

Abstract
Voltage-gated ether à go-go (EAG) K+ channels are expressed in various types of cancer cells and also in the central nervous system. Aberrant overactivation of human EAG1 (hEAG1) channels is associated with cancer and neuronal disorders such as Zimmermann-Laband and Temple-Baraitser syndromes. Although hEAG1 channels are recognized as potential therapeutic targets, regulation of their functional properties is only poorly understood. Here, we show that the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) is a potent inhibitory gating modifier of hEAG1 channels. PIP2 inhibits the channel activity by directly binding to a short N-terminal segment of the channel important for Ca2+/calmodulin (CaM) binding as evidenced by bio-layer interferometry measurements. Conversely, depletion of endogenous PIP2 either by serotonin-induced phospholipase C (PLC) activation or by a rapamycin-induced translocation system enhances the channel activity at physiological membrane potentials, suggesting that PIP2 exerts a tonic inhibitory influence. Our study, combining electrophysiological and direct binding assays, demonstrates that hEAG1 channels are subject to potent inhibitory modulation by multiple phospholipids and suggests that manipulations of the PIP2 signaling pathway may represent a strategy to treat hEAG1 channel-associated diseases.
The human ether à go-go channel (hEAG1, also known as Kv10.1, encoded by the gene KCNH1) is a voltage-gated K+ channel mainly expressed in neuronal and cancer cells1. Besides the well-established roles in tumor development2,3,4,5, the importance of hEAG1 channels in the nervous system is now increasingly appreciated. For instance, recent genetic studies have demonstrated that EAG1 channels are critical for shaping the action potential in mice6, and gain-of-function mutations of the channel are associated with Zimmermann-Laband and Temple-Baraitser syndromes, two severe neurological and developmental disorders7,8. The hEAG1 channel has promising therapeutic and diagnostic potential, and development of the channel inhibitors is one therapeutic strategy for treating cancer and neurological disorders7,8,9. However, regulation of the hEAG1 channel function is only beginning to be understood. Although a section of the C-terminal area of hEAG1 shares high sequence similarity with the cyclic nucleotide binding domain in cyclic nucleotide-gated (CNG) channels and hyperpolarization-activated cyclic nucleotide-modulated (HCN) channels10, several lines of evidence have demonstrated that the hEAG1 channel fails to bind cyclic nucleotides11,12,13. Only a few endogenous regulators of the hEAG1 channel have been identified12,14. Among them, Ca2+/calmodulin (CaM) potently inhibits the EAG1 channel by binding to up to three discrete intracellular areas located in its N and C termini15,16,17.
Phosphatidylinositol 4,5-bisphosphate (PIP2), a phospholipid composed of one negatively charged head group and two fatty acid tails, serves as a structural cofactor for many membrane proteins and it is the precursor of two important second messengers, diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). Acting as a multifunctional molecule, PIP2 plays pivotal roles in normal and pathological cellular functions18,19,20. For instance, PIP2 is implicated in cell proliferation and neurological diseases21,22,23. Consequently, the lipid kinases including phosphatidylinositol 4-kinases (PI4Ks) and phosphatidylinositol-4-phosphate 5-kinases (PIP5KIs) responsible for PIP2 synthesis are considered to be potential therapeutic targets for various disorders involving altered neuronal excitability such as chronic pain19,22,24,25,26. In addition, the degradation of PIP2 is subject to dynamic regulation by many important neuronal transmitters via G protein-coupled receptor (GPCR)-induced phospholipase C (PLC) activation, which in turn affects many important downstream targets including a wide array of ion channels19,27,28. The mechanisms of ion channel regulation by PIP2 appear diverse. A crystal structure of an inward-rectifier K+ channel with PIP2 bound shows a direct interaction between the negatively charged head group of the lipid and a cluster of positively charged residues of the channel29. Besides the direct interaction mechanism, PIP2 also exerts its modulatory influences on ion channels indirectly by binding to numerous channel-associated inositol-binding proteins30,31,32. Moreover, modulation of ion channel function by PIP2 may involve alteration of the physical and chemical properties of the plasma membrane, which could then change the channel structure and function33. For instance, membrane lipid rafts enriched in PIP234 may influence ion channels contained therein35,36. Despite the existence of these direct and indirect mechanisms, many studies investigating modulation of ion channels by PIP2 relied solely on electrophysiological measurements and mutagenesis, and failed to clarify the exact mechanism of the PIP2 action.
The potential importance of both hEAG1 channels and PIP2 in neurological and developmental disorders7,8,9 prompted us to investigate whether hEAG1 channels are regulated by PIP2. In this study, we show that gating of the hEAG1 channel is potently inhibited by exogenous PIP2 and that physiological depletion of endogenous PIP2 in the plasma membrane enhances the channel activity. Moreover, the bio-layer interferometry (BLI) assay demonstrates that PIP2 directly binds to the hEAG1 channel complex. Removal of a short domain located in the N terminus of the channel abolished its interaction with PIP2 as well as the lipid-induced ionic current inhibition. Our findings show that the hEAG1 channel is directly regulated by PIP2 and that this regulation may contribute to normal human physiology and pathology.
Results
Inhibition of hEAG1 channels by PIP2
The effect of PIP2 on heterologously expressed hEAG1 channels was studied by direct application of exogenous PIP2 to the intracellular side of excised patches from Chinese hamster ovary (CHO) cells transiently expressing hEAG1 channels. Acute application of 3 μM brain-derived PIP2 with 18 to 20 carbon tail groups31 to the intracellular side caused a rapid and potent current inhibition (Fig. 1A,B). Because currents through hEAG1 channels frequently underwent progressive rundown after patch excision, our analysis was typically limited to the results obtained within 5–8 min of patch excision. We also tested the effect of PIP2 on the whole-cell current by perfusing PIP2 into the HEK293T cells stably expressing hEAG1 channels using a fast intracellular perfusion system and observed a similar inhibitory effect (Fig. 1C,D). The development of the inhibitory effect was slower with lower concentrations of PIP2 (Fig. 1E) and the results obtained with 200-s PIP2 application epochs showed that the fractional decrease in current size was also concentration dependent with an apparent half-maximal inhibitory concentration (IC50) of 0.35 ± 0.01 μM and the Hill coefficient of 0.92 ± 0.32 (Fig. 1F). By contrast, dioctanoyl-PIP2 (diC8- PIP2) and its analogs with different numbers of negative charges (3 μM), which have 8-carbon tails and higher water solubility27 than brain-derived PIP2, showed less overall potency in inhibiting hEAG1 channels (Fig. 1F–H). The IC50 value for diC8-PIP2 was 18.6 ± 0.6 μM, suggesting that the inhibitory potency of PIP2 depends on the length of the fatty acid chains.
