

Abstract
6,6″-bis(2,4,6-trimethylanilido)terpyridine (H2TpyNMes) was prepared as a rigid, tridentate pincer ligand containing pendent anilines as hydrogen bond donor groups in the secondary coordination sphere. The coordination geometry of (H2TpyNMes)copper(I)-halide (Cl, Br and I) complexes is dictated by the strength of the NH-halide hydrogen bond. The Cu(I)Cl and Cu(II)Cl complexes are nearly isostructural, the former presenting a highly unusual square planar geometry about Cu(I). The geometric constraints provided by secondary interactions are reminiscent of blue copper proteins where a constrained geometry, or entatic state, allows for extremely rapid CuI/II electron transfer self-exchange rates. Cu(H2TpyNMes)Cl shows similar fast electron transfer (~105 M−1s−1) which is the same order of magnitude as biological systems.
Keywords: hydrogen bonds, secondary sphere, square planar, copper, entatic state
Graphical Abstract
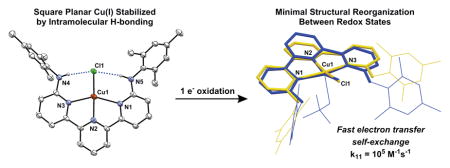
Entatic state: Hydrogen bonds constrain the geometry of Cu(I) and Cu(II) complexes. A highly unusual square planar geometry about Cu(I) is shown to be nearly isostructural to the Cu(II) core. The minimal reorganization energy between redox states allows for extremely rapid CuI/II electron transfer self-exchange rates
The secondary coordination sphere of blue copper proteins (BCPs) serves a key structural role to regulate the primary geometry of the Type-I copper center.[1] Importantly, the imposed coordination geometry is inextricably tied to rapid electron transfer (ET) processes (104–106 M−1s−1), which facilitate electron mobility within the protein.[2] In contrast to the fast ET rates found in copper proteins, synthetic systems generally feature much slower ET rates[3], likely as a result of the large structural differences preferred by Cu(I) and Cu(II) centers. Cu(I) favors tetrahedral coordination while Cu(II) generally adopts distorted octahedral, square planar, or square pyramidal geometries. These differences in the preferred primary coordination geometry naturally impart large barriers for reorganization during ET. To lower the reorganizational barrier and maximize associated ET rates in BCPs, the geometry at copper is regulated by a rich network of non-covalent interactions, which include hydrogen bonds (H-bonds). The surrounding protein scaffold, dubbed the ‘rack’, stabilizes copper in an intermediate geometry such that structural reorganization is minimized, which allows for fast ET.[4]
This strained geometric state of copper has been referred to as the ‘entatic’ state, coined by Vallee and Williams,[5] or ‘rack-induced’ state, coined by Malmström.[4a] The primary coordination sphere electronic environment, as well as noncovalent secondary interactions both contribute to the entatic state.[4b–e] The effects of secondary sphere residues on primary geometry, redox potential, and subsequent ET in BCPs have been examined by numerous research groups.[1b, 6] Concomitant with mutagenesis studies on BCPs, other groups have investigated the entatic state hypothesis by synthesizing small molecule complexes that exhibit minimal structural reorganization upon oxidation and reduction.[3, 7] Typically, the ligand frameworks for these studies exploit steric interactions or a macrocyclic ligand to achieve fast ET rates.[8] In contrast to the covalent coordination strategies most commonly employed by small molecule models, synthetic frameworks incorporating secondary sphere H-bonding interactions as a means to stabilize entatic states are exceedingly rare, yet are critical to the ‘rack’ in BCPs.[9]
Metal-ligand constructs featuring pendent H-bond donors or acceptors are ideal systems in which to probe how H-bonding interactions can impart a given geometric structure in copper complexes. Metal complexes with appended H-bond donors have been used to stabilize reactive intermediates and to enhance reactivity in catalytic reactions.[10] As an alternative, we sought to use H-bonding interactions as the principal design criterion to stabilize an otherwise uncommon square planar Cu(I) geometry. By directing H-bonds toward a metal-bound substrate within a rigid pincer framework, herein we demonstrate the stabilization of square planar copper(I) geometries.
The terpyridine framework functionalized in the 6 and 6″ positions allows for directed H-bonding interactions with an equatorially-coordinated substrate. Our lab[11] and others[12] have shown that ligands featuring the 2-hydroxypyridine (2-hp) fragment can be employed for cooperative substrate binding, proton-coupled electron transfer, and catalysis.[13] The combination of a metal halide H-bond acceptor[14] with a rigid planar ligand featuring pendent hydroxyl H-bond donors was hypothesized to bind Cu(I) in a square planar geometry. Cu(I) complexes with the ligand, 6,6″-dihydroxyterpyridine (dhtp),[11a] which incorporates two 2-hp fragments, was initially explored. Unfortunately, rather than affording the desired square planar compounds, metalations using CuCl with dhtp resulted in the formation of multinuclear clusters (Figure 1, top). To mitigate the challenges associated with limited steric protection, 6,6″-bis(2,4,6-trimethylanilido)terpyridine (H2TpyNMes), with pendent secondary anilines, was prepared to provide directed H-bonding interactions within an extended steric environment. Notably, in these systems, both the –OH and –NHAr groups present potent H-bond donors to a metal-coordinated substrate.[10d, 15]
Figure 1.
