity’, refractory anemia with ring sideroblasts and thrombocytosis (RARS-T), and a ‘by exclusion’ subcategory, MDS/MPN unclassified (MDS/MPN-U) (Figure 1).3,4 Currently there is a paucity of published registry data on the precise incidence of the various subtypes, though there is a perception that the relative incidence of MDS/MPN is quite low. The current classification defines distinct biological entities with myelodysplastic and myeloproliferative features, considerable molecular heterogeneity, and the lack of specific genotypic markers.5 While monocytosis or eosinophilia foster recognition of CMML/JMML or chronic eosinophilic leukemia (CEL), respectively, the differentiation between aCML, MDS/MPN-U and MPN-U is often difficult. Candidate molecular pathways include JAK-STAT, mTOR, PI3K/AKT, MEK signaling cascades and epigenetic changes, most of which are of interest for developing targeted agents.6
Figure 1.
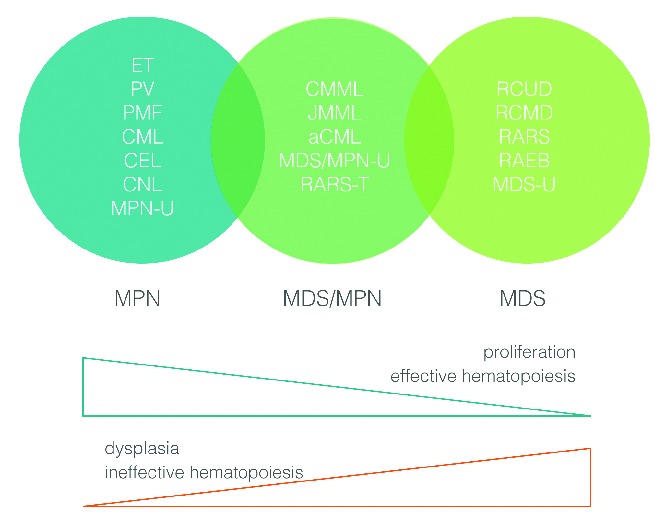
Myeloproliferative neoplasms and myelodysplastic syndromes.
To address some of the current challenges related to MDS/MPN, a panel comprised of laboratory and clinical experts in MDS/MPN was established, and four independent academic MDS/MPN workshops. These were held in Miami, Florida, USA (9th March 2013), in New Orleans, Louisiana, USA (6th December 2013), in Milan, Italy (13th June 2014), and in San Francisco, USA (5th December 2014), under the aegis of the MDS Foundation. In addition, several conference calls involving deliberations and discussions amongst the panellists took place between June 2013 and December 2014. A concise perspective and recommendations on molecular pathogenesis, diagnosis, clinical characterization and management of adult onset MDS/MPN based on the result of this collaborative initiative is summarized here; recommendations for uniform response in MDS/MPN have been submitted in a separate report.
MDS/MPN: cytogenetic, molecular genetics and signaling abnormalities
Chromosome analysis using conventional cytogenetics and high-resolution single nucleotide polymorphism array karyotyping (SNP-A) reveals chromosome abnormalities in 70% of MDS/MPN patients.7 Most of these are aneuploidies (trisomy 8, monosomy 7) or deletions (del7q, del13q, del20q); a minority have reciprocal translocations involving diverse tyrosine kinase (TK) fusion genes.8,9 Some of these fusions are listed separately within the current WHO classification: ‘myeloid and lymphoid neoplasms with eosinophilia’ (MLN-eo) and abnormalities of PDGFRA, PDGFRB and FGFR1. Fusions involving other kinases are also seen in patients with MDS/MPN or MPNs.10 Fusions involving PDGFRA, PDGFRB and ABL1 are important to recognize as they confer sensitivity to TK inhibitors (TKIs), such as imatinib.11 Other fusions involving FGFR1 or JAK2 are insensitive to imatinib but may respond to ponatinib or ruxolitinib, respectively.12–16
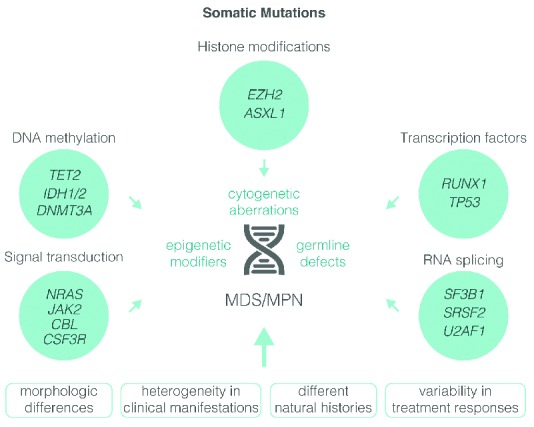
Most mutant genes fall into four functional classes: signaling, epigenetic, splicing and transcription (Figure 2).17–20 Signaling mutations result in aberrant activation of proliferative and anti-apoptotic pathways normally induced by growth factors (GFs). In addition to the TK gene fusions mentioned above, mutations have been described in GF receptors (CSF3R), downstream cytokine receptor signaling intermediates (JAK2, NRAS, KRAS) and negative regulators of signaling pathways (PTPN11, CBL, NF1).21–27 Mutations involving RAS are demonstrable in 90% of JMML cases and may emerge as a defining feature of this condition.28 Signaling mutations occur in approximately 50% of CMML patients and correlate with a myeloproliferative phenotype and enhancement of in vitro sensitivity to GM-CSF.29 Up to 80% of patients with RARS-T have activated JAK-STAT signaling as a consequence of the presence of JAK2V617F or mutations in MPL [encoding for the thrombopoietin receptor (Tpo-R)].30 In mice, abrogation of Notch signaling leads to a MDS/MPN phenotype, but its relevance in humans is unknown.31
Figure 2.
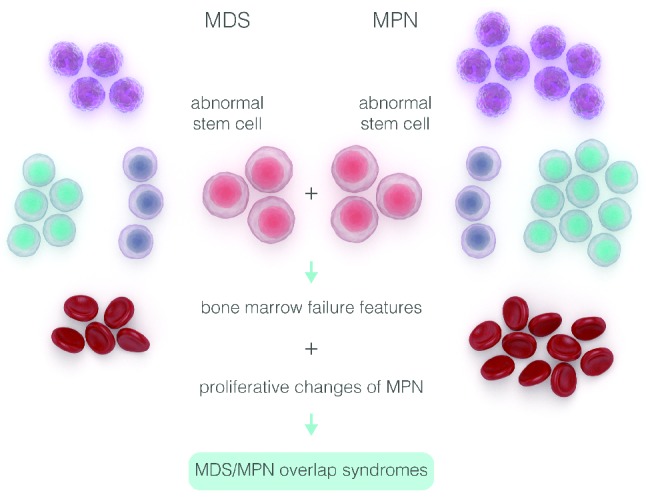
A schematic description of genotypic diversity in patients with myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN).
MDS/MPN: nuclear events - epigenetics, spliceosomes and transcription factors
Mutations in genes encoding epigenetic regulators are common in MDS/MPN.32–35 The most frequently mutated genes are TET2 and ASXL1, followed by SRSF2, IDH1/2, EZH2, SUZ12, EED and UTX.36 The interaction between epigenetic mutations is complex, and apart from the general mutual exclusivity of TET2 and IDH1/2 mutations, no clear patterns have emerged.37,38
Mutations in elements involved in the recognition and processing of 3′-mRNA splice sites are also common in MDS/MPN.39 Around 50% of CMML patients have mutations involving SRSF2, with a further 20% exhibiting mutations in other splicing complex genes (SF3B1, U2AF35, U2AF65 and SF3A1).34,35,40,41 In addition, SF3B1 mutations are present in 72% of patients with RARS-T.42,43 These SF3B1 mutations are not always mutually exclusive and may be accompanied by DNMT3, JAK2, ASXL1 and TET2 mutations. Functionally, disruption of SF3B1 function leads to the formation of ring sideroblasts; however, its exact role in malignant transformation remains unclear.20,44–47 Studies of mutant U2AF35 in model systems indicate global impairment of splicing induction of mRNA surveillance pathways and impairment of growth. Nevertheless, it is not known if the critical effect of such mutations is indeed global or whether they impact only a small subset of genes.
