

ABSTRACT
The envelope of Staphylococcus aureus is comprised of peptidoglycan and its attached secondary polymers, teichoic acid, capsular polysaccharide, and protein. Peptidoglycan synthesis involves polymerization of lipid II precursors into glycan strands that are cross-linked at wall peptides. It is not clear whether peptidoglycan structure is principally determined during polymerization or whether processive enzymes affect cell wall structure and function, for example, by generating conduits for protein secretion. We show here that S. aureus lacking SagB, a membrane-associated N-acetylglucosaminidase, displays growth and cell-morphological defects caused by the exaggerated length of peptidoglycan strands. SagB cleaves polymerized glycan strands to their physiological length and modulates antibiotic resistance in methicillin-resistant S. aureus (MRSA). Deletion of sagB perturbs protein trafficking into and across the envelope, conferring defects in cell wall anchoring and secretion, as well as aberrant excretion of cytoplasmic proteins.
IMPORTANCE Staphylococcus aureus is thought to secrete proteins across the plasma membrane via the Sec pathway; however, protein transport across the cell wall envelope has heretofore not been studied. We report that S. aureus sagB mutants generate elongated peptidoglycan strands and display defects in protein secretion as well as aberrant excretion of cytoplasmic proteins. These results suggest that the thick peptidoglycan layer of staphylococci presents a barrier for protein secretion and that SagB appears to extend the Sec pathway across the cell wall envelope.
INTRODUCTION
Staphylococcus aureus, a Gram-positive bacterial pathogen, replicates via septal assembly of membranes and peptidoglycan into the cross wall compartment (1, 2). The peptidoglycan of the cross wall is split by murein hydrolases, separating daughter cells that assume a spherical shape (3). Earlier work identified three murein hydrolases with cross-wall-splitting activities: Atl (autolysin), Sle1, and LytN (3–5). Atl and Sle1 are secreted into the extracellular milieu and subsequently cleave septal peptidoglycan at the cross wall but not elsewhere as access is restricted by teichoic acid modification of peptidoglycan (6–8). LytN, on the other hand, is secreted into the cross wall compartment (5). S. aureus Atl is synthesized as a preproenzyme with an N-terminal signal peptide and prodomain (9, 10). Secreted pro-Atl is processed to generate Atl N-acetylmuramoyl-l-Ala-amidase (AtlAM) and Atl N-acetylglucosaminidase (AtlGL), and each binds via GW domains to lipoteichoic acids (10–12). Earlier work demonstrated that Atl functions as an endo-β-N-acetylglucosaminidase (12, 13). Although initially designated autolysin (Atl), the S. aureus atl mutant does not display an autolysis phenotype yet forms large clusters of incompletely separated bacteria and is defective for penicillin-induced killing (14). The LysM domains of Sle1 promote its binding to cross wall peptidoglycan, and sle1 mutants also form clusters of incompletely separated bacteria (3, 7).
Murein sacculi are composed of peptidoglycan, a single large macromolecule with glycan strands and cross-linked wall peptides (15). In S. aureus, glycan strands are polymers of [-4(-N-acetylmuramic acid-β(1-4)-N-acetylglucosamine-β)1-]n, 4 to 6 N-acetylmuramyl (MurNAc)-GlcNAc disaccharides in length (16, 17). Each MurNAc residue is tethered to wall peptide, l-Ala-d-iGln-(Gly5)-l-Lys-d-Ala, where the amino group of the pentaglycine cross bridge (Gly5) is amide linked to the carboxyl group of d-Ala within wall peptide from another glycan strand (18–20). Peptidoglycan synthesis involves a bactoprenol-linked intermediate, lipid II [C55-(PO4)2-MurNAc(-l-Ala-d-iGln-(NH2-Gly5)-l-Lys-d-Ala-d-Ala)-β(1-4)-GlcNAc] (21, 22), that is polymerized into glycan strands by penicillin binding protein 2 (PBP2), as well as the monofunctional glycosyltransferases MGT and SgtA (23–25). Wall peptides of newly assembled glycan strands are cross-linked by transpeptidases (26, 27), i.e., penicillin binding proteins (PBP1, PBP2, and PBP4), and at low frequency (<1%) are trimmed of their terminal d-Ala (28–31).
When analyzed by electron microscopy of thin-sectioned staphylococci or isolated murein sacculi, the peptidoglycan layer of S. aureus has a diameter of 20 to 40 nm (1). Boiling staphylococci in suspension with ionic detergent does not lead to cell lysis or leakage of cytoplasmic protein, suggesting that staphylococcal murein sacculi are impenetrable for proteins (32). Nevertheless, during exponential growth, S. aureus secretes at least 59 proteins processed from signal peptide-bearing precursors into the culture medium in addition to excreting 53 polypeptides that apparently do not travel via the Sec pathway (33–35). S. aureus genes for cell wall synthesis and peptidoglycan processing were heretofore not reported to contribute to protein secretion.
By analyzing N-acetylglucosaminidases of S. aureus Newman, we observed that mutations in sagB (Staphylococcus aureus glucosaminidase B) perturb protein secretion. Further, sagB mutants display growth and cell-morphological defects that are caused by the exaggerated lengths of peptidoglycan strands, and purified SagB cleaves glycan strands to generate the physiological structure of S. aureus cell wall. During the preparation of the manuscript, Wheeler et al. reported on S. aureus SH1000 sagB mutants and associated defects in bacterial growth and peptidoglycan structure without analyzing protein secretion or antibiotic resistance (36).
MATERIALS AND METHODS
Bacterial strains, bacterial growth, and reagents.
Escherichia coli DH5α was used for cloning. E. coli XL-1 Blue was used for expression and purification of glutathione S-transferase (GST) fusions, and E. coli BL21(DE3) was used for the purification of histidine-tagged proteins. E. coli cultures were grown in Luria broth (LB) or on LB agar supplemented with ampicillin at a concentration of 100 μg/ml and with isopropyl 1-thio-β-d-galactopyranoside (IPTG) when indicated. S. aureus strains were grown in tryptic soy broth (TSB) or on tryptic soy agar (TSA) supplemented with appropriate antibiotics. Erythromycin and chloramphenicol were used at a concentration of 10 μg/ml, kanamycin was used at 50 μg/ml, and spectinomycin was used at 200 μg/ml. To examine the effect of the glucosaminidase gene deletions on bacterial growth, stationary-phase cultures normalized to an A600 of 3 were diluted (1:50) into 100 μl of fresh TSB, and growth at 37°C was monitored every 15 min for 12 h in a Synergy HT plate reader (BioTek) by measuring the optical density at 600 nm. Bacterial strains and plasmids utilized in this study are listed in Table 1.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference(s) or source |
|---|---|---|
| Strains | ||
| S. aureus Newman | Wild type, clinical isolate | 42, 75 |
| atl mutant | Δatl::Sp | This work |
| sagB mutant | ΔsagB::aphA | This work |
| sagA mutant | ΔsagA | This work |
| scaH mutant | scaH::erm | This work |
| atl sagB mutant | Δatl::Sp ΔsagB::aphA | This work |
| atl sagA mutant | Δatl::Sp ΔsagA | This work |
| atl scaH mutant | Δatl::Sp scaH::erm | This work |
| sagAB mutant | ΔsagB::aphA ΔsagA | This work |
| sagB scaH mutant | ΔsagB::aphA scaH::erm | This work |
| sagA scaH mutant | ΔsagA scaH::erm | This work |
| atl sagAB mutant | Δatl::Sp ΔsagB::aphA ΔsagA | This work |
| atl sagB scaH mutant | Δatl::Sp ΔsagB::aphA scaH::erm | This work |
| atl sagA scaH mutant | Δatl::Sp ΔsagA scaH::erm | This work |
| sagAB scaH mutant | ΔsagB::aphA ΔsagA scaH::erm | This work |
| atl sagAB scaH mutant | Δatl::Sp ΔsagB::aphA ΔsagA scaH::erm | This work |
| atl sagAB scaH lytP2 lytP4 mutant | atl::erm ΔsagAB ΔscaH lytP2::Cm lytP4::Sp | Lab collection |
| USA300 LAC | Wild type, clinical MRSA isolate | 55 |
| USA300 sagB | USA300 ΔsagB::aphA | This work |
| Plasmids | ||
| pΔatl::Sp | pKOR1 with Δatl::Sp for allelic replacement | This work |
| pΔsagB::aphA | pKOR1 with ΔsagB::aphA for allelic replacement | This work |
| pΔsagA | pKOR1 with ΔsagA for allelic replacement | This work |
| pOS1 | E. coli/S. aureus shuttle vector | 32 |
| psagB | sagB ORF and 852 bp upstream cloned into pOS1 | This work |
| pET15b-AtlGL | atl3103–3771 cloned into the BamHI site of pET-15bb | This work |
| pET15b-sagB91–855 | sagB91–855 cloned into the NdeI and BamHI sites of pET-15b | This work |
| pGEX-AtlAM | gst-atlAM fusionc | 76 |
| pGEX-AtlGL | gst-atlGL fusiond | 76 |
ORF, open reading frame.
atl3103–3771, atl gene encoding residues 3103 to 3771 of Atl.
atlAM, the atl gene encoding the Atl N-acetylmuramoyl-l-Ala-amidase.
atlGL, the atl gene encoding Atl N-acetylglucosaminidase.
Deletion strains were created by amplifying 1 kb upstream and downstream of the gene of interest from S. aureus Newman chromosomal DNA. Products were combined by spliced overlap extension (SOE)-PCR and cloned into pKOR1. Allelic replacement followed a previously described protocol (37). The atl::Sp strain was generated by amplification of the up- and downstream regions of atl using the upstream primer pair 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTACCATTTTCATGGGTATATGATGAAAATGGC-3′ and 5′-CGAACGAAAATCGATCGCCATTCTATTTATTACTCCTAACATTTATTAATTATTAC-3′ and the downstream pair 5′-CCCTTGCATATAAGCAACATGAACATAGGATCAAAAGTCATCC-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGTATGGTTTATCAATATTTTTCGCGAAATAACC-3′. The spectinomycin resistance cassette was amplified using 5′-GTAATAAATAGAATGGCGATCGATTTTCGTTCGTGAATACATGTTAT-3′ and 5′-GTTCATGTTGCTTATATGCAAGGGTTTATTGTTTTCTAAAATCT-3′ from pJRS312 (38). Similarly, sagB::aphA was generated by amplification of sagB upstream regions using the primer pair 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTGAGGATAAAGATTGCTTGCTTGAGGG-3′ and 5′-CCTCAAATGGTTCCATATCCACACCTCTTAGGTCATTG-3′, and the downstream region was amplified using 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGTCCAAAAATTACACCGATAGGCTCTT-3′ and 5′-GGAATTTGTATCGCATTTGAATAAGTAATTTGATAAGCTACGAG-3′; the aphA kanamycin resistance marker from pJK4 (39) was amplified using 5′-GGTGTGGATATGGAACCATTTGAGGTGATAGGTAAG-3′ and 5′-CTTATTCAAATGCGATACAAATTCCTCGTAGGC-3′. The sagA deletion was generated by amplification of the upstream region of sagA using the primer pair 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTTAAATAATGATGCGATGGAAAATGGAG-3′ and 5′-CACACTCAGAATCACGATGAGTAATACAGCAAAAACAAC-3′ and the downstream pair 5′-GTTGTTTTTGCTTCCTTTAGCGCATTCTGAGTGTG-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCCCATATTAGGCGTTGTCG-3′. The scaH::erm mutant was obtained from the Phoenix transposon library and transduced by bacteriophage ϕ85 into Newman wild-type background (40). Combinatorial mutants of the glucosaminidase genes were generated by bacteriophage transduction and selection with the appropriate antibiotic.
