

ABSTRACT
Toll-like receptors (TLRs) can be expressed by tumor cells, and each TLR exhibits different biological functions. Evidences showed the activation of some certain TLRs could promote tumor progression. One of which TLR4 has been found to promote hepatocellular carcinoma (HCC) cells proliferation, but the detailed mechanism is still unknown. In the present study, we verified that TLR4 was functionally expressed on HCC cells, and TLR4 agonist lipopolysaccharide (LPS) could stimulate the proliferation and clone formation of HCC cells. Most importantly, we found a COX-2/PGE2/STAT3 positive feedback loop exists in HCC cells, which could be provoked by TLR4 activation. Consistently, the expression of TLR4, COX-2 and p-STAT3Y705 was positively correlated with each other in liver tumor tissues from patients with primary HCC. Further investigation demonstrated this loop played a dominant role in TLR4-induced HCC cell proliferation and multidrug resistance (MDR) to chemotherapy. Inhibition of TLR4 or COX-2/PGE2/STAT3 loop would attenuate LPS-induced inflammation and proliferation of HCC cells, and enhance the sensitivity of HCC cells to chemotherapeutics in vitro. By using a primary HCC model, we observed COX-2/PGE2/STAT3 loop was significantly blocked in TLR4−/− mice compared to wild type mice, and there was no obvious tumorgenesis sign in TLR4−/− mice. Therefore, these findings provided the precise molecular mechanism of TLR4 signaling pathway involved in HCC progress, and suggested that TLR4 may be a promising target for HCC treatment.
KEYWORDS: HCC, proliferation, multidrug resistance, STAT3, TLR4
Abbreviations
- TLR
toll like receptor
- HCC
hepatocellular carcinoma
- LPS
lipopolysaccharide
- PRR
pattern recognition receptor
- MDR
multidrug resistance
- PAMP
pathogen-associated molecular patterns
- STAT3
signal transducer and transcription 3
- COX-2
cyclooxygenase-2
- mPGEs-1
microsomal PGE synthase 1
- PGE2
prostaglandin E2
- DEN
diethylnitrosamine
- CCl4
carbontetrachloride
- AFP
alpha fetoprotein
- VEGF
vascular endothelial growth factor
- MDR
multidrug resistance
- P-gp
P-glycoprotein
- DOX
doxorubicin
- 5-FU
5-fluorouracil
Introduction
As the important pattern recognition receptors (PRRs) family members, Toll-like receptors (TLRs) are expressed by a variety of cell types, especially immune cells. TLRs mediate the recognition of conserved molecular patterns of microbial origin, and regulate both innate and adaptive immune responses.1,2 Due to the pivotal role of TLRs in immune responses, TLR-based immune therapeutic strategies have been accepted broadly and adopted in the treatment of infectious diseases, cancer and allergic diseases.3-5 However, TLRs expression is not confined to immune cells. Increasing evidences demonstrate that TLRs are also expressed in many tumor tissues and cell lines.6 Further research indicates TLRs expression is closely associated with the biological characterizations of tumor cells, and various TLRs may work diversely on different tumor types. For instance, TLR4 expressed by human head and neck squamous cell carcinoma induces tumor growth and facilitates tumor escape from immune surveillance 7; the activation of TLR9 by CpG-oligonucleotides (CpG-ODNs) stimulates prostate cancer invasion.8 In addition, the increased expression of TLR5 and TLR9 may play a significant role in cervical neoplastic progression and is considered as promising marker for malignant transformation of cervical squamous cell.9,10 In contrast, the activation of some certain TLRs in tumor cells, such as TLR3 and TLR9, can induce cell apoptosis and growth inhibition.11,12 Because of the diversity of TLRs, the functions and precise mechanisms of each TLR in tumor pathogenesis need to be clarified completely.
Hepatocellular carcinoma (HCC) is the fifth common cancer and ranks second among all the primary cancer-related mortalities worldwide. It has been well recognized that HCC is a long-term sequence of chronic inflammatory liver diseases, and 80% cases develop from fibrotic and cirrhotic livers.13,14 Clinical research demonstrates that increased translocation of intestinal bacteria is commonly observed in patients with chronic liver disease, which is accompanied by high level of plasmatic endotoxin,15 indicating liver is inevitably exposed to the attack of intestinal bacteria. Consequently, intestinal bacteria components termed as pathogen-associated molecular patterns (PAMPs) contribute to liver inflammation via activating TLRs, which aggravates liver fibrosis and cirrhosis.16 A recent in vivo study observed that LPS from gut microbiota contributed to HCC promotion by activating TLR4 signaling, inducing proliferative and anti-apoptotic signals in non-bone marrow-derived resident liver cells; and gut sterilization at late stages could efficiently prevent HCC promotion.17 These observations indicate there is a close link between gut dysbacteriosis and HCC progression, in which TLR4 displays critical roles. Nevertheless, the exact underlying molecular mechanism of the complex biological process is still undiscovered.
In the present research, we found a COX-2/PGE2/STAT3 positive feedback loop could be provoked by LPS stimulation, which plays a central role in HCC cell growth and chemoresistance. These findings suggest that blocking TLR4 signaling in HCC cells might be an efficient treatment option.
Results
TLR4 was functionally expressed on HCC cells
As we observed previously, TLR1-TLR10 was expressed in HCC cells at different levels, of which TLR4 was expressed at a comparatively high level.12 To investigate whether TLR4 was functional in HCC cells, LPS was used to treat HCC cell lines HepG2 and H7402. We observed that TLR4 expression was increased at both mRNA level (Fig. 1A) and protein level (Fig. 1B). Meanwhile, NF-κB signaling pathway was activated in HCC cells (Fig. 1C), accompanied with the increase of downstream inflammatory cytokine genes, including IL-6, IL-8 and TNF-α (Fig. 1D). These results confirmed that TLR4 was functionally expressed in HCC cells and responsive to LPS stimulation, indicating TLR4 may be involved in HCC biological properties.
Figure 1.

