

Silva et al. identify Mte1 as a novel interactor of the Mph1 helicase in Saccharomyces cerevisiae. Their data indicate that Mte1 plays a role in regulation of crossover recombination, response to replication stress, and telomere maintenance.
Keywords: homologous recombination, telomere maintenance, genome integrity, DNA repair, Mph1, Mte1
Abstract
Mph1 is a member of the conserved FANCM family of DNA motor proteins that play key roles in genome maintenance processes underlying Fanconi anemia, a cancer predisposition syndrome in humans. Here, we identify Mte1 as a novel interactor of the Mph1 helicase in Saccharomyces cerevisiae. In vitro, Mte1 (Mph1-associated telomere maintenance protein 1) binds directly to DNA with a preference for branched molecules such as D loops and fork structures. In addition, Mte1 stimulates the helicase and fork regression activities of Mph1 while inhibiting the ability of Mph1 to dissociate recombination intermediates. Deletion of MTE1 reduces crossover recombination and suppresses the sensitivity of mph1Δ mutant cells to replication stress. Mph1 and Mte1 interdependently colocalize at DNA damage-induced foci and dysfunctional telomeres, and MTE1 deletion results in elongated telomeres. Taken together, our data indicate that Mte1 plays a role in regulation of crossover recombination, response to replication stress, and telomere maintenance.
Several human genome instability diseases are caused by mutations in DNA helicases. For example, mutations in FANCM cause Fanconi anemia (FA), which is a condition associated with bone marrow failure, physical abnormalities, organ defects, and cancer predisposition. FA has been ascribed to defects in repair of DNA interstrand cross-links, protein–DNA adducts, and DNA double-strand breaks (DSBs) (for review, see Duxin and Walter 2015). Another example is RTEL1, where mutations give rise to Hoyeraal-Hreidarsson syndrome, which is associated with bone marrow failure, microcephaly, immunodeficiency, telomere fragility, and predisposition to cancer (for review, see Vannier et al. 2014). Mph1 appears to be the functional homolog of FANCM and RTEL1 in the budding yeast model system (Whitby 2010; Luke-Glaser et al. 2012; Vannier et al. 2014; Xue et al. 2015b), where it serves as a mutation suppressor and a negative regulator of crossover (CO) recombination (Prakash et al. 2005, 2009).
The canonical homologous recombination (HR) pathway for repairing DSBs in yeast is initiated by Rad52-mediated loading of Rad51 on the 3′ ssDNA tails generated by resection of a DSB that consequently facilitates displacement of replication protein A (RPA). The Rad51 nucleoprotein filament is subsequently responsible for the crucial homology search and strand invasion into a homologous donor duplex to form a characteristic displacement loop (D loop) (Sung 1994; Symington et al. 2014). After initial extension of the invading 3′ end, the extended D loop can be processed by several alternative pathways. In mitotic cells, breaks are primarily repaired by synthesis-dependent strand annealing (SDSA), in which the extended D loop is displaced to yield non-CO (NCO) products (Symington et al. 2014). If the extended strand is instead stabilized, second end capture can ensue, giving rise to a double Holliday junction (dHJ) that can be either resolved or dissolved to yield CO or NCO events, respectively (see the Discussion for details; Symington et al. 2014). CO events are a potential threat to genome integrity because they can result in genomic rearrangements and loss of heterozygosity (LOH). One important negative regulator of CO events is the 3′–5′ helicase Mph1. Mph1 has the ability to disrupt D loops formed by Rad51 and catalyze migration of branched DNA structures (Prakash et al. 2005, 2009; Whitby 2010; Zheng et al. 2011; Mitchel et al. 2013). Independent of its role in D-loop unwinding, the Mph1 helicase promotes fork regression and branch migration during replication-associated recombinational repair (Prakash et al. 2005, 2009; Mankouri et al. 2009; Panico et al. 2010; Zheng et al. 2011; Xue et al. 2014). The role of Mph1 in replication fork rescue is negatively regulated by the Smc5/6 complex by direct binding to the regulatory domain of Mph1 at its C terminus. Furthermore, the inhibition of Mph1 by the Smc5/6 complex is attenuated by the Mhf1–Mhf2 histone fold complex (MHF) (Xue et al. 2015a). In mutants of the Smc5/6 complex, Mph1 is responsible for accumulation of replication intermediates that lead to increased sensitivity to replication stress and genomic instability (Chen et al. 2009, 2013; Qiu et al. 2013; Xue et al. 2014).
Mph1 has recently been implicated in telomere maintenance, promoting telomere uncapping and accelerating senescence when overexpressed in telomerase-negative cells (Luke-Glaser and Luke 2012). In yeast, telomeres consist of 250- to 300-base-pair (bp) arrays of TG1–3 repeats with a 3′ ssDNA overhang of 12–14 nucleotides (G-tail) that is specifically recognized and bound by Cdc13 together with Stn1 and Ten1 (CST complex) (Wellinger and Zakian 2012). The yeast telomeres are also bound in the double-stranded region by the Ku complex and by Rap1 and its binding partners, Rif1–Rif2. The association of these protein complexes with telomeric DNA forms a protective capping structure that accounts for two crucial outcomes: the full replication of telomeres and protection of telomeres from recognition by the recombination and checkpoint machineries and associated nuclease and helicase activities (Bartsch et al. 2000; Raschle et al. 2004; Lisby and Geli 2009; Dewar and Lydall 2012). In the absence of telomerase, telomeres shorten progressively with each round of replication. Within ∼60–80 cell divisions, telomeres become critically short, rendering chromosomes unstable, and cells lose viability in a process called replicative senescence (Lundblad and Szostak 1989; Abdallah et al. 2009). Telomere shortening leads to the accumulation of ssDNA at chromosome ends, which elicits the activation of the DNA damage checkpoint and recruitment of repair factors that are usually excluded from telomeres, such as Rad52 (d'Adda di Fagagna et al. 2003; Takai et al. 2003; Abdallah et al. 2009; Khadaroo et al. 2009; Lin et al. 2009). Despite the loss of telomerase, some cells are able to use a RAD52-dependent HR mechanism to elongate their telomeres, thereby bypassing senescence and surviving (Sugiyama et al. 1998; Le et al. 1999).
In this study, we identify and characterize the YGR042W-encoded protein Mte1 (Mph1-associated telomere maintenance protein 1) as a novel factor involved in the DNA damage response. Mte1 has homology with the human zinc finger protein ZGRF1 (C4ORF21; implicated in HR and DNA interstrand cross-link repair) (Smogorzewska et al. 2010; Adamson et al. 2012) and the fission yeast Dbl2 (Yu et al. 2013). The Mte1-GFP fusion protein has been shown to form nuclear foci in response to replication stress (Huh et al. 2003; Tkach et al. 2012; Yimit et al. 2015). Moreover, high-throughput screens have implicated Mte1 in telomere length maintenance (Askree et al. 2004) and indicated that it can interact with Cmr1 (Gilmore et al. 2012), a chromatin-binding protein associated with replication stress response (Choi et al. 2012; Tkach et al. 2012; Gallina et al. 2015). In this report, we show that Mte1 is a D-loop-binding protein that interacts with the Mph1 helicase and has an antagonizing role to Mph1 in recombination outcomes by enhancing CO formation. Furthermore, we show that Mte1 is involved in telomere length maintenance and associates with dysfunctional telomeres together with Mph1.
Results
The Mph1 helicase interacts with Mte1
To identify new factors involved in genome maintenance and elucidate the molecular function of Mph1, we searched for Mph1-interacting proteins by a stable isotope labeling by amino acids in cell culture (SILAC)-based mass spectrometry (MS) protocol (de Godoy et al. 2006). To enrich for interaction partners relevant to DNA damage response, we performed the Mph1 pull-down after treatment of cells for 1 h with the DNA-alkylating agent methyl methanesulfonate (MMS), which induces base modifications and replication-associated DSBs upon fork collapse and leads to robust accumulation of YFP-tagged Mph1 into foci in the nucleus (Fig. 1A; Chen et al. 2009). Using this approach, we confirmed previously reported physical interactions of Mph1, such as Mgm101 (Ward et al. 2012), Rim1 (Kucejova and Foury 2003), Rfa1 (Gavin et al. 2002), and Fkh1 (Ho et al. 2002) (Fig. 1B; Supplemental Table S1). In addition, we observed Rad52 and subunits of RPA (Rfa2 and Rfa3), which had not previously been reported. Finally, under these experimental conditions, the strongest scoring hit corresponded to the uncharacterized putative protein Ygr042W, which we name in this report as Mte1.
Figure 1.
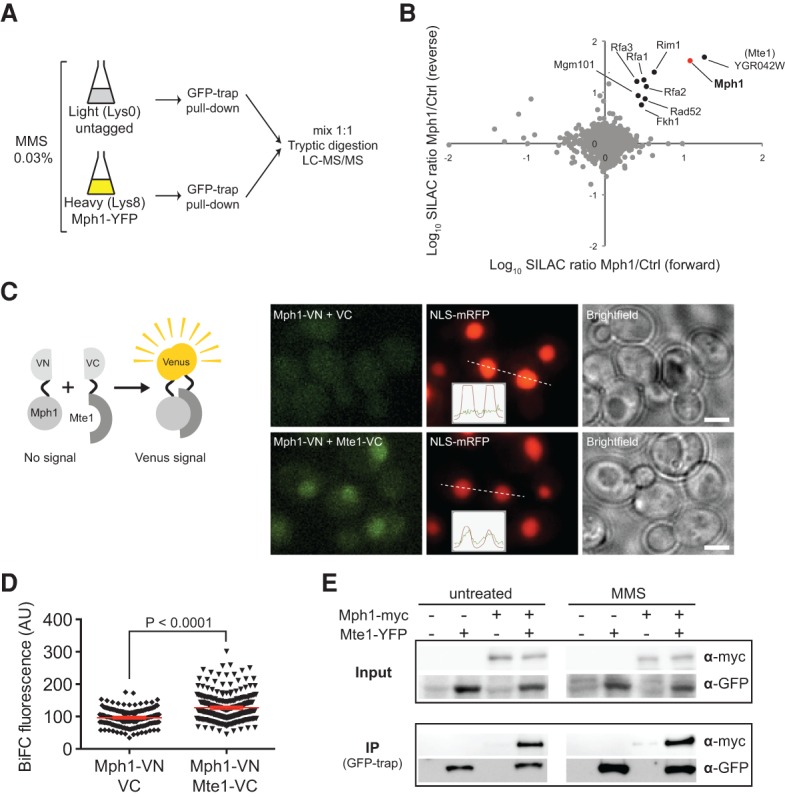
Mte1 interacts with the Mph1 helicase. (A) SILAC experimental workflow. Yeast strains expressing YFP-tagged Mph1 (SS30-3C) or untagged (IG45-8A) were cultured in SILAC medium and harvested in log phase after treatment with 0.03% MMS for 1 h. Protein complexes from SILAC lysates were affinity-purified using GFP-Trap. Proteins were trypsin-proteolysed, and peptides were identified by liquid chromatography-tandem MS (LC-MS/MS). (B) Mph1-interacting proteins. The plot shows log10 SILAC ratios from YFP-tagged Mph1 versus control from forward and reverse (SILAC label swap) experiments. Each point represents a different identified protein. Bait protein Mph1 is highlighted in red. Mte1 (YGR042W) and other top-scoring interacting factors identified in this screen are highlighted in black. (C) Mte1 and Mph1 interact in vivo. Bimolecular fluorescence complementation (BiFC) between VN-tagged Mph1 and VC-tagged Mte1 (ML961-1A) or VC alone (ML961-2A) was assayed in cells expressing NLS-RFP as a nuclear marker. (Left) Schematic representation of the BiFC assay. (Right) Representative images of cells. The insets show intensity plots along the dashed lines. Bar, 3 µm. (D) Quantification of BiFC fluorescence. The nuclear fluorescence of cells from the experiment in C was quantified using Volocity software. Each point represents one cell measurement, with the mean value and 95% confidence interval bars represented in red. P-value was calculated using the unpaired t-test. n > 200. (AU) Arbitrary units. (E) Mph1 coimmunoprecipitates with Mte1 independently of DNA. Mte1-YFP was immunoprecipitated using GFP-Trap from DNase-treated extracts of cells expressing Mte1-YFP and Mph1-13myc (SS329-6C), Mte1-YFP (SS71-11A), Mph1-13myc (SS288), or no tag (ML8-9A). Whole-cell extract (input) and pull-downs from untreated cells and cells treated with 0.05% MMS for 1 h were analyzed by immunoblotting using anti-myc and anti-GFP antibodies (top and bottom panels, respectively).