Figure 1. Inhibitory effects of PIP2 on hEAG1 channels.

(A) Representative current traces recorded before and after application of brain-derived PIP2 (3 μM). Pulses to 40 mV were applied from the holding potential of −80 mV. (B) Representative time course of inhibition of normalized hEAG1 current by 3 μM PIP2 at 40 mV. Representative whole-cell hEAG1 currents (C) and normalized hEAG1 channel peak current (D) elicited by pulses to 40 mV from −80 mV using an automated patch-clamp instrument (Patchliner, Nanion) before and after intracellular perfusion of PIP2 (3 μM). **P < 0.01 compared to control levels before lipid applications. (E) Time course of normalized peak hEAG1 current at 40 mV after application of different concentrations of PIP2 (n = 5–13). (F) Concentration-dependent effects of PIP2 (open circles, n = 5–9) and diC8-PIP2 (closed circles, n = 3–7) (200 s applications) on the peak hEAG1 current size at 40 mV. The smooth curves were obtained by the Hill equation fitting. (G) Structures of long-chain and short-chain (S) phospholipids with the hydrophobic tails (long chain) framed. (H) Summarized normalized peak current of hEAG1 channels recorded at 3 min after application of diC8-PIP2 and its analogs (3 μM). The current size was normalized to that before lipid application in each patch. (I) Fractional peak hEAG1 current at 40 mV in the presence of the lipids indicated (3 μM). The numbers of independent measurements are shown in parentheses.
In addition to PIP2, other types of phosphatidylinositol (PIs) are also involved in cellular signaling and they may show specificities in ion channel regulation37,38. We therefore tested whether these PI lipids inhibited the hEAG1 channel. When applied at 3 μM, PI, PI(4)P, PI(4,5)P2, and PI(3,5)P2 differed noticeably in their abilities to alter the hEAG1 current, from no effect by PI to the strongest inhibition by PIP2 and PI(3,5)P2 (Fig. 1I), showing that the number of negative charges of the head group is also critical for the channel inhibition.
PIP2 (3 μM) virtually annihilated currents through hEAG1 at 40 mV at which the channel was maximally activated (Fig. 1A). However, even with PIP2 present (2 μM), depolarization to an extremely positive voltage (e.g., 200 mV) did elicit a robust outward current and the peak tail current size following the depolarization was similar to that after depolarization to 40 mV without PIP2 (Fig. 2A,B). This observation suggests that hEAG1 remained functional even in the presence of 3 μM PIP2 (Fig. 1A) but the voltage dependence of activation was shifted markedly to the positive direction. Consistent with this possibility, the conductance-voltage (GV) curve obtained with a lower concentration of PIP2 (0.1 μM) showed a clear positive shift; the half-activation voltage (V0.5) changed from −16.1 ± 4.0 to 40.8 ± 6.1 mV (Fig. 2E,F). Additionally, the GV curve became less steep; the slope factor (k) increased from 14.7 ± 1.2 to 44.2 ± 9.3 mV (Fig. 2C–F).
Figure 2. Voltage dependence of hEAG1 channel inhibition by PIP2.

(A) Representative current traces were recorded at −80 mV following depolarization to different voltages indicated. PIP2 was applied at 2 μM. (B) Normalized conductance under the conditions shown in (A). The results obtained in the presence of PIP2 (2 μM) were normalized to the maximal conductance inferred from the tail currents before PIP2 application in each patch. The normalized conductance values were 0.94 ± 0.02, 0.51 ± 0.04 and 0.81 ± 0.04, respectively (n = 6). (C) Illustrative current traces elicited by pulses from −80 to 50 mV in 10 mV increments before and after application of 0.1 μM PIP2. (D) Peak current-voltage curves from the results shown in (C). (E) Normalized conductance-voltage (GV) curves constructed from hEAG1 channel tail currents before and 3–5 min after application of 0.1 μM PIP2. The curves were fitted with a Boltzmann function from the results obtained using the same protocol as in (C). The fit parameters, half-activation voltage (V0.5) and slope factor (k), of voltage-dependent activation of hEAG1 channels before and after PIP2 application (0.1 μM) are shown in (F). **P < 0.01 compared to the control before PIP2 application and ††P < 0.01 compared to after PIP2 application at 40 mV.
hEAG1 channels interact directly with PIP2
To test whether PIP2 directly interacts with the hEAG1 channel, we used bio-layer interferometry (BLI) to measure the kinetics of binding of PIP2 to the purified hEAG1 channel complex. BLI is a novel methodology that has emerged recently for detecting protein-protein interactions as well as those between proteins and small molecules. Binding events between a protein and its ligands can be measured by changes in optical interference (Fig. 3A). The quality and the specificity of purified biotinylated hEAG1 channel protein was confirmed by Western blot (Fig. 3B). Addition of PIP2 to the protein anchored to the BLI sensor tip rapidly increased the optical interference signal (Fig. 3C). The kinetics of the BLI signal resembled those observed in the patch-clamp measurements in the excised patches (Fig. 1B). The kinetics of the increase in the BLI signal by PIP2 was concentration dependent, becoming faster with greater concentrations, and the BLI signal increase was partially reversible after 3 min of wash (Fig. 3C,D). Using a 200-s application epoch at each concentration as utilized in the electrophysiological experiments, the cumulative concentration dependence suggested a dissociation constant (KD) value of 0.35 ± 0.04 μM (Fig. 3E), similar to the IC50 value obtained from the electrophysiological measurements (Fig. 1F).
Figure 3. hEAG1 channels directly interact with PIP2.