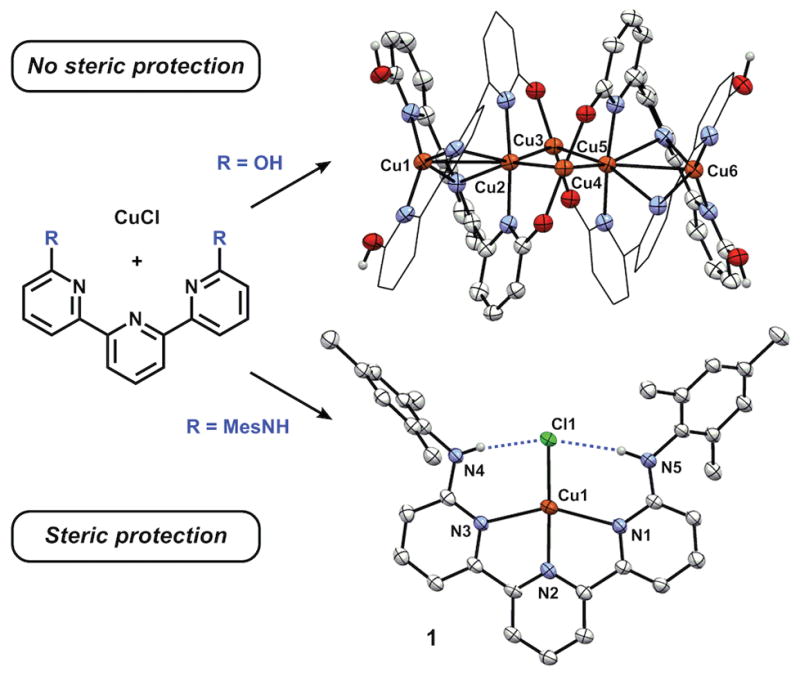
Synthesis and X-ray crystal structure of Cu6(dhtp)4 (top), and 1 (bottom) (30% and 50% ellipsoids respectively, H atoms not involved in H-bonding omitted for clarity).
The H2TpyNMes ligand was synthesized in a single step from 6,6″-dibromoterpyridine via nucleophilic substitution with potassium 2,4,6-trimethylanilide (See SI). Following an aqueous workup and purification by passage through an alumina plug, the reaction proceeded in 70% yield and can be scaled to gram quantities. Similar 6,6″-substituted terpyridines containing weaker pendent H-bond donor amine groups have been reported previously with R = NH2, NH(CH3), NH(C2H5).[16] However, their associated coordination chemistry is limited to three reports using Pt and Pd.[16b, 16c, 17]
To evaluate the salient structural features imparted by H2TpyNMes, Cu(I) complexes were prepared. The synthesis of Cu(H2TpyNMes)Cl (1) occurs over 18 hours by allowing H2TpyNMes to react with CuCl in benzene, affording a dark purple precipitate in 83% yield (Figure 1). Crystals suitable for an X-ray diffraction experiment were grown from diffusion of pentane into toluene. The solid-state structure of complex 1 revealed a remarkably square planar (SP) geometry, affording a τ4 value of 0.303 (τ4=0 for perfectly SP, τ4=1 for tetrahedral)[18] with the pendent anilines engaged in directed H-bonding interactions with the chloride ligand (Avg. Cl–N=3.3 Å). This τ4 value is highly unusual for a mononuclear Cu(I) complex. We have found only one example that exhibits smaller τ4 values, and in that case, the geometry is likely enforced by a rigid macrocyclic ligand.[19] To the best of our knowledge, 1 is the most SP mononuclear Cu(I) supported by an acyclic ligand scaffold.[20] The ability of H-bonds to influence the planarity of the N–Cu–X bond angle was evaluated by preparing analogous bromide and iodide complexes, which are weaker H-bond acceptors.[21] Cu(H2TpyNMes)Br (2) and Cu(H2TpyNMes)I (3) were obtained as black and brown crystals respectively from their appropriate copper(I)-halide salts.
The solid state structures of 2 and 3 revealed significant deviations from the square planarity observed in 1 (Figure 2A). For example, the iodide ligand in 3 is distorted away from the terpyridine plane, affording a τ4 value of 0.548. This structural distortion is consistent with a weakened NH–I H-bond interaction in 3 (Avg. I–N=3.8 Å) as compared to the NH–Cl interaction in 1. The H-bond acceptor ability of halide congeners decreases down the group,[22] which parallels the empirical trend noted above and suggests the H-bonding interactions are crucial contributors to the observed geometry in 1. Non-covalent intermolecular interactions may also serve to modify the geometry about copper. We note that several π-π interactions are present in the structures of 1, 2, and 3. Of particular note, 1 and 2 crystallize in the same space group (C2/c), and also feature similar τ4 values (0.303 and 0.309, for 1 and 2) in contrast to 3 (P21/c space group), which presents a significantly different packing orientation. Thus, we propose that the crystal packing forces (of similar strength to weak/medium H-bonds)[23] can present an alternative, albeit less predictive, driving force for a given geometrical preference, if two limiting structures have similar ground state energies.