To validate the interaction between Mte1 and Mph1, we first took advantage of bimolecular fluorescence complementation (BiFC). This assay is based on the reconstitution of a fluorescence signal upon reassembly of two halves of the YFP variant Venus (VN and VC) that are fused separately to two query proteins (Sung and Huh 2007). Using Mph1 fused to the N-terminal fragment of Venus (VN) and Mte1 fused with the C-terminal complementary fragment (VC), we found that Mph1 and Mte1 directly interact in the nucleus (Fig. 1C,D). Second, in a strain coexpressing YFP-tagged Mte1 and myc-tagged Mph1, the two proteins coimmunoprecipitated in both untreated and MMS-treated conditions (Fig. 1E), indicating that the interaction is independent of DNA damage. Furthermore, the interaction is likely to be independent of DNA, as treatment of extracts with DNase prior to immunoprecipitation did not reduce the amount of Mph1 retrieved by pull-down of Mte1 (cf. Fig. 1E and Supplemental Fig. 1A,B).
DNA damage-induced Mte1 and Mph1 foci are interdependent
The formation of nuclear foci has been observed for many factors involved in DNA damage response, and HR and is commonly used as marker for sites of DNA damage and ongoing repair (Lisby and Rothstein 2015). To gain further understanding of the relationship between Mph1 and Mte1, we monitored Mte1 and Mph1 focus formation and colocalization between the two proteins in untreated cells and after exposure to various types of DNA damaging agents. For this purpose we generated YFP-tagged Mte1 at the C terminus, which showed the expected nuclear localization (Huh et al. 2003; Srikumar et al. 2013; Yimit et al. 2015). In untreated cells, only a subset of Mte1 foci colocalized with Mph1, which significantly increased when cells were treated with replication stress-inducing agents MMS and, to a lesser extent, hydroxyurea (HU) and 4-nitroquinoline 1-oxide (4NQO) (Fig. 2A,B). MMS induced Mte1 foci specifically in the S/G2-phase cells (Fig. 2C). In contrast, the topoisomerase inhibitor camptothecin (CPT) and the radiomimetic agent zeocin (ZEO), which causes DSBs, did not induce a significant increase in Mte1 foci. Interestingly, when MPH1 is deleted, the ability of Mte1 to form foci is abrogated, suggesting that Mte1 is enriched at sites of DNA damage by Mph1 (Fig. 2D).
Figure 2.

Mte1 and Mph1 colocalize at foci in response to DNA damage. (A) Quantitative analysis of Mte1 and Mph1 foci. Cells expressing Mte1-YFP and Mph1-CFP (SS82-15C) were untreated (ctrl) or incubated with DNA damage-inducing agents (0.05% MMS, 100 mM HU, 0.2 μg/mL 4NQO, 5 μg/mL CPT, 200 μg/mL ZEO), and focus formation was analyzed. Error bars represent the 95% confidence interval. Significant differences (P < 0.05) observed between untreated and treated cells are marked with an asterisk. n > 150. (B) Representative images for Mte1- and Mph1-colocalizing foci in the nucleus. Yellow arrowheads indicate Mte1-YFP foci, blue arrowheads indicate Mph1-CFP foci, and green arrowheads indicate colocalization between the two proteins. Bar, 3 µm. (C) MMS induces Mte1 foci in S/G2 cells. Cell cycle distribution of Mte1 foci was assessed by microscopy in asynchronously growing cultures of cells expressing YFP-tagged Mte1 (SS71-11A) before and after treatment with 0.05% MMS for 1 h. Cell cycle stage classification is illustrated. Error bars represent the 95% confidence interval. n > 200. (D) Mte1 focus formation is abrogated in mph1 mutant cells. Mte1 focus formation was analyzed in mph1Δ (SS262-4A) and wild-type (SS331-7C) cells. Cells were treated with 0.05% MMS for 1 h. (E) DNA damage induction of Mph1 foci is dependent on Mte1. Mph1 foci were quantitated in mte1Δ (SS260-24B) and wild-type (ML66-11A) cells before and after treatment with 0.05% MMS for 1 h. (F) Estimation of the number of Mte1 molecules per cell. Total nuclear YFP fluorescence intensities were measured using Volocity software in haploid and diploid cells expressing Mte1-YFP (SS71-11A; ML905), Rad52-YFP (W5094-1C; ML191), Rdh54-YFP (ML130-3C; ML906), Mrc1-YFP (IG147; ML907), and Mph1-YFP (ML66-11A; ML919) from the native loci. Red numbers indicate the mean number of molecules per cell based on normalization to Rad52 in haploid cells (Lisby et al. 2003). Error bars represent 95% confidence intervals. n = 100–200. (G) Estimation of the number of Mte1 and Mph1 molecules per focus. Focus and total nuclear YFP fluorescence intensities were measured at the single-cell level using Volocity software in haploid cells expressing Mte1-YFP (SS71-11A) or Mph1-YFP (ML66-11A) for spontaneous and MMS-induced foci (0.05% MMS). The number of molecules per focus was estimated using the ratio of focus/total nuclear fluorescence × estimated total number of molecules per nucleus, calculated in F. The mean value ± SEM is indicated. All fluorescence intensity values were background-corrected. n = 18–69 cells. Error bars represent 95% confidence intervals.
To test whether Mph1 foci are likewise dependent on Mte1, we examined Mph1-YFP localization in mte1Δ cells. Despite observing a small number of spontaneous Mph1 foci, mte1Δ cells failed to induce additional Mph1 foci in response to MMS (Fig. 2E). Protein levels of Mte1, as measured by whole-nuclear fluorescence signal, were also reduced in mph1Δ cells in comparison with wild-type expression levels (Supplemental Fig. 1C). Reciprocally, Mph1 protein levels within the nucleus were also reduced in mte1Δ cells (Supplemental Fig. 1D). However, the reduced level of Mph1 protein is unlikely to be a consequence of increased Mph1 turnover in the absence of Mte1 (Supplemental Fig. 1E). These results suggest that the two proteins are likely functioning together, possibly forming a heteromeric complex, and thus deleting one of the components might affect the other component's localization and ability to respond to DNA damage.
To estimate the abundance of Mte1 and Mph1 protein in vivo, we quantitated the total nuclear YFP fluorescence of the tagged proteins in both haploid and diploid cells using Rad52-YFP as a reference (Fig. 2F; Lisby et al. 2003). Based on this method, there were slightly more than 300 molecules of Mte1 in a haploid nucleus and roughly twice that amount in diploid cells. By comparison, Mph1 is twice as abundant as Mte1 in haploid cells, while, in diploid cells, the two proteins are present in equal amounts. By quantitating the fraction of fluorescence within foci, we found that both spontaneous and MMS-induced foci contain approximately four molecules of either protein (Fig. 2G), indicating that Mte1 and Mph1 are present at stoichiometric amounts at sites of DNA damage.
Deletion of MTE1 suppresses the MMS sensitivity of mph1Δ mutants
We next tested the DNA damage sensitivity of mte1Δ cells and epistasis to mph1Δ. As expected, deletion of MPH1 in both haploid and diploid cells increases cellular sensitivity primarily to drugs leading to replication stress, such as MMS and 4NQO (Fig. 3; Supplemental Fig. 2; Scheller et al. 2000; Schurer et al. 2004; Panico et al. 2010). MTE1 deletion did not increase cell sensitivity to DNA-damaging agents. Interestingly, we observed a modest but reproducible rescue of MMS sensitivity of mph1Δ by mte1Δ in haploid cells (Fig. 3A). Conversely, overexpression of Mte1 from a galactose-inducible promoter enhanced the sensitivity of cells to genotoxic stress (Fig. 3B; Supplemental Fig. 2B). This suggests that Mte1 produces or stabilizes a repair intermediate acted upon by Mph1.
Figure 3.