The RUNX1 gene is mutated in 15%–30% of CMML patients. RUNX1 encodes core-binding factor alpha (CBFα), which plays a fundamental role for definitive commitment of hematopoiesis. NPM1 and TP53 are mutated in only a small percentage of cases. SET binding protein 1 (SETBP1) was recently identified as a novel oncogene mutated in 25% of aCML cases, and less frequently in other MDS/MPN.48 The precise downstream consequences of SETBP1 mutations are unknown, but they may attenuate the activity of the tumor suppressor phosphatase, PP2A though abrogation of a ubiquitination site (functionally equivalent to overexpression). A small minority of MDS/MPN have calreticulin (CALR) mutations, which are more commonly associated with JAK2 and MPL unmutated MPN.49 Although somatically acquired in myeloid malignancies, mutations in SETBP1, ASXL1, EZH2 and other genes are also found in rare congenital developmental disorders of variable phenotypic severity. A likely unifying factor is that these mutations alter the expression of HOX genes that are important for both embryonic development and adult hematopoiesis.48,50
MDS/MPN: subtypes
Chronic myelomonocytic leukemia
The annual incidence of chronic myelomonocytic leukemia (CMML) is 1/100,000 adults, with a median age of 70 years and a male predominance.51 The BCR-ABL1 gene and rearrangements of either PDGFRA, PDGFRB or FGFR1 are absent. The JAKV617F mutation occurs in less than 10% of patients with CMML, in particular those with proliferative, rather than dysplastic features.52 Rarely, CMML can be therapy-related or a secondary neoplasm, arising in the background of MDS or as a progression of myelofibrosis (MF), in particular in the presence of an SRSF2 mutation.53,54
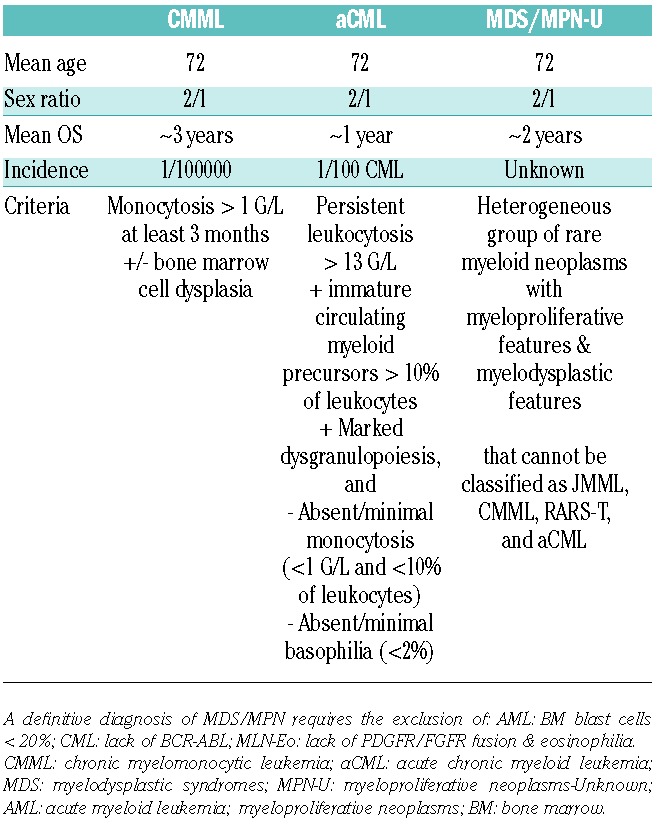
Although the diagnosis of CMML is based on laboratory, morphological and clinical parameters, the incorporation of molecular data is now recognized, with the notable presence of somatic mutations in TET2 (50%–60%), SRSF2 (40%–50%), ASXL1 (35%–40%) and RUNX1 (15%). Indeed, several investigators have noted that over 90% of CMML patients studied exhibited one or more mutations and that concurrent mutations in TET2 and SRSF2 appear to be highly specific for this entity.34,55,56 Other mutations include those affecting cytosine methylation (DNMT3A, IDH2, IDH1), RNA splicing (SF3B1, U2AF35, ZRSR2), chromatin remodeling (UTX, EZH2), and signaling pathways (NRAS, KRAS, CBL, JAK2, FLT3, CSF3R), whereas TP53 mutations are rare.33,55–58 A cardinal feature is persistent peripheral blood monocytosis more than 1×109/L, with a WBC percentage of monocytes of more than 10%. Morphologically, these monocytes demonstrate an abnormal appearance with bizarre nuclei and cytoplasmic granules.59 In some patients, blood cells identified as monocytes are later recognized to be dysplastic and immature granulocytes endowed with immunosuppressive properties.60 Clinical features include splenomegaly, skin and lymph node infiltration, and serous membrane effusions. The diagnostic criteria for CMML versus aCML versus MDS/MPN-U are shown in Table 1; RARS-T is a provisional entity that remains apart.
Table 1.
A potential diagnostic approach for patients suspected to have myelodysplastic/myeloproliferative neoplasms.
The current WHO classification divides CMML into two risk groups, CMML-1 and CMML-2, based on the number of blasts and promonocytes in the peripheral blood and bone marrow (BM) (Figure 3A–D).3 The BM is hypercellular with dysplasia and an increase in the ‘paramyeloid cells’; some patients may also have reticulin fibrosis.61 Recent data from the Dusseldorf registry also suggest the notion of a poorer outcome in ‘proliferative’ compared to ‘dysplastic’ CMML.62 Cytogenetic abnormalities include trisomy 8, monosomy 7, del(7q), and rearrangements with a 12p breakpoint.
Figure 3.
A photomicrograph from a patient with chronic myelomonocytic leukemia (CMML)-1. (A) Peripheral blood smear showing three abnormal monocytes and one neutrophil. (B and C) Bone marrow aspirate and the corresponding napthyl butyrate esterase image of the aspirate. (D) Bone marrow trephine biopsy.
Clonal architecture analysis in CMML has demonstrated linear acquisition of candidate mutations with limited branching through loss of heterozygosity.56 The principal CMML characteristics seem to be early clonal dominance arising within the CD34(+)/CD38(−) cells, and the subsequent granulo-monocytic differentiation skewing of progenitors. Based on this, a unique causal linkage between early clonal dominance and skewed granulo-monocytic differentiation has been proposed (Figure 4).63
Figure 4.
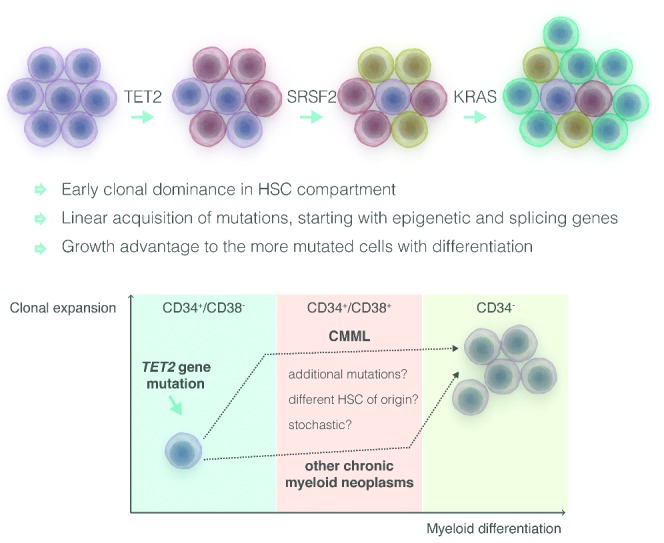
Early clonal dominance (CD34+/CD38−cells) in chronic myelomonocytic leukemia (CMML) compared to myeloproliferative neoplasms (MPN). Adapted from Itzykson et al.56
Another important biological feature is the unique hypersensitivity to GM-CSF, as measured by hematopoietic colony formation and GM-CSF-dependent phosphorylation of STAT5.29,64 This STAT5 pathway convergence is supported by transgenic models of mutated genes in CMML. Mouse models recapitulating mutations in TET2, JAK2, CBL, and NRAS have also been reported to up-regulate the STAT5 pathway and/or increase hematopoietic colony formation in a cytokine-dependent fashion. These novel observations support the candidacy of Janus kinase (JAK) inhibitors and other novel treatment strategies in future CMML clinical trials.
Most, if not all, of the prognostic tools in CMML have been derived from studies focused on MDS and preceded the use of hypomethylating agents (HMAs) (Table 2).65–71 Recent efforts include genetic information and clinical features.55,72 Solary and colleagues sequenced ASXL1 and other genes, including epigenetic (TET2, EZH2, IDH1, IDH2, DNMT3A), splicing (SF3B1, SRSF2, ZRSR2, U2AF1), transcription (RUNX1, NPM1, TP53), and signaling (NRAS, KRAS, CBL, JAK2, FLT3) regulators in 312 patients with CMML. They noted that ASXL1 mutations, age, hemoglobin, WBC, and platelet counts defined three prognostically distinct patient subsets with varied overall survival (Figure 5). Such and colleagues proposed a ‘CMML-specific prognostic scoring system’ (CPSS), based on cytogenetics and red blood cell (RBC) transfusion dependence, which divides patients into four risk groups for survival and risk of AML transformation.73 A third group of authors identified absolute monocyte count, presence of circulating immature myeloid cells, anemia and thrombocytopenia, but not the spliceosome complex nor ASXL1 mutations, as independent variables for survival.74 These investigators also confirmed the independent prognostic value of the SETBP1 mutation in CMML, initially reported by the Solary group.60,75
Table 2.
Principal historical CMML risk models.
Figure 5.