The psagB complementation vector was generated by PCR amplification of the sagB open reading frame using 5′-NNNGGATCCGTAATCGGGAGGTAACAATGGATTACGCAC-3′ and 5′-NNNGGATCCTTACTTATTCAAATGTTTACTGTCATCTTTATAC-3′, followed by BamHI digestion and insertion into pOS1. pET15b-sagB91–855, encoding the N-terminally His6-tagged SagB glucosaminidase domain (residues 91 to 855), was generated by PCR amplification using the primer pair 5′-AAAACATATGTCCGATCAGATATTTTTCAAACA-3′ and 5′-AAAAGGATCCTTACTTATTCAAATGTTTACTGTCAT-3′, followed by directional cloning into pET-15b using the NdeI and BamHI restriction sites. pET15b-AtlGL, encoding an N-terminal His6-tagged glucosaminidase domain of Atl, was PCR amplified using primers 5′-NNNGGATCCGGGTTTACAATATAAACCACAAGTACAACGTG-3′ and 5′-AAAAGGATCCTTATTTATATTGTGGGATGT-3′, cloned into the BamHI site of pET-15b, and screened for directionality. All constructs and mutants were verified by sequencing.
Protein and antibody production.
Bacterial cultures were grown to an A600 of 0.6, and protein expression was induced with 1 mM IPTG for 4 h at 37°C for His6-tagged fusions or with 0.3 mM IPTG for 16 h at room temperature for GST-AtlAM. Bacterial cells were sedimented by centrifugation (10,000 × g, 10 min) and suspended in phosphate-buffered saline (PBS; pH 7) for the purification of GST fusions and in PBS (pH 7.4) containing 20 mM imidazole for His6-tagged proteins. Cells were lysed by two passages in a French pressure cell at 15,000 lb/in2. Crude lysates were cleared by centrifugation (100,000 × g, 30 min), and the supernatant was loaded by gravity flow onto glutathione-Sepharose beads (GE Healthcare) or nickel-nitrilotriacetic acid beads (Qiagen) preequilibrated in their respective lysis buffers. Columns were washed with 20 volumes of lysis buffer, and bound proteins were eluted with either 10 mM reduced glutathione or 500 mM imidazole. Proteins in the eluates were dialyzed into PBS (pH 7), quantified by bicinchoninic acid assay (Pierce), and stored at 4°C for immediate use or stored frozen at −80°C.
For production of polyclonal antibodies, rabbits (6-month-old female New Zealand White rabbits; Charles River Laboratories) were immunized with purified recombinant SagB (rSagB) as described earlier (41). Polyclonal serum was stored at −80°C.
Lysostaphin susceptibility.
Stationary-phase cultures of staphylococci were washed, suspended in ice-cold Tris-HCl (pH 7.5) to an A600 of 6.0, and distributed in quadruplicate into a 96-well microtiter plate. Buffer alone or 40 μg of lysostaphin/ml of buffer was added to each well. Turbidity of cells was monitored every 15 min for 3 h at 37°C with agitation in a Synergy HT plate reader (BioTek) by measuring the A600. Relative turbidity was determined as the ratio of the average A600 of lysostaphin samples to that in buffer-only controls.
Peptidoglycan extraction.
Staphylococci from mid-exponential-growth cultures (A600 of 0.6) were suspended in 4% sodium dodecyl sulfate (SDS) and boiled for 30 min. Cells were subsequently washed five times in water to remove detergent and then broken in a bead-beating instrument (MP Biomedicals). Cellular material was collected by centrifugation (7,500 × g, 10 min), washed two times with water, and suspended in 50 mM Tris-HCl (pH 7.5), 10 mM CaCl2, and 20 mM MgCl2 for digestion with amylase (100 μg/ml), DNase (10 μg/ml), and RNase (50 μg/ml) for 2 h at 37°C and then with trypsin (100 μg/ml) for 16 h at 37°C. The cell wall material was sedimented by centrifugation (3,300 × g, 15 min), suspended in 1% SDS, boiled for 15 min to inactivate enzymes, and then washed two times with water, once with 8 M LiCl, once with 100 mM EDTA, two times with water, once with acetone, and two times with water. Murein sacculi were suspended in water, normalized to an A600 of 10, and stored at −20°C until further use. Murein sacculi were further processed to remove acetyl groups and phosphodiester-linked cell wall polymers, first by drying samples under speed vacuum and then by suspension in 49% hydrofluoric acid (HF; 2.5 mg/ml) for 48 h at 4°C. Peptidoglycan was recovered by centrifugation (33,000 × g for 45 min), washed two times with water, once with 100 mM Tris-HCl (pH 7.5), and two times with water. The pellet was then suspended in 100 mM (NH4)2CO3 and treated with alkaline phosphatase (250 μg/ml) for 16 h at 37°C. The enzyme was inactivated by boiling for 5 min, and peptidoglycan washed two times with water. The purified material was suspended in water, normalized to an A600 of 10, and stored at −20°C.
Peptidoglycan digestion and high-pressure liquid chromatography (HPLC) analysis.
Peptidoglycan (A600 of 10; 0.3 ml) was suspended in 12.5 mM sodium phosphate buffer (pH 7.4) and digested with lysostaphin (0.1 mg/ml) for 16 h at 37°C. To assess recombinant glucosaminidase protein activity, peptidoglycan (A600 of 10; 0.3 ml) suspended in 100 mM sodium phosphate buffer (pH 5.0) was treated with 0.5 mg/ml AtlGL or rSagB and incubated for 6 h at 37°C. Thereafter, the reaction mixture was adjusted to pH 7.0 with sodium hydroxide and subjected to digestion with lysostaphin (0.1 mg/ml) alone or with AtlAM (50 μg/ml) and incubated for 12 h at 37°C. The digestion reaction was quenched by boiling the mixture for 10 min at 95°C, and supernatants were collected after centrifugation (10 min at 23,000 × g).
For purification and analysis of lysostaphin and AtlAM-cleaved glycan chains, we used the procedure of Boneca et al. with minor modifications. The muropeptide-containing supernatants were diluted to 10 ml with water and adjusted to pH 2.0 with phosphoric acid. Glycan strands were separated from stem peptides on a MonoS column (GE Healthcare). Samples were applied over the MonoS column, washed with 10 mM sodium phosphate buffer, pH 2.0 (buffer A), at 0.7 ml/min, and eluted with 1 M NaCl in buffer A. Sample detection was followed at 202 nm and 215 nm. Glycan chains eluted in the void volume; the corresponding fractions were lyophilized and then suspended to approximately 500 μl in water (17). Muropeptides and/or glycan chains were reduced as previously described by addition of 0.5 M sodium borate (pH 9) and NaBH4 (28).The reaction was quenched by addition of 20% H2PO4 to reduce the pH to 2. Precipitate was sedimented by centrifugation (23,000 × g, 10 min). The reduced material was collected and stored at −20°C until further analysis. Separation of muropeptides by reverse-phase high-pressure liquid chromatography (RP-HPLC) was performed as previously described (28). Samples were applied to a 250- by 4.6-mm reversed-phase C18 column (ODS-Hypersil, 3 μm; Thermo Scientific). The column was eluted at a flow rate of 0.5 ml/min with a linear gradient starting 5 min after injection of 5% (vol/vol) methanol in 100 mM NaH2P04, (pH 2.5) to 30% (vol/vol) methanol in 100 mM NaH2P04 (pH 2.8) in 150 min. Column temperature was maintained at 52°C. The eluted compounds were detected by absorption at 206 nm. Desalted muropeptides were dried under vacuum and suspended in 20 μl of 30% acetonitrile, and 0.5 μl was cospotted with 0.5 μl of matrix, α-cyano-4-hydroxycinnamic acid, at 10 mg/ml in 50% acetonitrile–0.1% trifluoroacetic acid (TFA). Samples were subjected to matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) using an Autoflex Speed Bruker MALDI instrument. Ions were detected in reflectron-positive mode.
Quantification of cell wall phosphate levels.
The phosphate content was determined by incubating 45 μl of murein sacculus sample (A600 of 10) with 5 μl of trichloroacetic acid (TCA) at 80°C for 16 h. Inorganic phosphate released by this treatment was quantified with a colorimetric assay in which a mix composed of 6 N H2SO4, water, 2.5% ammonium molybdate, 10% ascorbic acid (in the ratio 1:2:1:1) was added at 1:1 (vol/vol) to TCA-treated preparations and incubated at 37°C for 90 min. Product formation corresponding to free phosphate was measured in a spectrophotometer at 820 nm (A820), and phosphate concentration in the samples was calculated from NaH2PO4 standards (concentration, 0 to 800 μM).
Transmission electron microscopy.
For transmission electron microscopy, bacterial cells were washed twice with 50 mM Tris-HCl (pH 7.5)–150 mM NaCl, bathed in fixative (2% glutaraldehyde, 4% paraformaldehyde [PFA], 0.1 M sodium cacodylate buffer) overnight at 4°C, and postfixed with 1% OsO4 in 0.1 M sodium cacodylate buffer for 60 min. Fixed samples were stained in 1% uranyl acetate in maleate buffer for 60 min, serially dehydrated with increasing concentrations of ethanol, embedded in Spurr resin for 48 h at 60°C, thin sectioned (90 nm) using a Reichert-Jung Ultracut device, and poststained in uranyl acetate and lead citrate. The samples were imaged on an FEI Tecnai F30 with a Gatan charge-coupled-device (CCD) digital micrograph. Quantification of cell separation defects was done on cultures grown to an A600 of 0.5. A separation defect was defined as a dividing cell displaying more than one septal plane. Three hundred to 500 dividing cells from at least 15 fields of images were enumerated. Statistical significance was determined by one-way analysis of variance (ANOVA), followed by Bonferroni's multiple comparison to the wild type.
Antibiotic resistance.
MICs of antibiotics for S. aureus strains were determined by measuring their growth in 96-well plates. Experiments were performed in duplicate and repeated at least three times. TSB (100 μl with or without antibiotic) was inoculated 1:50 with stationary-phase culture (2 μl) normalized to an A600 of 3, plates were incubated at 37°C with agitation for 16 h, and the A600 was measured. MIC determinations tested flavomycin (0.5 to 40 μg/ml), oxacillin (0.005 to 20 μg/ml), vancomycin (0.078 to 10 μg/ml), bacitracin (3.125 to 400 μg/ml), and nisin (0.97 to 125 μg/ml). Statistical significance was calculated by one-way ANOVA followed by Dunnett's test (S. aureus Newman) or with an unpaired t test (USA300 LAC).
Cellular fractionation and immunoblotting.