(A) A schematic diagram showing the BLI binding assay protocol. (B) Western blot detection of hEAG1 channel protein from purified protein samples. The anti-FLAG antibody used recognizes a single protein band of ~110 kDa consistent with full-length of FLAG-tagged hEAG1 channel. (C) Changes in optical interference showing the dynamic association and dissociation processes between the channel protein and PIP2 at the concentrations indicated. The dashed line denotes the time at which the BLI sensors were transferred to the control buffer. The raw traces of association and dissociation were fitted well using double-exponential functions. For the association component, the time constants for the fast and slow component were 7.90 ± 0.04 s and 81.7 ± 1.2 s, respectively. For the dissociation segment, the time constants for the fast and slow component were 4.53 ± 0.67 s and 128.8 ± 3.5 s, respectively. (D) Changes in optical interference in different concentrations (0.03–3 μM) of PIP2 in a representative assay. (E) Curve fit with Hill equation obtained from the peak value of the optical interference signal measured at different PIP2 concentrations for determination of the equilibrium dissociation constants (KD) of the interaction between the hEAG1 channel protein and PIP2 (n = 3). (F) Representative time courses of the interference signals elicited by PI(3,5)P2 and diC8-PIP2. Each was applied at 3 μM. Data are representative of three separate experiments. (G) Optical interferences signal changes by 3 μM of different species of phospholipids. Numbers of independent measurements are shown in parentheses. **P < 0.01 compared to control before PIP2 application.
The close correspondence between the result from the BLI and electrophysiological measurements was also observed in the ligand selectivity. PI(3,5)P2 (3 μM), which effectively inhibited the hEAG1 channel electrophysiologically (Fig. 1I), elicited robust BLI signals (Fig. 3F,G). In contrast, diC8-PIP2 (3 μM), which failed to inhibit the hEAG1 channel in our electrophysiological experiments (Fig. 1G,H), also failed to elicit any noticeable BLI signal (Fig. 3F,G).
Depletion of endogenous PIP2 increases hEAG1-mediated K+ currents
Some inward-rectifier K+ channels require PIP2 for function, indicating that these channels are subject to basal tonic modulation by PIP220. We investigated whether a similar tonic modulation of hEAG1 channels exists by selectively depleting PIP2 in HEK293T cells stably expressing hEAG1 channels. We used the plasma membrane-targeted, rapamycin-induced translocation system based on inducible dimerization of PM-FRB-CFP and 5-phosphatase-FKBP-mRFP to acutely deplete PIP2 by a specific dephosphorylation of PIP2 at the 5′ position39,40. PIP2 depletion in the plasma membrane was monitored by PLCδ1PH-GFP, which binds to PIP2 at the plasma membrane40. When hEAG1-expressing cells were transfected with PLCδ1PH-GFP alone, the GFP fluorescence was found primarily near the plasma membrane (Fig. 4A top and middle), suggesting an appreciable level of PIP2 in the plasma membrane at rest. Application of 100 nM rapamycin to these cells changed neither the location of GFP fluorescence distribution nor the electrophysiologically measured whole-cell hEAG1 current size (Fig. 4A bottom, C). By contrast, the same concentration of rapamycin diminished the GFP signal when 5-phosphatase-FKBP-mRFP and PM-FRB-CFP constructs were cotransfected with PLCδ1PH-GFP (Fig. 4B top and middle), indicative of a decrease in the level of plasma membrane PIP2. Rapamycin also increased the whole-cell hEAG1 current (Fig. 4B bottom). The fractional increase in current was especially noticeable at more negative voltages (Fig. 4C), consistent with the idea that PIP2 depletion shifted the current-voltage (IV) relationship to the negative direction.
Figure 4. Enhancement of hEAG1 channel current by PIP2 hydrolysis induced by rapamycin and HTR2A activation.

(A) Representative GFP fluorescence (top), normalized GFP fluorescence at the plasma membrane (middle), and whole-cell current traces (bottom) in hEAG1-expressing cells transfected with PLCδ1PH-GFP in the absence and presence of rapamycin (100 nM) for 30 s as indicated. (B) Representative fluorescence (top), the normalized GFP fluorescence at the plasma membrane (middle), and the whole-cell current (bottom) in hEAG1-expressing cells transfected with PLCδ1PH-GFP, 5-phosphatase-FKBP-mRFP (FKBP), and PM-FRB-CFP (FRB) before and after incubation with rapamycin (100 nM) for 30 s. (C) Rapamycin (100 nM)-induced fractional augmentation of hEAG1 current at different voltages from hEAG1-expressing cells transfected with PLCδ1PH-GFP alone (n = 5) and with PLCδ1PH-GFP, 5-phosphatase-FKBP-mRFP, and PM-FRB-CFP (n = 3). (D) Representative GFP fluorescence (top), the normalized GFP fluorescence at the plasma membrane (middle), and the whole-cell current in hEAG1-expressing cells transfected with PLCδ1PH-GFP before and after incubation with serotonin (100 μM) for 1 min. (E) Representative fluorescence (top), the normalized GFP fluorescence at the plasma membrane (middle), and whole-cell current traces (bottom) in hEAG1-expressing cells transfected with PLCδ1PH-GFP (PH) and the serotonin receptor HTR2A (HTR2A) in the absence and presence of serotonin (100 μM) for 1 min. (F) Serotonin-induced fractional increase of hEAG1 current at different voltages from hEAG1-expressing cells transfected with PLCδ1PH-GFP alone (n = 5) and PLCδ1PH-GFP and HTR2A together (n = 6). *P < 0.05 and **P < 0.01 compared to the control. Scale bars in (A,B,D,E) 20 μm. ∆F/F0 represents the normalized GFP fluorescence intensity in the plasma membrane after PIP2 depletion. The currents were elicited by 1 s-ramps from −80 to 20 mV every 5 s.