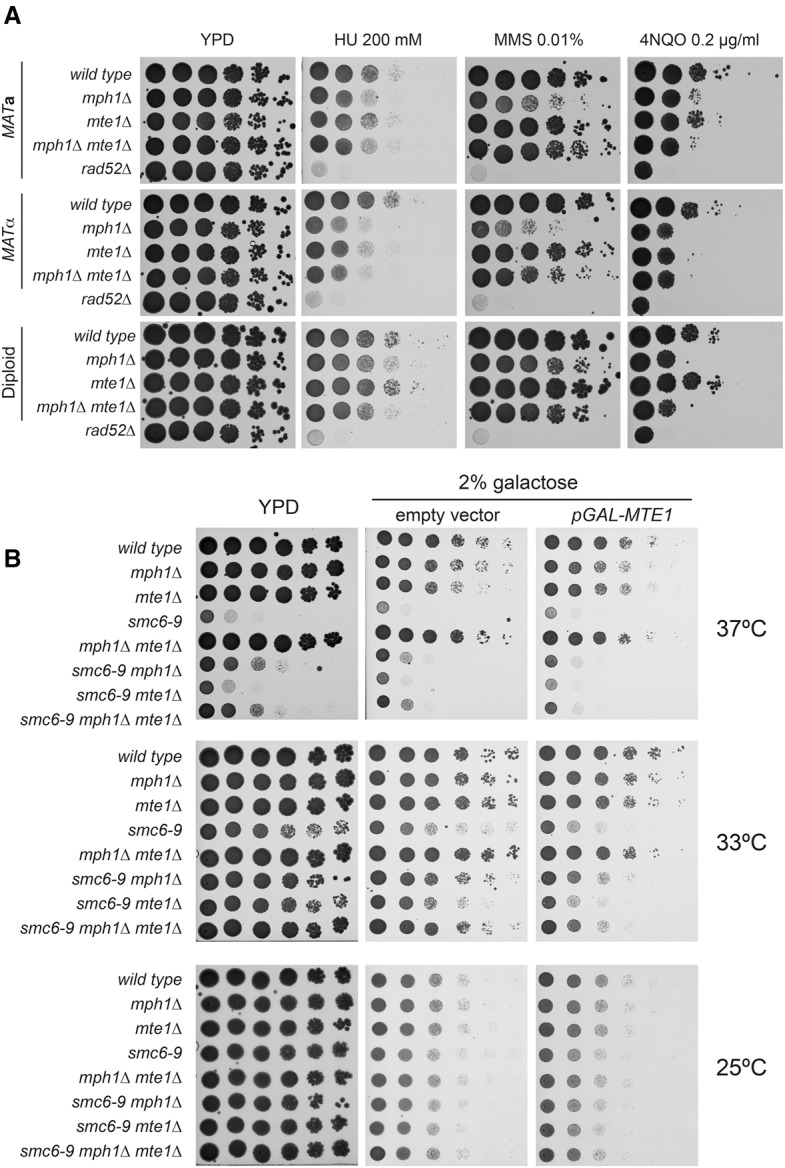
Mte1 regulates the response to replication stress. (A) mte1Δ cells are resistant to DNA damage and suppress the MMS sensitivity of mph1Δ. Tenfold serial dilutions were prepared from haploid MATa cells: wild type (LSY2202-15D), mph1Δ (LSY2362-6A), mte1Δ (ML859-1C), mph1Δ mte1Δ (ML860-1C), and rad52Δ (SMG259-11B); haploid MATα cells: wild type (ML857-9B), mph1Δ (LSY2361-6D), mte1Δ (ML860-7C), and mph1Δ mte1Δ (ML856-2A); and diploid wild-type (ML861), mph1Δ (ML862), mph1Δ mte1Δ (ML863), and mte1Δ (ML864) cells. Cells were spotted onto YPD or YPD containing one of the following DNA-damaging agents: 0.01% MMS, 0.2 μg/mL 4NQO, and 200 mM HU and incubated for 2–4 d at 30°C prior to imaging. (B) Overexpression of MTE1 exacerbates the temperature sensitivity of smc6-9 cells. Tenfold serial dilutions were prepared from haploid MATa cells: wild-type (ML8-9A), mph1Δ (SS346-4A), mte1Δ (SS346-6D), smc6-9 (SS347-13B), mph1Δ mte1Δ (SS346-4C), smc6-9 mph1Δ (SS337-22D), smc6-9 mte1Δ (SS346-5C), and smc6-9 mph1Δ mte1Δ (SS346-3D) cells were spotted onto YPD or strains transformed with empty vector (pRS415) or a vector for galactose-induced overexpression of MTE1 (pSS17) plated on synthetic complete (SC)-Leu containing 2% galactose and incubated for 2–4 d at 25°C, 33°C, or 37°C prior to imaging.
Mte1 binds branched DNA structures in vitro, stimulates Mph1 helicase and fork regression activities, and inhibits Mph1-catalyzed D-loop displacement
Our observation that Mte1 interacts with Mph1 prompted us to test whether purified Mte1 affects Mph1 catalytic activities in vitro. First, we tested the 3′-to-5′ helicase activity of Mph1 on a fork-like structure (Fig. 4A). Mph1 dissociated the labeled strand from this substrate in a reaction that was stimulated threefold to fivefold with increasing concentrations of Mte1. Similarly, Mte1 stimulated Mph1-catalyzed regression of a mobile replication fork twofold (Fig. 4B). Importantly, on its own, Mte1 did not catalyze these reactions. Second, we tested the impact of Mte1 on the ability of Mph1 to displace D loops (Prakash et al. 2009; Panico et al. 2010; Zheng et al. 2011). A D loop was formed by Rad51-catalyzed invasion of a 5′-radiolabeled 90-mer oligo D1 into a homologous negatively supercoiled dsDNA plasmid (pBSC) in the presence of Rad54 (Sebesta et al. 2011). The D loop was subsequently extended by addition of RPA, Pol δ, RFC, and PCNA (Fig. 4C, schematic). Finally, D-loop displacement was assayed by addition of Mph1 with or without prior addition of Mte1. This experiment revealed that Mte1 is able to completely inhibit Mph1-catalyzed D-loop displacement.
Figure 4.

Mte1 stimulates Mph1 helicase and fork regression activities but inhibits D-loop displacement. (A) Mte1 stimulates Mph1 helicase activity. A fluorescently labeled fork structure (4 nM) consisting of oligos 2, 5, 6, and 7 was incubated with 7 nM Mph1 for 15 min at 30°C in the absence or presence of the indicated concentrations of Mte1. Following deproteinization, the reaction products were resolved in a 10% native polyacrylamide gel. The percentage of the labeled strand that is dissociated from the fork is plotted in the graph. (B) Mte1 stimulates Mph1 fork regression activity. A fluorescently labeled mobile fork structure (4 nM) consisting of oligos 8, 9, 10, and 11 was incubated with 0.8 nM Mph1 for 15 min at 30°C in the absence or presence of the indicated concentrations of Mte1. Following deproteinization, the reaction products were resolved in a 10% native polyacrylamide gel. The percentage of the fork converted to linear duplexes is plotted in the graph. (C) Mte1 inhibits Mph1-catalyzed D-loop displacement. A schematic illustration of the D-loop displacement assay is shown at the right. The extended D loop was incubated with Mph1 in the absence or presence of the indicated amounts of Mte1. (Left) The reaction products were separated on a 0.8% agarose gel. A quantification of the band corresponding to the extended D loop is shown below the schematic. Error bars indicate SEM for three replicates. (D) Mte1 binds branched DNA molecules. The indicated amounts of Mte1 protein were incubated with 4 nM fluorescently labeled DNA substrates for 15 min at 30°C. The reaction products were resolved in 7.5% or 10% native polyacrylamide gels. The DNA substrates were assembled as follows: ssDNA: oligo 1; dsDNA: oligos 1 and 2; D loop: oligos 15, 16, 17, and 18; fork: oligos 2, 5, 6, and 7; mobile HJ: oligos 8, 12, 13, and 14; and 5′-flap: oligos 2, 3, and 4. The constants of dissociation (KD) were calculated using SigmaPlot software.
To test whether Mte1 can interact directly with DNA, we conducted electrophoretic mobility shift assays (EMSAs) with linear ssDNA, dsDNA, D-loop, fork, mobile HJ, and 5′-flap DNA structures (Fig. 4D; Supplemental Fig. 3). This analysis demonstrated that Mte1 binds DNA with a preference for D loops and forks. This preference was confirmed in pairwise competition experiments (Supplemental Fig. 4).
Taken together, the biochemical analyses indicate that Mte1 stimulates Mph1 helicase and fork regression activities while inhibiting its D-loop displacement activity through interaction with Mph1 and/or its DNA substrate.
Mte1 promotes CO recombination in vivo
Since Mph1 suppresses COs associated with mitotic recombination (Prakash et al. 2009; Mazon and Symington 2013), we measured the association of COs with gene conversion between homologs in mte1Δ diploid strains using a previously described assay (Ho et al. 2010). Briefly, the assay consists of ade2 heteroalleles (red colonies), one of which harbors an I-SceI endonuclease cut site and flanking heterozygous markers to discriminate between CO recombination and chromosome loss among Ade+ recombinants (white colonies) (Fig. 5A). Recombination was induced by I-SceI expression for 1.5 h after addition of galactose to the medium. To classify recombination events, cells were plated on YPAD medium, incubated for 2–4 d, and then replica-plated onto YPAD/Hyg/Nat, synthetic complete (SC)-Ade, SC-Met, SC-Ura, and SCR-Ade+Gal medium. The plating efficiency of the strains on medium containing galactose (constitutive I-SceI expression) relative to growth on glucose-containing medium was used to normalize the fractions of recombination events. Recombination events were classified as NCO, CO, or break-induced replication (BIR) based on LOH for the Nat and Hph markers. Half-sectored colonies (red/white) were generated by a recombination event and could be scored with less ambiguity than solid white or solid red colonies (Ho et al. 2010). Analysis of the red/white-sectored colonies revealed that mte1Δ cells had a significantly lower CO frequency than the wild type (4% and 9%; P = 0.02), and the mph1Δ mte1Δ double mutant reverted the higher frequency of COs seen in the mph1Δ single mutant to wild-type levels (8% and 18%; P = 0.002) (Fig. 5B).
Figure 5.

Mte1 promotes CO formation during interhomolog recombination. (A) Schematic representation of the assay used to infer CO frequency showing the positions of genetic markers on chromosome XV. The arrow indicates the I-SceI cut site within the ade2-I allele. The asterisk notes the location of the ade2-n mutation present on the homolog. Hph and Nat cassettes are inserted at the HIS3 locus, 150 kb downstream from the ade2 locus. The solid circle indicates the centromere. Not drawn to scale. (B) Mte1 promotes CO formation. (Left) Distribution of colony colors after I-SceI induction and plating on YPD. (Right) Distribution of NCO, CO, and BIR products for red/white-sectored colonies for wild-type (ML861), mph1Δ (ML862), mph1Δ mte1Δ (ML863), and mte1Δ (ML864) strains. Plating efficiency (PE) is indicated in parentheses. (C) Schematic representation of the assay used for measuring spontaneous mitotic direct repeat recombination between leu2-ΔEcoRI and leu2-ΔBstEII heteroalleles. (D) Mte1 and Mph1 synergistically suppress single-strand annealing (SSA)/unequal sister chromatid exchange (USCE). Recombination rates were calculated using the MSS-MLE method via the FALCOR Web tool for wild-type (ML829-13A), mph1Δ (ML829-12B), mte1Δ (ML829-9B), and mph1Δ mte1Δ (ML829-11C) cells. For each strain, the fluctuation analysis was based on nine trials. Error bars represent 95% confidence intervals. (*) Fraction of Leu+ Ura− recombinants significantly different from the wild type; P = 0.02, Student's t-test. (E) mte1Δ cells exhibit the wild-type mutation rate. Forward mutation at the CAN1 locus was determined by fluctuation analysis of 10 trials for wild type (ML904-14C), mph1Δ (ML904-6D), mte1Δ (ML904-14A), and mph1Δ mte1Δ (ML904-7C) strains.
We also tested whether Mte1 affected spontaneous mitotic direct-repeat recombination in haploid cells. The rate of direct-repeat recombination was largely unaffected by the deletion of mte1Δ (Fig. 5C,D). The mph1Δ mte1Δ double mutant showed a significant decrease in Leu+Ura− recombinants, which could reflect either increased single-strand annealing (SSA) or unequal sister chromatid exchange (USCE).
MTE1 overexpression exacerbates the replication stress sensitivity of the smc6 mutant
Deleting MPH1 relieves the replication stress sensitivity of smc5/6 mutant cells by reducing the formation of recombination intermediates (Chen et al. 2009, 2013; Choi et al. 2010; Chavez et al. 2011). To explore a possible participation of Mte1 in the formation or stabilization of such recombination intermediates together with Mph1, we tested the effect of mte1Δ on the replication stress sensitivity of the smc6-9 mutants. In cells grown at the semipermissive temperature, accumulation of recombination intermediates leads to genomic instability and increased cell death. Contrary to mph1Δ, we observed no phenotypic rescue of smc6-9 by mte1Δ (Fig. 3B). However, overexpression of MTE1 further enhanced the temperature sensitivity of smc6-9 cells similar to the increased DNA damage sensitivity upon MTE1 overexpression (Supplemental Fig. 2B).