TLR4 was functionally expressed on HCC cells. A. H7402 and HepG2 cells were collected after stimulated with LPS for 24 h, and then mRNA levels of TLR4 were detected by RT-PCR. B. After being treated with LPS for 2 h or 5 h, protein levels of TLR4 in H7402 and HepG2 cells were measured by western blot. C. Total proteins were extracted from H7402 and HepG2 cells treated with LPS for different time, and then p-NF-κB and NF-κB levels were analyzed by western blot. D. H7402 and HepG2 cells were treated with LPS for different time after starving for 4 h, then inflammatory cytokines IL-6, IL-8, and TNF-α were measured by qPCR. One representative data from three independent experiments was presented. Values are means ± S.D. of three independent experiments. *p < 0.05, **p < 0.01 compared with untreated group.
TLR4 promoted HCC proliferation in a STAT3 signaling pathway dependent manner
To investigate the effects of TLR4 on HCC characterizations, we analyzed the effect of LPS on HCC cell growth and colony formation abilities. As shown in Fig. S1A and B, HCC cells displayed significantly augmented proliferation ability under LPS stimulation in a dose- and time-dependent manner, especially H7402 cells. And, more clones were formed by HCC cells exposed to LPS for 7 d than untreated cells (Fig. S1C). Generally, the deregulated proliferation and inhibition of apoptosis are intimately coupled and involved in tumor development.18 However, LPS treatment did not affect the apoptosis of HCC cells significantly, as well as apoptosis-related genes (data not shown). Here, we found the cell-cycle related gene Cyclin D1 was elevated by LPS stimulation (Fig. S1D) accompanied with increased number of S phase HCC cells (Fig. S1E). These results indicated that TLR4 activation could augment HCC cell viability and proliferation via accelerating cell cycle.
NF-κB signaling, the most important downstream pathway of TLRs, actively participates in mediating inflammatory response and acts as a central link between hepatic injury, fibrosis and HCC.19,20 In addition, STAT3 (signal transducer and transcription 3) constitutively activated in a wide range of malignancies such as HCC cells,21 has been generally demonstrated to be associated with inflammation as well as tumorigenesis.22 Most importantly, a cross-talk between NF-κB and STAT3 has been demonstrated.23 Thus, we wanted to understand whether TLR4 activation could initiate STAT3 signaling in HCC cells. As shown in Fig. 2A, STAT3 signaling was activated under the treatment of LPS, and the activation status could last for more than 6 h. Furthermore, the luciferase reporter assay showed the transcriptional activity of STAT3 in HCC cells was obviously enhanced by LPS treatment (Fig. 2B). To further verify whether STAT3 is involved in LPS-induced HCC proliferation, STAT3-decoy ODN was used to block STAT3 signaling in HCC cells (25). We observed that the growth of H7402 cells transfected with STAT3 decoy-ODN was significantly decreased in response to LPS compared to scramble-ODN treated cells (Fig. 2C). Similarly, S3I-201, an inhibitor of STAT3 pathway functioning by blocking the phosphorylation and dimerization events necessary for activation, also suppressed LPS-induced HCC cells proliferation (Fig. 2D). These findings indicated that STAT3 activation was involved in LPS-induced HCC cells proliferation.
Figure 2.
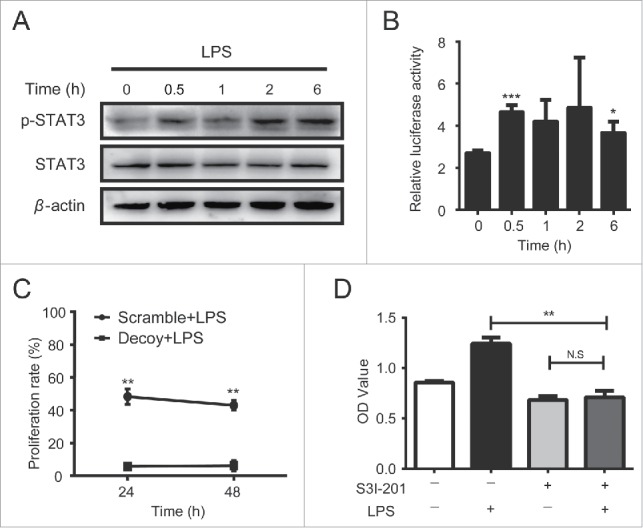
TLR4 promoted HCC proliferation in a STAT3 signaling pathway dependent manner. A. H7402 cells were treated with LPS and harvested at different time points. Then total proteins of these cells were extracted, and p-Tyr705-STAT3 and total STAT3 levels were detected by western blot. B. H7402 cells were co-transfected with pGL3-STAT3-TK-luciferase or pGL3-TK-luciferase and pRL-TK plasmid. After 12 h, cells were treated with LPS for 0.5 h, 1 h, 2 h or 6 h, and then the luciferase activity was measured. C. H7402 cells were transfected with STAT3-decoy ODN or scramble ODN with Lipofectamin™ 2000. After 12 h, these cells were treated with LPS for 24 h and 48 h, and WST-1 was used to assess the proliferation rate. D. H7402 cells were pre-treated with S3I-201 (100 μM) for 12 h, and then LPS was added into the culture. After 24 h, the proliferation rate was assessed by using WST-1. Values are means ± S.D. of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with untreated group.
TLR4 drived a COX-2/PGE2/STAT3 positive feedback loop in HCC cells
STAT3 signaling can be activated by cytokines such as IL-6, IL-10 and VEGF, depending on receptor or non-receptor tyrosine kinases like Jak or Src.22 However, LPS could not up-regulate the expression of IL-10 and VEGF in HCC cells (data not show), and IL-6 expression was only elevated in LPS-treated H7402 cells but not in HepG2 cells (Fig. 1D). Therefore, it was suggested that some other factors induced by NF-κB act as stimuli for STAT3 during this process. Publications reported that both NF-κB and STAT3 could regulate the expression of cyclooxygenase-2 (COX-2),24 a key mediator in chronic inflammation-related development of HCC.25 Here, we found COX-2 expression was significantly increased by LPS treatment (Fig. 3A), and TLR4 inhibitor TAK242 could inhibit this response (Fig. 3B). Moreover, the expression of microsomal PGE synthase 1 (mPGEs-1), the downstream enzyme in COX-2 dependent prostaglandin E2 (PGE2) production, was also elevated (Fig. 3C). Simultaneously, the secretion of PGE2 was increased, as well as PGE2 specific G-protein-coupled receptors EP1-4 (Fig. 3D). These findings demonstrated that TLR4 could trigger the activation of COX-2/PGE2 axis in HCC cells.