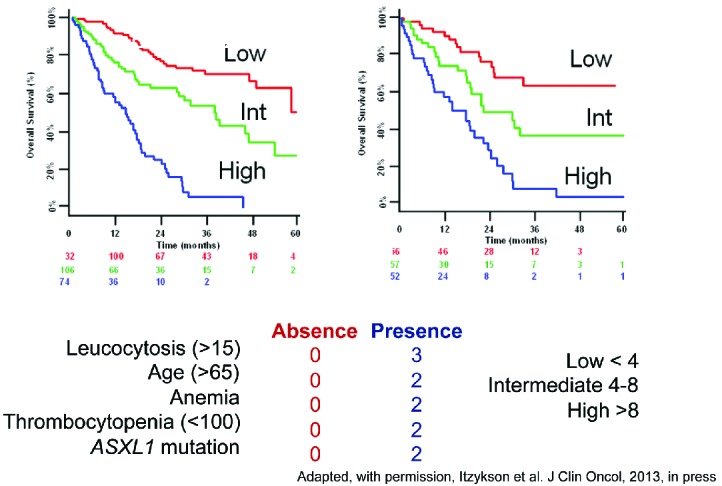
A simplified prognostic score for chronic myelomonocytic leukemia (CMML) that includes ASXL1 mutations. Adapted from Itzykson et al.55
The clinical management of patients with CMML can often be a challenge since some patients have a relatively indolent disorder with median survival in excess of ten years, whilst others progress rapidly to secondary AML, which is often difficult to treat. Allogeneic stem cell transplantation (allo-SCT) remains the only treatment modality associated with long-term remissions and potential cure; for transplant ineligible patients, there is no firm consensus with regards to the optimal treatment. The French registry data suggest a 3-year overall survival (OS) of 32% in a cohort of CMML patients allografted in chronic phase.76 Survival was negatively influenced by the presence of splenomegaly. Similar results have been reported by other groups, though few focused exclusively on patients with CMML.77 The Seattle group reported a 10-year OS of approximately 40%.78 Factors associated with favorable outcomes appear to be CMML risk group (CMML1 vs. CMML2), pre-transplant hematocrit, cytogenetic risk category, comorbidity index, and age. Intriguingly, in this series there appeared to be a gender influence on risk of relapse, with female-female transplants faring the worst. It was worthy of note that neither the type of pre-conditioning regimen [reduced intensity conditioning (RIC) vs. myeloablative] nor the type of pre-transplant therapy appeared to influence allo-SCT outcomes significantly. There was, however, a tendency for a lower relapse and a better survival with fludarbine and targeted busulfan conditioning. Most published results suggest disease relapse as a principal cause for transplant failure.
The historical results of conventional allo-SCT have been confounded by the substantial non-relapse transplant-related mortality (NRM). This is probably due, at least in part, to the older age of patients with CMML and the increasing presence of significant co-morbid conditions. Efforts to improve these results have led to general improvements in allo-SCT technology. These include strategies to enhance the graft-versus-leukemia (GvL) effects, which account for probable cure in those who achieve long-term remission, and an increased use of RIC preparative regimens. At present, the very considerable advances in the understanding of the genomic landscape in CMML, with the notable exception of ASXL1, appear not to have been validated sufficiently for adaptation in treatment algorithms to assess candidacy for allo-SCT compared to conventional therapy. Results of treatments for CMML patients who are either in frank AML transformation, or at high risk of transformation, remain suboptimal, with a median survival of 2.4 months for those who fail to achieve a complete remission following induction chemotherapy; for patients who achieve a complete remission following induction and then receive an allo-SCT, survival is about 28 months.79
Hypomethylating agents (HMAs) are currently the preferred non-transplant treatment option, though the response rates are relatively low, with no important impact on overall survival.80–83 Furthermore, even when responses are achieved, most tend to be short-lived. It is of interest that, in a recent study, ASXL1, RUNX1 and TET2 mutations portended a better response to decitabine, whereas MYB and JUN expression negatively affected outcome.84 Current efforts are investigating diverse agents, including JAK and MEK inhibitors, BCL-XL and BCL-2 inhibitors, clofarabine, next generation HMAs, and other novel agents.
The notion of using HMAs in order to improve the performance status and allo-SCT eligibility appears attractive. What is not known is the impact of HMAs on short and long-term outcomes following allo-SCT. There are no CMML transplant-specific risk scores other than the time-tested Gratwohl methodology for transplant recipients in general.85 An interesting compromise would be to offer a transplant only to patients with high-risk disease or patients with low-risk disease in whom parameters start to deteriorate. This may allow patients responding to HMAs to continue the therapy until resistance/intolerance, or indeed, progression of disease, are noted. The disadvantages with such an approach are the potential risks for leukemic transformation and the prolonged use of a therapy that has yet to demonstrate a durable survival benefit.
Atypical chronic myeloid leukemia
Atypical chronic myeloid leukemia (aCML) is an extremely rare subtype of MDS/MPN with an estimated incidence of 1% that of typical BCR-ABL1-positive CML.86 It was initially described as a subtype of myeloid neoplasm resembling CML, but with the notable absence of the BCR-ABL1 fusion gene. Diagnosis of aCML requires the exclusion of not only BCR-ABL1, but also rearrangement of PDGFRA, PDGFRB or FGFR1.3,87 Patients tend to have severe anemia, thrombocytopenia, neutrophilic leukocytosis with granulocytic dysplasia, and splenomegaly; monocytosis and basophilia are not prominent in the peripheral blood.88 In the clinic, aCML patients can be difficult to distinguish from those with another very rare MDS/MPN subtype, known as MDS/MPN-U.
Orazi and colleagues recently analyzed a series comprising 69 patients with aCML and 65 with MDS/MPN-U, in an effort to define clinical, histological and genetic characteristics which would help distinguish these two rare entities.89 They identified aCML patients to have an aggressive disease course, with a poor prognosis and an overall survival of 12.4 months, compared with 21.8 months for patients with MDS/MPN-U (P=0.004). They attempted to subclassify the study cohort by the presence of leukocytosis more than 13×109/L, peripheral blood myeloid precursors more than 10%, and dysgranulopoiesis more than 10% in patients with aCML (Figure 6). Median leukocytes for aCML was 40.8×109/L, compared to 19.4×109/L for those with MDS/MPN-U (P<0.001). Bone marrow (BM) samples revealed hypercellularity and dysgranulopoiesis in all patients with aCML, compared to about half of MDS/MPN-U patients; there was variable fibrosis and osteosclerosis, and non-specific recurrent complex cytogenetic abnormalities and i(17q) appeared to be slightly more frequent in aCML. aCML patients were also found to have increased LDH, splenomegaly, severe anemia, thrombocytopenia less than 100×109/L, higher peripheral blood myeloid precursors, and less than 2% basophils.
Figure 6.
Hematologic parameters in a cohort of 121 patients with atypical chronic myeloid leukemia (aCML).
Although no specific molecular abnormality has been described in aCML, recurrent mutations in SETBP1, located on chromosome 18q21.1, have been observed in 25% of aCML, 6%–15% of CMML and less than 3% of JMML cases.58,90–93 The functional significance of these mutations are not yet fully understood. Recurrent somatic mutations in JAK2, NRAS, IDH2, CBL, CSF3R and ETNK1 can also be detected in aCML, although at a much lower frequency; anecdotal cases with fusion genes such as BCR-JAK2 or NUP98-HOXA9 have also been detected.94–97 Future studies should provide insights into the potential impact of such analyses on precision-medicine therapeutic approaches. In this regard, the recent proposal of considering the reactivation of PP2A as a therapeutic strategy in SETBP1-mutated cells is of interest.98
There are also clinical and morphological similarities between aCML and chronic neutrophilic leukemia (CNL), a rare subtype of MPN. The genomic landscape, however, appears to be quite distinct. A seminal observation by Maxson and colleagues demonstrated the presence of mutated CSF3R in about 90% of patients with CNL and 40% of those with aCML; subsequent studies confirmed the high frequency in CNL but were unable to confirm the mutations in aCML.99–101 This gene encodes the receptor for colony-stimulating factor 3 (G-CSF).27 Somatic CSF3R mutations, together with ELANE, HAX1, and G6PC3 mutations have previously been described in severe congenital neutropenia (SCN).102 A germ-line T640N CSF3R mutation has also been identified in hereditary neutrophilia. Interestingly, a homologous CSF3R somatic mutation affecting the extracellular domain and conferring autonomous signaling properties has been found in sporadic transformed SCN and de novo AML.103 In sporadic cases, the most common CSF3R mutation is CSF3RT618I, which strongly activates the JAK/STAT pathway; however, CSF3R truncating mutations were also observed and these predominantly signal through SRC family kinases.104 Recently, a CALR mutation was reported in a case of CSF3R-positive CNL.105
Allo-SCT appears to be the only treatment that can accord aCML patients a long-term remission, though there is no firm consensus due to the extremely low incidence of this rare disease. Most of the published series, including registry data, include aCML as part of a more general series of myeloid malignancies. A recent report of 2 aCML patients with a heterozygous CSF3RT618I mutation is of some interest as it highlights the candidacy of this mutation to be used as a disease-specific biomarker of residual disease.106
Patients not suitable for allo-SCT often receive HMAs with some demonstrating transient improvements in some of the clinical and pathological features. Other treatments used include hydroxyurea and lenalidomide. It is best, therefore, to offer these patients suitable clinical trials. The notion of the CSF3R mutation activating the JAK/STAT pathway and, in some instances, the SRC kinases, provides some support for clinical trials to assess JAK inhibitors, such as ruxolitinib, and SRC inhibitors, such as dasatinib, respectively. A recent case report of a CSF3RT618I -positive-aCML patient treated with ruxolitinib showed a significant improvement in his constitutional symptoms and splenomegaly, providing additional support for such trials.107
Juvenile myelomonocytic leukemia
Juvenile myelomonocytic leukemia (JMML) is an uncommon WHO-defined MDS/MPN with an incidence of 0.12 per 100,000 children, a median age of two years, and a disproportionate male preponderance. It carries a poor prognosis108,109 and shares some clinical and molecular features with CMML. Congenital JMML predisposition syndromes exist, particularly neurofibromatosis and Noonan syndrome, which converge on RAS signaling abnormalities and markedly increase the risk of developing JMML110,111 JMML is a heterogeneous clinical entity in that some patients, particularly those with Noonan syndrome, have spontaneous resolution of their disease despite identification of clonal hematopoiesis, while others can have a fulminant course refractory to allo-SCT.112,113 Although leukemic transformation is seen in JMML, it is uncommon in comparison with adult myeloid malignancies.114
Clinically JMML is characterized by an overproduction of monocytes that infiltrate liver, spleen lung, intestine and other organs, which may also lead to considerable morbidity and mortality. The cardinal clinical features also include fever, thrombocytopenia, monocytosis, splenomegaly, hepatomegaly, hemoglobin F elevations, and failure to thrive. Despite a readily apparent diagnostic marker of disease (peripheral monocytosis), the diagnosis of JMML is not straightforward due to the extreme rarity of disease and confounding clinical characteristics in common with more common entities (such as viral infections).