Figure 2.
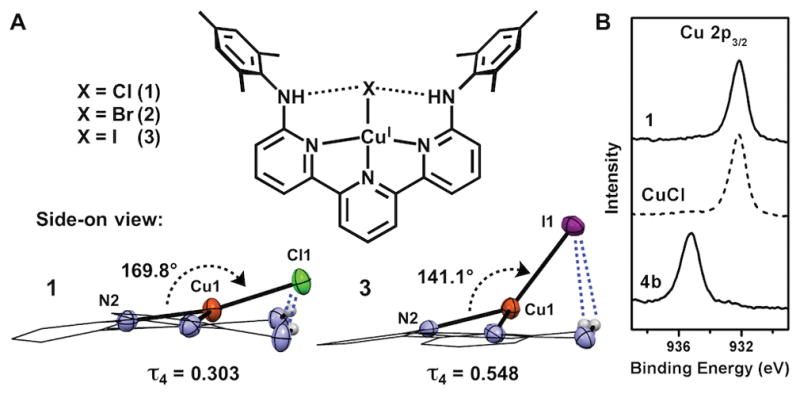
Side-on views of X-ray crystal structures 1 and 3 ((A) 50% ellipsoids, H atoms not involved in H-bonding and mesityl groups omitted for clarity; terpyridine backbone shown in wireframe style) and XPS spectra of the Cu 2p3/2 peak for 1, CuCl and 4b.
Due to the unusually dark color of these complexes, as well as solid state geometries that are more reminiscent of Cu(II), we interrogated whether the true oxidation state of complexes 1–3 are best described as Cu(I) or Cu(II). Weighardt and coworkers showed that metal-coordinated terpyridines can be redox non-innocent, a situation where an electron resides on the ligand, rather than the metal.[24–25] Importantly, reduction at the terpyridine ligand and oxidation of Cu can be confirmed by careful analysis of the bond distances in the crystal data, X-ray photoelectron spectroscopy (XPS), as well as by the visualization of intraligand π* to π* transitions in the low-energy region of the electronic absorption spectrum.[25] The terpyridine ligand in 1 displays no structural distortions that could be ascribed to ligand-based reduction. For example, the two Cpy-C′py bonds are equivalent at 1.490(4) Å and 1.483(5) Å, which fall in the reported range of 1.48 ± 0.01 Å for a single bond between two sp2-hybridized carbon atoms. Furthermore, the average pyridine C-C (1.387(5) Å) and C-N (1.346(4) Å) bond lengths are normal (typical values: 1.38 ± 0.01 Å and 1.35 ± 0.01 Å, respectively). [25]
UV-Vis and near-IR spectroscopy were used to assess the nature of the ground state in 1. In the visible region (400–1000 nm) one-electron reduced terpyridines contain bands that exhibit ε > 2,000 M−1cm−1.[25] The electronic absorption spectrum of 1, however, does not exhibit bands with ε > 425 M−1cm−1 between 400–1000 nm, and thus is inconsistent with a reduced terpyridine unit. Furthermore, the electronic spectrum of 1 in the near-IR region (1000–3000nm) displays no IVCT bands as would be expected for a ligand-reduced terpyridine (Figure S18).[25]
To further clarify the oxidation state of the Cu center in 1, XPS was performed. This technique can distinguish between the binding energies of electrons in Cu 2p orbitals in Cu(I) and Cu(II) complexes, and thus can be used to assign oxidation states.[26] A solid sample of 1 exhibits Cu 2p binding energies (932.2 eV for Cu 2p3/2) consistent with a Cu(I) oxidation state, which was further confirmed by comparison with an authentic sample of CuCl (Figure 2B).[26] In contrast to these Cu(I) complexes, the analogous Cu(II) complex 4b (see below) displays a Cu 2p3/2 binding energy of 935.2 eV, in addition to Cu 2p satellite peaks typical for Cu(II) compounds.[26b] Complementary DFT calculations also confirmed that a closed-shell singlet electronic state is the lowest energy state.[27] Collectively, these experimental and theoretical results provide strong evidence for a Cu(I) oxidation state in 1.