Figure 3.

TLR4 drived a COX-2/PGE2/STAT3 positive feedback loop in HCC cells. A. H7402 cells were treated with LPS for 24 h, and then COX-2 mRNA was measured by qPCR (left). Western blot was used to detect the protein levels of COX-2 after H7402 treated with LPS for 24 h and 48 h (right). B. H7402 cells pre-treated with TAK-242 (3 μM) for 1 h were incubated with LPS for 24 h, and then the protein levels of COX-2 were measured by western blot. C. H7402 cells were treated with LPS for 4 h, and qPCR was used to measure the mRNA level of mPGEs-1 (upper). Supernatant was collected from H7402 cells cultured with or without LPS for 12 h, and ELISA was used to measure the production of PGE2 (lower). D. H7402 cells were treated with LPS for 12 h, and qPCR was used to measure the mRNA levels of EP1, EP2, EP3 and EP4. E. The total proteins were extracted from H7402 cells treated with LPS for different time, and COX-2, p-Tyr705-STAT3 and total STAT3 were detected by western blotting. F. H7402 cells were incubated with NS398 at the concentration of 100 μM and 200 μM for 48 h, and then COX-2 expression was detected by western blot (left). H7402 cells were treated with NS398 (200 μM) for 48 h, and Western blot was used to detect the p-Tyr705-STAT3 and total STAT3 (right). G. H7402 cells pre-treated with/without NS398 (200 μM) for 48 h were incubated with/without LPS for 24 h, and then p-Tyr705-STAT3 and total STAT3 levels were detected by western blot. H. H7402 cells were incubated with selective EP receptors antagonists for 24 h (AH6809, L-798,106 and L-161,982 at the concentration of 10 μM, 1 μM and 10 μM, respectively), and Western blot was used to detect the levels of p-Tyr705-STAT3 and total STAT3 (left). H7402 cells pre-treated with/without EP receptors antagonists for 1 h were incubated with/without LPS for 24 h, and then P-Tyr705-STAT3 and total STAT3 levels were detected by western blot (right). I. COX-2 in H7402 cells was detected by western blot after incubated with S3I-201 (100 μM) for 12 h. Values are means ± S.D. of three independent experiments. *p < 0.05, **p < 0.01 compared with untreated group.
To confirm whether COX-2/PGE2 axis was associated with LPS-induced STAT3 activation, H7402 cells were treated with LPS, and we observed a positive relationship between COX-2 expression level and STAT3 activation level (Fig. 3E). Furthermore, the COX-2 selective inhibitor NS398 could significantly inhibit the expression of COX-2 at the dose of 200 μM (Fig. 3F left) and the phosphorylation level of STAT3 (Fig. 3F right), resulting in low responsive ability of STAT3 signaling pathway to LPS stimulation (Fig. 3G). Similarly, EP receptor (EP1-4) antagonists that selectively bind to EP receptors and block PGE2 function also inhibited the phosphorylation of STAT3 in H7402 cells, and blunted the responsiveness of STAT3 signaling to LPS stimulation (Fig. 3H). On the other hand, COX-2 expression could be downregulated by STAT3 activation inhibitor S3I-201 (Fig. 3I). Consistent with these, LPS stimulated the expression of COX-2 and activation of STAT3, as well as PGE2 production in HepG2 cells (Fig. S2). These data demonstrated that there was a COX-2/PGE2/STAT3 positive feedback loop in HCC cells, which can be promoted by TLR4 activation.
TLR4, COX-2 and p-STAT3Y705 displayed positive relationships in HCC tumor tissues
To identify the relationship among TLR4, COX-2 and p-STAT3Y705 in clinical specimens, a tissue microarray containing matched pairs of tumor and peritumoral liver tissues from 13 patients with primary HCC was used. Immunohistochemical results showed that the tumor sites exhibited significantly higher levels of all these three proteins compared to the adjacent non-tumor sites (Fig. 4A and B). Further data analysis showed the expression of TLR4, COX-2 and p-STAT3Y705 was positively correlated with each other (Fig. 4C). These results further verified the existence of TLR4/COX-2/PGE2/STAT3 loop in HCC.
Figure 4.

TLR4, COX-2 and p-STAT3Y705 displayed positive relationships in HCC tumor tissues A. Immunohistochemical staining of TLR4, COX-2 and p-STAT3 in HCC tumor and peritumor tissues (200×). B. The density mean of each section calculated by Image-pro Plus 6.0 software (average value of 4 random fields). C. Correlation analysis of TLR4, COX-2 and p-STAT3 expression in HCC tumor and peritumor samples. Data are represented as means ± S.D. *p < 0.05, **p < 0.01, ***p < 0.001.