The above notwithstanding, JMML is arguably considered the most well understood hematologic malignancy after CML, at least in children. Most, if not all, children with JMML harbor either a somatic or germ-line unique mutation in the Ras pathway (PTPN11> NF1>NRAS/KRAS>CBL).115,116 In rare cases, additional mutations in SETBP1 or JAK3 have been identified; these appear to confer a poorer prognosis.117 However, the mutational landscape of JMML distinguishes it from various adult myeloid malignancies in that the genetic abnormalities appear to be restricted to a limited set of genes, particularly excluding epigenetic and alternative splicing modifiers that are enriched in adults.118,119 It is also of some interest that most patients with JMML exhibit an increased in vitro sensitivity to GM-CSF.120 Despite the virtually universal dysregulation of RAS, this hypersensitivity appears to augment signaling of other downstream effectors, particular JAK/STAT.121,122 This has been demonstrated in human samples and murine models of NRAS-derived JMML.29
Allo-SCT remains the principal treatment for JMML, with an event-free 5-year survival of 52%.123 The principal cause for failure is relapse, which approaches 50%, though 50% of these patients can be rescued with a second allograft.124,125 It has been speculated that the high relapse rate might be related to an underlying fundamental immune defect or incomplete eradication of resistant disease prior to myeloablation. Strategies to rescue children post relapse remain suboptimal, with limited success of donor lymphocyte infusions (DLI).126
Current non-transplant alternatives are limited, and many efforts to target underlying driver mutations are in progress. Efforts to target the RAS proteins, which is involved in the vast majority of JMML patients, have met little success so far.127 The first generation of farnesyltransferase inhibitors (FTIs) have now been tested, but in view of unacceptable toxicities and poor efficacy they are no longer under development. Clinical trials are now in progress with MEK inhibitors, such as trametinib, JAK inhibitors, such as ruxolitinib, as well as SRC inhibitors and HMAs.128–130
RARS-T
Considerable debate remains as to how RARS-T is best characterized among chronic myeloid malignancies. It was provisionally defined by the WHO to be part of the MDS/MPN in 2001, since patients had MDS features of refractory anemia with ring sideroblasts (RARS) in addition to thrombocytosis and megakaryocyte cytological features resembling essential thrombocythemia (ET).131,132 By the time of the 2008 revision of the WHO classification, several reports indicated the presence of clonal JAK2V617F and MPLW515 gene mutations in RARS-T, favoring the notion that this sub-category should be considered an MPN akin to ET. Nonetheless, RARS-T displays poor in vitro colony forming capacity, a recognized feature of MDS.3 Distinction between RARS-T and RARS with moderate thrombocytosis has become more difficult following the WHO 2008 revisions that lowered the platelet threshold for RARS-T and ET from more than 600×109/L to more than 450×109/L. Therefore, RARS-T currently remains a ‘provisional’ member of the MDS/MPN family, in which entity mutations in SF3B1 (60%–80% of cases) may be responsible for mitochondrial iron overload in sideroblasts, ineffective erythropoiesis, and anemia (myelodysplastic features), while mutations in JAK2 or MPL are thought to be responsible for thrombocytosis (myeloproliferative features) (Figure 7).133,134 Furthermore, the notion of secondary RARS-T developing in RARS has also been suggested.135
Figure 7.
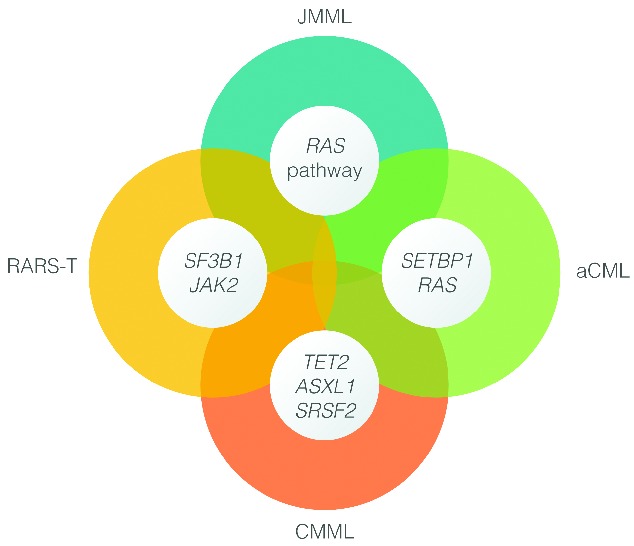
Emerging molecular fingerprints of myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN).
As in the case of the other MDS/MPN, there is no firm consensus regarding optimal clinical management and supportive care remains the cornerstone of treatment. Since the thrombotic risk in RARS-T appears to be low, there is no recommendation for platelet-suppressive therapy or aspirin prophylaxis. However, a recent report noted an increased rate of thrombotic events in RARS-T patients carrying SF3B1 mutations.136 There are anecdotal reports of ‘partial remission’ following the use of imatinib or lenalidomide.137–139 In the 60%–80% of RARS-T patients who harbor a JAK2 or MPL mutation, it is reasonable to consider a JAK inhibitor.
MDS/MPN-Unclassified
MDS/MPN-Unclassified (MDS/MPN-U) is quite possibly the most heterogeneous subgroup of MDS/MPN and includes patients who lack defining characteristics of the other MDS/MPN subtypes. Some patients with MDS/MPN-U may be phenotypically similar to those with aCML, but lack isolated granulocytic dysplasia and may have basophilia and megakaryocytic hyperplasia accompanied by intense BM fibrosis.89,140 MDS/MPN-U probably accounts for less than 5% of all myeloid malignancies. The recent MD Anderson Cancer Center (MDACC) series of 85 WHO-defined MDS/MPN-U patients is arguably the largest published series so far.141 These investigators elected to apply both MDS and MPN prognostic scoring systems to allow for the defining dysplastic and proliferative features. This, together with the recent work of Orazi and colleagues, which included patients from multiple institutions including the MDACC, allows us to have a better understanding of the pertinent clinical and biological features of MDS/MPN-U.89
Both series of WHO-defined MDS/MPN-U patients showed a median age of 71 years, a male predominance (around 2:1), presence of splenomegaly, low monocyte counts, 20%–30% JAK2V617F-positivity, and non-specific cytogenetic findings, with the exception of trisomy 8, which was the sole cytogenetic abnormality in 15% of the MDACC cohort. The principal differences in the series were the proportion of patients with thrombocytosis more than 450×109/L: 18% vs. 32%, and the median overall survival, which was considerably worse for the MDACC series: 12.4 months vs. 21.8 months. It is possible, but not certain, that the greater number of patients with thrombocytopenia (<100×109/L) in the MDACC series might reflect a biologically more aggressive disease resulting in the poorer survival. It would have been of interest to assess the AML-free survival of both series, which was 18.9 months in the Orazi series and not reported in the MDACC series. It was interesting that the MDS-IPSS model allocated 68% of the MDACC cohort as ‘low-risk’ despite the poor survival; conversely the MDA global model appeared to be a useful prognostic tool.142
Currently, there is no optimal treatment consensus for MDS/MPN-U patients who are ineligible for an allo-SCT. In the MDACC series, the majority of patients received HMA and the overall survival was better compared to ‘other’ approaches (16.4 months vs. 11.5 months). The other non-transplant treatments included interferon alpha, cyclosporine, thalidomide, lenalidomide and anti-thymocyte globulin.141 There is much interest in combining HMA with JAK inhibitors in the context of a clinical trial, given the moderate frequency of JAK2 mutations.
Transformation to AML in MDS/MPN
Transformation to acute myeloid leukemia (AML), which is often refractory to conventional treatment, is a challenging complication in MDS/MPN, as it is in MDS and MPN. The rate and incidence of AML transformation in MDS/MPN is unknown, with the exception of CMML and RARS-T. Most estimates are based on MPN patients who transform into AML. A French trial in PV estimated the risk of transformation of 24% at 15 years in patients treated with hydroxyurea or pipobroman; smaller series suggest a risk of 3%–40%.143–146
The incidence of CMML transformation (AML-M5) is 15%–52%, with higher white blood counts, marrow cellularity, karyotype risk score, and revised IPSS score associated with greater risk.147,148 The presence of ASXL1 or RUNX1 may also increase the transformation risk.149–152 Transformation in patients with RARS-T appears comparable to RARS patients (1.8 and 2.4 per 100 patient-years, respectively) and higher than that in ET.153 Collectively, MDS/MPN appears to have a higher risk of transformation compared to MPN, akin to that in MDS. It is, therefore, imperative to better characterize the incidence and potential for transformation risk in MDS/MPN.
Other candidate genetic events that have been linked to AML risk in MPN include TET2, IDH1/2, DNMT3A and EZH2 mutations.154,155 Cytogenetic progression, often involving abnormalities in chromosomes 7 (target genes EZH2, IKZF1), 8 (MYC), 17p (p53), 21 (ERG, RUNX1), and 12 (ETV6), is commonly observed at transformation. MDS/MPN with an isolated isochromosome (i)17p (leading to TP53 haploinsufficiency) may be a distinct disease entity with further increased risk of AML progression.156,157 It is possible that some patients may harbor sub-clones with mutations in TP53, which are only detected by next generation sequencing (NGS). Clearly this is important in view of the associated high risk of transformation, and perhaps an early consideration for allo-SCT.