In addition to solid-state and spectrophotometric characterization, both NMR and vibrational spectroscopy provided complementary analyses of the H-bonding in 1–3. The room temperature 1H NMR spectrum of 1 in CD2Cl2 revealed symmetric ligand resonances with a single downfield peak at δ = 8.90, corresponding to two N-H protons (confirmed by H/D exchange in the presence of D2O; Figure S13) engaged in H-bonding interactions. The terpyridine and mesitylene resonances were unchanged upon halide substitution. In contrast, the NH peak position varied significantly between 1–3 (δ = 8.55 and 7.82 for 2 and 3, respectively), consistent with persistent H-bonding interactions in solution that weaken with decreasing H-bond accepting ability of the halide. Variable temperature NMR experiments revealed static solution structures of 1 and 2 between −80 to 60°C, while the NMR spectrum of 3 resolved new peaks at −80°C, indicative of a fluxional structure at room temperature. This halide-dependent fluxionality further supports a weakened NH–halide interaction in 3 as compared to 1 and 2.
IR spectroscopy was used to approximate the NH–halide H-bonding strength in 1–3. CH2Cl2 solutions of 1, 2, and 3 feature a single νNH band at 3234, 3242, and 3256 cm−1, respectively. The shift to higher energy is consistent with an increased covalency of the N-H bond and decrease in the H-bond strength, when moving down the halide series. In addition to this qualitative description, the shift of the νNH bands in the Cu-X complexes, relative to the free ligand (3406 cm−1) in CH2Cl2 solvent, was used to evaluate the strength of the H-bonding interactions. The H-bond enthalpy[28] was calculated to be 4.0 kcal/mol for 1, 3.9 kcal/mol for 2, and 3.7 kcal/mol for 3 per H-bond. These data clearly delineate the energetic differences in the H-bonding interactions imposed by the varied halide ligands.
The square planar geometry of 1 is persistent upon oxidation to Cu(II). Oxidation of 1 with ferrocenium hexafluorophosphate affords [Cu(H2TpyNMes)Cl]PF6 (4a) in 89% yield. Alternatively, treating CuCl2 with H2TpyNMes in the presence of AgClO4 similarly afforded [Cu(H2TpyNMes)Cl]ClO4 (4b). An X-ray diffraction experiment using 4b revealed a square-based pyramidal structure. Other than the presence of an additional axial ClO4− ligand in 4, complexes 1 and 4 are nearly isostructural, as noted by an overlay of the two structures that feature similar τ values (τ4=0.303 vs τ′ = 0.358 for complex 1 and 4, respectively; Figure 4). Note that while 4 is five-coordinate, the τ′ value was calculated by omitting the ClO4 ligand and considering the copper as four-coordinate. Much like the active site of BCPs, an interplay of small ligand distortions coupled with a stabilizing effect from H-bonding interactions allows the Cu(II) geometry to remain relatively unchanged.
Figure 4.
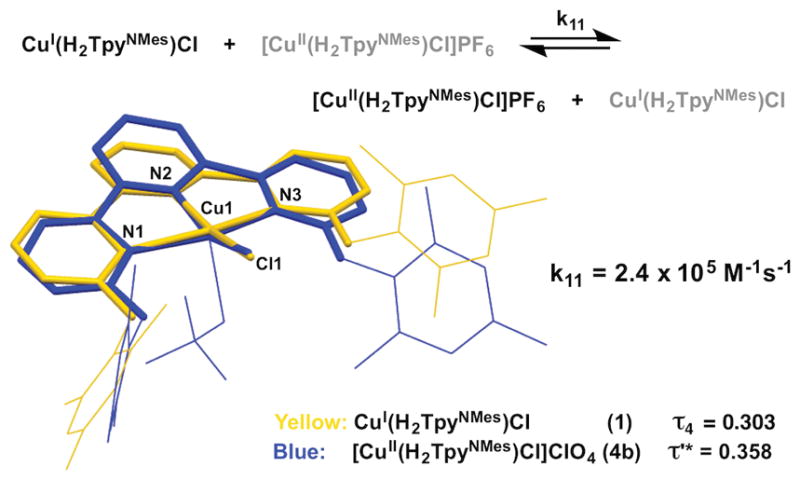
Crystal structure overlap of 1 and 4b from the primary sphere atoms (3N, Cu and Cl) and the experimentally determined ET self-exchange rate (structures shown in capped stick/wireframe view, H atoms omitted for clarity).
The requirement for H-bonding interactions as the main contributor to the square planar geometry in 1 was assessed with a control ligand devoid of H-bond donors. The ligand, 6,6″-bis(2,4,6-trimethylphenoxy)terpyridine, (TpyOMes), features an isosteric environment to H2TpyNMes, without containing H-bond donor groups, and was synthesized via an Ullmann-type coupling. Under the same synthetic conditions as those used to generate 4b, [Cu(TpyOMes)Cl]ClO4 (5) was obtained, and an X-ray quality crystal was grown from diffusion of diethyl ether into a THF solution (Figure 3A). The absence of H-bonding interactions allows 5 to adopt a geometry that is less square pyramidal (τ5=0.36) than 4b (τ5=0.11). These structural differences between 5 and 4b provide clear support that the H-bonding interactions can impart structural distortions in these copper complexes, and allude to H-bonding as the key contributor to the observed geometries in 1–4.
Figure 3.