One of the physiological ways by which PIP2 is hydrolyzed is by the action of phospholipase C (PLC) following activation of GPCRs such as serotonin receptors41, which are in turn implicated in tumorigenesis42 and also in neuronal signaling. We thus examined if serotonin increases the hEAG1 channel activity by depleting PIP2. We transfected the hEAG1-expressing HEK293T cells with human 5-hydroxytryptamine (serotonin) receptor 2A (HTR2A)43 and PLCδ1PH-GFP. Hydrolysis of PIP2 by PLC leads to IP3 release and increases the intracellular Ca2+ concentration ([Ca2+]i), which may confound any effect of PIP2 depletion16. To avoid this potential complication, the “fast” Ca2+ chelator 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 10 mM) was used in the whole-cell recording solution to rapidly control [Ca2+]i. Serotonin (100 μM) had no detectable effect on the GFP signal or the current size in the control hEAG1-expressing cells (Fig. 4D,F). However, when the channels were co-expressed with the serotonin receptor HTR2A, serotonin dimmed the GFP signal reporting the PIP2 level (Fig. 4E top and middle) and noticeably increased the whole-cell current (Fig. 4E bottom, F). The fractional increase in current was greater at more negative voltages (Fig. 4F), suggesting that the IV curve shifted to the negative direction. The results collectively suggest that endogenous PIP2 exerts a detectable tonic inhibitory influence on hEAG1 channels in intact cells.
Molecular determinants of hEAG1 channel inhibition by PIP2
PIP2-binding sites typically consist of positively charged amino acid residues44. To identify the molecular locus required for the PIP2-dependent regulation of hEAG1, we first investigated whether the positively charged residues in S4 of hEAG1 are involved in PIP2-induced channel inhibition in part because PIP2 alters the GV characteristics of the hEAG1 channel (Fig. 2E). The hEAG1 S4 segment, like those in other voltage-gated K+ channels45,46, contains multiple positively charged residues that move within the electric field (Fig. 5A). If these mobile charges interact with PIP2 applied to the intracellular side, some voltage/S4 state dependence of the effect of PIP2 on hEAG1 may be expected as observed in KCNQ channels47. PIP2 quickly inhibited the hEAG1 channel when the membrane potential was held at 40 mV (Fig. 5B,D). At this voltage, assuming that hEAG1 and Kv1.2/2.146 activate in a similar fashion, the voltage sensors are expected to be fully activated, placing hEAG1 K327 through R336 on the extracellular side and R339 interacting with negatively charged residues in S2 and S3. A similar inhibition was observed when the membrane potential was held at −80 mV at which R330 through K340 electrically should face the intracellular side (Fig. 5C,D). Furthermore, neutralization of the positively charged residues at R333, R336, R339, and/or K340 had no appreciable effect on the PIP2-induced channel inhibition (Fig. 5E).
Figure 5. Positive charges in the S4 voltage sensor are not essential for the inhibitory effect of PIP2.

(A) A cartoon showing the positively charged amino acids in hEAG1 S4. (B) Representative current trace (top) and the normalized time course (bottom) of hEAG1 channel currents in an excised patch before and after application PIP2 (3 μM) when the membrane potential was held at 40 mV. The arrow denotes the start of PIP2 application. (C) Representative current traces recorded sequentially at −80 mV, 40 mV, −80 mV, and 40 mV (top), and the normalized time course (bottom) at the membrane voltages indicated. (D) Averaged time constants of PIP2-induced hEAG1 current inhibition at 40 and −80 mV according to the protocols in (B,C) respectively. (E) Summary of the normalized peak current and averaged time constants (Tau) of PIP2-induced current inhibition at 40 mV in wild-type and mutant hEAG1 channels. The time constants were obtained from single-exponential fits to the normalized current inhibition by PIP2.
Amino-acid sequences of hEAG1, hELK1, and hERG1 show considerable similarity (~40%). A recent study has shown that PIP2 inhibits hELK1 by interacting with some of the positively charged residues in the S4–S5 linker, S6, and EAG domain48. However, these residues are not well conserved in hEAG1 channels. By contrast, PIP2 increases currents through hERG1 and a short region rich in positively charged residues (883RQRKRKLSFRRR894) in the intracellular C terminus of hERG1 has been implicated49. The distal C terminus of hEAG1 also contains a similar segment with multiple positively charged residues (C3 806KRKSWARFK814, Fig. 6A). Deletion of this segment did not alter the inhibitory effect of exogenous PIP2 (∆C3, Fig. 6B). In many ion channels such as KCNQ50, TRPC51, SK252, and CNG channels53, the putative PIP2 interaction sites overlap with CaM binding sites. Therefore, we hypothesized that the CaM interaction domains in the hEAG1 channel15,16,17 may be involved in the inhibitory effect of PIP2. Up to three potential CaM binding segments in hEAG1 have been reported: CaM-N in the N terminus and CaM-C1 and CaM-C2 in the C terminus (Fig. 6A)15,16,17. We deleted these CaM binding sites individually (∆CaM-N, ∆CaM-C1 and ∆CaM-C2). Currents through hEAG1 ∆CaM-C1 and hEAG1 ∆CaM-C2 maintained wild-type-like sensitivity to PIP2; however, those from hEAG1 ∆CaM-N were virtually unaltered by PIP2, up to 3 μM (Fig. 6B,C). Moreover, no BLI signals were observed from the purified hEAG1 ∆CaM-N in response to PIP2 (3 μM; Fig. 6D,E). In addition to hEAG1, we also found that PIP2 significantly inhibited hEAG2 (KCNH5, Kv10.2) channels, which have virtually the same sequence near the CaM-N area as hEAG1 (Supplementary Fig. S1).
Figure 6. Deletion of the N-terminal CaM binding site impairs the PIP2-mediated inhibition of hEAG1.

(A) Structural organization of one hEAG1 subunit. The putative PIP2 binding domain, CaM-N, CaM-C1, CaM-C2, and C3 are illustrated. (B) Representative current traces recorded at 40 mV before (black) and after application (gray) of 3 μM PIP2 in wild-type and the mutant channels. (C) Summary of the normalized peak current at 40 mV after application of PIP2 (3 μM) in wild-type and mutant hEAG1 channels. **P < 0.01 compared to before PIP2 application. (D) Kinetics of PIP2 (3 μM) interaction with wild-type (gray) and ∆CaM-N (red) hEAG1 channels using the BLI assay. (E) Mean optical interference responses obtained 300 s after application of PIP2 (3 μM) for wild-type and ∆CaM-N hEAG1 channel proteins. **P < 0.01 compared to WT. Numbers of independent measurements are shown in parentheses.