Results of treatments for AML transformation in MDS/MPN, including allo-SCT, remain suboptimal, with a median survival of less than five months.158 Management is empiric and often is comprised of conventional cytotoxic combinations (used in de novo AML) or novel induction regimens, of both higher and lower intensity.79,159,160 Current efforts are investigating diverse agents, including hypomethylating agents, JAK and MEK inhibitors, BCL-XL and BCL-2 inhibitors, and clofarabine.161–164
Impact of symptom burden in patients with MDS/MPN
There has been considerable interest on the pathobiology of MDS and MPN-related symptoms and the potential impact of associated abnormalities, such as cytokine abnormalities and inflammation, on the overall prognosis. Furthermore, prospective assessment of disease-specific symptom burden and its impact on quality of life (QOL) has been found to be useful in clinical trials to assess benefit.165,166 Symptoms for MPN and MDS are similar, but disparate. For MPN patients, thromboembolic (macrovascular) and metabolic/catabolic symptoms are more prominent.167 MPN patients, particularly those with myelofibrosis (MF), frequently suffer from fever, night sweats, pruritus, bone pain, profound fatigue, weight loss, cachexia, as well as abdominal pain and distension.168,169 MDS patients sustain debilitating fatigue, infections, and cardiovascular complications in addition to significant age-associated co-morbidities.170
Symptom burden assessment has not been studied in MDS/MPN, which is likely to depict symptoms of both MDS and MPNs. We recommend a prospective symptom assessment study using the MPN-SAF TSS, EORTC-QOL-C30 and EQ-5D as initial symptom scales/questionnaires, along with an open-ended cognitive feedback tool capturing patients’ answers to specific symptom questions. Such efforts should lead to the collation of candidate questions from which a refined MDS/MPN specific symptom assessment tool could be developed and then prospectively validated.171
Novel strategies and future clinical trial designs for the treatment of MDS/MPN
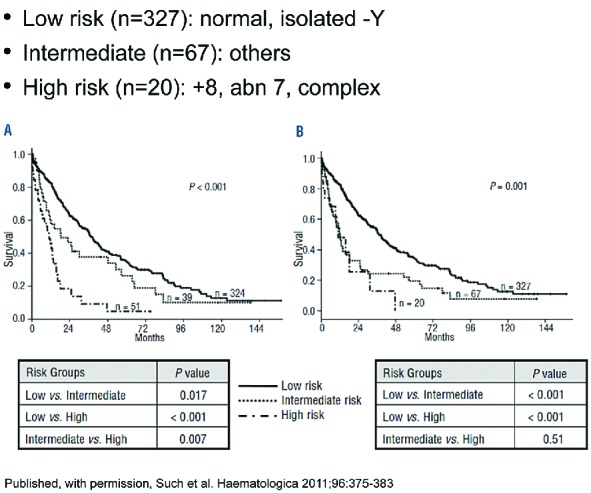
Since there is no treatment consensus for patients with MDS/MPN, strategies to improve outcomes must focus on rationally developed predictive and prognostic biomarkers based on molecular and clinical perspectives.172 Questions remain as to whether eligibility for future clinical studies should be restricted to WHO subtypes of MDS/MPN, such as a study for CMML patients alone versus broad inclusion of MDS/MPN-U patients, or whether the focus should be based on clinical disease phenotype or proliferative versus non-proliferative features. As we move forward with clinical studies based on targeted molecular pathways, these should ideally determine patient selection based upon ‘founder’ tyrosine kinase signaling pathway mutations or those with transcription factor mutations. The underlying molecular complexity of these diseases will be a significant challenge. It is critical to identify MDS/MPN specific therapeutic response criteria and suitable end points correlated with survival and AML risk for uniform assessment of treatment benefit. Moreover, criteria for disease progression or stability while on therapy ought to be introduced and vetted among experts.
The high frequency of SF3B1 mutations in RARS-T subtypes suggests that spliceosome inhibition may offer the prospect of selective synthetic lethality. Trials are currently evaluating the benefit of the JAK1/JAK2 inhibitor ruxolitinib in CMML patients, either as monotherapy or in combination with 5-azacytidine. Based on the pre-clinical data suggestive of GM-CSF dependent STAT-5 hypersensitivity in CMML, it would be reasonable to design trials assessing GM-CSF neutralizing antibodies (KB003) or JAK inhibitors. Other putative targets include small-molecule inhibitors directed against STAT3/5, MAPK, AKT, MEK and PI3K-mTORC pathways.173–175
Conclusion
The MPN/MDS group is a very heterogeneous group defined by WHO mainly on morphological grounds, especially the concomitance of cytopenia(s) and at least one “cytosis”. Current studies suggest considerable genetic complexity and heterogeneity in MDS/MPN.176 Most patients with JMML, and up to 50% of cases with other subtypes, have mutations that directly activate proliferative signaling pathways. Over 30 recurrent gene mutations have now been identified, whereas in the case of CMML, there may be 5–20 such gene mutations per case, suggesting a multi-step and highly variable molecular pathogenesis. Collectively, TET2, ASXL1 and SRSF2 represent the most commonly mutated genes.
Importantly, at present there are no specific mutations in MDS/MPN that stringently define particular subtypes. Nevertheless, some clear associations have emerged, including SF3B1 and JAK2 mutations in RARS-T, and SETBP1 aCML. Understanding clonal hierarchies should serve as a cornerstone for development of a robust molecular classification of MDS/MPN, as well as molecular predictors of prognosis and therapeutic response. An immediate initiative to consider is the set-up of large registries for these rare hematologic malignancies, along with collaborative efforts to define risk models and suitable end points for clinical trials. Outside of clinical trials, allo-SCT remains the most viable treatment options for the eligible patients with about one-third of patients achieving long-term remission and probable cure. For those who are not transplant candidates and who have no recourse to a clinical trial, it appears reasonable to consider HMAs in the first instance, except for those who have JAK2-mutant disease, for whom a trial of a JAK inhibitor might be indicated.
Acknowledgments
The members of this MDS/MPN International Working Group wish to thank the MDS International Foundation and the Alpine Oncology Foundation, in particular Dr. Alpa Parmar, who helped to organize the various workshops and discussion groups. They are grateful for research funding from Incyte Corporation and Celgene International towards this project. The content is the sole responsibility of the authors.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Vardiman J, Brunning RD, Arber DA, et al. Introduction and overview of the classification of myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. World Health Organization Classification of tumours of haematopoietic and lymphoid tissue. 4th ed. IARC Press: Lyon; 2008: p. 18–30. [Google Scholar]
- 2.Vardiman J, Melo J, Baccarani M, Thiele J. Chronic myelogenous leukaemia, BCR-ABL1-positive. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. World Health Organization Classification of tumours of haematopoietic and lymphoid tissue. 4th ed. Lyon: IARC Press; 2008: p. 32–37. [Google Scholar]
- 3.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. [DOI] [PubMed] [Google Scholar]
- 4.Orazi A, Germing U. The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia. 2008;22(7):1308–1319. [DOI] [PubMed] [Google Scholar]
- 5.Hebeda KM, Fend F. Changed concepts and definitions of myeloproliferative neoplasms (MPN), myelodysplastic syndromes (MDS) and myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in the updated 2008 WHO classification. J Hematopath. 2009;2(4):205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radich J. Genetically informed therapy in leukemia. N Engl J Med. 2013; 368(19):1838–1839. [DOI] [PubMed] [Google Scholar]
- 7.Tiu RV, Gondek LP, O’Keefe CL, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011; 117(17):4552–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cazzola M, Malcovati L, Invernizzi R. Myelodysplastic/myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2011;264–72. [DOI] [PubMed] [Google Scholar]
- 9.Delhommeau F, Pisani DF, James C, et al. Oncogenic mechanisms in myeloproliferative disorders. Cell Mol Life Sci. 2006; 63(24):2939–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steensma DP, Dewald GW, Lasho TL, et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and the myelodysplastic syndrome. Blood. 2005;106(4):1207–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. [DOI] [PubMed] [Google Scholar]
- 12.James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 muttaion leading to constitutive signalling causes polycthemia vera. Nature. 2005;434(7037):1144–1148. [DOI] [PubMed] [Google Scholar]
- 13.Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes is a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003; 348(13):1201–1214. [DOI] [PubMed] [Google Scholar]
- 14.Chase A, Bryant C, Score J, Cross NC. Ponatinib as targeted therapy for FGR1 fusions associated with the 8p11 myeloproliferative syndrome. Haematologica. 2003;98(1):103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lierman E, Selleslag D, Smits S, Bilet J, Vandenberghe P. Ruxolitinib inhibits transforming JAK2 fusion proteins in vitro and induces complete remission in t(8;9)(p22;p24)/PCM1-JAK2-positive chronic eosinophilic leukemia. Blood. 2012; 120(7):1529–1531. [DOI] [PubMed] [Google Scholar]
- 16.Chase A, Bryant C, Score J, et al. Ruxolitinib as potential targeted therapy for patients with JAK2 rearrangements. Haematologica. 2013;98(3):404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohlmann A, Grossmann V, Haferlach T. Integration of next-generation sequencing into clinical practice: are we there yet? Semin Oncol. 2012;39(1):26–36. [DOI] [PubMed] [Google Scholar]