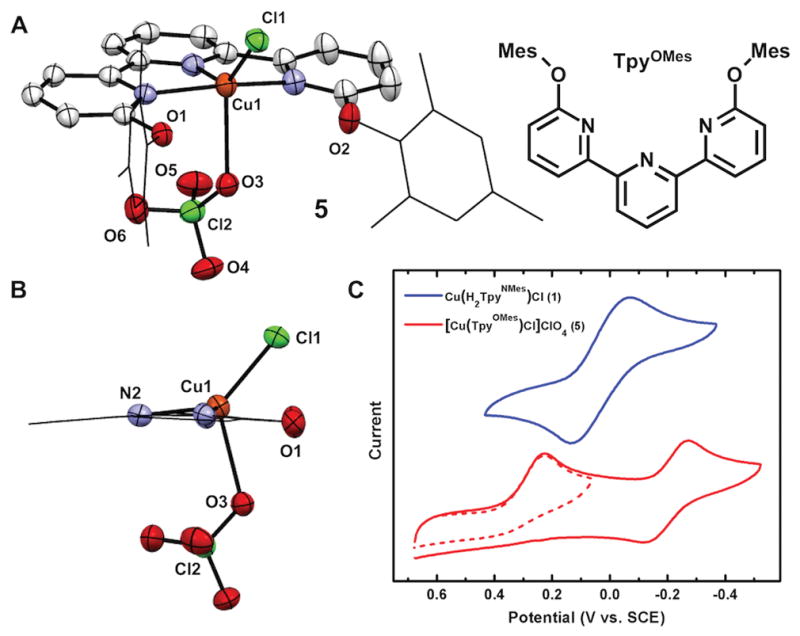
X-ray crystal structure of 5 ((A) 30% ellipsoids, H atoms omitted for clarity). A side view of 5 (B) and a comparison of the cyclic voltammograms of 1 and 5 at 100mV s−1 scan rate in 0.1M [nBu4N][ClO4] CH2Cl2 (C).
In addition to stabilizing a distinct ground state geometry, there are marked differences in the stability between 4 and 5. In contrast to solutions of 4, which are stable in THF solution for at least 30 days, 5 is unstable in solution (THF or CH2Cl2) and undergoes a disproportionation reaction to afford the homoleptic complex, [Cu(TpyOMes)2](ClO4) (6), and free CuCl2 in solution. When a THF solution of 5 was subjected to an ESI mass spectrometry experiment, both 5 and 6 were identified (m/z = 599.1 and 532.7). Furthermore, complex 6 was independently synthesized to establish the spectroscopic features contained in solutions of 5. The difference in solution stability between 4 and 5 was also visualized using cyclic voltammetry. In [Bu4N][ClO4] electrolyte, the voltammogram of 5 features an irreversible reduction at +272 mV (vs SCE; Figure 4C). Additionally, the disproportionation products of 5 were also observed at −223 mV and +790 mV for 6 and CuCl2, respectively (See SI). In contrast to this behavior, the voltammogram of either 1 or 4 exhibits a single redox couple at 47 mV (vs SCE). This peak is electrochemically reversible, as established by the linear relationship between the peak current and the square root of the scan rate over three orders of magnitude, the ratio of ipa/ipc = 1, and a peak-to-peak separation similar to that of ferrocene under identical conditions.[29] The differences in solution stabilities, in addition to the irreversible reduction of 5 clearly demonstrates the instability of related Cu(II) and importantly, Cu(I) species without the aid of H-bonding interactions.
We hypothesized that the minimal structural reorganization required for the interconversion between 1 and 4 would allow for extremely facile electron transfer (ET) rates, similar to BCPs. We evaluated the self-exchange ET rate using an NMR line broadening experiment, where the line width of a diamagnetic peak in the 1H NMR spectra of 1 was measured as a function of the concentration of 4.[3, 30] A plot of [CuII] vs πΔW (W=line width in Hz) provided a slope that correlates to an ET self-exchange rate of 2.4 × 105 M−1s−1 for 1 in THF at room temperature (see SI), which is the same order of magnitude as the fasted, reported synthetic systems as well as BCPs (104–106 M−1s−1).[8a, 8b, 31]
In conclusion, we have demonstrated the ability to tune a Cu(I) complex’s primary coordination geometry through the use of highly directed, intramolecular H-bonding. The geometry of Cu(I) was reliant on the H-bond strength to the respective metal-bound halides (Cl, Br, and I) with the Cu(I)Cl complex exhibiting a highly unusual square planar geometry. Just as the ‘rack’ helps stabilize the entatic state in BCPs, oxidation to Cu(II) complexes results in primary geometries at copper that are nearly identical to Cu(I), indicative of an H-bond stabilized entatic state. ET self-exchange reactions between 1 and 4 show extremely fast rates of ET self-exchange (105 M−1s−1). Although BCPs maintain a pseudo-tetrahedral geometry for both Cu(I) and Cu(II), the fast rates of ET are thought to stem from a minimization of structural reorganization, rather than the specific stabilized geometry. In 1, the presence of H-bonds are absolutely critical to the structure and stability, which employ highly directed H-bonds to stabilize a square planar Cu(I) geometry.