Functional independence between PIP2 and Ca2+/CaM in the regulation of hEAG1 channels
The observation that the potential binding sites for PIP2 and Ca2+/CaM in the N terminus overlap suggests that these two inhibitory factors may interfere with each other. Furthermore, both Ca2+/CaM and PIP2 may be present together under physiological conditions. The exposure of inside-out membrane patches to 200 nM CaM in 1 μM [Ca2+]i resulted in a complete inhibition of hEAG1 currents and this effect, unlike that by PIP2, was rapidly and fully reversible (Fig. 7A). The second exposure to Ca2+/CaM at the same concentrations together with PIP2 (3 μM) inhibited the current as rapidly as the first exposure. However, no recovery was observed after wash out of Ca2+/CaM (Fig. 7A), an indication of the long-lasting action of PIP2 (see Fig. 1). To assess whether the presence of PIP2 alters its sensitivity to Ca2+/CaM, we utilized the following protocol. We first verified that Ca2+/CaM (30 nM CaM in 1 μM [Ca2+]i) inhibited the current in a rapidly reversible fashion (Fig. 7B, first “CaM”). Following wash out of Ca2+/CaM, application of PIP2 (0.5 μM) decreased the current by 50% (Fig. 7B, “PIP2”). Subsequent application of Ca2+/CaM results in a near 100% inhibition (Fig. 7B, second “CaM”), suggesting that Ca2+/CaM and PIP2 may have additive effects on hEAG1 channel inhibition.
Figure 7. CaM- and PIP2-induced hEAG1 channel inhibition.

(A) Representative current traces (top), the time course of the normalized peak current (middle) and the summarized normalized peak current (bottom) of hEAG1 channels recorded at 40 mV in excised patches before and after sequential applications of CaM (200 nM) and PIP2 (3 μM) as indicated (n = 4). (B) Representative current traces (top), the time course of the normalized peak current (middle) and the summarized current inhibition (bottom) of hEAG1 channels recorded at 40 mV in excised patches before and after sequential applications of CaM (30 nM) and PIP2 (0.5 μM), both of which induced around 50% current inhibition as shown by the dotted line (n = 3). The internal Ca2+ was buffered to 1 μM in both (A,B).
Discussion
Increasing evidence suggests that hEAG1 K+ channels are closely linked with tumorigenesis and developmental diseases7,8,9. However, the regulation of the hEAG1 channel function is only beginning to be elucidated and only a few physiologically relevant regulators have been identified12,14,16,54 . Here we have demonstrated that PIP2 profoundly shifts the voltage dependence of activation of the hEAG1 channel to the positive direction and acts as a potent inhibitory modulator of the channel. Further, we have identified that a region near the channel’s N-terminal CaM interaction site is essential for both the electrophysiological effects and binding of PIP2.
Membrane lipids may be an important class of modulators of EAG channels. A recent study has shown that mouse neurons have two populations of EAG channels, one of which is sensitive to the alteration of lipid raft integrity such as cholesterol depletion54. In our study, temporally-controlled depletion of PIP2 by rapamycin-induced translocation of 5-phosphatase55 in whole cells noticeably increased hEAG1 currents especially at negative voltages. Thus, PIP2-mediated inhibition of hEAG1 channels is well suited to influence action potential firing in neurons. Physiological relevance of the tonic inhibitory influence of PIP2 as an endogenous modulator of hEAG1 is further suggested by the finding that activation of serotonin HTR2A receptors, known to activate PLC43 to promote hydrolysis of PIP2, also increases hEAG1 currents at negative voltages. However, the magnitude of hEAG1 current increase in vivo mediated by GPCR activation is difficult to predict. Under such conditions, GPCR activation leads to generation of multiple signaling molecules capable of affecting hEAG1 channels in distinct ways. For example, PIP2 depletion by PLC produces IP3, which in turn can promote Ca2+ release from the endoplasmic reticulum. Ca2+/CaM could then inhibit hEAG1 channels, dampening the direct stimulatory effect of PIP2 depletion on the channels described here. In addition, activation of many GPCRs including serotonin receptors can stimulate phospholipases A2 (PLA2) thereby inducing the release of arachidonic acid (AA), a lipid messenger which has been reported to activate hEAG channels in human melanoma cells14. Thus, depending on the comparative potencies and kinetics of these direct and indirect effects, complex regulation of hEAG1 channels by PIP2 under physiological conditions is possible.
Numerous ion channels have been reported to be PIP2 sensitive20. However, elucidation of the exact mode of action of PIP2, for example direct binding vs. indirect effects, has been hampered by the lack of appropriate PIP2 binding assays with a good time resolution that detect physiologically relevant binding events. Here, we used bio-layer interferometry (BLI), a novel assay capable for measuring the kinetics of binding between proteins and their ligands56, to detect binding of PIP2 to the isolated hEAG1 protein. The BLI measurements showed the existence of two kinetic components in association and dissociation (Fig. 3C). The fast BLI association and slow dissociation components especially well correspond to the electrophysiological results in their kinetics and concentration dependence. The slow BLI association component and the fast dissociation signals, which became more prominent at high concentrations of PIP2, may be electrophysiologically silent. Furthermore, the deletion of the N-terminal Ca2+/CaM binding area in hEAG1, which eliminates the electrophysiological effect of PIP2, also abolishes the BLI signal. A Hill coefficient (~1) of the concentration dependence curve of the current inhibition by PIP2 hints that binding of one PIP2 is capable of exerting a full inhibitory effect; this idea needs further investigation. The close correspondence between the electrophysiological and BLI results demonstrates that BLI, which requires no labeling and has a time resolution comparable to that of the patch-clamp method, could be a very promising method for studying the pharmacology of ion channels, thus complementing the established electrophysiological assays.