MPH1 was originally identified on the basis of elevated spontaneous mutation rates of haploid cells (Scheller et al. 2000). We therefore tested whether MTE1 is related to the mutator phenotype of mph1Δ cells by monitoring forward mutation at the CAN1 locus, which leads to canavanine resistance. This analysis showed that mph1Δ leads to an eightfold increase in the rate of CAN1 mutation independently of MTE1 (Fig. 5E). Taken together, we conclude that Mte1 promotes CO recombination in both the presence and absence of Mph1, and mte1Δ does not relieve either replication stress of smc5/6 cells or the mutator phenotype of mph1Δ.
MTE1 deletion increases telomere length
In a previous genome-wide screen, deletion of MTE1 was found to reduce telomere length (Askree et al. 2004). To examine telomere length in mte1Δ and mph1Δ mutants in the W303 background, we analyzed XhoI-generated telomeric fragments of genomic DNA by Southern blotting using a probe for TG1–3 tracts (Hardy et al. 1992). As previously reported, the mph1Δ single mutant did not affect telomere length (Askree et al. 2004; Luke-Glaser and Luke 2012). Surprisingly, we found that mte1Δ cells show a significant increase in telomere length, opposite to what has been previously reported (Askree et al. 2004), and mte1Δ was epistatic to mph1Δ for telomere length (Fig. 6A). For comparison with the previous report, we also tested the effect of mte1Δ in the S288c background and observed the same effect on telomere length as for the W303 background strains (Supplemental Fig. 5A). Hence, we conclude that Mte1 contributes to telomere length maintenance in an Mph1-independent manner.
Figure 6.

Mte1 affects telomere homeostasis. (A) mte1Δ mutants exhibit long telomeres. Genomic DNA was isolated from wild-type (ML829-13A), mph1Δ (ML829-12B), mte1Δ (ML829-9B), and mte1Δ mph1Δ (ML829-11C) cells; digested with XhoI; and subjected to Southern blotting using a telomeric TG1–3 repeat probe. Terminal restriction fragment analysis was used to calculate the average telomere length difference between the wild type and the deletion mutants. (B) Mte1 and Mph1 colocalize at dysfunctional telomeres. Quantitative analysis of Mte1, Mph1, and Cdc13 foci in cdc17-1 cells. Cells with the temperature-sensitive cdc17-1 mutant allele and expressing fluorescently tagged Mte1-RFP, Mph1-YFP, and Cdc13-CFP (SS303-6A) were pregrown at the permissive temperature (25°C) and subsequently transferred to the semipermissive temperature (30°C) and examined by microscopy at 0, 1.5, and 5 h after temperature shift. Error bars represent 95% confidence intervals. n > 200 cells. Asterisks mark values significantly different from measurements at the permissive temperature. P < 0.05. (n.s.) Not significantly different from the permissive temperature. (C) Mte1 and Mph1 colocalize with the telomeric single-stranded binding protein Cdc13. The panels show representative images for cells at the semipermissive temperature. Red arrowheads mark Mte1-yEmRFP foci, yellow arrowheads indicate Mph1-YFP foci, blue arrowheads indicate Cdc13-CFP foci, and green arrowheads mark RFP–YFP–CFP-colocalizing foci. Bar, 3 µm. (D) Mte1 colocalizes with Cdc13 foci during senescence. Mte1-YFP and Cdc13-CFP foci were monitored during senescence and formation of survivors in telomerase-negative MPH1 (SS261-12C) and mph1Δ (SS272-13D) (est2Δ) cells. Senescence was induced by loss of the pAP81 plasmid carrying EST2 and propagation in liquid medium at 25°C. Population doubling times were monitored by measuring OD600, and a representative growth curve for each strain is presented at the top right corner of each panel. Red dots and numbers represent the points at which the microscopy images were acquired for each culture.
Mph1 and Mte1 are recruited to dysfunctional telomeres
Prompted by our findings that mte1Δ mutants have longer telomeres, we proceeded to study the colocalization of Mte1 and Mph1 with telomere markers under different telomere stress conditions. We took advantage of a temperature-sensitive mutant of DNA polymerase α, cdc17-1. In this mutant, long G-rich single-stranded telomeric overhangs are generated in a telomerase-dependent manner when cells are propagated at the semipermissive temperature (30°C) (Carson and Hartwell 1985; Adams and Holm 1996; Adams Martin et al. 2000). The progressive accumulation of single-stranded telomeric overhangs can be visualized as foci of Cdc13 (Khadaroo et al. 2009), which specifically binds telomeric ssDNA (Lin and Zakian 1996; Nugent et al. 1996; Hughes et al. 2000). In cells grown at the permissive temperature (25°C), there was no change in the number of Mte1 or Mph1 foci, and we rarely observed colocalization between Mte1 and Cdc13, while the majority of the observed Mte1 foci colocalized with Mph1 (Fig. 6B). Upon shifting the cells to 30°C, the number of Cdc13 foci progressively increased as expected (Khadaroo et al. 2009), and we observed a similar behavior for both Mte1 and Mph1. This increase in the number of foci was accompanied by increased colocalization between all three proteins (Fig. 6B,C).
To determine whether Mte1 also responds to telomere shortening during senescence in telomerase-negative mutants (est2Δ), we followed the formation of Mte1 foci and the colocalization with Cdc13 through successive generations after loss of telomerase. Interestingly, Mte1 showed a trajectory of focus formation very similar to Cdc13 with a marked increase in foci as cells approached crisis and a decrease after formation of survivors (Fig. 6D, inset panel). A substantial degree of Cdc13 and Mte1 colocalization was observed during senescence and crisis. Deleting MPH1 resulted, again, in the complete loss of Mte1 focus formation while not significantly affecting the rate of senescence or the response of Cdc13 to the shortening of telomeres (Fig. 6D, bottom panel). Likewise, deleting MTE1 did not significantly change the rate of senescence (Supplemental Fig. 5B). We further investigated whether mte1Δ cells were impaired in recruiting Cdc13 and Mph1 to ssDNA at telomeres in the cdc17-1 mutant. Consistent with the interdependency of Mte1 and Mph1 foci (Fig. 2D,E), we observed that Mph1 recruitment to these dysfunctional telomeres was significantly reduced in the absence of MTE1, while Cdc13 recruitment was largely unaffected (Supplemental Fig. 5C). Taken together, the data show that Mte1 and Mph1 are recruited to dysfunctional telomeres in an interdependent manner, but loss of MTE1 does not prevent formation of survivors in telomerase-negative cells.
Mte1 is recruited to telomeres independently of Cdc13
Since the pattern of recruitment of Mte1 to dysfunctional telomeres shared a striking resemblance to Cdc13, we asked whether Cdc13 is responsible for recruiting Mte1 to eroded telomeres. To test this, we used a conditional mutant of Cdc13 (cdc13-1), which leads to telomere uncapping, accumulation of extensive ssDNA at telomeres, and telomere shortening when shifted to the restrictive temperature (>30°C) (Nugent et al. 1996; Lydall and Weinert 1997). Upon shifting to 37°C, we observed a significant increase in Mte1 foci in cdc13-1 cells (Supplemental Fig. 6A [top panel], B). As expected, the majority of these Mte1 foci colocalized with Rad52 (Supplemental Fig. 6A; Abdallah et al. 2009; Khadaroo et al. 2009). To also test whether resection of uncapped telomeres is required for recruitment of Mte1 to foci, we analyzed the effect of exo1Δ and pif1Δ on Mte1 focus formation. The Exo1 5′-to-3′ exonuclease and Pif1 helicase have a prominent role in resection of uncapped telomeres (Maringele and Lydall 2002; Vega et al. 2007; Dewar and Lydall 2010). Recruitment of Mte1 and Rad52 to foci at 37°C was significantly reduced in exo1Δ and pif1Δ mutant cells, suggesting that resection and the formation of ssDNA at telomeres promote Mte1 recruitment rather than Cdc13 itself.
Defective HR and telomere dysfunction induce Mte1 focus formation
To further characterize the genetic requirements for Mte1 focus formation, we tested factors involved in DNA repair and telomere maintenance: Rad52, the major recombination mediator in yeast (for review, see Symington et al. 2014), and Mre11, a component of the Mre11–Rad50–Xrs2 complex involved in the initial recognition and short-range resection of DSBs and in telomere length maintenance (Goudsouzian et al. 2006; Sabourin et al. 2007; Longhese et al. 2010). In both the rad52Δ and mre11Δ cells, we observed an increase in both spontaneous and MMS-induced Mte1 foci (Supplemental Fig. 7A), indicating that Mte1 responds to a defective HR pathway. We also tested a knockout of the Ku complex (yKu70–yKu80), which binds and regulates telomeres and promotes nonhomologous end joining (NHEJ) as an alternative DSB repair pathway (Milne et al. 1996; Gravel et al. 1998). The yku70Δ cells showed Mte1 foci at levels comparable with the wild type. The final factor tested was Rif1, a negative regulator of telomere length that binds Rap1 together with Rif2 at telomeres to promote gene silencing and end protection (Hardy et al. 1992; Marcand et al. 1997; Ghaemmaghami et al. 2003). In rif1Δ cells, which have longer telomeres than wild type (Teixeira et al. 2004), we observed a marked increase in Mte1 foci, with a significant percentage of the cells exhibiting multiple foci. Taken together, these results indicate that Mte1 responds to defective HR and dysfunctional telomeres but is unaffected by NHEJ defects.
Mte1 colocalizes with persistent DSBs and Mps3 at the nuclear periphery
We noticed that Mte1 foci often localize at the nuclear periphery, which is reminiscent of uncapped telomeres and persistent DSBs. These have been shown to relocalize to the nuclear periphery to promote alternative repair pathways (Nagai et al. 2008; Kalocsay et al. 2009; Khadaroo et al. 2009; Oza et al. 2009). To examine the localization of Mte1 to a persistent DNA lesion, we introduced an inducible I-SceI endonuclease cut site on chromosome V adjacent to an array of Tet repressor (TetR)-binding sites in cells expressing mRFP-tagged TetR (Fig. 7A). We used cells expressing the nuclear pore complex (NPC) component CFP-Nup49 to infer the position of the break and Mte1 foci relative to the nuclear periphery (Fig. 7B). The position of Mte1 foci relative to the nuclear periphery and the marked DSB were analyzed before and at different time points after induction of I-SceI expression by galactose (Fig. 7C). Notably, the spontaneous Mte1 foci in the uninduced condition localized almost exclusively at the nuclear periphery (95%). Interestingly, a significant degree of relocalization of Mte1 to the marked DSB occurred only after 3 h of continuous I-SceI induction, and less than half of those Mte1-recognized DSBs localized at the nuclear periphery (Fig. 7C, top panel). The colocalization of Mte1 with the I-SceI cut site was largely independent of Rad52 (Fig. 7C, bottom panel). In mre11Δ mutant cells that are defective in short-range resection of DSBs (Mimitou and Symington 2008), the number of Mte1 foci was dramatically increased even prior to cleavage of the I-SceI site, but localization of Mte1 to the I-SceI cut site was reduced.
Figure 7.