S. aureus Newman strains were diluted from overnight cultures and grown to mid-exponential phase (A600 of 0.5). For comparative analyses of proteins using immunoblotting, bacteria from 2 ml of culture were sedimented, and the supernatant (S) was collected and concentrated by TCA precipitation. The sediment was suspended in 1 ml of TSM buffer (50 mM Tris-HCl [pH 7.5], 0.5 M sucrose, 10 mM MgCl2) supplemented with 10 μg/ml lysostaphin and incubated for 30 min at 37°C to generate protoplasts. Protoplasts were sedimented by centrifugation (23,000 × g, 5 min), and the solubilized cell wall material (CW) in the supernatant was collected and TCA precipitated. Protoplasts were lysed and separated into membrane (M) and cytoplasm (C) fractions by repeated freeze-thaw cycles (four times) in an ethanol–dry-ice bath and a 56°C heat block. Membranes were sedimented and separated from cytoplasmic material by centrifugation (23,000 × g, 60 min). Cytoplasmic fractions were concentrated by TCA precipitation. All TCA-precipitated samples were reconstituted in 50 μl of 0.5 M Tris-HCl (pH 8.0)–4% SDS and heated at 90°C for 10 min. Proteins were separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride membrane (Millipore) for immunoblot analysis with appropriate rabbit polyclonal antibodies. Immunoreactive signals were revealed by a secondary antibody conjugated to horseradish peroxidase and enhanced chemiluminescent substrate.
For MALDI-TOF mass spectrometry of secreted proteins, 100 ml of S. aureus culture grown to mid-exponential phase (A600 of 0.5) was centrifuged to sediment bacteria. Supernatants were passed through a 0.2-μm-pore-size filter and concentrated 1,000-fold by TCA precipitation, followed by methanol-chloroform extraction and protein quantification via a bicinchoninic acid (BCA) assay. Protein (3.5 μg) was resolved on a 12% SDS-PAGE gel stained with Coomassie. Bands with different intensities between the wild type and sagB mutant were excised for protein identification and semiquantitative analysis at the Harvard University Taplin Mass Spectrometry Facility. The identity of each band (Table 2) was predicted by assessment of the most abundant species (total peptides) in each excised region; the top three candidates are shown for each. Protein species identified by MALDI-TOF MS in wild-type and sagB supernatant samples were compiled for pairwise comparison of sum intensity. Proteins were grouped into those that displayed at least a 10-fold increase or decrease in sum intensity in the sagB sample relative to that in the wild type. Protein species fitting these threshold parameters were subsequently analyzed using SignalP, version 4.1, and TMHMM, version 2.0, servers to predict cellular localization.
TABLE 2.
Mass spectrometry identification of proteins with diminished secretion in sagB mutant S. aureus
| Slice | Protein | Localizationa | Mass (kDa) | Total no. of peptides | % coverage | Intensityb |
|---|---|---|---|---|---|---|
| 1 | vWbp | S | 59.24 | 541 | 77.95 | 11.6:1 |
| Coa | S | 71.67 | 324 | 75 | 6:1 | |
| NWMN_0401 | S | 56.51 | 288 | 86.5 | 16:1 | |
| 2 | PrsA | TM | 35.62 | 170 | 75.94 | 1:6.6 |
| PhdB | C | 35.22 | 86 | 77.3 | 1:7.4 | |
| GapA | C | 36.25 | 44 | 62.8 | 1:7.8 | |
| 3 | Ssl11 | S | 25.35 | 271 | 86.22 | 8.7:1 |
| Ssl7nm | S | 26.15 | 194 | 74.46 | 104:1 | |
| Ssl1nm | S | 25.63 | 125 | 72.12 | 17:1 | |
| 4 | NWMN_2203 | S | 17.39 | 101 | 54.22 | 1:7.3 |
| NWMN_0364 | C | 21.29 | 59 | 62.63 | 1:14.5 | |
| 5 | Fib | TM | 18.75 | 345 | 78.18 | 5.5:1 |
| Chp | S | 17.05 | 271 | 81.88 | 6.7:1 | |
| 6 | Scn | S | 13.06 | 213 | 61.21 | 1.25:1 |
| NWMN_1066 | S | 12.59 | 165 | 65.14 | 2.2:1 |
Cellular localization was deduced by analysis of amino acid sequences using SignalP, version 4.1, and TMHMM, version 2.0, servers. S, secreted; TM, transmembrane; C, cytoplasmic.
Data represent the ratio of values for the wild type to those for the sagB strain.
Triton X-100-induced autolysis.
S. aureus Newman and its variants diluted from overnight cultures were grown to mid-exponential phase (A600 of 0.5). Cells were washed in 50 mM sodium phosphate buffer (pH 7.5) and suspended in the same buffer with or without 0.05% Triton X-100 to an A600 of 6.0. Cells were aliquoted into a 96-well microtiter plate in quadruplicate and incubated at 37°C for 3 h with agitation, with A600 measurements taken at 15-min intervals. Percent autolysis was calculated for each corresponding time point (t) as follows: (A600 in Triton X-100)/(A600 in buffer)/(A600 at t = 0) × 100.
Membrane permeability measurements.
Overnight cultures of S. aureus were diluted 1:100 into TSB and grown at 37°C to an A600 of 0.5. One milliliter of culture was centrifuged, and bacteria were washed twice with PBS and fixed for 20 min with 4% paraformaldehyde. Cells were labeled for 15 min in PBS containing 5 μM SYTO 9 (Invitrogen), which stains nucleic acids in live and in dead cells, and 1 μg/ml propidium iodide (Invitrogen), a nucleic acid dye that selectively permeates membrane-compromised cells, and then washed twice and suspended in PBS for flow cytometric analyses. Flow cytometric analyses were performed using a BD-LSR-II cytometer. For analysis of membrane integrity, SYTO 9-positive cells captured under the fluorescein isothiocyanate (FITC) parameter were analyzed for propidium iodide staining in the phycoerythrin (PE) parameter. The parameters for negative propidium iodide staining were determined using unstained controls.
RESULTS
N-Acetylglucosaminidases of Staphylococcus aureus Newman.
The endo-β-N-acetylglucosaminidase domain of S. aureus Atl, AtlGL (residues 1096 to 1256), was used as a query for blastp searches against the genome of S. aureus Newman (42). Six genes were identified: NWMN_0922 (Atl), NWMN_1667 (SagB), NWMN_2207 (SagA), NWMN_2543 (ScaH), NWMN_1035 (LytP2), and NWMN_0309 (LytP4) (Fig. 1A). LytP2 and LytP4 are encoded by S. aureus Newman prophages ϕNM2 and ϕNM4, respectively (43). Topology predictions using Psort and TMMH suggested that ScaH is synthesized as a precursor protein with a cleavable N-terminal signal peptide, whereas SagB and SagA were predicted as type II membrane proteins with their N termini in the cytoplasm, followed by a transmembrane segment and the C-terminal glucosaminidase domains outside the plasma membrane. As expected, LytP2 and LytP4 were predicted not to harbor topogenic sequences (Fig. 1B). Four staphylococcal gene products (SagB, SagA, LytP2, and LytP4) harbor lysozyme subfamily 2 domains (LYZ2/COG4193) also found in Atl (Fig. 1A). ScaH harbors an FlgJ-type muramidase domain (44), which in Listeria monocytogenes Auto and Lactococcus lactis AcmA displays N-acetylglucosaminidase activity (45, 46). Of note, the genome of S. aureus Newman does not encode an N-acetylmuramidase, indicating that all processing of glycan strands must be accomplished either by Atl, SagA, SagB, or ScaH.
FIG 1.

N-Acetylglucosaminidases of S. aureus Newman. (A) Alignment of the COG4193/glucosaminidase domains of S. aureus Newman gene products identified by blastp query with the endo-β-N-acetylglucosaminidase domain of Atl (residues 1096 to 1256). Asterisks identify identical residues; colons identify conserved residues; dots identify similar amino acid residues. (B) Domain structures of Atl and the staphylococcal glucosaminidases SagA, SagB, and ScaH and the prophage-encoded LytP2 and LytP4.
S. aureus sagB mutants synthesize peptidoglycan with elongated glycan strands.
We generated mutants in the atl, sagA, sagB, and scaH genes of S. aureus Newman (see Fig. S1 in the supplemental material). Strains deficient in two (atl sagB, atl sagA, atl scaH, sagAB, sagB scaH, or sagA scaH), three (atl sagAB, atl sagB scaH, or atl sagA scaH), or all four (atl sagAB scaH) glucosaminidase genes were also constructed (see Fig. S1). We asked whether the mutants displayed differences in peptidoglycan integrity and measured susceptibility to lysostaphin-induced lysis. Lysostaphin is a glycyl-glycyl endopeptidase that hydrolyzes staphylococcal peptidoglycan at pentaglycine cross bridges (47, 48). S. aureus wall peptides are 70 to 90% cross-linked (49), and, due to the small size of glycan strands, lysostaphin cleavage rapidly degrades the cell wall and causes bacterial lysis (17). Suspensions of S. aureus wild-type and glucosaminidase variants were adjusted to the same optical densities, treated with lysostaphin, and monitored for lysis by measuring decreased turbidity at A600. The atl and sagB mutant strains, but not sagA and scaH mutants, displayed delayed lysis relative to the wild type (Fig. 2A). Deletion of atl and sagB in double and quadruple mutant strains generated increased lysostaphin resistance, whereas deletion of sagA and scaH had no effect (Fig. 2B and C). Lysostaphin resistance of the sagB mutant was complemented by transformation with psagB, for plasmid-borne expression of wild-type sagB, but not by the vector control (Fig. 2D, pOS1).
FIG 2.
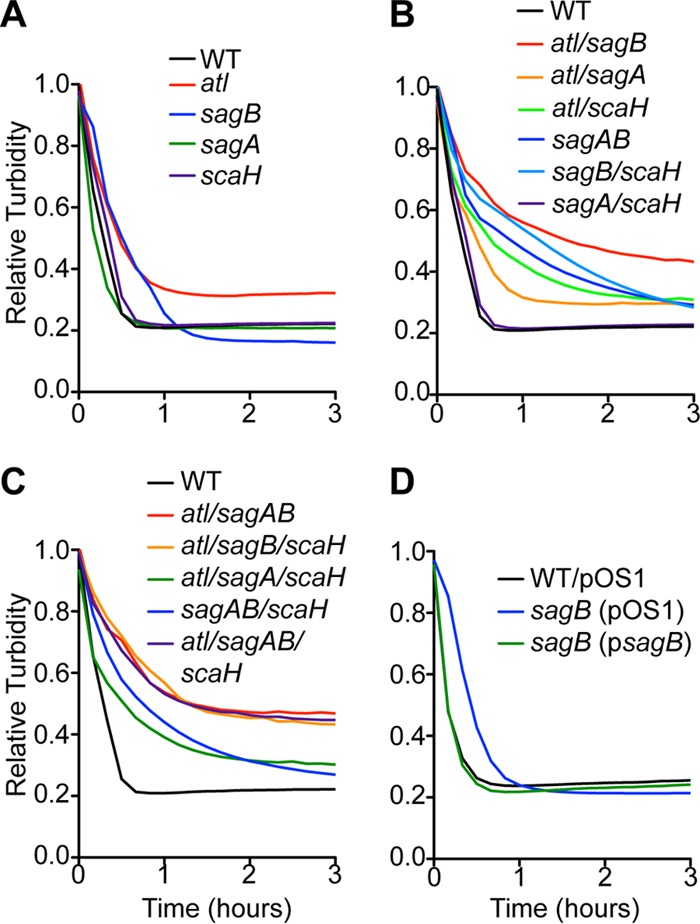
S. aureus glucosaminidase mutants resistant to lysostaphin. Lysostaphin (40 μg/ml) or buffer alone was added to stationary-phase cultures of S. aureus strains suspended in 50 mM Tris-HCl (pH 7.5). Turbidity at A600 was monitored over 3 h. Relative turbidity was calculated as the A600 of lysostaphin-inoculated staphylococci compared to that of staphylococci in buffer alone at each time point. Lysostaphin susceptibilities of the glucosaminidase single (A), double (B), and triple and quadruple (C) mutants and of the sagB-complemented strains (D) were compared to wild-type (WT) levels.