Our electrophysiological results show that PIP2 functions as a potent inhibitory gating modifier of the hEAG1 channel by shifting the overall voltage dependence of activation to the positive direction. We did not measure the exact shift at high concentrations of PIP2; however, the observation that a functionally saturating concentration of PIP2 (3 μM) essentially eliminates the current at 40 mV where the normalized conductance without PIP2 is near unity suggests that PIP2 may induce a >100 mV shift. Such a large shift in the voltage dependence of activation may partially explain the profound current inhibition. How binding of PIP2 to the N terminus of hEAG1 leads to this marked shift in the overall voltage dependence can only be speculated. Studies show that the N-terminal region of the members of the KCNH channel family (KV10, KV11 and KV12) including hEAG1 (KV10.1) plays pivotal roles in channel gating57. An area termed the EAG domain in the N terminus directly interacts with an area in the C terminus called the cyclic nucleotide-binding homology domain (CNBHD), which in turn regulates behavior of the ion conduction gate in S6 via the C-linker domain13. Downstream of the EAG domain close to S1, the N terminus contains one of the CaM binding sites, CaM-N, which is involved in Ca2+/CaM-induced inhibition of hEAG117. Our results show that the amino-acid residues involved in PIP2 binding partially overlap with the CaM-N site, suggesting that PIP2 and Ca2+/CaM may interact. Such an interaction is in fact seen in many other ion channels such as TRPC6 in which PIP2 binding disrupts the association of CaM with the channel protein51. A structural study shows that PIP2 activates type 2 small-conductance Ca2+-dependent K+ channels (SK2) by binding to the positively charged residues located at the interface of the SK2-CaM complex and that phosphorylation of CaM reduces the affinity of PIP2 for the channel complex52. However, in hEAG1, the long-lasting inhibitory effect of PIP2 is unaltered by Ca2+/CaM while we cannot formally exclude the possibility that binding of PIP2 somehow renders the interaction of the CaM-N domain and CaM essentially irreversible. Thus, PIP2 and Ca2+/CaM with vastly different kinetics are capable of regulating the channel in each other’s presence, expanding the modulatory versatility of the hEAG1 channel. The N terminus of the EAG channel has been suggested to interact directly with CNBHD13. Perhaps, this interaction is modified by PIP2 to alter the voltage dependence of activation (Fig. 8).
Figure 8. Proposed mechanism of PIP2 inhibition in hEAG1 channels.

A cartoon of two subunits of a tetrameric hEAG1 channel complex shown in the open (left) and closed (right) states. PIP2 (red) binding to the CaM-N segment (blue) may cause a conformational change of the complex formed by EAG domain and CNBHD and then close the channel gate via the C-linker region as described in Haitin et al.13.
In summary, we have demonstrated that PIP2 is a potent endogenous gating modulator of the hEAG1 channel that exerts its tonic inhibitory influence by direct binding to the N terminus of the channel protein. Gain-of-function mutations in the gene coding for hEAG1 have been associated with developmental and neurological disorders such as Zimmermann-Laband and Temple-Baraitser syndromes7,8. Thus, the tonic inhibition of the hEAG1 channel by PIP2 may be beneficial to the aforementioned disorders and dysregulation of the PIP2 signaling pathway may contribute to disease through hEAG1 channels.
Materials and Methods
Channel expression
hEAG1 (NM_002238.3) and hEAG2 (NM_139318.4) in the expression vector pcDNA3.1 were transiently expressed in CHO cells using X-tremeGENE 9 (Roche Diagnostics). The mutant channels were constructed by using the QuickChange Site-Directed Mutagenesis kit (Agilent) following the manufacturer’s instruction.
Purification and biotinylation of FLAG-tagged channel proteins
Wild-type and mutant channels with a FLAG tag at the distal C terminus were prepared in the pCDH lentiviral expression plasmid (System Bioscience). To generate cells stably expressing hEAG1 or the mutant channels, ~2.45 μg of the DNA construct was transfected into 5 × 105 HEK293T cells using the calcium phosphate transfection method in a 35 mm diameter tissue-culture dish. The cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) and 5 μg/ml puromycin (Invitrogen). After 2 days of culture, cells were trypsinized and seeded onto 10 cm diameter tissue-culture plates. The cells were passaged every 2–3 days and supplied with fresh puromycin 5 μg/ml contained selection medium for 12–14 days. Puromycin-resistant cells were trypsinized and seeded onto 96-well tissue-culture dishes at a density of ~1 cell per 2 wells. Monoclonal puromycin-resistant cells were cultured and expanded to prepare the seed stocks for future use and concomitantly to electrophysiologically examine the channel expression. To purify hEAG1 wild-type and the mutant channel proteins, cells stably expressing the channel of interest were harvested from DMEM containing 2 μg/ml puromycin, and were solubilized in a lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM NaCl, 1% NP-40, pH 7.0) for 30 min followed by sedimentation by centrifugation (15,000 × g, 30 min). The supernatant was collected and mixed with anti-FLAG beads (Sigma) overnight. The beads-captured protein was collected based on the manufacturer’s instruction. All steps were performed at 4 °C or on ice and all buffers contained Complete Protease Inhibitor (Roche Diagnostics). The purified proteins were quantified using BCA kit (Biyuntian, China) and labeled with biotin (Pierce). The biotinylated membrane proteins were stored in the buffer (PBS with 0.02% Tween-20 and 0.1% BSA, pH 7.4) for the bio-layer interferometry (BLI) assay.
Western blot
The purified hEAG1 protein samples (2 μg) were separated by 8% SDS-polyacrylamide gel and then transferred to nitrocellulose membranes. After blocking with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at room temperature, the transferred membranes were incubated overnight at 4 °C with M2 monoclonal anti-FLAG antibody (1: 1000; Sigma), followed by a goat anti-mouse HRP-conjugated secondary antibody (1: 5000; Santa Cruz Biotechnology). The ECL chemiluminescence was visualized and captured by GE Amersham Imager 600 Imaging System.
Bio-layer interferometry (BLI)
Binding of phospholipids to the channel protein was measured and analyzed on an Octet Red96 instrument (FortéBio) at room temperature. The buffer-equilibrated streptavidin biosensors were loaded with 100 μg/ml protein. A duplicate set of sensors was incubated in the buffer without protein for a background binding control. The assay was performed in black 96-well plates (Thermo Fisher Scientific) with the total working volume of 0.21 ml per well. The signal was analyzed using a double reference subtraction protocol to subtract the non-specific binding, background, and signal drift caused by sensor variability. The binding event between the protein and the lipids was quantified by the shift of interference pattern of the light58.