- 18.Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–726. [DOI] [PubMed] [Google Scholar]
- 19.Score J, Cross NC. Acquired uniparental disomy in myeloproliferative neoplasms. Hematol Oncol Clin North Am. 2012;26(5):981–991. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida K. Sanada M, Shiraishi Y, et al. Frequent pathways mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. [DOI] [PubMed] [Google Scholar]
- 21.Jones AV, Kreil S, Zoi K, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106(6):2162–2168. [DOI] [PubMed] [Google Scholar]
- 22.Savage N, George TI, Gotlib J. Myeloid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, and FGFR1: a review. Int J Lab Hematol. 2013;35(5):491–500. [DOI] [PubMed] [Google Scholar]
- 23.Kratz CP, Niemeyer CM, Castleberry RP, et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood. 2005;106(6):2183–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bumm TG, Elsea C, Corbin AS, et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res. 2006;66(23):11156–11165. [DOI] [PubMed] [Google Scholar]
- 25.Grand FH, Hidalgo-Curtis CE, Ernst T, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009;113(24):6182–6192. [DOI] [PubMed] [Google Scholar]
- 26.Haferlach C, Grossmann V, Kohlmann A, et al. Deletion of the tumor-suppressor gene NF1 occurs in 5% of myeloid malignancies and is accompnaied by a muttaion in the remaining allele in half of the cases. Leukemia. 2012;26(4):834–839. [DOI] [PubMed] [Google Scholar]
- 27.Maxson JE, Gotlib J, Pollea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flotho C, Steinemann D, Mullighan CG, et al. Genome-wide single nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene. 2007;26(39):5816–5821. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Liu Y, Li Z, et al. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood. 2010;116(26):5991–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sattler M, Durstin MA, Frank DA, et al. RARS-T & Tpo-R The thrombopoietin receptor c-MPL activates JAK2 and TYK2 tyrosine kinases. Exp Hematol. 1995; 23(9):1040–1048. [PubMed] [Google Scholar]
- 31.Klinakis A, Lobry C, Abdel-Wahab O, et al. A novel tumor-suppressor function for Notch pathway in myeloid leukemia. Nature. 2011;473(7346):230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid. Nat Rev Cancer. 2012; 12(9):599–612. [DOI] [PubMed] [Google Scholar]
- 33.Jankowska AM, Makishima H, Tiu RV, et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118(14):3932–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meggendorfer M, Roller A, Haferlach T, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120(15):3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makishima H, Visconte V, Sakaguchi H, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gelsi-Boyer V, Trouplin V, Adelaide J, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145(6):788–800. [DOI] [PubMed] [Google Scholar]
- 37.Smith AE, Mohamedali AM, Kulasekararaj A, et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood. 2010;116(19):3923–3932. [DOI] [PubMed] [Google Scholar]
- 38.Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pandit S, Zhou Y, Shiue L, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013;50(2):223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kar SA, Jankowska A, Makishima H, et al. Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia. Haematologica. 2013;98(1):107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Przychodzen B, Jerez A, Guinta K, et al. Patterns of missplicing due to somatic U2AFimutations in myeloid neoplasms. Blood. 2013;122(6):999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Visconte V, Makishima H, Jankowska A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ringed sideroblasts. Leukemia. 2012;26(3):542–545. [DOI] [PubMed] [Google Scholar]
- 44.Hirabayashi S, Flotho C, Moetter J, et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood. 2012;119(11):e96–99. [DOI] [PubMed] [Google Scholar]
- 45.Visconte V, Rogers HJ, Singh J, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012;120(16):3173–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanada M, Suzuki T, Shih LY, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460(7257):904–908. [DOI] [PubMed] [Google Scholar]
- 47.Li Z, Cai X, Cai CL, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118(17):4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piazza R, Valletta S, Winkelmann N, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45(1):18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emanuel PD. Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia. Leukemia. 2008;22(7):1335–1342. [DOI] [PubMed] [Google Scholar]
- 51.Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood. 2008;112(1):45–52. [DOI] [PubMed] [Google Scholar]
- 52.Patnaik MM, Parikh SA, Hanson CA, Tefferi A. Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol. 2014; 165(3):273–286. [DOI] [PubMed] [Google Scholar]
- 53.Wang SA, Galili N, Cerny J, et al. Chronic myelomonocytic leukemia evolving from preexisting myelodysplasia shares many features with de novo disease. Am J Clin Pathol. 2006;126(5):789–797. [DOI] [PubMed] [Google Scholar]
- 54.Boiocchi L, Espinal–Witter R, Geyer JT, et al. Development of monocytosis in patients with primary myelofibrosis indicates an accelerated phase of the disease. Mod Pathol. 2013;26(2):204–212. [DOI] [PubMed] [Google Scholar]
- 55.Itzykson R, Kosmider O, Renneville A, et al. Prognostic Score Including Gene Mutations in Chronic Myelomonocytic Leukemia. J Clin Oncol. 2013;31(19):2428–2436. [DOI] [PubMed] [Google Scholar]
- 56.Itzykson R, Kosmider O, Renneville A, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186–2198. [DOI] [PubMed] [Google Scholar]
- 57.Damm F, Itzykson R, Kosmider O, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27(6):1401–1403. [DOI] [PubMed] [Google Scholar]
- 58.Kosmider O, Itzykson R, Chesnais V, et al. Mutation of the Colony-Stimulating Factor-3 Receptor gene is a rare event with poor prognosis in chronic myelomonocytic leukemia. Leukemia. 2013;27(9):1946–1949. [DOI] [PubMed] [Google Scholar]
- 59.Itzykson R, Solary E. An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia. 2013;27(7):1441–1450. [DOI] [PubMed] [Google Scholar]
- 60.Orazi A, Bennett J, Germing U, Brunning RD, Bain BJ, Thiele J. Chronic myelomonocytic leukaemia. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. World Health Organization Classification of tumours of haematopoietic and lymphoid tissue. 4th ed. Lyon: IARC Press; 2008. p. 76–79. [Google Scholar]
- 61.Orazi A, Chiu R, O’Malley DP, et al. Chronic myelomonocytic leukemia: the role of bone marrow biopsy immunohistology. Mod Pathol. 2006;19(12):1536–1545. [DOI] [PubMed] [Google Scholar]
- 62.Droin N, Jacquel A, Hendra JB, et al. Alpha-defensins secreted by dysplastic granulocytes inhibit the differentiation of monocytes in chronic myelomonocytic leukemia. Blood. 2010;115(1):78–88. [DOI] [PubMed] [Google Scholar]
- 63.Schuler E, Schroeder M, Neukirchen J, et al. Refined medullary blast and white blood cell count based classification of chronic myelomonocytic leukemia. Leuk Res. 2014;38(12):1413–1419. [DOI] [PubMed] [Google Scholar]
- 64.Padron E, Painter JA, Kunigal S, et al. GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood. 2013;121(25):5068–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bennett JM, Catovsky D, Daniel MT, et al. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol. 1994;87(4):746–754. [DOI] [PubMed] [Google Scholar]
- 66.Padron E, Abdel-Wahab O. The importance of genetics in the clinical management of chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2374–2376. [DOI] [PubMed] [Google Scholar]
- 67.Onida F, Kantarjian HM, Smith TL, et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99(3):840–849. [DOI] [PubMed] [Google Scholar]
- 68.Worsley A, Oscier DG, Stevens J, et al. Prognostic features of chronic myelomonocytic leukaemia: a modified Bournemouth score gives the best prediction of survival. Br J Haematol. 1988;68(1):17–21. [DOI] [PubMed] [Google Scholar]
- 69.Gonzalez-Medina I, Bueno J, Torrequebrada A, et al. Two groups of chronic myelomonocytic leukaemia: myelodysplastic and myeloproliferative. Prognostic implications in a series of a single center. Leuk Res. 2002;26(9):821–824. [DOI] [PubMed] [Google Scholar]
- 70.Germing U, Kundgen A, Gattermann N. Risk assessment in chronic myelomonocytic leukemia (CMML). Leuk Lymphoma. 2004;45(7):1311–1318. [DOI] [PubMed] [Google Scholar]
- 71.Such E, Cervera J, Costa D, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96(3):375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008; 113(6):1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Such E, Germing U, Malcovati L, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013; 121(15):3005–3015. [DOI] [PubMed] [Google Scholar]
- 74.Patnaik MM, Padron E, Laborde RR, et al. Mayo prognostic model for WHO-Defined chronic myelomonocytic leukemia-ASXL1 and spliceosome component mutations and outcomes. Leukemia. 2013;27(7):1504–1510. [DOI] [PubMed] [Google Scholar]