Peptidoglycan was isolated from mid-exponential-phase cultures of wild-type and mutant strains, purified, digested with lysostaphin, and analyzed by RP-HPLC on a C18 column (Fig. 3A). Compared to the wild type (black trace), lysostaphin treatment did not affect the elution profile of peptidoglycan cleavage products from atl (blue), sagA (red), and scaH (purple) strains, with the bulk of peptidoglycan chains eluting within 120 min. In contrast, lysostaphin treatment of a sagB mutant peptidoglycan (green) generated few muropeptides or short-chain peptidoglycan species (peaks eluting through 90 min), with the bulk of peptidoglycan eluting after 130 min, indicative of elongated peptidoglycan strands. These structural changes in the peptidoglycan of the sagB mutant were restored to wild-type levels by psagB complementation but not by empty vector (Fig. 3B). A similar analysis of the peptidoglycan chain length was performed with the atl sagAB scaH variant lacking all four glucosaminidases (Fig. 3C). Similar to the sagB mutant, the atl sagAB scaH mutant also displayed elongated peptidoglycan lengths that were complemented by psagB.
FIG 3.

Structure of peptidoglycan in S. aureus glucosaminidase mutants. RP-HPLC analysis of the soluble muropeptides released from S. aureus peptidoglycan after incubation with lysostaphin. Digested muropeptides were reduced with sodium borohydride and applied to a C18 column. Peptides were eluted with a gradient of 100 mM sodium phosphate (pH 2.5)–5% methanol to 100 mM sodium phosphate buffer (pH 2.8)–30% methanol in 150 min. Peaks were detected by absorbance at 206 nm. (A) Comparison of HPLC traces of the wild type (WT) and single-glucosaminidase mutant strains (atl, sagA, sagB, and scaH) reveals that the sagB and atl sagAB scaH mutant peptidoglycans contain elongated glycan strands compared to those of the wild-type peptidoglycan. (B) RP-HPLC analyses of the sagB mutant with (psagB) and without complementation (pOS1). (C) RP-HPLC analysis of peptidoglycan length in the atl sagAB scaH mutant carrying pOS1 or psagB compared to the length in the wild-type (pOS1) strain. (D) Glycan strand size analysis was conducted by enzymatic digestion of peptidoglycan with lysostaphin and AtlAM, followed by removal of wall peptides during cation exchange chromatography and RP-HPLC of glycan strands. (E) The abundance of glycan strands from samples used for the experiment shown in panel D was calculated as the percent area under the curve for each 25-min interval of the total area over the course of 26 to 150 min.
Peptidoglycan from wild-type (pOS1), sagB(pOS1), and sagB(psagB) strains was digested with lysostaphin and Atl amidase (AtlAM), which cleaves the wall peptide off glycan strands. Peptide cleavage products were separated from glycan strands by cation exchange chromatography. Glycan strands were reduced and analyzed by RP-HPLC (Fig. 3D). Approximately 90% of glycan strands of wild-type S. aureus eluted within 100 min, whereas the value for the sagB mutant was 55% (Fig. 3E). About 30% of glycan strands from the sagB mutant eluted between 101 to 125 min, compared to fewer than 10% from the wild type. The elongated-strand phenotype of the sagB mutant was complemented by psagB. These data demonstrate that expression of sagB but not of the other staphylococcal glucosaminidases is necessary for controlling the glycan chain length of S. aureus peptidoglycan.
Purified SagB cleaves elongated glycan strands.
Atl glucosaminidase (AtlGL) and recombinant SagB (rSagB), truncated of its N-terminal topogenic sequence, were expressed in E. coli and purified (Fig. 4A). When incubated with purified peptidoglycan of the sagB mutant strain together with lysostaphin and analyzed by RP-HPLC, AtlGL generated predominantly disaccharides, in agreement with earlier reports that AtlGL functions as an endo-β-N-acetylglucosaminidase (9, 13) (Table 3; see also Fig. S2 in the supplemental material). In contrast, rSagB did not yield disaccharide products but reduced the length of sagB mutant glycan strands (red trace) to a size similar to that observed in wild-type peptidoglycan (gray trace) (Fig. 4B). Treatment of sagB peptidoglycan with higher concentrations of rSagB enzyme or for prolonged periods of time or switching the sequence of enzymatic digestion with lysostaphin did not affect the muropeptide profile on RP-HPLC. Of note, rSagB displayed optimal cleavage activity at pH 5.0 and was inactive at pH 7.0 (data not shown). Taken together, these data suggest that in contrast to AtlGL, rSagB cleaves glycan strands intermittently at regular intervals, thereby generating the physiological length of wild-type peptidoglycan.
FIG 4.
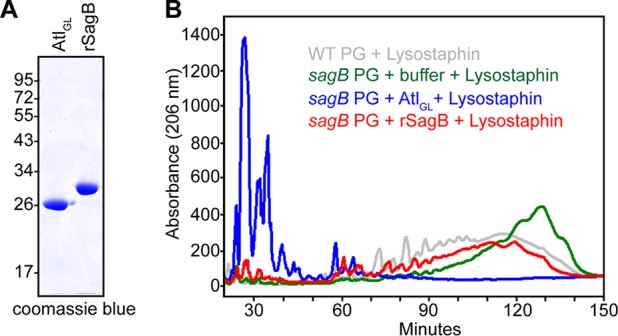
rSagB cleaves the glycan strands in sagB mutant peptidoglycan to their physiological sizes.(A) Coomassie-stained SDS-PAGE of purified AtlGL and rSagB. (B) sagB peptidoglycan (PG) was incubated sequentially with buffer, AtlGL, or rSagB, followed by lysostaphin, and then separated on a C18 RP-HPLC column.
TABLE 3.
Mass spectrometry of AtlGL and lysostaphin-digested sagB mutant peptidoglycan
| Peak | HPLC elution time (min) | Observed m/z | Calculated m/z | Proposed structure |
|---|---|---|---|---|
| 1 | 21–24 | 1,009.5,004 | 1,009.471 | MN-GN-AQKA-G2 |
| 2 | 25–28 | 1,066.561 | 1,066.4,925 | MN-GN-AQKA-G3 |
| 1,123.5,883 | 1,123.514 | MN-GN-AQKA-G4 | ||
| 1,180.647 | 1,180.5,355 | MN-GN-AQKA-G5 | ||
| 1,237.693 | 1,237.557 | MN-GN-AQKA-G6 | ||
| 3 | 29–34 | 1,080.5,666 | 1,080.5,081 | MN-GN-AQKA(A)-G2 |
| 1,137.5,811 | 1,137.5,296 | MN-GN-AQKA(A)-G3 | ||
| 1,194.6,266 | 1,194.5,511 | MN-GN-AQKA(A)-G4 |
S. aureus sagB mutants display defects in growth and in antibiotic susceptibility.
Growth of S. aureus mutants defective for sagA, sagB, scaH, or atl expression, either alone or as combinations of two, three, and four mutations, was analyzed in liquid culture in comparison with the wild type. For S. aureus variants with mutations in single genes, only the sagB mutant—not the sagA, scaH, or atl mutant—exhibited delayed growth (Fig. 5A.) For S. aureus variants with lesions in two, three, or four genes, only strains harboring the sagB mutation exhibited reduced growth. Further, deletion of any one glucosaminidase gene (atl, sagA, and/or scaH) in addition to sagB exacerbated the growth defect imparted by the sagB mutation (Fig. 5B and C). The sagB phenotype was complemented by plasmid-borne expression of sagB (Fig. 5D). These data suggest that although Atl, SagA, and ScaH exhibit roles distinctive from the role of SagB, further genetic loss of glucosaminidase activity aggravates the sagB mutant phenotype. Of note, Wheeler and colleagues, studying S. aureus SH1000, a laboratory strain, reported that expression of glucosaminidase was essential for S. aureus growth and that this requirement was mostly dependent on sagB (36).
FIG 5.
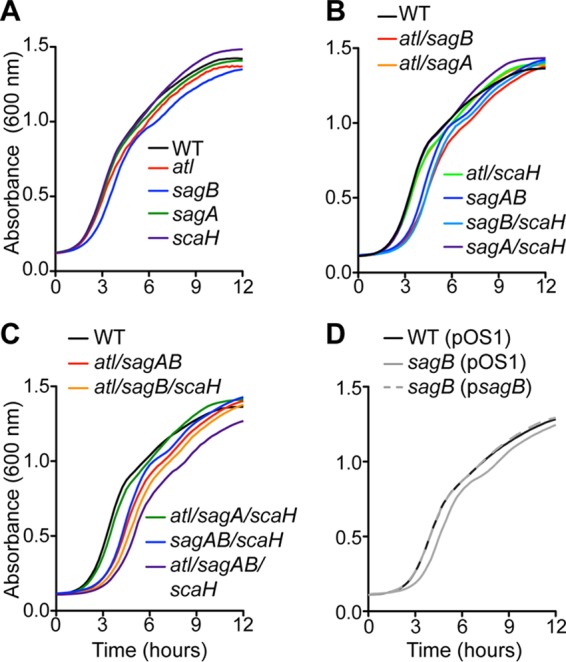
Growth attributes of S. aureus glucosaminidase mutants. Growth curves of S. aureus Newman (WT) and single (A), double (B), and triple and quadruple glucosaminidase mutants (C) and the complemented single sagB mutant (D). Overnight cultures were normalized to an A600 of 3.0, diluted 1:50, and monitored by recording optical density over time.