Electrophysiology and analysis
Macroscopic currents were recorded in the inside-out patch-clamp configuration from CHO cells transiently expressing hEAG1 channels by an EPC-10 amplifier (HEKA Electronics) controlled by PatchMaster software (HEKA Electronics). Patch pipettes pulled from borosilicate glass (Warner) had a typical initial resistance of 1 to 3 MΩ when filled with the pipette solution described below and ~60% of the initial input resistance was electronically compensated in the macroscopic current measurements. Leak and capacitative currents were subtracted with a P/6 protocol. The external solution contained (in mM): 140 KCl, 2 MgCl2, 10 HEPES, 15 glucose, pH 7.2 with N-methyl-D-glucamine (NMG). The internal solution without Ca2+ contained (in mM): 140 KCl, 10 EGTA, 10 NaCl, 1 MgCl2 and 10 HEPES, pH 7.2 with NMG. Internal solutions with higher concentrations of free Ca2+ were prepared using various amounts of EGTA as described previously59. The current was elicited to different voltages from the holding potential of −80 mV every 2 s. The lipids were applied to the intracellular side when the current reached a stable level after patch excision. Half-activation potential (V0.5) was obtained by fitting the maximal peak tail current versus membrane voltage with a Boltzmann function,
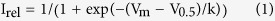 |
where Vm is the membrane potential, and k is the slope factor.
Whole-cell currents were recorded from HEK293T cells stably expressing wild-type or mutant hEAG1 channels using an EPC10 amplifier and in some experiments using an automated planar patch-clamp instrument (Nanion) as described previously60. The external solution contained (in mM): 134 NaCl, 6 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, pH 7.4 (with NMG). The intracellular solution contained (in mM): 110 K aspartate, 30 KCl, 10 NaCl, 2 MgCl2, 10 HEPES, pH 7.2 (with NMG).
Automated whole-cell electrophysiological measurements were conducted according to Nanion’s standard procedure with 8-channel Patchliner (Nanion) equipped with an EPC-10 quadro patch-clamp amplifier (HEKA Electronics). Single-use borosilicate glass chips with medium resistance (1.8 to 3 MΩ, NPC-16, Nanion) were used for all recordings. The PatchControlHT (Nanion) and PatchMaster (HEKA Electronics) were used for cell capture, seal formation, whole-cell access, and subsequent recording in the voltage-clamp configuration. The internal solution contained (in mM): 50 KCl, 60 KF, 10 NaCl, 20 EGTA, and 10 HEPES, pH 7.2 (KOH). The external solution contained (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 5 glucose, and 10 HEPES, pH 7.4 (NaOH). Currents were elicited by voltage steps from the holding potential of −80 mV to 40 mV every 2 s. All electrophysiological experiments were performed at room temperature. Data analysis and curve fitting were done using FitMaster (HEKA Electroncis) and figures were prepared with Origin 8.5 (OriginLab).
Confocal imaging
HEK293T cells were cultured on glass coverslips and transfected with the various constructs, PH(PLCδ1)-GFP, 5-phosphatase-FKBP-mRFP and PM-FRB-CFP55 (generous gifts from T. Balla) using X-tremeGENE 9 (Roche Diagnostics) and cultured for 24 h. Live-cell dual-color measurements were performed on a spinning-disc confocal microscope (Nikon). Excitation lasers were processed with appropriate filter sets for GFP and RFP to capture cellular fluorescence images. Images were captured at room temperature and processed by the NIS elements imaging software (Nikon).
Reagents
Brain-derived PIP2 was from Sigma. All other phospholipids were obtained from Avanti. Serotonin, rapamycin, and calmodulin were purchased from Sigma. Stock solutions of phospholipids (1 mM) were prepared in deionized H2O and stored in glass vials at −20 °C and diluted to the final concentrations immediately before experiments by vigorous vortexing as described previously38. Unless otherwise noted, brain-derived PIP2 was used in the study.
Statistical analysis
Data are expressed as mean ± SEM. Statistical significance was evaluated using either an unpaired t-test or one-way ANOVA followed by Student-Newman-Keuls’s multiple comparisons test as appropriate. Statistical significance of P < 0.05 and P < 0.01 is indicated by single and double asterisks or daggers, respectively.
Additional Information
How to cite this article: Han, B. et al. Human EAG channels are directly modulated by PIP2 as revealed by electrophysiological and optical interference investigations. Sci. Rep. 6, 23417; doi: 10.1038/srep23417 (2016).
Supplementary Material
Acknowledgments
We thank Dr. Tamas Balla (National Institutes of Health, Bethesda, USA) for kindly providing the 5-phosphatase-FKBP-mRFP and PM-FRB-CFP constructs and Drs. Ling Bai and Xiaowei Li (Shanghai Jiao Tong University) for their technical support in protein purification and confocal imaging. This work was supported by National Natural Science Foundation of China (31271217), National Basic Research Program of China (2014CB910304), Shanghai Science and Technology Commission (11JC1406400, 12PJ1404200), and State Key Laboratory of Oncogenes and Related Genes Grant (90-12-03), the National Institutes of Health (T.H., R01GM057654), and the German Research Foundation (S.H.H., T.H., DFG FOR 1738 and S.H.H. RTG 1715). S.H. is a Pujiang Investigator.
Footnotes
Author Contributions B.H. and S.H. designed research; B.H., L.Y., K.H., C.C. and Y.T. performed research; B.H. and S.H. analyzed data; and B.H., S.H.H., T.H. and S.H. wrote the paper.