Loss of TLR4 blocked COX-2/PGE2/STAT3 positive feedback loop in vivo
To confirm the role of COX-2/PGE2/STAT3 loop in HCC development, we subjected wide-type (WT) and TLR4−/− mice to a combination of diethylnitrosamine (DEN), hepatotoxin carbontetrachloride (CCl4) and 10% alcohol drinking to establish the primary HCC model (Fig. 5A). During this process, the levels of IL-6 and PGE2 in serum were measured at different time points. As shown in Fig. 5B, serum PGE2 levels of TLR4−/− mice were significantly lower than WT mice. However, serum IL-6 levels exhibited no significant difference between TLR4−/− and WT groups, which was consistent with in vitro data (Fig. 1D). At 18.5 weeks post of the first DEN injection, the mice were sacrificed and the livers were excised. As shown in Fig. 5C, no abnormal sign was observed in the livers from TLR4−/− mice, while the livers from WT mice were rough and exhibited tissue necrosis to some extent. Accordantly, H&E staining showed that the livers from WT mice were characterized by large nucleus, nuclear size inhomogeneity and dense cytoplasm; and the lobules of WT liver were disordered with a pile of deposition of fibrous tissue, demonstrating an obvious tendency to hepatocarcinogenesis. Furthermore, the phosphorylation level of STAT3 in liver tissues of TLR4−/− mice was obviously lower than WT mice (Fig. 5D). At the same time, the expression of COX-2, Cyclin D1, proliferation markers Ki67 and PCNA (Fig. 5E) was obviously down regulated in livers of TLR4−/− mice compared to WT control. Importantly, a widely used tumor biomarker α fetoprotein (AFP)26 was dramatically decreased in TLR4−/− mice, along with the vascular endothelial growth factor (VEGF), a marker for predicting the angiogenesis, invasion and metastasis of HCC.27 These data demonstrated that TLR4-induced COX-2/PGE2/STAT3 loop functioned as an important accelerator in carcinogen-induced tumor progression; silence or blockage of TLR4 might alleviate the process.
Figure 5.

Loss of TLR4 blocked COX-2/PGE2/STAT3 positive feedback loop in vivo. A. Wide type (WT) (n = 4) and TLR4−/− mice (n = 4) were i.p. injected with DEN (100 mg/kg) at the age of 5 weeks followed by two i.p. injections of CCl4 (0.5 mL/kg) and DEN (100 mg/kg) in the next two weeks, respectively. Then the mice were intragastric administrated with CCl4 (0.5 ml/kg) once a week for totally 15 weeks, accompanied with 10% alcohol drinking. B. Serum PGE2 and IL-6 levels in WT and TLR4−/− mice were determined at the indicated time points after the first DEN injection. C. Gross and histology appearance (H&E staining) of livers from WT and TLR4−/− mice 18 weeks after the first DEN injection. Scale bar, 50 μm. D. Western blot was used to detect the p-Tyr705-STAT3 and total STAT3 levels in these liver tissues. E. qPCR was conducted to detect the mRNA levels of Cyclin D1, Ki67, PCNA, AFP, COX-2 and VEGF in these liver tissues. Data are represented as means ± S.D. *p < 0.05, **p < 0.01.
COX-2/PGE2/STAT3 loop was involved in TLR4-induced multidrug resistance
It has been reported that COX-2 was closely associated with drug resistance,28 and STAT3 was identified as a transcriptional factor positively regulating MDR1 (multidrug resistance) gene expression.29 In accordance with these, we found LPS obviously promoted MDR1 gene (Fig. 6A) and P-glycoprotein (P-gp) (Fig. 6B) expression in HCC cells. Inhibiting COX-2 or STAT3 would downregulate P-gp expression (Fig. 6C), and silence P-gp expression in HCC cells exposured to LPS stimulation (Fig. 6D). Furthermore, luciferase reporter assay was used to confirm whether STAT3 participated in LPS induced MDR. As shown in Fig. 6E, the MDR1 transcriptional activity was significantly enhanced by LPS stimulation, while the MDR1 plasmid with the mutation at STAT3 binding site exhibited no response to LPS. These data demonstrated that LPS induced MDR in HCC cells through the COX2/PGE2/STAT3 loop.
Figure 6.

COX-2/PGE2/STAT3 loop was involved in TLR4-induced multidrug resistance. A. H7402 cells were incubated with LPS at the dose of 1 μg/mL or 10 μg/mL for 4 h, then mRNA level of MDR1 was detected by qPCR. B. Western blot was used to detect P-gp expression in HCC cells treated with LPS for indicated time. C. H7402 cells were incubated with NS398 (200 μM) for 48 h (upper) or with S3I-201 (100 μM) (lower) for 12 h, and then P-gp expression was detected by western blot. D. H7402 cells were pre-treated with/without NS398 (200 μM) for 12 h, and then incubated with/without LPS for 24 h. P-gp was detected by western blot. E. The pGL3-mdr1-promoter luciferase plasmid or the indicated mutated plasmid, pGL3-TK-luciferase and pRL-TK plasmid were co-transfected into H7402 cells. After 12 h, the cells were treated with LPS for 6 h, and then the luciferase activity was measured. F. H7402 cells were pre-treated with LPS for 24 h, and then cells were incubated with DOX (0.125 μg/mL) for another 24 h. After three washes with ice-cold PBS, the cells were re-suspended and detected with flow cytometer. G. H7402 cells were pre-treated with LPS for 24 h followed by incubation with DOX (5 μg/mL) for 24 h. WST-1 assay was used to detect the proliferation rate of these cells. H. After pre-treated with LPS for 24 h, H7402 cells were incubated with 5-FU (100 μg/mL) alone, or combined with TAK-242 (3 μM) for 24 h, and then WST-1 assay was used to detect the proliferation rate of these cells. Data are represented as means ± S.D. of three independent experiments. *p < 0.05, **p < 0.01.
Based on the observations above, we subsequently evaluate the influence of TLR4 on the susceptibility of HCC cells to chemotherapeutics. First, we analyzed the effect of LPS on the accumulation of antitumor drug Doxorubicin (DOX) in H7402 cells. As shown in Fig. 6F, pre-treatment of LPS obviously decreased the accumulation of DOX in H7402 cells and inhibited DOX-mediated antitumor effect (Fig. 6G). Furthermore, pre-treatment of LPS markedly attenuated the antitumor efficacy of 5-fluorouracil (5-FU), which would be reversed by inhibiting TLR4 activity (Fig. 6H). These results suggested that TLR4 activation impaired the efficacy of chemotherapeutics via promoting proliferation and inhibiting drug accumulation, which can be reversed by TLR4 inhibition.