Mte1 localizes to persistent DSBs at the nuclear periphery. (A) Schematic illustration of a construct to visualize an I-SceI-inducible DSB. Red boxes represent the relative positions of tandem lacO sites on the left arm of chromosome V. (Blue circle) I-SceI cut site (I-SceIcs); (solid circle) centromere. (B) Mte1 foci localize to the nuclear periphery. The panels show YFP, CFP, and RFP and combinations of the merged images as well as a bright-field image of representative cells (SS142-23B) after I-SceI induction. Arrowheads indicate two peripheral Mte1 foci (yellow), a colocalizing TetR focus (red), and the NPCs (blue). (C) Localization of Mte1 to a DSB is delayed in mre11Δ. Wild-type (SS142-23B), rad52Δ (SS159-1B), and mre11Δ (SS160-6D) cells carrying the inducible DSB and expressing Mte1-YFP were transformed with the plasmids pWJ1320, expressing the I-SceI endonuclease under the control of a galactose-inducible promoter, and pNEB21, expressing the fluorescently tagged NPC subunit CFP-Nup49. Cells were grown in synthetic medium supplemented with 2% raffinose and lacking adenine prior to I-SceI induction. To induce expression of the nuclease, galactose was added to a final concentration of 2%. (Bottom panel) Mte1 focus formation and relative localization to the DSB and NPC was monitored by fluorescence microscopy prior to adding galactose and at 90, 120, and 180 min after I-SceI induction. Error bars represent 95% confidence intervals. (D) Model for promotion of CO recombination by Mte1. The homologous DNA donor strand is shown in red. (E) Model for regulation of Mph1 by Mte1 during replication stress. (F) Model for regulation of telomere extension by Mte1. Red strands indicate subtelomeric DNA or telomeric TG1–3 repeats.
To examine the localization of MMS-induced Mte1 foci relative to telomere-anchoring sites at the nuclear periphery, we fluorescently tagged Mps3, an inner nuclear membrane protein of the SUN family (Sad1/UNC-84 homology) that tethers telomeres to the nuclear periphery and suppresses their recombination (Bupp et al. 2007; Schober et al. 2009). The majority (>80%) of both spontaneous and MMS-induced Mte1 foci colocalized with peripheral Mps3 (Supplemental Fig. 7B–D), suggesting either that MMS preferentially induces Mte1 recruitment to Mps3-anchored telomeres or that Mte1 recognizes a type of DNA damage that relocalizes to Mps3.
Discussion
In this study, we identify Mte1 (YGR042W) as a novel interactor of the Mph1 helicase and show that the two proteins interdependently colocalize at DNA damage-induced foci. The interaction appears to be constitutive and independent of DNA. In vitro, Mte1 stimulates the helicase and fork regression activities of Mph1 while inhibiting the ability of Mph1 to dissociate D loops. Mte1 also binds directly to DNA with a preference for branched molecules, such as D loops and fork structures. We note that similar conclusions were reached in an independent study by Xue et al. (2016). Deletion of MTE1 reduces CO recombination and suppresses the MMS sensitivity of mph1Δ mutant cells, suggesting that Mte1 acts upstream of Mph1 to promote a DNA repair pathway that requires Mph1. Deletion of MTE1 alone does not cause pronounced sensitivity to DNA damage but results in elongation of telomeres by ∼40 bp. Consistently, Mph1 and Mte1 colocalize at dysfunctional telomeres.
Despite the physical interaction and interdependency of Mte1 and Mph1 foci, the epistasis analysis indicates that Mte1 and Mph1 are not strictly dependent on each other. For example, MTE1 is epistatic to MPH1 for telomere length regulation, while MPH1 is epistatic to MTE1 for mutation avoidance. This complex relationship may be rationalized by the fact that both Mph1 and Mte1 can interact directly with DNA and thereby potentially act in DNA repair independently of each other. Hence, the physical interaction of the two proteins may primarily serve to stabilize their coordinated recruitment to sites of DNA damage. We also note that the epistasis analysis indicated that focus formation of Mph1 and Mte1 is not strictly required for function. Based on our data, we propose a model in which Mte1 stabilizes D loops either directly or through inhibition of Mph1-catalyzed dissociation, thereby promoting CO recombination (Fig. 7D). This model is consistent with the reduced CO recombination observed in mte1Δ mutants, the biochemical inhibition of D-loop displacement by Mte1, and the late recruitment of Mte1 to DSBs. We further propose that Mte1 acts upstream of or in parallel to Mph1 to overcome replication stress (Fig. 7E). This notion is supported by the recruitment of Mte1 and Mph1 to MMS-induced foci, the stimulation of Mph1-catalyzed fork regression and helicase activities, and the suppression of MMS sensitivity of mph1Δ mutants by mte1Δ. Hence, Mte1 may stabilize fork intermediates that require Mph1 regression or helicase activity to be resolved. Finally, Mte1 may also promote replication fork restart through template switching by inhibiting Mph1-catalyzed D-loop displacement.
Telomeres appear to be the preferred substrate for Mte1, since almost all (>95%) spontaneous Mte1 foci colocalize with Mps3, which is an anchoring site for telomeres at the nuclear periphery (Bupp et al. 2007; Schober et al. 2009), and telomere dysfunction induced by telomere uncapping, overextension, or erosion leads to recruitment of Mte1 to multiple foci that colocalize with the telomere ssDNA-binding protein Cdc13. For example, loss of RIF1, which leads to long telomeres but only slightly increases single-stranded telomeric overhangs (Bonetti et al. 2010), increased dramatically the number of spontaneous Mte1 foci, suggesting that the increased Mte1 foci in rif1Δ could be due to replication stress associated with longer telomeres rather than ssDNA at telomeres per se. Furthermore, mre11Δ and rad52Δ also elicited a substantial increase in NPC-colocalizing Mte1 foci. In the context of telomere maintenance, this finding may be rationalized by reports that Mre11 is involved in G-tail formation and the recruitment of telomerase (Nugent et al. 1998; Chamankhah and Xiao 1999; Chamankhah et al. 2000), and 20% of spontaneous Rad52 colocalize with Cdc13 (Khadaroo et al. 2009), suggesting that both Mre11 and Rad52 play an active role in telomere maintenance even in telomerase-proficient cells. Interestingly, MMS treatment also preferentially induced Mte1 foci that colocalized with Mps3 at the nuclear periphery, where telomeres are anchored (Bupp et al. 2007; Schober et al. 2009), suggesting that telomeres are particularly sensitive to replication stress. Contrary to persistent DSBs and collapsed replication forks, stalled replication forks have been shown not to relocalize to Mps3 (Kalocsay et al. 2009; Oza et al. 2009; Horigome et al. 2014), so it is unlikely that MMS-induced Mte1 foci represent stalled replication forks. It is interesting to note that yeast telomeres have been proposed to form T loops (Luke-Glaser and Luke 2012), where telomeric 3′ overhangs invade telomeric dsDNA, similar to what has been reported for mammalian telomeres (Griffith et al. 1999). However, the telomere fold-back structures reported so far in yeast appear to involve interaction of the telomere end with the subtelomere region of the chromosome (Strahl-Bolsinger et al. 1997; Pryde and Louis 1999; de Bruin et al. 2000, 2001; Robin et al. 2014). Irrespective of the precise configuration of yeast fold-back structures, Mte1 might exert its effect on telomere length by preventing their dissociation (Fig. 7F). Notably, in human cells, the RTEL1 helicase and putative ortholog of Mph1 provide T-loop-unwinding activity (Sarek et al. 2015).
The putative homologs to MTE1 in fission yeast and humans are Dbl2 and ZGRF1, respectively. The three proteins share a domain of unknown function designated DUF2439 (Pfam domain PF10382) (Punta et al. 2012). Similar to our findings, Dbl2 colocalizes with Rad22 (Rad52) and Fml1 (Mph1), and the ability of Fml1 to form nuclear foci in response to replicative stress requires the presence of Dbl2 (Yu et al. 2013). Further evidence for a conserved function of Mte1/Dbl2 comes from a genome-wide screen that identified the dbl2Δ deletion mutant as having elongated telomeres (Liu et al. 2010). The putative human ortholog of Mte1, ZGRF1, was identified in two large-scale studies for genes required for resistance to cross-linking agents (Smogorzewska et al. 2010) and genes required for recombinational repair of an endonuclease-induced DSB (Adamson et al. 2012). Although ZGRF1 awaits further characterization, it is interesting to note that ZGRF1 harbors a C-terminal extension, which is predicted to encode a helicase domain. Thus, it is possible that, in human cells, the Mte1–Mph1 function is encoded within a single polypeptide, and it will be important to investigate the relationship of ZGRF1 with FANCM and FA or other cancer predisposition syndromes.
Materials and methods
Yeast strains and plasmids
Standard procedures for yeast strain construction and cell growth were used (Sherman et al. 1986). Strains generated for this study were isogenic RAD5 derivatives of W303 (Supplemental Table S2; Thomas and Rothstein 1989). Strains from the gene disruption collection (Invitrogen) were derivatives of S288C. An MTE1 deletion strain was generated by homology targeted replacement of the MTE1 ORF by a fragment containing the NatMX resistance cassette using primers NatMX-YGR-Fw and NatMX-YGR-Rv. Fluorescently tagged proteins were constructed for expression from their native chromosomal loci with a four-alanine linker as described using primers YGR042W-Fw, YGR042W-YFP-Rv, YGR042W-YFP-Fw, and YGR042W-down-Rv (Mte1-YFP); YGR042W.Fw, YGR042W_cherrystart.Rv, YGR042W_cherrystop.Fw, and YGR042W-down Rv (MTE1-yEmRFP); and Mph1fwd, Mph1-Tup(-stop), Mph1-Tdown, and Mph1down (Mph1-YFP) (Reid et al. 2002; Silva et al. 2012). Myc-tagged Mph1 was generated in the W303 background by transfer of an MPH1-13xMyc::HIS3 allele from strain BL295 after PCR amplification using primers Mph1fwd and Mph1-rv2. BiFC assay strains were generated as described previously (Sung and Huh 2007). Mte1-VC was constructed using primers YGR042W-F2 and YGR042W-R1 to amplify the VC fragment from pML103 containing VC155-tADH1-NatMX6. To generate Mph1-VN, the primer pair MPH1-F2 and MPH1-R1 was used to amplify a fragment containing VN173-tADH1-Kl.URA3 from genomic DNA of the strain from the VN collection library (Bioneer) harboring RAD59-VN173-tADH1-Kl.URA3 as template. Plasmid pML103 was constructed by subcloning an FspI fragment containing the NatMX cassette from p4339 (Goldstein and McCusker 1999) into FspI-digested pFA6a-VC-KanMX6 (Sung and Huh 2007). Mph1-mCherry-sfGFP was constructed using primers MPH1-S3-Fnew and MPH1-S2-R to amplify mCherry-sfGFP::HphNT1 from pMaM60 (Khmelinskii et al. 2012). Cells expressing VC from the MTE1 promoter were constructed using primers pMTE1-F2 and YGR042W-R1 to amplify the VC fragment from pML103 with 5′ ends designed to replace MTE1 upon genomic integration.
All strains and plasmids were verified by sequencing. All primer pairs used in strain construction are in Supplemental Table S3. The plasmid for bacterial expression of Mte1 was generated by cloning of the MTE1 gene into the EcoRI and SalI sites of the pET28a vector. The plasmid for galactose-induced overexpression of MTE1 was constructed by first replacing the URA3 gene in pML174 (Sabatier 2014)—harboring the inducible GAL1-10 promoter and the ADH1 termination sequence separated by SalI and NotI cloning sites—with a LEU2 containing the NaeI/SwaI fragment from pRS415 (Sikorski and Hieter 1989) to produce pSS15. Next, the MTE1 gene was PCR-amplified from genomic DNA using SalI/NotI adapted primers Gal1-Mte1-Fw (SalI) and Gal1-Mte1-Rv (NotI) and cloned into the SalI/NotI site of pSS15 to yield pSS17.