Experimental Section
General experimental details and characterization data for all of the reported compounds are included in the Supporting Information. CCDC 1428280, 1428281, 1428282, 1428283, 1428284, 1428285 contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Supplementary Material
Acknowledgments
This work was supported by the University of Michigan Department of Chemistry, the NIH (GM 111486), NSF grant CHE-0840456 for X-ray instrumentation, and through computational resources and services provided by Advanced Research Computing at UM. NKS is a Dow Corning Assistant Professor and Alfred P. Sloan Research Fellow. We thank Dr. Jeff W. Kampf for X-ray assistance, Prof. Paul Zimmerman for computational insight, and Dr. Cameron M. Moore and Jacob B. Jeri for helpful discussions.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.(a) Gray HB, Malmström BG, Williams RJ. J Biol Inorg Chem. 2000;5:551–559. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]; (b) Machczynski MC, Gray HB, Richards JH. J Inorg Biochem. 2002;88:375–380. doi: 10.1016/s0162-0134(02)00364-1. [DOI] [PubMed] [Google Scholar]; (c) Warren JJ, Lancaster KM, Richards JH, Gray HB. J Inorg Biochem. 2012;115:119–126. doi: 10.1016/j.jinorgbio.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dennison C. Coord Chem Rev. 2005;249:3025–3054. [Google Scholar]
- 3.Rorabacher DB. Chem Rev. 2004;104:651–698. doi: 10.1021/cr020630e. [DOI] [PubMed] [Google Scholar]
- 4.(a) Malmström BG. Eur J Biochem. 1994;223:711–718. doi: 10.1111/j.1432-1033.1994.tb19044.x. [DOI] [PubMed] [Google Scholar]; (b) Solomon EI, Baldwin MJ, Lowery MD. Chem Rev. 1992;92:521–542. [Google Scholar]; (c) Guckert JA, Lowery MD, Solomon EI. J Am Chem Soc. 1995;117:2817–2844. [Google Scholar]; (d) Ryde U, Olsson MH, Pierloot K, Roos BO. J Mol Biol. 1996;261:586–596. doi: 10.1006/jmbi.1996.0484. [DOI] [PubMed] [Google Scholar]; (e) Comba P. Coord Chem Rev. 2000;200:217–245. [Google Scholar]
- 5.Vallee BL, Williams RJ. Proc Natl Acad Sci USA. 1968;59:498–505. doi: 10.1073/pnas.59.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Marshall NM, Garner DK, Wilson TD, Gao YG, Robinson H, Nilges MJ, Lu Y. Nature. 2009;461:113–116. doi: 10.1038/nature08551. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) New SY, Marshall NM, Hor TSA, Xue F, Lu Y. Chem Commun. 2012;48:4217–4219. doi: 10.1039/c2cc30901g. [DOI] [PubMed] [Google Scholar]; (c) Van Pouderoyen G, Mazumdar S, Hunt NI, Hill AO, Canters GW. Eur J Biochem. 1994;222:583–588. doi: 10.1111/j.1432-1033.1994.tb18900.x. [DOI] [PubMed] [Google Scholar]; (d) Libeu C, Kukimoto M, Nishiyama M, Horinouchi S, Adman ET. Biochemistry. 1997;36:13160–13179. doi: 10.1021/bi9704111. [DOI] [PubMed] [Google Scholar]; (e) Carrell CJ, Sun D, Jiang S, Davidson VL, Mathews FS. Biochemistry. 2004;43:9372–9380. doi: 10.1021/bi049634z. [DOI] [PubMed] [Google Scholar]
- 7.(a) Hoffmann A, Binder S, Jesser A, Haase R, Flörke U, Gnida M, Salomone-Stagni M, Meyer-Klaucke W, Lebsanft B, Grünig LE, Schneider S, Hashemi M, Goos A, Wetzel A, Rübhausen M, Herres-Pawlis S. Angew Chem Int Ed. 2013;53:299–304. doi: 10.1002/anie.201306061. [DOI] [PubMed] [Google Scholar]; (b) Chaka G, Sonnenberg JL, Schlegel HB, Heeg MJ, Jaeger G, Nelson TJ, Ochrymowycz LA, Rorabacher DB. J Am Chem Soc. 2007;129:5217–5227. doi: 10.1021/ja068960u. [DOI] [PubMed] [Google Scholar]; (c) Yu Q, Salhi CA, Ambundo EA, Heeg MJ, Ochrymowycz LA, Rorabacher DB. J Am Chem Soc. 2001;123:5720–5729. doi: 10.1021/ja002995r. [DOI] [PubMed] [Google Scholar]