Changes in the structure of the cell envelope have been described to affect bacterial susceptibility to antibiotics (50). For example, mutations in E. coli mltG, which encodes a lytic transglycosylase, gives rise to peptidoglycan with elongated glycan strands and causes increased susceptibility to β-lactam antibiotics (51, 52). S. aureus glucosaminidase variants as well as the atl sagAB scaH mutant strain were tested for susceptibility to flavomycin, a glycosyltransferase inhibitor (53), and oxacillin, a β-lactamase-resistant β-lactam antibiotic that, unlike the related antibiotic methicillin, displays peroral drug activity (54). The sagB and atl sagAB scaH mutants displayed modest increases in resistance to flavomycin. The sagB mutant displayed diminished resistance to oxacillin, whereas the atl sagAB scaH mutant exhibited increased resistance to oxacillin (Table 4). USA300 LAC is a methicillin (oxacillin)-resistant S. aureus (MRSA) clone that is responsible for the American epidemic of community-associated MRSA infections (55, 56). Compared to wild-type USA300 LAC, the isogenic sagB variant exhibited increased resistance to flavomycin and diminished resistance to oxacillin (Table 4). Further, resistance to nisin, an inhibitor of MurG, and vancomycin, an antibiotic that binds lipid II and is used for the therapy of USA300 infections, was also increased in the sagB variant, while sensitivity to bacitracin (bactoprenol recycling inhibitor) was not affected (Table 4). In S. aureus Newman, the sagB mutation also increased the MIC for vancomycin, whereas sensitivity was increased for bacitracin and not altered for nisin (Table 4).
TABLE 4.
Antibiotic susceptibility of sagB mutant staphylococci
| S. aureus strain | MIC (μg/ml)a of: |
||||
|---|---|---|---|---|---|
| Flavomycin | Oxacillin | Vancomycin | Bacitracin | Nisin | |
| Newman strains | |||||
| Wild type | 6.250 | 0.2083 | 2.083 | 100.0 | 26.04 |
| atl strain | 8.125 | 0.5* | 2.188 | 166.7* | 23.44 |
| sagA strain | 6.563 | 0.2083 | 1.875 | 100.0 | 23.44 |
| sagB strain | 13.75† | 0.1042† | 3.750* | 50.00‡ | 23.44 |
| scaH strain | 6.250 | 0.25 | 1.875 | 100.0 | 26.04 |
| atl sagAB scaH strain | 30.00* | 0.4167* | 3.750* | 50.00‡ | 23.44 |
| USA300 strains | |||||
| Wild type | 6.667 | 5.416 | 2.500 | >1,000 | 11.72 |
| sagB strain | 8.611‡ | 3.125* | 4.583* | >1,000 | 15.63† |
The MIC of each antibiotic is the mean determined from at least three experiments performed in duplicate. Statistical significance was calculated by one-way ANOVA followed by Dunnett's test (Newman) or unpaired t test (USA300), as follows: †, P < 0.05; ‡, P < 0.01; *, P < 0.001.
Mutations in atl or sagB affect cell separation but not WTA synthesis.
Mutations in genes for muralytic enzymes can cause morphological defects during the S. aureus cell cycle (3, 5, 57). For example, atl mutants display both aberrant septum formation and delayed cell separation, where cells initiate a second septum prior to completion of cell division (57). To analyze the impact of mutations in the other glucosaminidase genes, bacteria from mid-exponential-phase cultures of the wild type and the atl, sagA, sagB, or scaH mutant were fixed, thin sectioned, stained with uranyl acetate, and analyzed by transmission electron microscopy (Fig. 6). As expected, wild-type staphylococci appeared round and uniform in size, and dividing cells manifested a single cross wall septum (Fig. 6B). The cell wall envelopes of wild-type staphylococci also comprised distinct interior and exterior electron-dense regions, an attribute of lipoteichoic acid and wall teichoic acid (WTA), bordering the central peptidoglycan layer. For the atl mutant, 4.4% of cells harboring a complete cross wall displayed either a nascent septum or a second septal plane (Fig. 6). Further, cell surfaces of atl mutant staphylococci exhibited a disordered structure, suggestive of sloughing of the wall material (Fig. 6A). The sagB mutant also displayed cell cycle defects as 4.8% of cells with completed cross wall compartments had either initiated or already completed a second septal plane, whereas the cellular architecture of sagA and scaH mutants appeared similar to that of the wild type (Fig. 6; see also Fig. S3 in the supplemental material). Deletion of both the atl and sagB genes caused an additive effect, with 9.6% of cells committing to premature assembly of cross wall peptidoglycan; this defect was increased to 20.5% in the atl sagAB scaH variant (Fig. 6).
FIG 6.

Cell separation defects in S. aureus glucosaminidase mutants. (A) S. aureus Newman strains were fixed, thin sectioned, uranyl acetate stained, and viewed by transmission electron microscopy. Images in the left-most column are representative low-magnification fields of cells for each corresponding frame. The three columns to the right show high-magnification images of representative cell morphologies. Arrowheads identify aberrantly formed septa. Gray scale bars, 2 μm; black scale bars, 0.2 μm. (B) Aberrant cell separation observed in the images shown in panel A was quantified. Approximately 300 to 500 dividing cells from at least 15 fields of images were enumerated. Separation defects were determined as the average percentage of cells exhibiting more than one septum or plane of division. Statistical significance was determined by one-way ANOVA, followed by Bonferroni's multiple comparison test, and P values were recorded.
Mutations in the synthesis pathway for wall teichoic acid (WTA), polyribitol phosphate modified with d-alanyl and N-acetylglucosaminyl and tethered via murein linkage units to peptidoglycan (58), also cause defects in the premature assembly of septal peptidoglycan (41, 59, 60). We asked whether S. aureus mutants lacking glucosaminidase genes are defective for WTA assembly. Murein sacculi isolated from wild-type and mutant strains were subjected to acid hydrolysis, and extracts were analyzed for the release of phosphate (Fig. 7A). Mutants with single gene deletions and the atl sagAB scaH variant all harbored similar amounts of cell wall phosphates as wild-type staphylococci (Fig. 7A). WTA was also released by alkaline lysis and analyzed by alcian blue-silver-stained PAGE to demonstrate that wild-type and mutant strains released polyribitol-phosphate from murein sacculi in similar abundances and sizes (Fig. 7B). As a control, hydrofluoric acid (HF) treatment of murein sacculi removed secondary cell wall polymers, thereby abolishing the subsequent release of phosphate or WTA via acid and base treatment (Fig. 7). These data indicate that mutations in S. aureus N-acetylglucosaminidase genes do not impact WTA synthesis or attachment to peptidoglycan.
FIG 7.
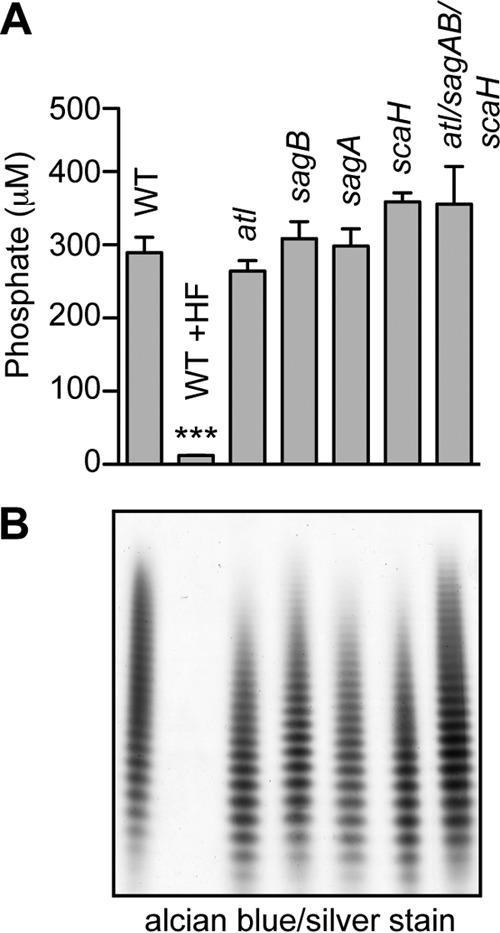
Wall teichoic acids of S. aureus glucosaminidase mutants. Murein sacculi of S. aureus Newman and its variants were isolated from mid-exponential-growth cultures (A600 of 0.6). (A) Cell wall phosphates were acid hydrolyzed from murein sacculi and quantified by colorimetric assay. Statistical significance of phosphate levels from at least three independent murein sacculus isolations was determined by one-way ANOVA, followed by Bonferroni's multiple-comparison test to wild-type levels (***, P < 0.001). (B) Wall teichoic acid was released from murein sacculi by 0.1 M NaOH treatment, separated by PAGE, and visualized with alcian blue-silver staining.
SagB is located in the membrane.
S. aureus wild type (pOS1) and a sagB(pOS1) or sagB(psagB) mutant strain were cultured to mid-exponential phase and fractionated into culture supernatant (S), cell wall (CW), membrane (M), and cytoplasm (C) (Fig. 8). Proteins in each fraction were analyzed by immunoblotting. SagB was found in the membrane (Fig. 8). As controls, immunoblotting of coagulase (Coa; supernatant), clumping factor A (ClfA; cell wall), sortase A (SrtA; membrane), and 50S ribosomal subunit L6 (L6; membrane and cytoplasm) showed that these proteins fractionated in their expected subcellular locations (Fig. 8). SagB immunoreactive signals were not detected in the sagB mutant strain; however, SagB expression and membrane localization were restored to wild-type levels when the mutant was transformed with psagB (Fig. 8).
FIG 8.
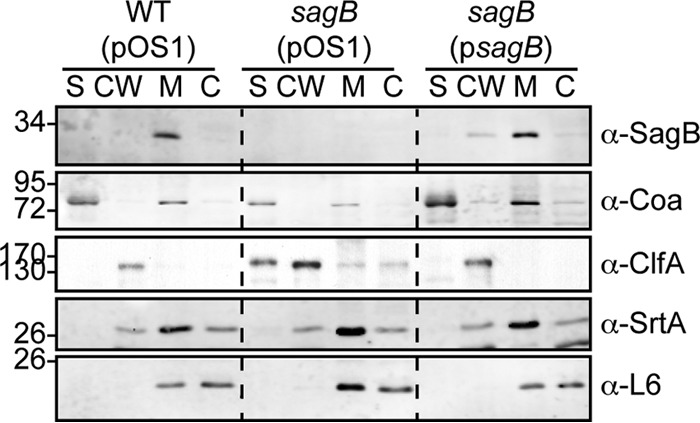
SagB localizes to the staphylococcal membrane. Cellular localization of SagB in fractionated cultures of the S. aureus wild-type (pOS1) and sagB(pOS1) strains and the complemented sagB(psagB) strain. Cultures were centrifuged to separate supernatant (S) from bacterial sediment. Staphylococci were treated with lysostaphin to generate protoplasts and solubilize cell wall-anchored proteins (CW). Protoplasts were lysed by repeated freeze-thaw cycles, and membrane (M) was separated from cytoplasmic (C) fractions. Fractionation controls for the blots include coagulase (Coa) for the supernatant, clumping factor A (ClfA) for the cell wall, sortase A (SrtA) for the membrane, and the ribosomal L6 subunit for the cytoplasm. Data shown are representative of three independent experiments.
The sagB mutant exhibits diminished secretion and increased release of cytoplasmic proteins.