SILAC and MS analysis
For identification of proteins interacting with Mph1, lysine auxotroph cells expressing Mph1-YFP fusion protein were grown in SC medium containing either Lys0 (12C6 and 14N2) or Lys8 (13C6 and 15N2) (Sigma, catalog no. 608041) for >10 generations. Cultures were treated with 0.03% MMS (Sigma) for 1 h prior to harvesting to induce DNA damage and allow Mph1-YFP to accumulate at foci. Whole-cell extracts were prepared in lysis buffer without EDTA (10 mM Tris-HCl at pH 7.5, 150 mM NaCl, 0.5% NP40, 1 mM PMSF, 1× complete protease inhibitor [Roche]), and immunoprecipitation of YFP-tagged Mph1 was performed using 25 μL of GFP-Trap_A beads (Chromotek) for 2 h at 4°C according to the manufacturer's instructions. After washing in dilution buffer (10 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1 mM PMSF, 1× complete protease inhibitor [Roche]), GFP-Trap_A beads incubated with heavy-labeled and light-labeled proteins were mixed and washed again. To release bound proteins from the beads, samples were incubated with 1 vol of 4× LDS sample buffer (NuPAGE) and 40 mM DTT for 10 min at 70°C followed by 35 min at room temperature. Chloroacetoamide was added to a final concentration of 110 mM, and samples were incubated for 45 min at room temperature. Proteins were resolved by SDS-PAGE, and each loaded gel lane was sliced into three or four slices containing an estimated equal amount of proteins. Gel pieces were in-gel-digested in trypsin protease solution (13 ng/µL trypsin, 20 mM NH4HCO3). In-gel trypsin digestion was carried out overnight at 37°C. Tryptic peptides were extracted by incubating with increasingly organic solutions for 20 min and collecting the resulting fractions. The gel-extracted peptides were loaded onto Stage Tips as described (Rappsilber et al. 2007). Samples were analyzed by liquid chromatography-tandem MS (LC-MS/MS) using the EASY-nLC HPLC (Thermo Scientific) in combination with LTQ-Orbitrap Velos or Q-Exactive (Thermo Scientific). For the Velos instrument, the top 10 most intense peaks were analyzed after each full scan, while the top 12 were analyzed for the Q Exactive. MS raw data files were analyzed with the MaxQuant software package (developer's version 1.2.2.9) (Cox and Mann 2008; Cox et al. 2009). Full scan peaks and fragment scan peaks were searched against the Saccharomyces Genome Database (SGD) release 63 containing 6717 putative protein sequences (http://downloads.yeastgenome.org). False discovery rate was estimated using a target–decoy approach, allowing a maximum of 1% false identifications from the reversed sequence database.
Immunoprecipitation and immunoblot analysis
Whole-cell extracts were prepared in lysis buffer without EDTA (10 mM Tris-HCl at pH 7.5, 150 mM NaCl, 0.5% NP40, 1 mM PMSF, 1× complete protease inhibitor [Roche]). For DNase treatment, extracts were incubated with 50 U of DNase I with mild rotation for 45 min at 16°C. Immunoprecipitation of YFP-tagged Mte1 was performed using 25 μL of GFP-Trap_M beads (Chromotek) for 2 h at 4°C according to the manufacturer's instructions. Pull-downs were washed with increasing salt concentrations (10 mM Tris-HCl at pH 7.5, 150–350 mM NaCl, 1 mM PMSF, 1× complete protease inhibitor [Roche]). Immunoblot analysis was performed following standard protocol. Briefly, protein samples were mixed with 2× SDS buffer, boiled, separated by SDS-PAGE, and electrotransferred onto nitrocellulose membranes. The membranes were first probed for Mte1-YFP using a mouse primary anti-GFP (Roche) and anti-mouse conjugated to HRP secondary antibody (Dako). For detection of myc-tagged Mph1, membranes were subjected to mild stripping according to Abcam protocol. Briefly, membranes were washed twice for 5 min with stripping buffer (0.2 M glycine, 0.001% SDS, 0.01% Tween 20 at pH 2.2), washed twice for 10 min in PBS, washed twice for 5 min in TBS-T, and finally blocked and reprobed using primary mouse anti-c-myc antibody (Santa Cruz Biotechnology, 9E10).
DNA-damaging agents
Unless otherwise stated, cells were grown overnight at 30°C to saturation in liquid YPD medium, and 10-fold serial dilutions were prepared for spotting on solid medium. DNA damage sensitivity was assayed by allowing cells to grow for up to 72 h at 30°C after spotting onto YPD or YPD containing ZEO (Invitrogen), MMS (Sigma), HU (Sigma), 4NQO (Sigma), or CPT (Sigma) to the final concentrations stated in the figures. To test cisplatin sensitivity, cultures were grown to OD600 = 0.4 and incubated for 3 h in the presence of 0.3 mM cisplatin. Cells were then serially diluted, plated onto solid YPD plates, and allowed to grow for 48 h until imaging. For MTE1 overexpression experiments, cells transformed with the control vector (pRS415) and the vector for galactose-induced overexpression of MTE1 (pSS17) were grown overnight at 25°C or 30°C to saturation in liquid SC-Leu supplemented with 2% raffinose medium, and 10-fold serial dilutions were prepared for spotting on solid medium containing 2% glucose or 2% galactose and allowed to grow up to 96 h until imaging.
I-SceI induction
Induction of a single fluorescently marked DNA DSB was carried out essentially as described (Lisby et al. 2003). Briefly, cells containing an I-SceI cut site integrated immediately upstream of a ura3::3xURA3-tetOx112 tandem array on chromosome V and expressing TetR-mRFP from the intergenic region iYGL119W were grown to an optical density (OD600) of ∼0.2–0.3 in synthetic medium containing 2% raffinose as a carbon source. For inducible expression of the I-SceI endonuclease, cells were transformed with the plasmid pWJ1320 harboring the I-SceI gene controlled by a GAL1-10 promoter and grown in medium lacking adenine. Expression was induced by adding galactose to the cultures to a final concentration of 2%.
Senescence induction in telomerase-deficient cells
Cells with EST2-deleted in the genome were propagated at 25°C in liquid medium lacking uracil to select for the plasmid pAP81 (for ectopic expression of EST2), and foci of Mph1-YFP, Cdc13-CFP, and Rad52-RFP (SS283-23D) were quantitated prior to telomerase loss. Loss of the telomerase-encoding plasmid was enabled by streaking cells on solid YPD medium and subsequently checking for loss of growth in SC-Ura by replica plating. We estimate that this procedure involved ∼20–30 generations from the point of colony formation on solid YPD until cells were analyzed by microscopy. Selected Ura− colonies were inoculated in liquid SC medium supplemented with 100 µg/mL adenine (SC+Ade) and propagated at 25°C for ∼100 generations to monitor senescence and formation of survivors. Samples were collected for monitoring population doubling time by measuring OD600 and for live-cell microscopy analysis at regular intervals. Cell cultures were kept at OD600 between 0.2 and 0.9 through the course of the experiment.
Live-cell imaging
Cells were grown shaking in liquid SC+Ade medium at 25°C to OD600 = 0.2–0.3 and processed for fluorescence microscopy as described previously (Silva et al. 2012) unless otherwise stated. For this study, the following fluorophores were used: CFP (clone W7) (Heim and Tsien 1996), YFP (clone 10C) (Ormo et al. 1996), and RFP (yEmRFP) (Campbell et al. 2002; Keppler-Ross et al. 2008). Fluorophores were visualized on a DeltaVision Elite microscope (Applied Precision, Inc.) equipped with a 100× objective lens (Olympus, U-Plan S-Apo, N.A. 1.4), a cooled Evolve 512 EMCCD camera (Photometrics), and an Insight solid-state illumination source (Applied Precision, Inc.). Images were acquired using softWoRx (Applied Precision, Inc.) software. Image analysis and fluorescence intensity quantification were done using Volocity software (PerkinElmer). Images were pseudocolored according to the approximate emission wavelength of the fluorophores.
Statistical analysis
For live-cell microscopy experiments, the significance of the differences observed between cell populations was determined using one-tailed Fisher's exact test. P-values <0.05 were considered significant. Statistical tests applied for determining the significance of the different assays used in this study are stated.
Protein expression and purification
The plasmid expressing Mte1 protein with (His)6 affinity tag was introduced into Escherichia coli strain BL21(DE3)RIPL. Protein expression was induced by 1 mM IPTG for 4 h at 37°C. Extract from 3 g of cell paste was prepared by sonication in 14 mL of cell breakage buffer containing 50 mM Tris-HCl (pH 7.5), 10% sucrose, 2 mM EDTA, 600 mM KCl, 0.01% NP40, 1 mM β-mercaptoethanol, and complete protease inhibitor cocktail (Roche). The lysate was clarified by ultracentrifugation, and the resulting supernatant was incubated with 700 µL of His-Select nickel affinity gel (Sigma) for 1 h at 4°C. The nickel beads with bound proteins were washed with 10 mL of buffer K (20 mM K2HPO4, 20% glycerol, 0.5 mM EDTA at pH 7.5, 0.01% NP40, 1 mM β-mercaptoethanol) containing 150 mM KCl and eluted in steps with 50, 150, 300, 500, and 1000 mM imidazole in buffer K containing 50 mM KCl. Fractions containing Mte1 protein (300–1000 mM imidazole) were applied onto a 1-mL heparin column (GE Healthcare) and eluted using a 9-mL gradient of 100–1000 mM KCl in buffer K. The peak fractions (350–650 mM KCl) were pooled, concentrated to 3.5 µg/µL in a Vivaspin-2 concentrator, and stored at −80°C.
Mph1 protein was purified as described in Prakash et al. (2005). Rad51, Rad54, and RPA were purified as described in Van Komen et al. (2006). The RFC complex was purified as described in Finkelstein et al. (2003). PCNA and Pol δ were purified according to Sebesta et al. (2011).
DNA substrates
Oligonucleotides used in this study were purchased from VBC Biotech or TAG Copenhagen and are in Supplemental Table S3. Synthetic DNA substrates were prepared as described (Matulova et al. 2009).
EMSA
The indicated amounts of Mte1 protein were incubated with 4 nM fluorescently labeled DNA substrates for 15 min at 30°C in buffer D (40 mM Tris-HCl at pH 7.5, 50 mM KCl, 1 mM DTT, 0.1 mg/mL BSA). After the addition of gel loading buffer (60% glycerol, 10 mM Tris–HCl at pH 7.4, 60 mM EDTA), the reaction mixtures were resolved in 7.5% or 10% native polyacrylamide gels in 0.5× TBE buffer (45 mM Tris-ultrapure, 45 mM boric acid, 1 mM EDTA). Gels were scanned using a FLA-9000 Starion (Fujifilm) and quantified by MultiGauge version 3.2 software (Fujifilm).
In the competition assay, two different DNA substrates (4 nM each) were incubated with the indicated concentrations of Mte1 for 15 min at 30°C. After adding loading buffer, the reaction mixtures were separated on a 7.5% native polyacrylamide gel in 0.5× TBE buffer and analyzed as described above.