Discussion
The relationship between inflammation and cancer has been widely accepted.30 The pivotal role of TLRs in regulating innate or adaptive immunity and inducing persistent low-level inflammation has been generally recognized.31 Due to the special physiological environment, liver receives approximately 70% of its blood supply from the intestinal venous outflow, making it the first line of defense against gut-derived antigens, especially for the conserved components of gut-derived microbiota.32 Bacterial PAMPs trigger inflammatory responses through TLRs, and emerging evidences showed a close link between gut microbiota and human liver diseases.15,32 A recent in-vivo study demonstrated that LPS derived from the intestinal microbiota induced activation of TLR4 signaling in resident liver cells, which contributed to injury- and inflammation-induced HCC promotion.17 In present study, we tried to discover the potential molecular mechanism during this process. First of all, we confirmed that TLR4 was functionally expressed in HCC cells and could be elevated by LPS stimulation (Fig. 1 A and B). Then we observed, similar to immune cells, TLR4 activation in HCC cells also excited NF-κB signaling, and resulted in the expression of inflammatory cytokines IL-8 and TNF-α (Fig. 1C and D). Meanwhile, LPS significantly promoted the growth and colony formation of human HCC cell lines via accelerating the cell-cycle progression (Fig. S1).
As a well-known transcriptional factor which has been defined crucial in oncogenesis, STAT3 regulates a range of genes contributing to cell growth, differentiation and angiogenesis, including Cyclin D1, Bcl-2, C-myc and IL-10. Constitutive activation of STAT3 has been observed in about 50% of human HCC specimens.33 Besides, a cross talk between NF-κB and STAT3 signaling was demonstrated, which was majorly dependent on an autocrine manner by cytokines IL-6 or IL-10.34,35 Here, we found STAT3 signaling in HCC cells could be activated by LPS, which mediated LPS-induced HCC proliferation (Fig. 2). However, STAT3-activating cytokines such as IL-6, IL-10 and VEGF did not exhibit significant change in response to LPS stimulation, indicating other factors might be responsible for LPS induced STAT3 activation.
Cyclooxygenase-2 (COX-2) is one of the cellular factors associated with tumorigenesis, which can be regulated by both NF-κB and STAT3.36,37 Dysregulation of COX-2 would lead to an increase of its principal metabolic product prostaglandin E2 (PGE2), and now the aberrant COX-2/PGE2 activation is regarded as a hallmark of cancer.38 Bae SH, et al demonstrated the abnormal COX-2 expression might be involved in HCC progression,25 and elevated level of PGE2 in HCC tumor sites has been observed in clinic.39 All these evidences indicate there is a strong link between PGE2 and STAT3 signaling. Based on these, therefore we tried to clarify the relationships among TLR4, COX-2 and STAT3 signaling in HCC. We found COX-2/PGE2 axis mediated LPS-induced the activation of STAT3 signaling, and in turn over-activated STAT3 was necessary for LPS-induced COX-2/PGE2 activation (Fig. 3). Moreover, the immunohistochemical staining results showed that TLR4, COX-2 and p-STAT3 were highly expressed in HCC tumor tissues and correlated positively among each other (Fig. 4). These findings suggested that a positive feedback loop was established between COX-2/PGE2 axis and STAT3 signaling, which acted as a key mechanism in LPS-induced HCC cell proliferation.
To investigate the role of TLR4-boosted COX-2/PGE2/STAT3 loop in tumorigenesis, we employed a modified HCC model by adding alcohol to the drinking water to inhibit intestinal peristalsis and promote intestinal bacterial overgrowth, which shared several features with the intestinal microecological disorders found in HCC cases and the microenvironment where most human HCCs arise. We found that wide type mice had aggravated liver injury, exhibiting early histological features of HCC and an obvious tendency to hepatocarcinogenesis (Fig. 5). Intriguingly, deficiency of TLR4 reduced PGE2 level in serum and phosphorylated-STAT3 in liver tissues, as well as COX-2, Cyclin D1, Ki67, PCNA, AFP and VEGF levels. These observations implied that TLR4-boosted COX-2/PGE2/STAT3 loop plays important roles in HCC development.
Multidrug resistance (MDR) is one of the major factors explaining chemotherapy failure in patients with cancer.40 Studies showed that the aberrant COX-2 expression is associated with P-gp mediated MDR.41,42 Our previous study also demonstrated a role of STAT3 in regulating P-gp expression in myeloid leukemia.29 These observations indicated there is a connection between MDR1 phenotype development and HCC cell growth, in which COX-2/PGE2/STAT3 loop may function as the linker. Indeed, we found COX-2/PGE2/STAT3 loop is crucial in LPS-induced MDR of HCC cells to chemotherapeutics (Fig. 6).
Taken together, this study further demonstrates that intestinal flora contributes to inflammation-driven HCC promotion. Importantly, we elucidate the potential molecular mechanisms as summarized in Fig. S3. We found TLR4 signaling could promote the proliferation and induce multidrug resistance of HCC cells to chemotherapeutics, in which COX-2/PGE2/STAT3 positive feedback loop played a determinant role. Furthermore, these findings provide new sights for HCC clinical treatment, such as the combination of chemotherapeutics and TLR4 inhibitors, or the adjunctive therapy via improving intestinal microecology, which may enhance the efficacy of chemotherapeutics and improve the prognosis.