Protein secretion was analyzed in the S. aureus wild-type strain and the 15 glucosaminidase mutants by separating proteins in culture medium on Coomassie-stained PAGE gels. Culture media of strains harboring the sagB deletion displayed discrete defects in protein secretion (see Fig. S4 in the supplemental material), which were restored to wild-type levels following plasmid complementation (Fig. 9A). SDS-PAGE slices for proteins whose secretion was affected by the sagB mutation were analyzed by trypsin digestion, mass spectrometric analysis, and database identification of tryptic peptides (Table 2). S. aureus secretion of Coa, von Willebrand factor binding protein (vWbp), staphylococcal complement inhibitor (SCIN), chemotaxis inhibitory protein of S. aureus (CHIPS), staphylococcal superantigen-like 1 (SSL1), SSL7, and SSL11 was reduced by the sagB mutant, whereas excretion of PhdB, GapA, and NWMN_0364 was increased (Fig. 9).
FIG 9.
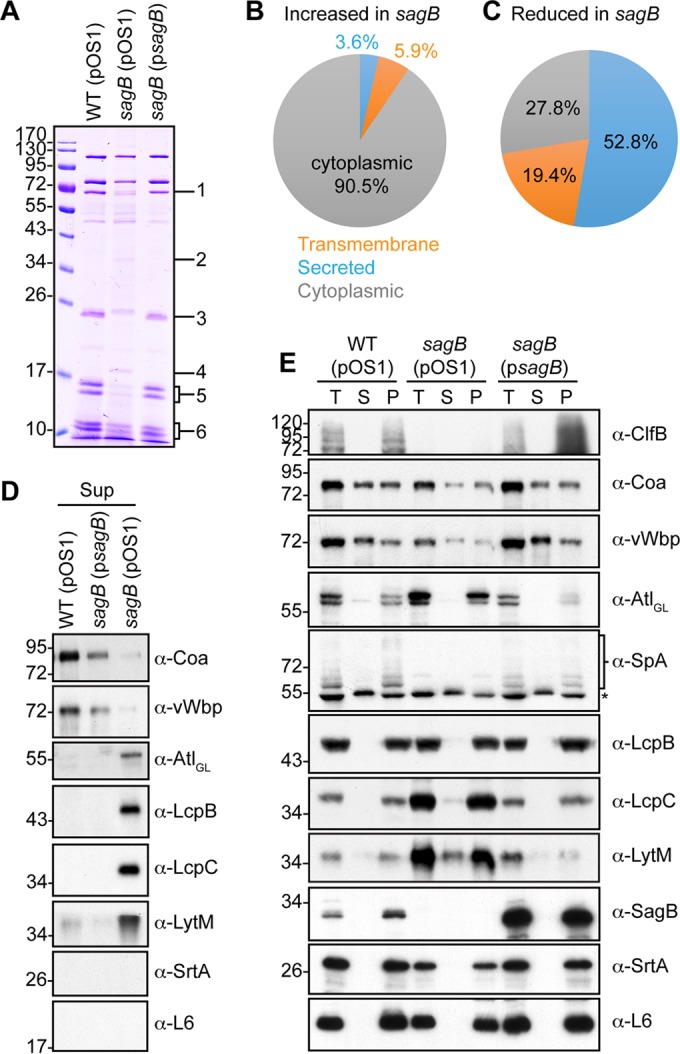
Mutations in the sagB mutant perturb protein secretion in S. aureus. (A) Supernatants of S. aureus wild-type, mutant, and complemented strains cultured to mid-exponential phase (A600 of 0.5) were analyzed for secreted protein on Coomassie-stained SDS-PAGE gels. Regions of difference in protein abundances in the gel, as indicated by the numbers on the right, were excised for the wild-type (pOS1) and sagB(pOS1) strains for analysis and protein identification by mass spectrometry (Table 2). Protein species in the sagB(pOS1) strain extract displaying at least a 10-fold increase (B) or decrease (C) relative to that of wild type (pOS1) were analyzed for predicted cellular localization using SignalP, version 4.1, and TMHMM, version 2.0, servers. (D) Immunoblots of supernatant samples prepared in the experiment shown in panel A for MALDI-TOF MS. Representative protein species (Coa, vWbp, Atl, LcpB, LcpC, and LytM) were determined by MS to exhibit a difference in abundances in the sagB mutant versus the wild type. Sortase A and ribosomal L6 blots serve as indicators of cell lysis. (E) One milliliter of mid-exponential-phase cultures (A600 of 0.5) was fractionated into total culture (T), supernatant (S), and cell pellet (P) and analyzed for protein content by immunoblotting. The asterisk in the α-SpA panel identifies Sbi.
To investigate the secretion defect of the sagB mutant, proteins detected in the excised gel slices were analyzed. A pairwise comparison of proteins identified between the wild-type and sagB strains was made, and the sum of their respective intensities was calculated. Protein species were categorized according to their enrichment or reduction in supernatants from the sagB mutant compared to the level for the wild type, using a threshold of 10-fold or greater. These species were subsequently analyzed for predicted cellular localization (see Tables S2 and S3 in the supplemental material). Compared to wild-type levels, 251 proteins meeting these criteria were enriched in sagB culture supernatants, of which 90.5% were predicted to be cytoplasmic (Fig. 9B). In contrast, 36 protein species were reduced in the sagB supernatants, 72.2% of which were predicted to be secreted or membrane localized (Fig. 9C). These data suggest that the sagB mutant not only fails to secrete many signal peptide-bearing precursor proteins but also excretes cytoplasmic proteins into culture supernatants. To validate these claims, some of the proteins detected by mass spectrometry were analyzed by immunoblot analysis. As predicted, Coa and vWbp secretion was decreased in the sagB mutant, whereas Atl, LcpB, LcpC, and LytM were secreted in greater abundance (Fig. 9C and D). Cell wall-anchored protein clumping factor B (ClfB) and protein A (SpA) were also diminished in the sagB mutant (Fig. 9E). SrtA, a membrane protein, and ribosomal subunit L6 were used as controls to assess cell lysis of wild-type and mutant strains.
Excretion of proteins without signal peptides is not attributable to autolysis or membrane leakage.
Studying S. aureus isolate SA113, Pasztor et al. reported that atl mutation caused defects in the excretion of 22 cytoplasmic proteins (61). We note that in S. aureus Newman, the sagB mutation causes increased expression of Atl (Fig. 9D and E). However, increased expression of atl was not associated with increased bacterial lysis as treatment of staphylococci with 0.05% Triton X-100, a known inducer of staphylococcal autolysis (62), did not trigger increased lysis of sagB mutant cells compared to levels of wild-type cells (Fig. 10A and B). To determine whether the observed excretion of cytoplasmic proteins in the sagB strain is attributable to defects in membrane integrity, staphylococci were stained with propidium iodide, a nucleic acid dye that requires ruptured membranes for penetrance into staphylococci. Flow cytometry of propidium iodide-stained staphylococci did not reveal significant differences in membrane rupture between sagB mutant and wild-type staphylococci (Fig. 10C).
FIG 10.

Autolysis and membrane permeability in S. aureus glucosaminidase mutants. Triton X-100 (0.05%) or buffer alone was added to mid-exponential-phase cultures (A600 of 0.5) of S. aureus suspended in 50 mM phosphate buffer (pH 7.5). Turbidity at A600 was monitored over 3 h and plotted as the A600 of lysostaphin-inoculated staphylococci of buffer alone at each time point. Autolysis, as determined by reduction of turbidity, was compared between the wild-type, atl, and sagB strains (A). (B) Autolysis in 0.05% Triton X-100 was assessed in the wild-type (pOS1), sagB(pOS1), and sagB(psagB) strains. (C) Propidium iodide staining of bacteria was used to assess membrane permeability. One milliliter of culture (A600 of 0.5) was fixed with paraformaldehyde and subsequently stained with SYTO 9 for total cells and with propidium iodide to assess membrane integrity. SYTO 9-positive cells were analyzed for propidium iodide positivity using flow cytometry. Data from triplicate samples of 10,000 cells are presented. Statistical significance was assessed by one-way ANOVA with Bonferroni's multiple comparison test. ns, not significant.
DISCUSSION
During peptidoglycan synthesis, glycan strands are polymerized from lipid II precursors through the processive activity of peptidoglycan glycosyltransferases (PGTs) (25, 63). Studying purified PGTs from E. coli (PBP1A), S. aureus (PBP2), and Enterococcus faecalis, Wang et al. observed that each PGT generated polymers with characteristic lengths, independently of enzyme/substrate ratios, and proposed that PGTs may rely on an intrinsic mechanism for controlling product length (64). Mutations in the structural gene for mltG, encoding a lytic transglycosylase of the YceG family (Pfam02618) in E. coli, increase the length of glycan strands in the peptidoglycan (52). As MltG is thought to associate with PBP1B, an E. coli PGT, Yunck and colleagues proposed an alternative model, namely, that lytic transglycosylases of the YceG family may terminate glycan chain polymerization during peptidoglycan synthesis (52). The genome of S. aureus does not encode lytic YceG-type transglycosylases, suggesting that S. aureus must have evolved another mechanism to generate the characteristically short glycan strands of its peptidoglycan layer (17). We show here that SagB cleaves the glycan strands of S. aureus peptidoglycan to its physiological length and that sagB mutant staphylococci assemble murein sacculi comprised of elongated glycan strands. The possibility that SagB associates with PBP2, MGT, or SgtA transglycosidases either in vitro or in vivo was also examined; however, physical associations between these polypeptides and SagB could not be detected (data not shown). We therefore propose that, following PBP2-mediated peptidoglycan synthesis, SagB alone may be responsible for cleaving staphylococcal glycan strands to their final sizes.
Earlier work used genetic approaches to identify the genetic determinants for methicillin resistance in S. aureus, for example, by focusing on mutations that confer both lysostaphin resistance and increased susceptibility to methicillin (oxacillin) (65–67). These studies identified several genes and biochemical reactions of peptidoglycan assembly, specifically, the catalysts for pentaglycine cross bridge synthesis (FemA, FemB, and FmhB) (68). Although not identified in screens for factors essential for methicillin resistance (fem), we note that sagB meets the criteria established by Berger-Bächi and Labischinski and colleagues for genes that contribute to β-lactam resistance in staphylococci (69, 70). The increased lengths of S. aureus glycan chains likely represent the underlying mechanism for lysostaphin resistance, as cleavage of pentaglycine cross bridges in the cell wall of sagB mutants, unlike cleavage of wild-type peptidoglycan, does not trigger rapid lysis of bacterial cells. A related mechanism likely underwrites the increased susceptibility of the sagB mutant USA300 LAC toward oxacillin. In the presence of β-lactam antibiotics, PBP2a, the methicillin resistance determinant (71), catalyzes the transpeptidation reaction of cell wall synthesis yet relies on PBP2, a bifunctional enzyme with PGT and β-lactam-sensitive transpeptidase activity, to polymerize glycan strands (23, 72). Alteration of peptidoglycan substrate, i.e., the exaggerated length of glycan chains in sagB mutant staphylococci, likely perturbs PBP2a substrate recognition and transpeptidation to generate cross-linked cell wall, thereby increasing the susceptibility of sagB mutant MRSA toward oxacillin. Of note, the sagB mutation also caused a modest increase in resistance of USA300 to vancomycin, an antibiotic frequently used for the therapy of MRSA infections. Because of associated toxicity, tissue concentration of vancomycin must be maintained at low levels, and MRSA strains with moderate increases in resistance (vancomycin-intermediate S. aureus [VISA]) cause therapeutic failures (73).