Helicase and fork regression assay
Fluorescently labeled fork (helicase assay) or mobile fork (fork regression assay) substrate (4 nM) was incubated with 7 nM or 0.8 nM Mph1 protein, respectively, and the indicated concentrations of Mte1 protein for 15 min at 30°C in buffer R (50 mM Tris-HCl at pH 7.5, 1 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA, 20 mM creatine phosphate, 20 µg/mL creatine kinase, 1 mM ATP). Following deproteinization by incubation with 0.05% SDS and 0.5 mg/mL proteinase K for 5 min at 37°C, the reactions were mixed with loading buffer and resolved in a 10% native polyacrylamide gel in TBE buffer. Gels were analyzed as described above.
D-loop extension assay
The D-loop extension assay was performed essentially as described previously (Sebesta et al. 2011). Briefly, radioactively labeled 90-mer ssDNA (oligo D1) was incubated for 5 min at 37°C with 0.8 µM Rad51 in 10 µL of buffer R1 (35 mM Tris-HCl at pH 7.5, 2.5 mM ATP, 2.5 mM MgCl2, 50 mM KCl, 1 mM DTT, 100 µg/mL BSA, an ATP-regenerating system consisting of 20 mM creatine phosphate and 20 µg/mL creatine kinase). Following addition of 1 µL of 150 nM Rad54, the mixtures were incubated for an additional 3 min at 23°C. The D-loop synthesis was initiated by adding pBluescript replicative form I (pBSC; 50 µM base pairs) in 1.5 µL, and the reaction was incubated for 5 min at 23°C. Next, 660 nM RPA, 6.66 nM PCNA, 10 nM RFC, and 15 nM Pol δ in buffer O (20 mM Tris–HCl at pH 7.5, 5 mM DTT, 0.1 mM EDTA, 150 mM KCl, 40 µg/mL BSA, 8 mM MgCl2, 5% glycerol, 0.5 mM ATP, 75 µM each dGTP, dTTP, dATP, and dCTP) were added, and the mixtures were incubated for 5 min at 30°C. Various amounts of Mte1 were added to the reactions and incubated for 5 min at 30°C followed by the addition of 56 nM Mph1. After additional incubation for 5 min at 30°C, the reactions were stopped with 0.5% SDS (final) and 0.5 mg/mL proteinase K for 15 min at 37°C and loaded onto a 0.8% agarose gel. After electrophoresis, the gel was dried on grade 3 chromosome paper (Whatman), exposed to a phosphorimager screen, and scanned using a FLA-9000 Starion (Fujifilm) followed by analysis with MultiGauge version 3.2 software (Fujifilm).
Supplementary Material
Acknowledgments
We thank Marek Sebesta, Martin Pacesa, and Victoria Marini for providing us with reagents; Ivona Necasova for the statistical analysis of KD constants; and Patrick Sung, Grant Brown, and Juro Gregan for sharing data before publication. This work was supported by The Danish Agency for Science, Technology, and Innovation, the Villum Foundation, the Lundbeck Foundation, and the European Research Council (ERC) to M.L. and I.G.; the Danish National Research Foundation (DNRF115) to M.L.; the Chinese Scholarship Council to X.Y.; Fundação para a Ciência e Tecnologia (FCT) to S.S.; and GACR13-26629S and GACR207/12/2323 and European Regional Development Fund (Project FNUSA-ICRC, no. CZ.1.05/1.1.00/02.0123) grants to L.K. and V.A. V.A. also was supported by funds from the Faculty of Medicine Masaryk University to junior researchers. C.C. was supported by the EMBO Young Investigator program. The Novo Nordisk Foundation Center for Protein Research is supported financially by the Novo Nordisk Foundation (grant agreement NNF14CC0001).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.276204.115.
References
- Abdallah P, Luciano P, Runge KW, Lisby M, Geli V, Gilson E, Teixeira MT. 2009. A two-step model for senescence triggered by a single critically short telomere. Nat Cell Biol 11: 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams AK, Holm C. 1996. Specific DNA replication mutations affect telomere length in Saccharomyces cerevisiae. Mol Cell Biol 16: 4614–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams Martin A, Dionne I, Wellinger RJ, Holm C. 2000. The function of DNA polymerase α at telomeric G tails is important for telomere homeostasis. Mol Cell Biol 20: 786–796.10629035 [Google Scholar]
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. 2012. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol 14: 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askree SH, Yehuda T, Smolikov S, Gurevich R, Hawk J, Coker C, Krauskopf A, Kupiec M, McEachern MJ. 2004. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proc Natl Acad Sci 101: 8658–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch S, Kang LE, Symington LS. 2000. RAD51 is required for the repair of plasmid double-stranded DNA gaps from either plasmid or chromosomal templates. Mol Cell Biol 20: 1194–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti D, Clerici M, Anbalagan S, Martina M, Lucchini G, Longhese MP. 2010. Shelterin-like proteins and Yku inhibit nucleolytic processing of Saccharomyces cerevisiae telomeres. PLoS Genet 6: e1000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bupp JM, Martin AE, Stensrud ES, Jaspersen SL. 2007. Telomere anchoring at the nuclear periphery requires the budding yeast Sad1-UNC-84 domain protein Mps3. J Cell Biol 179: 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. 2002. A monomeric red fluorescent protein. Proc Natl Acad Sci 99: 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Hartwell L. 1985. CDC17: an essential gene that prevents telomere elongation in yeast. Cell 42: 249–257. [DOI] [PubMed] [Google Scholar]
- Chamankhah M, Xiao W. 1999. Formation of the yeast Mre11–Rad50–Xrs2 complex is correlated with DNA repair and telomere maintenance. Nucleic Acids Res 27: 2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamankhah M, Fontanie T, Xiao W. 2000. The Saccharomyces cerevisiae mre11(ts) allele confers a separation of DNA repair and telomere maintenance functions. Genetics 155: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Agrawal V, Johnson FB. 2011. Homologous recombination-dependent rescue of deficiency in the structural maintenance of chromosomes (Smc) 5/6 complex. J Biol Chem 286: 5119–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Choi K, Szakal B, Arenz J, Duan X, Ye H, Branzei D, Zhao X. 2009. Interplay between the Smc5/6 complex and the Mph1 helicase in recombinational repair. Proc Natl Acad Sci 106: 21252–21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Szakal B, Castellucci F, Branzei D, Zhao X. 2013. DNA damage checkpoint and recombinational repair differentially affect the replication stress tolerance of Smc6 mutants. Mol Biol Cell 24: 2431–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Szakal B, Chen YH, Branzei D, Zhao X. 2010. The Smc5/6 complex and Esc2 influence multiple replication-associated recombination processes in Saccharomyces cerevisiae. Mol Biol Cell 21: 2306–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DH, Kwon SH, Kim JH, Bae SH. 2012. Saccharomyces cerevisiae Cmr1 protein preferentially binds to UV-damaged DNA in vitro. J Microbiol 50: 112–118. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, Mann M. 2009. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc 4: 698–705. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature (London) 426: 194–198. [DOI] [PubMed] [Google Scholar]
- de Bruin D, Kantrow SM, Liberatore RA, Zakian VA. 2000. Telomere folding is required for the stable maintenance of telomere position effects in yeast. Mol Cell Biol 20: 7991–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin D, Zaman Z, Liberatore RA, Ptashne M. 2001. Telomere looping permits gene activation by a downstream UAS in yeast. Nature (London) 409: 109–113. [DOI] [PubMed] [Google Scholar]
- de Godoy LM, Olsen JV, de Souza GA, Li G, Mortensen P, Mann M. 2006. Status of complete proteome analysis by mass spectrometry: SILAC labeled yeast as a model system. Genome Biol 7: R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, Lydall D. 2010. Pif1- and Exo1-dependent nucleases coordinate checkpoint activation following telomere uncapping. EMBO J 29: 4020–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, Lydall D. 2012. Similarities and differences between ‘uncapped’ telomeres and DNA double-strand breaks. Chromosoma 121: 117–130. [DOI] [PubMed] [Google Scholar]
- Duxin JP, Walter JC. 2015. What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol 37: 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein J, Antony E, Hingorani MM, O'Donnell M. 2003. Overproduction and analysis of eukaryotic multiprotein complexes in Escherichia coli using a dual-vector strategy. Anal Biochem 319: 78–87. [DOI] [PubMed] [Google Scholar]
- Gallina I, Colding C, Henriksen P, Beli P, Nakamura K, Offman J, Mathiasen DP, Silva S, Hoffmann E, Groth A, et al. 2015. Cmr1/WDR76 defines a nuclear genotoxic stress body linking genome integrity and protein quality control. Nat Commun 6: 6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature (London) 415: 141–147. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. 2003. Global analysis of protein expression in yeast. Nature (London) 425: 737–741. [DOI] [PubMed] [Google Scholar]
- Gilmore JM, Sardiu ME, Venkatesh S, Stutzman B, Peak A, Seidel CW, Workman JL, Florens L, Washburn MP. 2012. Characterization of a highly conserved histone related protein, Ydl156w, and its functional associations using quantitative proteomic analyses. Mol Cell Proteomics 11: M111.011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15: 1541–1553. [DOI] [PubMed] [Google Scholar]
- Goudsouzian LK, Tuzon CT, Zakian VA. 2006. S. cerevisiae Tel1p and Mre11p are required for normal levels of Est1p and Est2p telomere association. Mol Cell 24: 603–610. [DOI] [PubMed] [Google Scholar]
- Gravel S, Larrivee M, Labrecque P, Wellinger RJ. 1998. Yeast Ku as a regulator of chromosomal DNA end structure. Science 280: 741–744. [DOI] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. 1999. Mammalian telomeres end in a large duplex loop. Cell 97: 503–514. [DOI] [PubMed] [Google Scholar]
- Hardy CF, Sussel L, Shore D. 1992. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev 6: 801–814. [DOI] [PubMed] [Google Scholar]
- Heim R, Tsien RY. 1996. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol 6: 178–182. [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature (London) 415: 180–183. [DOI] [PubMed] [Google Scholar]
- Ho CK, Mazon G, Lam AF, Symington LS. 2010. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell 40: 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM. 2014. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell 55: 626–639. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Weilbaecher RG, Walterscheid M, Lundblad V. 2000. Identification of the single-strand telomeric DNA binding domain of the Saccharomyces cerevisiae Cdc13 protein. Proc Natl Acad Sci 97: 6457–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. 2003. Global analysis of protein localization in budding yeast. Nature (London) 425: 686–691. [DOI] [PubMed] [Google Scholar]
- Kalocsay M, Hiller NJ, Jentsch S. 2009. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol Cell 33: 335–343. [DOI] [PubMed] [Google Scholar]
- Keppler-Ross S, Noffz C, Dean N. 2008. A new purple fluorescent color marker for genetic studies in Saccharomyces cerevisiae and Candida albicans. Genetics 179: 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadaroo B, Teixeira MT, Luciano P, Eckert-Boulet N, Germann SM, Simon MN, Gallina I, Abdallah P, Gilson E, Geli V, et al. 2009. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat Cell Biol 11: 980–987. [DOI] [PubMed] [Google Scholar]
- Khmelinskii A, Keller PJ, Bartosik A, Meurer M, Barry JD, Mardin BR, Kaufmann A, Trautmann S, Wachsmuth M, Pereira G, et al. 2012. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol 30: 708–714. [DOI] [PubMed] [Google Scholar]
- Kucejova B, Foury F. 2003. Search for protein partners of mitochondrial single-stranded DNA-binding protein Rim1p using a yeast two-hybrid system. Folia Microbiol (Praha) 48: 183–188. [DOI] [PubMed] [Google Scholar]