- 75.Laborde RR, Patnaik MM, Lasho TL, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013;27(10): 2100–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park S, Labopin M, Yakoub-Agha I, et al. Allogeneic stem cell transplantation for chronic myelomonocytic leukemia: a report from the Societe Francaise de Greffe de Moelle et de Therapie Cellulaire. Eur J Haematol. 2013;90(5):355–364. [DOI] [PubMed] [Google Scholar]
- 77.Kroger N. Allogeneic stem cell transplantation for elderly patients with myelodysplastic syndrome. Blood. 2012;119(24):5632–5639. [DOI] [PubMed] [Google Scholar]
- 78.Eissa H, Gooley TA, Sorror ML, et al. Allogeneic Hematopoietic Cell Transplantation for Chronic Myelomonocytic Leukemia: Relapse-Free Survival is Determined by Karyotype and Co-morbidities. Biol Blood Marrow Transplant. 2011;17(6):908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gonsalves W, Gangat N, Gupta V, et al. The role of induction chemotherapy and allogeneic stem cell transplantation in patients with chronic myelomonocytic leukemia who have undergone leukemia transformation. Biol Blood Marrow Transplant. 2014;20:S151–S164, abstract 216. [Google Scholar]
- 80.Wiermans PW, Rüter B, Baer MR, Slack JL, Saba HI, Lübbert M. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). Leuk Res. 2008;32(4):587–591. [DOI] [PubMed] [Google Scholar]
- 81.Wong E, Seymour J, Kenealy M, Westerman D, Herbert K, Dickenson M. Treatment of chronic myelomonocytic leukemia with azacytidine. Leuk Lymphoma. 2013;54(4): 878–880. [DOI] [PubMed] [Google Scholar]
- 82.Ades L, Sekeres MA, Wolfromm A, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37(6):609–613. [DOI] [PubMed] [Google Scholar]
- 83.Durairaj S, Keenan N, Hyslop A, Groves MJ, Bowen DT, Tauro S. Azacitidine-eligibility in higher-risk myelodysplastic syndromes and chronic myelomonocytic leukaemia: a registry-based study. Br J Haematol. 2013;161(2):280–282. [DOI] [PubMed] [Google Scholar]
- 84.Braun T, Itzykson R, Renneville A, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118(14):3824–3831. [DOI] [PubMed] [Google Scholar]
- 85.Gratwohl A, Stern M, Brand R. Risk score for outcome after allogeneic hematopoietic stem cell transplantation. Cancer. 2009;115(20):4715–4726. [DOI] [PubMed] [Google Scholar]
- 86.Fend F, Horn T, Koch I, Vela T, Orazi A. Atypical chronic myeloid leukemia as defined in the WHO classification is a JAK2 V617F negative neoplasm. Leuk Res. 2008;32(12):1931–1935. [DOI] [PubMed] [Google Scholar]
- 87.Hernandez JM, del Canizo MC, Cuneo A, et al. Clinical, hematological, cytogenetic characteristics of atypical chronic myeloid leukemia. Ann Oncol. 2000;11(4):441–444. [DOI] [PubMed] [Google Scholar]
- 88.Breccia M, Biondo F, Latagliata R, Carmosino I, Mandelli F, Alimena G. Identification of risk factors in atypical chronic myeloid leukemia. Haematologica. 2006;91(11):1566–1568. [PubMed] [Google Scholar]
- 89.Wang SA, Hasserjian RP, Fox PS, et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood. 2014;123(17):2645–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoischen A, van Bon BW, Gilissen C, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42(6):483–485. [DOI] [PubMed] [Google Scholar]
- 91.Oakley K, Han Y, Vishwakarma BA, et al. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood. 2012;119(25):6099–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Trimarchi T, Ntziachristos P, Aifantis I. A new player SETs in myeloid malignancy. Nat Genet. 2013;45(8):846–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu Y, Yin J, Pan J, et al. A BCR-JAK2 fusion gene from ins(22;9)(q11;p13p24) in a patient with atypical chronic myeloid leukemia. Leuk Lymphoma. 2013;54(10):2322–2324. [DOI] [PubMed] [Google Scholar]
- 95.Bellesso M, Santucci R, Dias DF, Centrone R, Elias RC. Atypical chronic myeloid leukemia with t(9;22)(p24,11.2), a BCR-JAK2 fusion gene. Rev Bras Hematol Hemoter. 2013;35(3):218–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Murayama H, Matsushita H, Ando K. Atypical chronic myeloid leukemia harboring NUP98-HOXA9. Int J Hematol. 2013;98(2):143–144. [DOI] [PubMed] [Google Scholar]
- 97.Gambacorti-Passerini C, Donadoni C, Parmiani A, et al. Recurrent ETNK1 mutations in aytipcal chronic myeloid leukemia. Blood. 2015;125(3):499–503. [DOI] [PubMed] [Google Scholar]
- 98.Cristóbal I, Garcia-Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia. 2011;25(4):606–614. [DOI] [PubMed] [Google Scholar]
- 99.Gotlib J, Maxson JE, George TI, Tyner JW. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013;122(10):1707–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pardanani A, Lasho TL, Laborde RR, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 2013;27(9):1870–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beekman R, Valkhof MG, Sanders MA, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood. 2012;119(22): 5071–5077. [DOI] [PubMed] [Google Scholar]
- 103.Plo I, Zhang Y, Le Couédic JP, et al. An activating mutation in the CSF3R gene induces a hereditary chronic neutrophilia. J Exp Med. 2009;206(8):1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fleischman AG, Maxson JE, Luty SB, et al. The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood. 20134;122(22):3628–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tefferi A, Thiele J, Vannucchi AM, Barbui T. An overview of CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia. 2014;28(7):1407–1413. [DOI] [PubMed] [Google Scholar]
- 106.Langabeer SE, McCarron SL, Haslam K, O’Donovan MT, Conneally E. The CSF3R T618I mutation as a disease-specific marker of atypical CML post allo-SCT. Bone Marrow Transplantat. 2014;49(6):843–84. [DOI] [PubMed] [Google Scholar]
- 107.Dao KH, Solti MB, Maxson JE, et al. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk Res Rep. 2014;3(2):67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chang TY, Dvorak CC, Loh ML. Bedside to bench in juvenile myelomonocytic leukemia: insights into leukogenesis from a rare pediatric leukemia. Blood. 2014;124(16): 2487–2497. [DOI] [PubMed] [Google Scholar]
- 109.Passmore SJ, Chessells JM, Kempski H, Hann IM, Brownbill PA, Stiller CA. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol. 2003;121(5): 758–767. [DOI] [PubMed] [Google Scholar]
- 110.Fernandez-Mercado M, Pellagatti A, Di Genua C, et al. Mutations in SETBP1 are recurrent in myelodysplastic syndromes and often coexist with cytogenetic markers associated with disease progression. Br J Haematol. 2013;163(2):235–239. [DOI] [PubMed] [Google Scholar]
- 111.Bader-Meunier B, Tchernia G, Mielot F, et al. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr. 1997;130(6):885–889. [DOI] [PubMed] [Google Scholar]
- 112.Miles DK, Freedman MH, Stephens K, et al. Patterns of hematopoietic lineage involvement in children with neurofibromatosis type 1 and malignant myeloid disorders. Blood. 1996;88(11):4314–4320. [PubMed] [Google Scholar]
- 113.Matsuda K, Shimada A, Yoshida N, et al. Spontaneous improvement of hematologic abnormalities in patients having juvenile myelomonocytic leukemia with specific RAS mutations. Blood. 2007;109(12):5477–5480. [DOI] [PubMed] [Google Scholar]
- 114.Bastida P, Garcia-Minaur S, Ezquieta B, Dapena JL, Sanchez de Toledo J. Myeloproliferative disorder in Noonan syndrome. J Pediatr Hematol Oncol. 2011;33(1): e43–45. [DOI] [PubMed] [Google Scholar]
- 115.Shiba N, Ohki K, Park MJ, et al. SETBP1 mutations in juvenile myelomonocytic leukaemia and myelodysplastic syndrome but not in paediatric acute myeloid leukaemia. Br J Haematol. 2014;164(1):156–159. [DOI] [PubMed] [Google Scholar]
- 116.Sakaguchi H, Okuno Y, Muramatsu H, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45(8):937–941. [DOI] [PubMed] [Google Scholar]
- 117.Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hirabayashi S, Flotho C, Moetter J, et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood. 2012;119(11):e96–99. [DOI] [PubMed] [Google Scholar]
- 119.Perez B, Kosmider O, Cassinat B, et al. Genetic typing of CBL, ASXL1, RUNX1, TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct from chronic myelomonocytic leukaemia. Br J Haematol. 2010;151(5):460–468. [DOI] [PubMed] [Google Scholar]
- 120.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77(5):925–929. [PubMed] [Google Scholar]
- 121.Kotecha N, Flores NJ, Irish JM, et al. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell. 2008;14(4):335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mughal TI, Girnius S, Rosen S, Kumar S, et al. Emerging therapeutic paradigms to target the deregulated JAK/STAT pathways in hematological malignancies. Leuk Lymphoma. 2014;55(9):1968–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maguire AM, Vowels MR, Russell S, et al. Allogeneic bone marrow transplant improves outcome for juvenile myelomonocytic leukaemia. J Paediatr Child Health. 2002;38(2):166–169. [DOI] [PubMed] [Google Scholar]
- 124.Woods WG, Barnard DR, Alonzo TA, et al. Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children’s Cancer Group. J Clin Oncol. 2002;20(2):434–440. [DOI] [PubMed] [Google Scholar]
- 125.Yoshimi A, Mohamed M, Bierings M, et al. Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia. Leukemia. 2007;21(3):556–560. [DOI] [PubMed] [Google Scholar]
- 126.Yoshimi A, Bader P, Matthes-Martin S, et al. Donor leukocyte infusion after hematopoietic stem cell transplantation in patients with juvenile myelomonocytic leukemia. Leukemia. 2005;19(6):971–977. [DOI] [PubMed] [Google Scholar]