- 8.(a) Doine H, Yano Y, Swaddle TW. Inorg Chem. 1989;28:2319–2322. [Google Scholar]; (b) Krylova K, Kulatilleke CP, Heeg MJ, Salhi CA, Ochrymowycz LA, Rorabacher DB. Inorg Chem. 1999;38:4322–4328. [Google Scholar]; (c) Kulatilleke CP. Polyhedron. 2007;26:1166–1172. [Google Scholar]; (d) Augustin MA, Yandell JK. Inorg Chem. 1979;18:577–583. [Google Scholar]
- 9.Garcia L, Cisnetti F, Gillet N, Guillot R, Aumont-Nicaise M, Piquemal J-P, Desmadril M, Lambert F, Policar C. J Am Chem Soc. 2015;137:1141–1146. doi: 10.1021/ja510259p. [DOI] [PubMed] [Google Scholar]
- 10.(a) Zhao M, Wang HB, Ji LN, Mao ZW. Chem Soc Rev. 2013;42:8360–8375. doi: 10.1039/c3cs60162e. [DOI] [PubMed] [Google Scholar]; (b) Borovik AS. Acc Chem Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]; (c) Natale D, Mareque-Rivas JC. Chem Commun. 2008:425–437. doi: 10.1039/b709650j. [DOI] [PubMed] [Google Scholar]; (d) Thammavongsy Z, LeDoux ME, Breuhaus-Alvarez AG, Seda T, Zakharov LN, Gilbertson JD. Eur J Inorg Chem. 2013:4008–4015. [Google Scholar]; (e) Matson EM, Park YJ, Fout AR. J Am Chem Soc. 2014;136:17398–17401. doi: 10.1021/ja510615p. [DOI] [PubMed] [Google Scholar]; (f) Tubbs KJ, Fuller AL, Bennett B, Arif AM, Berreau LM. Inorg Chem. 2003;42:4790–4791. doi: 10.1021/ic034661j. [DOI] [PubMed] [Google Scholar]; (g) Widger LR, Davies CG, Yang T, Siegler MA, Troeppner O, Jameson GNL, Ivanović-Burmazović I, Goldberg DP. J Am Chem Soc. 2014;136:2699–2702. doi: 10.1021/ja410240c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Cheruzel LE, Cecil MR, Edison SE, Mashuta MS, Baldwin MJ, Buchanan RM. Inorg Chem. 2006;45:3191–3202. doi: 10.1021/ic051280s. [DOI] [PubMed] [Google Scholar]; (i) Feng G, Mareque-Rivas JC, Williams NH. Chem Commun. 2006:1845–1847. doi: 10.1039/b514328d. [DOI] [PubMed] [Google Scholar]; (j) Shook RL, Borovik AS. Chem Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wada A, Honda Y, Yamaguchi S, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Inorg Chem. 2004;43:5725–5735. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]; (l) Berreau LM. Eur J Inorg Chem. 2006;2006:273–283. [Google Scholar]; (m) Rakowski DuBois M, DuBois DL. Chem Soc Rev. 2009;38:62–72. doi: 10.1039/b801197b. [DOI] [PubMed] [Google Scholar]; (n) Hart JS, Nichol GS, Love JB. Dalton Trans. 2012;41:5785–5788. doi: 10.1039/c2dt30539a. [DOI] [PubMed] [Google Scholar]; (o) Sickerman NS, Park YJ, Ng GKY, Bates JE, Hilkert M, Ziller JW, Furche F, Borovik AS. Dalton Trans. 2012;41:4358–4364. doi: 10.1039/c2dt12244h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Rosenthal J, Nocera DG. Acc Chem Res. 2007;40:543–553. doi: 10.1021/ar7000638. [DOI] [PubMed] [Google Scholar]
- 11.(a) Moore CM, Szymczak NK. Chem Commun. 2013;49:400–402. doi: 10.1039/c2cc36927c. [DOI] [PubMed] [Google Scholar]; (b) Moore CM, Szymczak NK. Chem Commun. 2015;51:5490–5492. doi: 10.1039/c4cc06832g. [DOI] [PubMed] [Google Scholar]; (c) Moore CM, Szymczak NK. Chem Sci. 2015;6:3373–3377. doi: 10.1039/c5sc00720h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Geri JB, Szymczak NK. J Am Chem Soc. 2015;137:12808–12814. doi: 10.1021/jacs.5b08406. [DOI] [PubMed] [Google Scholar]
- 12.(a) Nieto I, Livings MS, Sacci JB, III, Reuther LE, Zeller M, Papish ET. Organometallics. 2011;30:6339–6342. [Google Scholar]; (b) Fujita K-i, Tanaka Y, Kobayashi M, Yamaguchi R. J Am Chem Soc. 2014;136:4829–4832. doi: 10.1021/ja5001888. [DOI] [PubMed] [Google Scholar]; (c) Hull JF, Himeda Y, Wang WH, Hashiguchi B, Periana R, Szalda DJ, Muckerman JT, Fujita E. Nat Chem. 2012;4:383–388. doi: 10.1038/nchem.1295. [DOI] [PubMed] [Google Scholar]
- 13.Moore CM, Dahl EW, Szymczak NK. Curr Opin Chem Biol. 2015;25:9–17. doi: 10.1016/j.cbpa.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 14.Aullón G, Bellamy D, Guy Orpen A, Brammer L, Bruton EA. Chem Commun. 1998:653–654. [Google Scholar]