S. aureus Newman variants lacking four glucosaminidase genes—atl, sagAB, and scaH—are viable and, compared to the wild type, replicate at a slightly reduced rate. In contrast, the S. aureus SH1000 atl sagA scaH mutant cannot replicate without sagB expression (36). Unlike SH1000, a laboratory strain that has been cured of bacteriophages (36), S. aureus Newman is lysogenized by four different phages, two of which encode muralytic enzymes with predicted N-acetylglucosaminidase activity (Fig. 1A) (43). These enzymes, designated LytP2 and LytP4, are, however, also dispensable for growth as S. aureus Newman lacking all six glucosaminidase genes remained viable (Fig. 6). As expected, the atl sagAB scaH lytP2 lytPG4 mutant displayed cell cycle defects with premature assembly of cross walls in bacteria that had not yet completed cell division (Fig. 6). The genetic requirements for staphylococcal replication are known to vary among different strains, which was previously observed for LytR-CpsA-Psr (LCP) enzymes immobilizing secondary cell polymers via murein linkage units to bacterial peptidoglycan (41, 74). Thus, N-acetylglucosaminidase enzymes may represent yet another example for genetic heterogeneity in S. aureus.
We posit that the peptidoglycan layer of S. aureus is impenetrable for proteins destined to travel across the cell wall envelope. Mutations that perturb protein secretion across the staphylococcal cell wall envelope have heretofore not been identified. We demonstrate that sagB mutations diminish the secretion of signal peptide-bearing precursors across the staphylococcal cell wall envelope while simultaneously increasing the excretion of cytoplasmic proteins, a class of proteins first described by Pasztor and colleagues (61). The mechanisms underlying these phenotypes are not yet understood, and we propose two models that may be useful for future experimental testing. First, SagB-mediated truncation of glycan strands may introduce perforations in the peptidoglycan structure, thereby enabling passive diffusion of proteins destined for secretion across the cell wall envelope. Second, SagB-mediated processing of peptidoglycan may enable the assembly of conduits that extend the Sec pathway for the catalyzed secretion of signal peptide-bearing precursors across the cell wall envelope. Whatever the mode of protein trafficking across the cell wall envelope, the identification of a protein secretion phenotype in sagB mutant staphylococci provides new opportunities for experimental exploration of its underlying mechanisms.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yimei Chen (University of Chicago) for transmission electron microscopy, Ross Tomaino (Taplin Mass Spectrometry Facility, Harvard University) for mass spectrometry data, and Andrea DeDent, Vilasack Thammavongsa, Lena Thomer, Wenqi Yu, Stephanie Willing, Hwan Keun Kim, Carla Emolo, and Fabiana Falugi for experimental advice and discussion.
Y.G.Y.C. acknowledges support from the American Heart Association (award 13POST16980091). M.B.F. was supported by a postdoctoral fellowship award from the National Institute of Allergy and Infectious Diseases (NIAID; F32AI085709). This work was supported by grants AI038897 and AI052474 from the NIAID, Infectious Diseases Branch, to O.S.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00983-15.
REFERENCES
- 1.Giesbrecht P, Kersten T, Maidhof H, Wecke J. 1998. Staphylococcal cell wall: morphogenesis and fatal variations in the presence of penicillin. Microbiol Mol Biol Rev 62:1371–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monteiro JM, Fernandes PB, Vaz F, Pereira AR, Tavares AC, Ferreira MT, Pereira PM, Veiga H, Kuru E, VanNieuwenhze MS, Brun YV, Filipe SR, Pinho MG. 2015. Cell shape dynamics during the staphylococcal cell cycle. Nat Commun 6:8055. doi: 10.1038/ncomms9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kajimura J, Fujiwara T, Yamada S, Suzawa Y, Nishida T, Oyamada Y, Hayashi I, Yamagishi J, Komatsuzawa H, Sugai M. 2005. Identification and molecular characterization of an N-acetylmuramyl-l-alanine amidase Sle1 involved in cell separation of Staphylococcus aureus. Mol Microbiol 58:1087–1101. doi: 10.1111/j.1365-2958.2005.04881.x. [DOI] [PubMed] [Google Scholar]
- 4.Yamada S, Sugai M, Komatsuzawa H, Nakashima S, Oshida T, Matsumoto A, Suginaka H. 1996. An autolysin ring associated with cell separation of Staphylococcus aureus. J Bacteriol 178:1565–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frankel MB, Hendrickx AP, Missiakas DM, Schneewind O. 2011. LytN, a murein hydrolase in the cross-wall compartment of Staphylococcus aureus, is involved in proper bacterial growth and envelope assembly. J Biol Chem 286:32593–32605. doi: 10.1074/jbc.M111.258863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komatsuzawa H, Sugai M, Nakashima S, Yamada S, Matsumoto A, Oshida T, Suginaka H. 1997. Subcellular localization of the major autolysin, ATL, and its processed proteins in Staphylococcus aureus. Microbiol Immunol 41:469–479. doi: 10.1111/j.1348-0421.1997.tb01880.x. [DOI] [PubMed] [Google Scholar]
- 7.Frankel MB, Schneewind O. 2012. Determinants of murein hydrolase targeting to cross-wall of Staphylococcus aureus peptidoglycan. J Biol Chem 287:10460–10471. doi: 10.1074/jbc.M111.336404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baba T, Schneewind O. 1996. Target cell specificity of a bacteriocin molecule: a C-terminal signal directs lysostaphin to the cell wall of Staphylococcus aureus. EMBO J 15:4789–4797. [PMC free article] [PubMed] [Google Scholar]
- 9.Oshida T, Sugai M, Komatsuzawa H, Hong YM, Suginaka H, Tomasz A. 1995. A Staphylococcus aureus autolysin that has an N-acetylmuramoyl-l-alanine amidase domain and an endo-β-N-acetylglucosaminidase domain: cloning, sequence analysis, and characterization. Proc Natl Acad Sci U S A 92:285–289. doi: 10.1073/pnas.92.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baba T, Schneewind O. 1998. Targeting of muralytic enzymes to the cell division site of Gram-positive bacteria: repeat domains direct autolysin to the equatorial surface ring of Staphylococcus aureus. EMBO J 17:4639–4646. doi: 10.1093/emboj/17.16.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zoll S, Schlag M, Shkumatov AV, Rautenberg M, Svergun DI, Götz F, Stehle T. 2012. Ligand-binding properties and conformational dynamics of autolysin repeat domains in staphylococcal cell wall recognition. J Bacteriol 194:3789–3802. doi: 10.1128/JB.00331-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugai M, Komatsuzawa H, Akiyama T, Hong Y-M, Oshida T, Miyake Y, Yamaguchi T, Suginaka H. 1995. Identification of endo-β-N-acetylglucosaminidase and N-acetylmuramyl-l-alanine amidase as cluster dispersing enzymes in Staphylococcus aureus. J Bacteriol 177:1491–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wadström T, Hisatsune K. 1970. Bacteriolytic enzymes from Staphylococcus aureus. Specificity of action of endo-β-N-acetylglucosaminidase. Biochem J 120:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi J, Komatsuzawa H, Yamada S, Nishida T, Labischinski H, Fujiwara T, Ohara M, Yamagishi J, Sugai M. 2002. Molecular characterization of an atl null mutant of Staphylococcus aureus. Microbiol Immunol 46:601–612. doi: 10.1111/j.1348-0421.2002.tb02741.x. [DOI] [PubMed] [Google Scholar]
- 15.Strominger JL, Ghuysen J-M. 1967. Mechanisms of enzymatic bacteriolysis. Science 156:213–221. doi: 10.1126/science.156.3772.213. [DOI] [PubMed] [Google Scholar]
- 16.Ghuysen J-M, Strominger JL. 1963. Structure of the cell wall of Staphylococcus aureus, strain Copenhagen. II. Separation and structure of the disaccharides. Biochemistry 2:1119–1125. [DOI] [PubMed] [Google Scholar]
- 17.Boneca IG, Huang ZH, Gage DA, Tomasz A. 2000. Characterization of Staphylococcus aureus cell wall glycan strands, evidence for a new β-N-acetylglucosaminidase activity. J Biol Chem 275:9910–9918. doi: 10.1074/jbc.275.14.9910. [DOI] [PubMed] [Google Scholar]
- 18.Ghuysen J-M, Tipper DJ, Birge CH, Strominger JL. 1965. Structure of the cell wall of Staphylococcus aureus strain Copenhagen. VI. The soluble glycopeptide and its sequential degradation by peptidases. Biochemistry 4:2245–2254. [DOI] [PubMed] [Google Scholar]
- 19.Tipper DJ, Ghuysen J-M, Strominger JL. 1965. Structure of the cell wall of Staphylococcus aureus, strain Copenhagen. III. Further studies of the disaccharides. Biochemistry 4:468–473. [DOI] [PubMed] [Google Scholar]
- 20.Tipper DJ, Strominger JL. 1965. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-d-alanyl-alanine. Proc Natl Acad Sci U S A 54:1133–1141. doi: 10.1073/pnas.54.4.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higashi Y, Strominger JL, Sweeley CC. 1967. Structure of a lipid intermediate in cell wall peptidoglycan synthesis: a derivative of C55 isoprenoid alcohol. Proc Natl Acad Sci U S A 57:1878–1884. doi: 10.1073/pnas.57.6.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson JS, Matsuhashi M, Haskin MA, Strominger JL. 1965. Lipid-phosphoacetylmuramyl-pentapeptide and lipid-phosphodisaccharide-pentapeptide: presumed membrane transport intermediates in cell wall synthesis. Proc Natl Acad Sci U S A 53:881–889. doi: 10.1073/pnas.53.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovering AL, de Castro LH, Lim D, Strynadka NC. 2007. Structural insight into the transglycosylation step of bacterial cell wall biosynthesis. Science 315:1402–1405. doi: 10.1126/science.1136611. [DOI] [PubMed] [Google Scholar]
- 24.Reed P, Veiga H, Jorge AM, Terrak M, Pinho MG. 2011. Monofunctional transglycosylases are not essential for Staphylococcus aureus cell wall synthesis. J Bacteriol 193:2549–2556. doi: 10.1128/JB.01474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lovering AL, Safadi SS, Strynadka NC. 2012. Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem 81:451–478. doi: 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]
- 26.Kozarich JW, Strominger JL. 1978. A membrane enzyme from Staphylococcus aureus which catalyzes transpeptidase, carboxypeptidase, and penicillinase activities. J Biol Chem 253:1272–1278. [PubMed] [Google Scholar]
- 27.Spratt BG, Strominger JL. 1976. Identification of the major penicillin-binding proteins of Escherichia coli as d-alanine carboxypeptidase IA. J Bacteriol 127:660–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Jonge BLM, Chang YS, Gage D, Tomasz A. 1992. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. J Biol Chem 267:11248–11254. [PubMed] [Google Scholar]
- 29.Sieradzki K, Pinho MG, Tomasz A. 1999. Inactivated pbp4 in highly glycopeptide-resistant laboratory mutants of Staphylococcus aureus. J Biol Chem 274:18942–18946. doi: 10.1074/jbc.274.27.18942. [DOI] [PubMed] [Google Scholar]