- 127.Downward J. Targeting RAS signaling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. [DOI] [PubMed] [Google Scholar]
- 128.Bunda S, Kang MW, Sybingco SS, et al. Inhibition of SRC corrects GM-SCF hypersensitivity that underlies juvenile myelomonocytic leukemia. Cancer Res. 2013;73(8):2540–2550. [DOI] [PubMed] [Google Scholar]
- 129.Kong G, Wunderlich M, Yang D, et al. Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm. J Clin Invest. 2014;124(6):2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Furlan I, Balz C, Flotho C, et al. Intriguing response to azacitidine in a patient with myelomonocytic leukemia and monosomy 7. Blood. 2009;113(12):2867–2868. [DOI] [PubMed] [Google Scholar]
- 131.Jaffe ES, Harris NL, Stein H, Vardiman JW. (eds). WHO Classification: Tumours of Hematopoeitic and Lymphoid Tissues. International agency For Research on Cancer (IARC) Press, Lyon: 2001. p. 9–14. [Google Scholar]
- 132.Wardrop D, Steensma DP. Is refractory anaemia with ring sideroblasts and thrombocytosis (RARS-T) a necessary or useful diagnostic category? Br J Haemtol. 2008;144(6):809–817. [DOI] [PubMed] [Google Scholar]
- 133.Szpurka H, Tiu R, Murugesan G, et al. Refrcatory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myloproliferative condition characterized by JAK2V617F mutation. Blood. 2006;108(7):2173–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ceesay MM, Lea NC, Ingram W, et al. The JAK2 V617F mutation is rare in RARS but common in RARS-T. Leukemia. 2006; 20(11):2060–2061. [DOI] [PubMed] [Google Scholar]
- 135.Malcovati L, Della-Porta MG, Pietra D, et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood. 2009;114(17):3538–3545. [DOI] [PubMed] [Google Scholar]
- 136.Visconte V, Makishima H, Jankowska A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012;26(3):542–545. [DOI] [PubMed] [Google Scholar]
- 137.Cannizzo E, Carulli G, Azzara A, et al. JAK2V617F mutation in RARS-t: a target for Imatinib therapy? Leuk Res. 2008;32(10): 1636–1637. [DOI] [PubMed] [Google Scholar]
- 138.Taylor G, Culligan D, Vickers MA. Case report: Refractory Anemia with Ringed Sideroblasts Associated with marked thrombocytosis complicated by massive splenomegaly treated with lenalidomide resulting in resolution of splenomegaly but severe and prolonged pancytopenia. Case Reports in Hematology 2013; e-pub . 10.1155/2013/718480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huls G, Mulder AB, Rosati S, et al. Efficacy of single-agent lenalidomide in patients with JAK2(V617F) mutated refractory anemia with ring sideroblasts and thrombocytosis. Blood. 2010;116(2):180–182. [DOI] [PubMed] [Google Scholar]
- 140.Hall J, Foucar K. Diagnosing myelodysplastic/myeloproliferative neoplasms: laboratory testing strategies to exclude other disorders. Int J Lab Haematol. 2010;32(6 Pt 2):559–571. [DOI] [PubMed] [Google Scholar]
- 141.DiNardo CD, Daver N, Jain N, et al. Myelodysplastic/myeloproliferative neoplasms, unclassifiable (MDS/MPN, U): natural history and clinical outcome by treatment strategy. Leukemia. 2014;28(4):958–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113(6):1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29(29):3907–3913. [DOI] [PubMed] [Google Scholar]
- 144.Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973–977. [DOI] [PubMed] [Google Scholar]
- 145.Tefferi A. Primary myelofibrosis: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2011;86(12): 1017–1026. [DOI] [PubMed] [Google Scholar]
- 146.Huang J, Li CY, Mesa RA, et al. Risk factors for leukemic transformation in patients with primary myelofibrosis. Cancer. 2008;112(12):2726–2732. [DOI] [PubMed] [Google Scholar]
- 147.Gangat N, Caramazza D, Vaidya R, et al. DIPPS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic informatic from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):393–397. [DOI] [PubMed] [Google Scholar]
- 148.Tefferi A, Pardanani A, Gangat N, et al. Leukemia risk models in primary myelofibrosis: An International Working Group Study. Leukemia. 2012;26(6):1439–1441. [DOI] [PubMed] [Google Scholar]
- 149.Brecqueville M, Rey J, Bertucci F, et al. Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes Chromosomes Cancer. 2012;51(8):743–755. [DOI] [PubMed] [Google Scholar]
- 150.Courville EL, Wu Y, Kourda J, et al. Clinicopathologic analysis of acute myeloid leukemia arising from chronic myelomonocytic leukemia. Mod Pathol. 2013;26(6):751–761. [DOI] [PubMed] [Google Scholar]
- 151.Gelsi-Boyer V, Trouplin V, Adelaide J, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukemia. Br J Haematol. 2010;15(4):365–375. [DOI] [PubMed] [Google Scholar]
- 152.Kuo MC, Liang DC, Huang CF, et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23(8):1426–1431. [DOI] [PubMed] [Google Scholar]
- 153.Broseus J, Florensa L, Zipperer E, et al. Clinical features and course of refractory anemia with ring sideroblasts associated with marked thrombocytosis. Haematologica. 2012;97(7):1036–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011;118(7):1723–1735. [DOI] [PubMed] [Google Scholar]
- 155.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612. [DOI] [PubMed] [Google Scholar]
- 156.Thoennissen NH, Krug UO, Lee DH, et al. Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms. Blood. 2010;115(14):2882–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kanagal-Shamanna R, Bueso-Ramos CE, Barkoh B, et al. Myeloid neoplasms with isolated isochromosome 17q represent a clinicopathologic entity associated with myelodysplastic/myeloproliferative features, a high risk of leukemic transformation, and wild-type TP53. Cancer. 2012;118(11):2879–2888. [DOI] [PubMed] [Google Scholar]
- 158.Alchalby H, Zabelina T, Stübig T, et al. Allogeneic stem cell transpalntation for myelofibrosis with leukemic transformation: A study from the Myeloproliferative Neoplasm Subcommittee of the CMWP of the European Group for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2014;20(2):279–287. [DOI] [PubMed] [Google Scholar]
- 159.Mesa RA, Tibes R. MPN blast phase: clinical challenge and assessing response. Leuk Res. 2012;36(12):1496–1497. [DOI] [PubMed] [Google Scholar]
- 160.Mascarenhas J, Navada S, Malone A, Rodriguez A, Najfeld V, Hoffman R. Therapeutic options for patients with myelofibrosis in blast phase. Leuk Res. 2010;34(9):1246–1249. [DOI] [PubMed] [Google Scholar]
- 161.Cherington C, Slack JL, Leis J, et al. Allogeneic stem cell transplantation for myeloproliferative neoplasm in blast phase. Leuk Res. 2012;36(9):1147–1151. [DOI] [PubMed] [Google Scholar]
- 162.Eghtedar A, Verstovsek S, Estrov Z, et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119(20):4614–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bogenberger JM, Shi C-X, Gonzales I, et al. RNAi Screening Identifies BCL-XL As An Erythroid Lineage-Specific 5-Azacytidine Sensitizer While the BCL-2/BCL-XL/BCL-W Inhibitor ABT-737 Results in More Universal Sensitization in Leukemia Cells. ASH Annual Meeting Abstracts 2011;118: Abstract 3513. [Google Scholar]
- 164.Borthakur G. PL, Kirschbaum M.H., et al. ,. Phase I/II trial of the MEK1/2 inhibitor GSK1120212 (GSK212) in patients (pts) with relapsed/refractory myeloid malignancies: Evidence of activity in pts with RAS mutation. J Clin Oncol 2011;29:Abstract 6506. [Google Scholar]
- 165.Bedard G, Zeng L, Lam H, et al. Meaningful change in oncology quality-of-life instruments: a systematic literature review. Expert Rev Pharmacoecon Outcomes Res. 2012;12(4):475–483. [DOI] [PubMed] [Google Scholar]
- 166.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30(33):4098–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118(2):401–408. [DOI] [PubMed] [Google Scholar]
- 169.Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007;109(1):68–76. [DOI] [PubMed] [Google Scholar]
- 170.Goldberg SL, Chen E, Corral M, et al. Incidence and clinical complications of myelodysplastic syndromes among United States Medicare beneficiaries. J Clin Oncol. 2010;28(17):2847–2852. [DOI] [PubMed] [Google Scholar]
- 171.Sekeres MA, Maciejewski JP, List AF, et al. Perceptions of disease state, treatment outcomes, and prognosis among patients with myelodysplastic syndromes: results from an internet-based survey. Oncologist. 2011;16(6):904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tibes R, Piper BF, Smith JA, et al. Patient willingness to undergo pharmacodynamic and pharmacokinetic tests in early phase oncology trials. Cancer. 2011;117(14):3276–3283. [DOI] [PubMed] [Google Scholar]
- 173.Bejanyan N, Tiu RV, Raza A, et al. A phase 2 trial of combination therapy with thalidomide, arsenic trioxide, dexamethasone, and ascorbic acid (TADA) in patients with overlap myelodysplastic/myeloproliferative neoplasms (MDS/MPN) or primary myelofibrosis (PMF). Cancer. 2012;118(16): 3968–3976. [DOI] [PubMed] [Google Scholar]
- 174.Chang T, Krisman K, Theobald EH, et al. Sustained MEK inhibition abrogates myloproliferativ disease in Nf1 mutant mice. J Clin Invest. 2013;123(1):335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Constantinescu SN, Vainchenker W. Small-molecule inhibitors in myeloproliferative neoplasms: are we aiming for the right targets? Hematology Am Soc Hematol Educ Program. 2012;2012:553–560. [DOI] [PubMed] [Google Scholar]
- 176.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013; 122(25): 4021–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]