- Le S, Moore JK, Haber JE, Greider CW. 1999. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152: 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJ, Zakian VA. 1996. The Saccharomyces CDC13 protein is a single-strand TG1–3 telomeric DNA-binding protein in vitro that affects telomere behavior in vivo. Proc Natl Acad Sci 93: 13760–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Chang CC, Wong CW, Teng SC. 2009. Recruitment of Rad51 and Rad52 to short telomeres triggers a Mec1-mediated hypersensitivity to double-stranded DNA breaks in senescent budding yeast. PLoS One 4: e8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Geli V. 2009. DNA damage response to eroded telomeres. Cell Cycle 8: 3617–3618. [DOI] [PubMed] [Google Scholar]
- Lisby M, Rothstein R. 2015. Cell biology of mitotic recombination. Cold Spring Harb Perspect Biol 7: a016535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R. 2003. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol 5: 572–577. [DOI] [PubMed] [Google Scholar]
- Liu NN, Han TX, Du LL, Zhou JQ. 2010. A genome-wide screen for Schizosaccharomyces pombe deletion mutants that affect telomere length. Cell Res 20: 963–965. [DOI] [PubMed] [Google Scholar]
- Longhese MP, Bonetti D, Manfrini N, Clerici M. 2010. Mechanisms and regulation of DNA end resection. EMBO J 29: 2864–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke-Glaser S, Luke B. 2012. The Mph1 helicase can promote telomere uncapping and premature senescence in budding yeast. PLoS One 7: e42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke-Glaser S, Poschke H, Luke B. 2012. Getting in (and out of) the loop: regulating higher order telomere structures. Front Oncol 2: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V, Szostak JW. 1989. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 57: 633–643. [DOI] [PubMed] [Google Scholar]
- Lydall D, Weinert T. 1997. Use of cdc13–1-induced DNA damage to study effects of checkpoint genes on DNA damage processing. Methods Enzymol 283: 410–424. [DOI] [PubMed] [Google Scholar]
- Mankouri HW, Ngo HP, Hickson ID. 2009. Esc2 and Sgs1 act in functionally distinct branches of the homologous recombination repair pathway in Saccharomyces cerevisiae. Mol Biol Cell 20: 1683–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcand S, Gilson E, Shore D. 1997. A protein-counting mechanism for telomere length regulation in yeast. Science 275: 986–990. [DOI] [PubMed] [Google Scholar]
- Maringele L, Lydall D. 2002. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Δ mutants. Genes Dev 16: 1919–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulova P, Marini V, Burgess RC, Sisakova A, Kwon Y, Rothstein R, Sung P, Krejci L. 2009. Cooperativity of Mus81.Mms4 with Rad54 in the resolution of recombination and replication intermediates. J Biol Chem 284: 7733–7745. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mazon G, Symington LS. 2013. Mph1 and Mus81–Mms4 prevent aberrant processing of mitotic recombination intermediates. Mol Cell 52: 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GT, Jin S, Shannon KB, Weaver DT. 1996. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol 16: 4189–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature (London) 455: 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchel K, Lehner K, Jinks-Robertson S. 2013. Heteroduplex DNA position defines the roles of the Sgs1, Srs2, and Mph1 helicases in promoting distinct recombination outcomes. PLoS Genet 9: e1003340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ. 2008. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322: 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent CI, Hughes TR, Lue NF, Lundblad V. 1996. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science 274: 249–252. [DOI] [PubMed] [Google Scholar]
- Nugent CI, Bosco G, Ross LO, Evans SK, Salinger AP, Moore JK, Haber JE, Lundblad V. 1998. Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr Biol 8: 657–660. [DOI] [PubMed] [Google Scholar]
- Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science 273: 1392–1395. [DOI] [PubMed] [Google Scholar]
- Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL. 2009. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 23: 912–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panico ER, Ede C, Schildmann M, Schurer KA, Kramer W. 2010. Genetic evidence for a role of Saccharomyces cerevisiae Mph1 in recombinational DNA repair under replicative stress. Yeast 27: 11–27. [DOI] [PubMed] [Google Scholar]
- Prakash R, Krejci L, Van Komen S, Anke Schurer K, Kramer W, Sung P. 2005. Saccharomyces cerevisiae MPH1 gene, required for homologous recombination-mediated mutation avoidance, encodes a 3′ to 5′ DNA helicase. J Biol Chem 280: 7854–7860. [DOI] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, et al. 2009. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23: 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde FE, Louis EJ. 1999. Limitations of silencing at native yeast telomeres. EMBO J 18: 2538–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. 2012. The Pfam protein families database. Nucleic Acids Res 40: D290–D301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Antony E, Doganay S, Koh HR, Lohman TM, Myong S. 2013. Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nat Commun 4: 2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y. 2007. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906. [DOI] [PubMed] [Google Scholar]
- Raschle M, Van Komen S, Chi P, Ellenberger T, Sung P. 2004. Multiple interactions with the Rad51 recombinase govern the homologous recombination function of Rad54. J Biol Chem 279: 51973–51980. [DOI] [PubMed] [Google Scholar]
- Reid R, Lisby M, Rothstein R. 2002. Cloning-free genome alterations in Saccharomyce cerevisiae using adaptamer-mediated PCR. Methods Enzymol 350: 258–277. [DOI] [PubMed] [Google Scholar]
- Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, Shay JW, Wright WE. 2014. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev 28: 2464–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier P. 2014. Intrabody engineering for monitoring of DNA damaged induced post-translational modifications. Master thesis, University of Copenhagen, Denmark. [Google Scholar]
- Sabourin M, Tuzon CT, Zakian VA. 2007. Telomerase and Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol Cell 27: 550–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarek G, Vannier JB, Panier S, Petrini JH, Boulton SJ. 2015. TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Mol Cell 57: 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller J, Schurer A, Rudolph C, Hettwer S, Kramer W. 2000. MPH1, a yeast gene encoding a DEAH protein, plays a role in protection of the genome from spontaneous and chemically induced damage. Genetics 155: 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober H, Ferreira H, Kalck V, Gehlen LR, Gasser SM. 2009. Yeast telomerase and the SUN domain protein Mps3 anchor telomeres and repress subtelomeric recombination. Genes Dev 23: 928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurer KA, Rudolph C, Ulrich HD, Kramer W. 2004. Yeast MPH1 gene functions in an error-free DNA damage bypass pathway that requires genes from Homologous recombination, but not from postreplicative repair. Genetics 166: 1673–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebesta M, Burkovics P, Haracska L, Krejci L. 2011. Reconstitution of DNA repair synthesis in vitro and the role of polymerase and helicase activities. DNA Repair (Amst) 10: 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F, Fink GR, Hicks JB. 1986. Methods in Yeast Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva S, Gallina I, Eckert-Boulet N, Lisby M. 2012. Live cell microscopy of DNA damage response in Saccharomyces cerevisiae. Methods Mol Biol 920: 433–443. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiacovo MP, et al. 2010. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell 39: 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikumar T, Lewicki MC, Costanzo M, Tkach JM, van Bakel H, Tsui K, Johnson ES, Brown GW, Andrews BJ, Boone C, et al. 2013. Global analysis of SUMO chain function reveals multiple roles in chromatin regulation. J Cell Biol 201: 145–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. 1997. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev 11: 83–93. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, New JH, Kowalczykowski SC. 1998. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci 95: 6049–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P. 1994. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 265: 1241–1243. [DOI] [PubMed] [Google Scholar]
- Sung MK, Huh WK. 2007. Bimolecular fluorescence complementation analysis system for in vivo detection of protein–protein interaction in Saccharomyces cerevisiae. Yeast 24: 767–775. [DOI] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M. 2014. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198: 795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T. 2003. DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556. [DOI] [PubMed] [Google Scholar]
- Teixeira MT, Arneric M, Sperisen P, Lingner J. 2004. Telomere length homeostasis is achieved via a switch between telomerase- extendible and -nonextendible states. Cell 117: 323–335. [DOI] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56: 619–630. [DOI] [PubMed] [Google Scholar]
- Tkach JM, Yimit A, Lee AY, Riffle M, Costanzo M, Jaschob D, Hendry JA, Ou J, Moffat J, Boone C, et al. 2012. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol 14: 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Komen S, Macris M, Sehorn MG, Sung P. 2006. Purification and assays of Saccharomyces cerevisiae homologous recombination proteins. Methods Enzymol 408: 445–463. [DOI] [PubMed] [Google Scholar]
- Vannier JB, Sarek G, Boulton SJ. 2014. RTEL1: functions of a disease-associated helicase. Trends Cell Biol 24: 416–425. [DOI] [PubMed] [Google Scholar]
- Vega LR, Phillips JA, Thornton BR, Benanti JA, Onigbanjo MT, Toczyski DP, Zakian VA. 2007. Sensitivity of yeast strains with long G-tails to levels of telomere-bound telomerase. PLoS Genet 3: e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward TA, Dudasova Z, Sarkar S, Bhide MR, Vlasakova D, Chovanec M, McHugh PJ. 2012. Components of a Fanconi-like pathway control Pso2-independent DNA interstrand crosslink repair in yeast. PLoS Genet 8: e1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger RJ, Zakian VA. 2012. Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. Genetics 191: 1073–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby MC. 2010. The FANCM family of DNA helicases/translocases. DNA Repair (Amst) 9: 224–236. [DOI] [PubMed] [Google Scholar]
- Xue X, Choi K, Bonner J, Chiba T, Kwon Y, Xu Y, Sanchez H, Wyman C, Niu H, Zhao X, et al. 2014. Restriction of replication fork regression activities by a conserved SMC complex. Mol Cell 56: 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Choi K, Bonner JN, Szakal B, Chen YH, Papusha A, Saro D, Niu H, Ira G, Branzei D, et al. 2015a. Selective modulation of the functions of a conserved DNA motor by a histone fold complex. Genes Dev 29: 1000–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Sung P, Zhao X. 2015b. Functions and regulation of the multitasking FANCM family of DNA motor proteins. Genes Dev 29: 1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Papusha A, Choi K, Bonner JN, Kumar S, Niu H, Kaur H, Zheng X-F, Donnianni RA, Lu L, et al. 2016. Differential regulation of the anti-crossover and replication fork regression activities of Mph1 by Mte1. Genes Dev (this issue). doi: 10.1101/gad.276139.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimit A, Riffle M, Brown GW. 2015. Genetic regulation of Dna2 localization during the DNA damage response. G3 (Bethesda) 5: 1937–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Ren JY, Zhang JM, Suo F, Fang XF, Wu F, Du LL. 2013. A proteome-wide visual screen identifies fission yeast proteins localizing to DNA double-strand breaks. DNA Repair (Amst) 12: 433–443. [DOI] [PubMed] [Google Scholar]
- Zheng XF, Prakash R, Saro D, Longerich S, Niu H, Sung P. 2011. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair (Amst) 10: 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.