- 30.Łeski TA, Tomasz A. 2005. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J Bacteriol 187:1815–1824. doi: 10.1128/JB.187.5.1815-1824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pereira SFF, Henriques AO, Pinho MG, de Lencastre H, Tomasz A. 2007. Role of PBP1 in cell division of Staphylococcus aureus. J Bacteriol 189:3525–3531. doi: 10.1128/JB.00044-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneewind O, Mihaylova-Petkov D, Model P. 1993. Cell wall sorting signals in surface protein of Gram-positive bacteria. EMBO 12:4803–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sibbald MJ, Ziebandt AK, Engelmann S, Hecker M, de Jong A, Harmsen HJ, Raangs GC, Stokroos I, Arends JP, Dubois JY, van Dijl JM. 2006. Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol Mol Biol Rev 70:755–788. doi: 10.1128/MMBR.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravipaty S, Reilly JP. 2010. Comprehensive characterization of methicillin-resistant Staphylococcus aureus subsp. aureus COL secretome by two-dimensional liquid chromatography and mass spectrometry. Mol Cell Proteomics 9:1898–1919. doi: 10.1074/mcp.M900494-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebner P, Prax M, Nega M, Koch I, Dube L, Yu W, Rinker J, Popella P, Flötenmeyer M, Götz F. 2015. Excretion of cytoplasmic proteins (ECP) in Staphylococcus aureus. Mol Microbiol 97:775–789. doi: 10.1111/mmi.13065. [DOI] [PubMed] [Google Scholar]
- 36.Wheeler R, Turner RD, Bailey RG, Salamaga B, Mesnage S, Mohamad SA, Hayhurst EJ, Horsburgh M, Hobbs JK, Foster SJ. 2015. Bacterial cell enlargement requires control of cell wall stiffness mediated by peptidoglycan hydrolases. mBio 6:e00660–15. doi: 10.1128/mBio.00660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Saile E, Koehler TM. 2002. Control of anthrax toxin gene expression by the transition state regulator abrB. J Bacteriol 184:370–380. doi: 10.1128/JB.184.2.370-380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kern J, Ryan C, Faull K, Schneewind O. 2010. Bacillus anthracis surface-layer proteins assemble by binding to the secondary cell wall polysaccharide in a manner that requires csaB and tagO. J Mol Biol 401:757–775. doi: 10.1016/j.jmb.2010.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. 2004. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci U S A 101:12312–12317. doi: 10.1073/pnas.0404728101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan YGY, Frankel MB, Dengler V, Schneewind O, Missiakas DM. 2013. Staphylococcus aureus mutants lacking the LytR-CpsA-Psr (LCP) family of enzymes release wall teichoic acids into the extracellular medium. J Bacteriol 195:4650–4659. doi: 10.1128/JB.00544-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes. J Bacteriol 190:300–310. doi: 10.1128/JB.01000-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bae T, Baba T, Hiramatsu K, Schneewind O. 2006. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol Microbiol 62:1035–1047. doi: 10.1111/j.1365-2958.2006.05441.x. [DOI] [PubMed] [Google Scholar]
- 44.Nambu T, Minamino T, Macnab RM, Kutsukake K. 1999. Peptidoglycan-hydrolyzing activity of the FlgJ protein, essential for flagellar rod formation in Salmonella typhimurium. J Bacteriol 181:1555–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bublitz M, Polle L, Holland C, Heinz DW, Nimtz M, Schubert WD. 2009. Structural basis for autoinhibition and activation of Auto, a virulence-associated peptidoglycan hydrolase of Listeria monocytogenes. Mol Microbiol 71:1509–1522. doi: 10.1111/j.1365-2958.2009.06619.x. [DOI] [PubMed] [Google Scholar]
- 46.Rolain T, Bernard E, Beaussart A, Degand H, Courtin P, Egge-Jacobsen W, Bron PA, Morsomme P, Kleerebezem M, Chapot-Chartier MP, Dufrêne YF, Hols P. 2013. O-Glycosylation as a novel control mechanism of peptidoglycan hydrolase activity. J Biol Chem 288:22233–22247. doi: 10.1074/jbc.M113.470716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindler CA, Schuhardt VT. 1964. Lysostaphin: a new bacteriolytic agent for the Staphylococcus. Proc Natl Acad Sci U S A 51:414–421. doi: 10.1073/pnas.51.3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Browder HP, Zygmunt WA, Young JR, Tavormina PA. 1965. Lysostaphin: enzymatic mode of action. Biochem Biophys Res Com 19:383–389. doi: 10.1016/0006-291X(65)90473-0. [DOI] [PubMed] [Google Scholar]
- 49.Snowden MA, Perkins HR, Wyke AW, Hayes MV, Ward JB. 1989. Cross-linking and O-acetylation of newly synthesized peptidoglycan in Staphylococcus aureus H. J Gen Microbiol 135:3015–3022. [DOI] [PubMed] [Google Scholar]
- 50.Pinho MG, Kjos M, Veening JW. 2013. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol 11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- 51.Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, Shales M, Lovett S, Winkler ME, Krogan NJ, Typas A, Gross CA. 2011. Phenotypic landscape of a bacterial cell. Cell 144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yunck R, Cho H, Bernhardt TG. 20 October 2015. Identification of MltG as a potential terminase for peptidoglycan polymerization in bacteria. Mol Microbiol doi: 10.1111/mmi.13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Heijenoort Y, Derrien M, van Heijenoort J. 1978. Polymerization by transglycosylation in the biosynthesis of the peptidoglycan of Escherichia coli K-12 and its inhibition by antibiotics. FEBS Lett 89:141–144. doi: 10.1016/0014-5793(78)80540-7. [DOI] [PubMed] [Google Scholar]
- 54.Simon HJ, Rantz LA. 1962. The newer penicillins. I. Bacteriological and clinical pharmacological investigations with methicillin and oxacillin. Ann Intern Med 57:335–343. [DOI] [PubMed] [Google Scholar]
- 55.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 56.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 57.Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Götz F. 2006. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett 259:260–268. doi: 10.1111/j.1574-6968.2006.00281.x. [DOI] [PubMed] [Google Scholar]
- 58.Xia G, Peschel A. 2008. Toward the pathway of S. aureus WTA biosynthesis. Chem Biol 15:95–96. doi: 10.1016/j.chembiol.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 59.Over B, Heusser R, McCallum N, Schulthess B, Kupferschmied P, Gaiani JM, Sifri CD, Berger-Bächi B, Stutzmann Meier P. 2011. LytR-CpsA-Psr proteins in Staphylococcus aureus display partial functional redundancy and the deletion of all three severely impairs septum placement and cell separation. FEMS Microbiol Lett 320:142–151. doi: 10.1111/j.1574-6968.2011.02303.x. [DOI] [PubMed] [Google Scholar]
- 60.Campbell J, Singh AK, Santa Maria JPJ, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S. 2011. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem Biol 6:106–116. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pasztor L, Ziebandt AK, Nega M, Schlag M, Haase S, Franz-Wachtel M, Madlung J, Nordheim A, Heinrichs DE, Götz F. 2010. Staphylococcal major autolysin (Atl) is involved in excretion of cytoplasmic proteins. J Biol Chem 285:36794–36803. doi: 10.1074/jbc.M110.167312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Jonge BLM, de Lencastre H, Tomasz A. 1991. Suppression of autolysis and cell wall turnover in heterogeneous Tn551 mutants of a methicillin-resistant Staphylcoccus aureus strain. J Bacteriol 173:1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perlstein DL, Zhang Y, Wang TS, Kahne DE, Walker S. 2007. The direction of glycan chain elongation by peptidoglycan glycosyltransferases. J Am Chem Soc 129:12674–12675. doi: 10.1021/ja075965y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang TS, Manning SA, Walker S, Kahne D. 2008. Isolated peptidoglycan glycosyltransferases from different organisms produce different glycan chain lengths. J Am Chem Soc 130:14068–14069. doi: 10.1021/ja806016y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maidhof H, Reinicke B, Blumel P, Berger-Bächi B, Labischinski H. 1991. femA, which encodes a factor essential for expression of methicillin resistance, affects glycine content of peptidoglycan in methicillin-resistant and methicillin susceptible Staphylococcus aureus strains. J Bacteriol 173:3507–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Henze U, Sidow T, Wecke J, Labischinski H, Berger-Bächi B. 1993. Influence of femB on methicillin resistance and peptidoglycan metabolism in Staphylococcus aureus. J Bacteriol 175:1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rohrer S, Ehlert K, Tschierske M, Labischinski H, Berger-Bächi B. 1999. The essential Staphylococcus aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation. Proc Natl Acad Sci U S A 96:9351–9356. doi: 10.1073/pnas.96.16.9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berger-Bächi B, Rohrer S. 2002. Factors influencing methicillin resistance in staphylococci. Arch Microbiol 178:165–171. doi: 10.1007/s00203-002-0436-0. [DOI] [PubMed] [Google Scholar]
- 69.Berger-Bächi B. 1994. Expression of resistance to methicillin. Trends Microbiol 2:389–309. doi: 10.1016/0966-842X(94)90617-3. [DOI] [PubMed] [Google Scholar]
- 70.Labischinski H, Ehlert K, Berger-Bachi B. 1998. The targeting of factors necessary for expression of methicillin resistance in staphylococci. J Antimicrob Chemother 41:581–584. doi: 10.1093/jac/41.6.581. [DOI] [PubMed] [Google Scholar]
- 71.Ubukata K, Nonoguchi R, Matsuhashi M, Konno M. 1989. Expression and inducibility in Staphylococcus aureus of the mecA gene, which encodes a methicillin-resistant S. aureus-specific penicillin-binding protein. J Bacteriol 171:2882–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pinho MG, Filipe SR, De Lencastre H, Tomasz A. 2001. Complementation of the essential peptidoglycan transpeptidase function of penicillin-binding protein 2 (PBP2) by the drug resistance protein PBP2A in Staphylococcus aureus. J Bacteriol 183:6525–6531. doi: 10.1128/JB.183.22.6525-6531.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chambers HF, DeLeo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dengler V, Stutzmann Meier P, Heusser R, Kupferschmied P, Fazekas J, Friebe S, Staufer SB, Majcherczyk PA, Moreillon P, Berger-Bächi B, McCallum N. 2012. Deletion of hypothetical wall teichoic acid ligases in Staphylococcus aureus activates the cell wall stress response. FEMS Microbiol Lett 333:109–120. doi: 10.1111/j.1574-6968.2012.02603.x. [DOI] [PubMed] [Google Scholar]
- 75.Duthie ES. 1954. Evidence for two forms of staphylococcal coagulase. J Gen Microbiol 10:427. doi: 10.1099/00221287-10-3-427. [DOI] [PubMed] [Google Scholar]
- 76.Chen C, Krishnan V, Macon K, Manne K, Narayana SV, Schneewind O. 2013. Secreted proteases control autolysin-mediated biofilm growth of Staphylococcus aureus. J Biol Chem 288:29440–29452. doi: 10.1074/jbc.M113.502039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.