Materials and methods
Cell lines and reagents
Human hepatoma cell lines HepG2 and H7402 were purchased from the Chinese Academy of Sciences (Shanghai, China) and conserved in our laboratory. Cells were cultured in DMEM supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 atmosphere. LPS isolated from Escherichia coli (0111:B4) was purchased from Sigma-Aldrich (L3023). If there is no special statement, 10 μg/ml of LPS was used to treat HCC cells in this study. NS398 (S1771) was purchased from Beyotime. TAK-242 (tlr1-cli095) was purchased from Invivogen. S3I-201 (573102) was purchased from Calbiochem. Selective EP1/2 receptor antagonist AH6809 (14050) and EP4 receptor antagonist L-161, 982 (10011565) were purchased from Cayman Chemical. Selective EP3 receptor antagonist L-798, 106 (sc-204047) was purchased from Santa Cruz Biotechnology. Doxorubicin was purchased from Huafeng United Technology, Beijing. 5-fluorouracil (F0151) was purchased from J&K Technology. STAT3 decoy ODN and scramble ODN were synthesized and purified as previously.43
Animals
Pathogen-free male C57BL/6 mice (5–6 weeks) were obtained from Beijing HuaFuKang biological technology Co., Ltd. C57BL/6-derived TLR4-knockout mice (TLR4−/−, male, 5–6 weeks) were kindly provided by S.B Sun (Sun Yat-Sen University). All animal experiments were performed in accordance with the Institutional Animal Care and Use Committee protocols of Shandong University.
Cell proliferation assay
Cell proliferation reagent WST-1 (11644807001, Roche Molecular Biochemical) was used to evaluate the cell growth according to the manufacturer's instruction. Briefly, 100 μl of cell suspension (containing 0.5–2 × 104 cells) were plated in each well of 96-well plates. After treated in indicated conditions, WST-1 (15 μl) was added into each well and the cells were incubated at 37°C for 1–3 h. A450nm was recorded by a scanning multiwall spectrophotometer (Bio-Rad, Hercules, CA).
Reverse transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells using TRIzol reagent (15596-018, Invitrogen). The first stranded cDNA was synthesized using 2 μg total RNA with M-MLV (AM2044, Invitrogen) according to the manufacturer's instructions. Standard PCRs were performed in a total reaction volume of 25 μl as previously described.44 The PCR products were electrophoresed in 2% agarose gels containing 5 μg/ml Nucleic acid dye. The expression analysis was conducted using AlphaEaseFC software (Genetic Technologies, Miami, FL, USA). Primers used to detect TLR1 to TLR10 were listed in Table S1.
Real-time quantitative PCR (qPCR)
The amplification of cDNA was performed by Real-time qPCR with SYBR Green Master Mix (QPK-201, Toyobo) on an iCycleriQ real-time PCR system (Bio-Rad, Hercules, CA). The primers used were shown in Table S2. The thermal cycle profile for PCR was as follows: 95°C for 5 min, 45 cycles for PCR (95°C for 25 s; 60°C for 20 s; 72°C for 30 s).
Western blot analysis
Tumor cells were lysed in lysis buffer with a protease inhibitor cocktail (310003, BestBio). Western blotting was performed as previously described.12 Antibodies were used as below: mouse anti-β-actin (sc-47778, Santa Cruz Biotechnology); rabbit anti-NF-κB p65 (#4764, Cell Signaling Technology); rabbit anti-p-NF-κB p65 (#3033, Cell Signaling Technology); mouse anti-STAT3 (#9139, Cell Signaling Technology); rabbit anti-p-Tyr705-STAT3 (#9145, Cell Signaling Technology); rabbit anti-CyclinD1 (BS1741, Bioworld Technology); rabbit anti-TLR4 (PL0402123) and anti-P-gp (PL0304068, PL Laboratories); rabbit anti-COX-2 (ab102005, Epitomics). Proteins were visualized using Immobilon Western Chemiluminescent HRP Substrate (WBKLS0500, Millipore) and detected with Alpha Ease FC software (Bio-Rad, Hercules, CA).
Luciferase reporter assay
HCC cells were transfected with the luciferase reporter plasmids by Lipofectamine 2000 (11668-019, Invitrogen) as previously described.29 The activity of luciferase was measured by using Dual-Glo TM Luciferase Assay System (E2920, Promega) according to the manufacturer's instruction. All experiments were done in triplicate wells and repeated separately at least three times.
Immunohistochemistry
Tissue microarray (TMA) containing matched pairs of tumor and peritumoral liver tissues from 13 patients with primary HCC was constructed by Shanghai Biochip Co., Ltd, as previously described.45 Briefly, the sections were dewaxed with xylene and dehydrated in a graded series of ethanol. After neutralization of endogenous peroxidase and microwave antigen retrieval, the sections were blocked with goat serum and incubated overnight in primary antibody against COX-2 (#12282, Cell Signaling Technology), p-Tyr705-STAT3 (#9145, Cell Signaling Technology) or TLR4 (sc-10741, Santa Cruz Biotechnology). After washing by PBS containing 0.05% Tween 20, the sections were incubated with second antibody for 30 min at room temperature, and then treated with HRP-conjugated streptavidin. The reaction products were visualized using 3-3-diamino-benzidine-tetrahydrochloride (DAB) followed by nuclei staining in hematoxylin. The stained area and Integrated Optical Density (IOD) of four random fields in each section were calculated using Image-pro Plus 6.0 software. The density mean, equal to (IOD SUM)/area, represented the exact protein expression.
Enzyme linked immunosorbent assay (ELISA)
PGE2 concentrations in supernatants of tumor cells were measured using the Human prostaglandin E2 Elisa Kit (CSB-E07965h, CUSABIO) according to the manufacturer's instructions. Interleukin-6 (IL-6) and PGE2 levels in mice serum were measured by using Mouse IL-6 ELISA Ready-SET-Go (88-7064, Ebioscience) and Mouse prostaglandin E2 Elisa Kit (CSB-E07966m, CUSABIO) according to the instructions respectively.
Statistical analysis
Statistical analysis was conducted by Prism Version 6.0 (GraphPad) software. All of the values are presented as the mean ± S.D. for three or more independent experiments. Difference between two groups was analyzed by the paired Student t test. A p value < 0.05 was considered to be statistically significant.
Disclosure of potential conflicts of interest
There are no potential conflicts of interest to disclose.
Funding
This study was supported by grants from National Basic Research Program of China (No. 2013CB531503), National Natural Science Foundation of China (No. 81172789, 81373222) and National Mega Project on Major Infectious Diseases Prevention and Treatment (No. 2012ZX10002006).