- 15.(a) Moore CM, Quist DA, Kampf JW, Szymczak NK. Inorg Chem. 2014;53:3278–3280. doi: 10.1021/ic5003594. [DOI] [PubMed] [Google Scholar]; (b) Rivas JCM, Hinchley SL, Metteau L, Parsons S. Dalton Trans. 2006:2316–2322. doi: 10.1039/b516234c. [DOI] [PubMed] [Google Scholar]
- 16.(a) Cheon JD, Mutai T, Araki K. Tetrahedron Lett. 2006;47:5079–5082. [Google Scholar]; (b) Petersen AR, Taylor RA, Vicente-Hernández I, Mallender PR, Olley H, White AJP, Britovsek GJP. J Am Chem Soc. 2014;136:14089–14099. doi: 10.1021/ja5055143. [DOI] [PubMed] [Google Scholar]; (c) Taylor RA, Law DJ, Sunley GJ, White AJP, Britovsek GJP. Angew Chem Int Ed. 2009;48:5900–5903. doi: 10.1002/anie.200806187. [DOI] [PubMed] [Google Scholar]
- 17.Petersen AR, Taylor RA, Vicente-Hernández I, Heinzer J, White AJP, Britovsek GJP. Organometallics. 2014;33:1453–1461. [Google Scholar]
- 18.Yang L, Powell DR, Houser RP. Dalton Trans. 2007:955–964. doi: 10.1039/b617136b. [DOI] [PubMed] [Google Scholar]
- 19.Gagne RR, Allison JL, Lisensky GC. Inorg Chem. 1978;17:3563–3571. [Google Scholar]
- 20.SP geometries about Cu(I) have also been observed in two multinuclear systems: Arnold PL, Scarisbrick AC, Blake AJ, Wilson C. Chem Commun. 2001:2340–2341. doi: 10.1039/b107855k.Shinoura M, Kita S, Ohba M, Ōkawa H, Furutachi H, Suzuki M. Inorg Chem. 2000;39:4520–4526.Note that Jacobi has reported a complex that may be viewed as either 4-coordinate SP Cu(I) or as 5-coordinate, with a κ2-carboxylate ligand (O-Cu = 2.56Å).: Balogh-Hergovich É, Kaizer J, Speier G, Huttner G, Jacobi A. Inorg Chem. 2000;39:4224–4229. doi: 10.1021/ic990521r.
- 21.Brammer L, Bruton EA, Sherwood P. Crystal Growth & Design. 2001;1:277–290. [Google Scholar]
- 22.Hobza P, Müller-Dethlefs K. Non-covalent Interactions. Royal Society of Chemistry; Cambridge: 2010. [Google Scholar]
- 23.Steed JW, Atwood JL. Supramolecular Chemistry. 2. John Wiley & Sons, Ltd; West Sussex: 2009. [Google Scholar]
- 24.Chirik PJ, Wieghardt K. Science. 2010;327:794–795. doi: 10.1126/science.1183281. [DOI] [PubMed] [Google Scholar]
- 25.Scarborough CC, Lancaster KM, DeBeer S, Weyhermüller T, Sproules S, Wieghardt K. Inorg Chem. 2012;51:3718–3732. doi: 10.1021/ic2027219. [DOI] [PubMed] [Google Scholar]
- 26.(a) Moulder JF, Stickle WF, Sobol PE, KD . In: Bomben in Handbook of X-Ray Photoelectron Spectroscopy. Chastain J, editor. Perkin-Elmer Corp., Phys. Electron. Div; Eden Prairie, MN: 1992. pp. 86–87. [Google Scholar]; (b) Frost DC, Ishitani A, McDowell CA. Mol Phys. 1972;24:861–877. [Google Scholar]; (c) Hui AK, Losovyj Y, Lord RL, Caulton KG. Inorg Chem. 2014;53:3039–3047. doi: 10.1021/ic402866e. [DOI] [PubMed] [Google Scholar]
- 27.Geometry optimizations performed on the crystal structure of 1 and subsequent stability calculations using spin-unrestricted functionals show the Cu(I) closed-shell singlet state to be an energy minimum. (See SI)
- 28.Iogansen AV, Kurkchi GA, Furman VM, Glazunov VP, Odinokov SE. Zh Prikl Spektrosk. 1980;33:460. [Google Scholar]
- 29.Note that under the identical conditions, ferrocene behaves similar to 1.
- 30.Due to the nearly diffusion-controlled ligand substitution rates typical of Cu(II) complexes, coordination number invariance is not required to access fast ET rates. For examples, see reference 8. The analogous ET self exchange experiment was not possible with 5, due to its facile disproportionation at room temperature and irreversible CuII/I reduction.
- 31.Pulliam EJ, McMillin DR. Inorg Chem. 1984;23:1172–1175. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.