Supplemental Material
Supplemental Material may be downloaded here: publisher's website
References
- 1.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004; 5:987-95; PMID:15454922; http://dx.doi.org/ 10.1038/ni1112 [DOI] [PubMed] [Google Scholar]
- 2.Beutler BA. TLRs and innate immunity. Blood 2009; 113:1399-407; PMID:18757776; http://dx.doi.org/ 10.1182/blood-2008-07-019307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meraldi V, Audran R, Romero JF, Brossard V, Bauer J, Lopez JA, Corradin G. OM-174, a new adjuvant with a potential for human use, induces a protective response when administered with the synthetic C-terminal fragment 242-310 from the circumsporozoite protein of Plasmodium berghei. Vaccine 2003; 21:2485-91; PMID:12744882; http://dx.doi.org/ 10.1016/S0264-410X(03)00093-8 [DOI] [PubMed] [Google Scholar]
- 4.Geisse J, Caro I, Lindholm J, Golitz L, Stampone P, Owens M. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol 2004; 50:722-33; PMID:15097956; http://dx.doi.org/ 10.1016/j.jaad.2003.11.066 [DOI] [PubMed] [Google Scholar]
- 5.Ma F, Zhang J, Zhang J, Zhang C. The TLR7 agonists imiquimod and gardiquimod improve DC-based immunotherapy for melanoma in mice. Cell Mol Immunol 2010; 7:381-8; PMID:20543857; http://dx.doi.org/ 10.1038/cmi.2010.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato Y, Goto Y, Narita N, Hoon DS. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenvir 2009; 2 Suppl 1:205-14; PMID: 19685283; http://dx.doi.org/ 10.1007/s12307-009-0022-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, Szyfter W, Zeromski J, Whiteside TL. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res 2009; 69:3105-13; PMID:19318560; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ilvesaro JM, Merrell MA, Swain TM, Davidson J, Zayzafoon M, Harris KW, Selander KS. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. The Prostate 2007; 67:774-81; PMID:17373717; http://dx.doi.org/ 10.1002/pros.20562 [DOI] [PubMed] [Google Scholar]
- 9.Lee JW, Choi JJ, Seo ES, Kim MJ, Kim WY, Choi CH, Kim TJ, Kim BG, Song SY, Bae DS. Increased toll-like receptor 9 expression in cervical neoplasia. Mol Carcinogen 2007; 46:941-7; PMID:17440926; http://dx.doi.org/ 10.1002/mc.20325 [DOI] [PubMed] [Google Scholar]
- 10.Kim WY, Lee JW, Choi JJ, Choi CH, Kim TJ, Kim BG, Song SY, Bae DS. Increased expression of Toll-like receptor 5 during progression of cervical neoplasia. Int J Gynecol Cancer 2008; 18:300-5; PMID:17587322; http://dx.doi.org/ 10.1111/j.1525-1438.2007.01008.x [DOI] [PubMed] [Google Scholar]
- 11.Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol 2006; 176:4894-901; PMID:16585585; http://dx.doi.org/24452201 10.4049/jimmunol.176.8.4894 [DOI] [PubMed] [Google Scholar]
- 12.Zhang YY, Lin A, Zhang C, Tian ZG, Zhang J. Phosphorothioate-modified CpG oligodeoxynucleotide (CpG ODN) induces apoptosis of human hepatocellular carcinoma cells independent of TLR9. Cancer Immunol Immun 2014; 63:357-67; PMID:24452201; http://dx.doi.org/ 10.1007/s00262-014-1518-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006; 6:674-87; PMID:16929323; http://dx.doi.org/doi: 10.1038/nrc1934 [DOI] [PubMed] [Google Scholar]
- 14.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004; 127:S35-S50; PMID:15508101; http://dx.doi.org/ 10.1053/j.gastro.2004.09.014 [DOI] [PubMed] [Google Scholar]
- 15.Almeida J, Galhenage S, Yu J, Kurtovic J, Riordan SM. Gut flora and bacterial translocation in chronic liver disease. World J Gastroenterol 2006; 12:1493-502; PMID:16570339; http://dx.doi.org/ 10.3748/wjg.v12.i10.1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut 2009; 58:704-20; PMID:19359436; http://dx.doi.org/ 10.1136/gut.2008.156307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R et al.. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012; 21:504-16; PMID:22516259; http://dx.doi.org/11357141 10.1016/j.ccr.2012.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature 2001; 411:342-8; PMID:11357141; http://dx.doi.org/ 10.1038/35077213 [DOI] [PubMed] [Google Scholar]
- 19.Luedde T, Schwabe RF. NF-kappa B in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastro Hepat 2011; 8:108-18; PMID:21293511; http://dx.doi.org/ 10.1038/nrgastro.2010.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carmody RJ, Chen YH. Nuclear factor-kappaB: activation and regulation during toll-like receptor signaling. Cell Mol Immunol 2007; 4:31-41; PMID:17349209 [PubMed] [Google Scholar]
- 21.Sun X, Zhang J, Wang L, Tian Z. Growth inhibition of human hepatocellular carcinoma cells by blocking STAT3 activation with decoy-ODN. Cancer Lett 2008; 262:201-13; PMID:18248786; http://dx.doi.org/ 10.1016/j.canlet.2007.12.009 [DOI] [PubMed] [Google Scholar]
- 22.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798-809; PMID:19851315; http://dx.doi.org/ 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res 2011; 21:159-68; PMID:21187858; http://dx.doi.org/11350912 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999; 18:7908-16; PMID:1063064311350912 [DOI] [PubMed] [Google Scholar]
- 25.Bae SH, Jung ES, Park YM, Kim BS, Kim BK, Kim DG, Ryu WS. Expression of cyclooxygenase-2 (COX-2) in hepatocellular carcinoma and growth inhibition of hepatoma cell lines by a COX-2 inhibitor, NS-398. Clin Cancer Res 2001; 7:1410-8; PMID:11350912 [PubMed] [Google Scholar]
- 26.Soresi M, Magliarisi C, Campagna P, Leto G, Bonfissuto G, Riili A, Carroccio A, Sesti R, Tripi S, Montalto G. Usefulness of alpha-fetoprotein in the diagnosis of hepatocellular carcinoma. Anticancer Res 2003; 23:1747-53; PMID:1282045210746978 [PubMed] [Google Scholar]
- 27.Li XM, Tang ZY, Qin LX, Zhou J, Sun HC. Serum vascular endothelial growth factor is a predictor of invasion and metastasis in hepatocellular carcinoma. J Exp Clin Canc Res 1999; 18:511-7; PMID:10746978 [PubMed] [Google Scholar]
- 28.Patel VA, Dunn MJ, Sorokin A. Regulation of MDR-1 (P-glycoprotein) by cyclooxygenase-2. J Biol Chem 2002; 277:38915-20; PMID:12138126; http://dx.doi.org/ 10.1074/jbc.M206855200 [DOI] [PubMed] [Google Scholar]
- 29.Zhang XL, Xiao WH, Wang LH, Tian ZG, Zhang J. Deactivation of signal transducer and activator of transcription 3 reverses chemotherapeutics resistance of leukemia cells via down-regulating P-gp. Plos One. 2011; 6(6):e20965; PMID:21677772; http://dx.doi.org/22151229 10.1371/journal.pone.0020965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morrison WB. Inflammation and cancer: a comparative view. J Vet Intern Med 2012; 26:18-31; PMID:22151229; http://dx.doi.org/ 10.1111/j.1939-1676.2011.00836.x [DOI] [PubMed] [Google Scholar]
- 31.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2001; 2:675-80; PMID:11477402; http://dx.doi.org/ 10.1038/90609 [DOI] [PubMed] [Google Scholar]
- 32.Son G, Kremer M, Hines IN. Contribution of gut bacteria to liver pathobiology. Gastroenterol Res Practice 2010; 2010; PMID:20706692; http://dx.doi.org/ 10.1155/2010/453563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang SF, Wang SN, Wu CF, Yeh YT, Chai CY, Chunag SC, Sheen MC, Lee KT. Altered p-STAT3 (tyr705) expression is associated with histological grading and intratumour microvessel density in hepatocellular carcinoma. J Clin Pathol 2007; 60:642-8; PMID:16901975; http://dx.doi.org/ 10.1136/jcp.2006.036970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benkhart EM, Siedlar M, Wedel A, Werner T, Ziegler-Heitbrock HW. Role of Stat3 in lipopolysaccharide-induced IL-10 gene expression. J Immunol 2000; 165:1612-7; PMID:10903771; http://dx.doi.org/ 10.4049/jimmunol.165.3.1612 [DOI] [PubMed] [Google Scholar]
- 35.Greenhill CJ, Rose-John S, Lissilaa R, Ferlin W, Ernst M, Hertzog PJ, Mansell A, Jenkins BJ. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J Immunol 2011; 186:1199-208; PMID:21148800; http://dx.doi.org/ 10.4049/jimmunol.1002971 [DOI] [PubMed] [Google Scholar]
- 36.Hsieh FC, Cheng G, Lin J. Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem Biophys Research Commun 2005; 335:292-9; PMID:16081048; http://dx.doi.org/ 10.1016/j.bbrc.2005.07.075 [DOI] [PubMed] [Google Scholar]
- 37.Ulivi V, Giannoni P, Gentili C, Cancedda R, Descalzi F. p38/NF-kB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes. J Cell Biochem 2008; 104:1393-406; PMID:18286508; http://dx.doi.org/ 10.1002/jcb.21717 [DOI] [PubMed] [Google Scholar]
- 38.Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009; 30:377-86; PMID:19136477; http://dx.doi.org/11915030 10.1093/carcin/bgp014 [DOI] [PubMed] [Google Scholar]
- 39.Yan W, Xiaoqian W, Xiujin L, Tang Nanhong H-BSIoFP, Union Hospital, Fujian Medical University. Correlation between hepatocellular carcinoma and the expressions of prostaglandin E2, mPGES-1 and HBx (in Chinese). J Clin Hepatol 2011; 27:174-7 [Google Scholar]
- 40.Gottesman MM, Pastan I, Ambudkar SV. P-glycoprotein and multidrug resistance. Curr Opin Genet Dev 1996; 6:610-7; PMID:893972711915030 [DOI] [PubMed] [Google Scholar]
- 41.Fantappie O, Masini E, Sardi I, Raimondi L, Bani D, Solazzo M, Vannacci A, Mazzanti R. The MDR phenotype is associated with the expression of COX-2 and iNOS in a human hepatocellular carcinoma cell line. Hepatology 2002; 35:843-52; PMID:11915030; http://dx.doi.org/ 10.1053/jhep.2002.32469 [DOI] [PubMed] [Google Scholar]
- 42.Sui H, Zhou S, Wang Y, Liu X, Zhou L, Yin P, Fan Z, Li Q. COX-2 contributes to P-glycoprotein-mediated multidrug resistance via phosphorylation of c-Jun at Ser63/73 in colorectal cancer. Carcinogenesis 2011; 32:667-75; PMID:21296766; http://dx.doi.org/ 10.1093/carcin/bgr016 [DOI] [PubMed] [Google Scholar]
- 43.Zhang XL, Jian Z, Wang LH, Wei HM, Tian ZG. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. Bmc Cancer 2007; 7:149; PMID:17683579; http://dx.doi.org/ 10.1186/1471-2407-7-149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Sun R, Wei HM, Zhang JH, Tian ZG. Characterization of interleukin-15 gene-modified human natural killer cells: implications for adoptive cellular immunotherapy. Haematologica 2004; 89:338-47; PMID:15020274 [PubMed] [Google Scholar]
- 45.Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 2007; 25:2586-93; PMID:17577038; http://dx.doi.org/ 10.1200/JCO.2006.09.4565 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.