References
- Occhiodoro T. et al. Cloning of a human ether-a-go-go potassium channel expressed in myoblasts at the onset of fusion. FEBS Lett 434, 177–182 (1998). [DOI] [PubMed] [Google Scholar]
- Downie B. R. et al. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J Biol Chem 283, 36234–36240 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo L. A. & Stuhmer W. Eag1: an emerging oncological target. Cancer Res 68, 1611–1613 (2008). [DOI] [PubMed] [Google Scholar]
- Hemmerlein B. et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol Cancer 5, 41 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo L. A. et al. Oncogenic potential of EAG K+ channels. EMBO J 18, 5540–5547 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen L. S. et al. KV 10.1 opposes activity-dependent increase in Ca2+ influx into the presynaptic terminal of the parallel fibre-Purkinje cell synapse. J Physiol 593, 181–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortum F. et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet 47, 661–667 (2015). [DOI] [PubMed] [Google Scholar]
- Simons C. et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet 47, 73–77 (2015). [DOI] [PubMed] [Google Scholar]
- Pardo L. A. & Stuhmer W. The roles of K+ channels in cancer. Nat Rev Cancer 14, 39–48 (2014). [DOI] [PubMed] [Google Scholar]
- Craven K. B. & Zagotta W. N. CNG and HCN channels: two peas, one pod. Annu Rev Physiol 68, 375–401 (2006). [DOI] [PubMed] [Google Scholar]
- Brelidze T. I., Carlson A. E., Sankaran B. & Zagotta W. N. Structure of the carboxy-terminal region of a KCNH channel. Nature 481, 530–533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson A. E., Brelidze T. I. & Zagotta W. N. Flavonoid regulation of EAG1 channels. J Gen Physiol 141, 347–358 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haitin Y., Carlson A. E. & Zagotta W. N. The structural mechanism of KCNH-channel regulation by the eag domain. Nature 501, 444–448 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilova-Ruch O., Schonherr R. & Heinemann S. H. Activation of hEAG1 potassium channels by arachidonic acid. Pflug Arch Eur J Phy 453, 891–903 (2007). [DOI] [PubMed] [Google Scholar]
- Goncalves J. T. & Stuhmer W. Calmodulin interaction with hEAG1 visualized by FRET microscopy. PloS One 5, e10873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonherr R., Lober K. & Heinemann S. H. Inhibition of human ether à go-go potassium channels by Ca2+/calmodulin. EMBO J 19, 3263–3271 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziechner U. et al. Inhibition of human ether à go-go potassium channels by Ca2+/calmodulin binding to the cytosolic N- and C-termini. FEBS J 273, 1074–1086 (2006). [DOI] [PubMed] [Google Scholar]
- Halstead J. R., Jalink K. & Divecha N. An emerging role for PtdIns(4,5)P2-mediated signalling in human disease. Trends Pharmacol Sci 26, 654–660 (2005). [DOI] [PubMed] [Google Scholar]
- Logothetis D. E. et al. Phosphoinositide control of membrane protein function: a frontier led by studies on ion channels. Annu Rev Physiol 77, 81–104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N. & Shapiro M. S. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci 8, 921–934 (2007). [DOI] [PubMed] [Google Scholar]
- Gericke A., Leslie N. R., Losche M. & Ross A. H. PtdIns(4,5)P2-mediated cell signaling: emerging principles and PTEN as a paradigm for regulatory mechanism. Adv Exp Med Biol 991, 85–104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo L., Wright B. D. & Zylka M. J. Lipid kinases as therapeutic targets for chronic pain. Pain 156 Suppl 1, S2–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K. E., Browne L., Selwood D. L., McAlpine D. & Jagger D. J. Phosphoinositide Modulation of Heteromeric Kv1 Channels Adjusts Output of Spiral Ganglion Neurons from Hearing Mice. J Neurosci 35, 11221–11232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright B. D. et al. The lipid kinase PIP5K1C regulates pain signaling and sensitization. Neuron 82, 836–847 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Zhang C. & Qin F. Functional recovery from desensitization of vanilloid receptor TRPV1 requires resynthesis of phosphatidylinositol 4,5-bisphosphate. J Neurosci 25, 4835–4843 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa N. A., Street S. E., Vihko P. & Zylka M. J. Prostatic acid phosphatase reduces thermal sensitivity and chronic pain sensitization by depleting phosphatidylinositol 4,5-bisphosphate. J Neurosci 30, 10282–10293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B., Dickson E. J., Kruse M., Vivas O. & Suh B. C. Phosphoinositides regulate ion channels. Biochim Biophys Acta 1851, 844–856 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N., Reznikov V., Yamada Y., Yang J. & Shapiro M. S. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci 24, 10980–10992 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S. B., Tao X. & MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A. Y. et al. Pirt, a phosphoinositide-binding protein, functions as a regulatory subunit of TRPV1. Cell 133, 475–485 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93, 1019–1137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decher N. et al. Structural determinants of Kvβ1.3-induced channel inactivation: a hairpin modulated by PIP2. EMBO J 27, 3164–3174 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S. & Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611 (2005). [DOI] [PubMed] [Google Scholar]
- Czech M. P. PIP2 and PIP3: complex roles at the cell surface. Cell 100, 603–606 (2000). [DOI] [PubMed] [Google Scholar]
- Dart C. Lipid microdomains and the regulation of ion channel function. J Physiol 588, 3169–3178 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinova R., Hobart E. A., Koeppe R. E. 2nd & Andersen O. S. Phosphoinositides alter lipid bilayer properties. J Gen Physiol 141, 673–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs T., Chen J., Prestwich G. D. & Logothetis D. E. Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J Biol Chem 274, 36065–36072 (1999). [DOI] [PubMed] [Google Scholar]
- Rohacs T. et al. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci USA 100, 745–750 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B. C., Inoue T., Meyer T. & Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314, 1454–1457 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P., Thyagarajan B., Rohacs T. & Balla T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J Cell Biol 175, 377–382 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garro M. A. et al. Regulation of phospholipase Cβ activity by muscarinic acetylcholine and 5-HT2 receptors in crude and synaptosomal membranes from human cerebral cortex. Neuropharmacology 40, 686–695 (2001). [DOI] [PubMed] [Google Scholar]
- Siddiqui E. J., Thompson C. S., Mikhailidis D. P. & Mumtaz F. H. The role of serotonin in tumour growth (review). Oncol Rep 14, 1593–1597 (2005). [PubMed] [Google Scholar]
- Saltzman A. G. et al. Cloning of the human serotonin 5-HT2 and 5-HT1C receptor subtypes. Biochem Biophys Res Commun 181, 1469–1478 (1991). [DOI] [PubMed] [Google Scholar]
- Suh B.-C. & Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 37, 175 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. O. et al. Mechanism of voltage gating in potassium channels. Science 336, 229–233 (2012). [DOI] [PubMed] [Google Scholar]
- Tao X., Lee A., Limapichat W., Dougherty D. A. & MacKinnon R. A gating charge transfer center in voltage sensors. Science 328, 67–73 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. et al. Dynamic PIP2 interactions with voltage sensor elements contribute to KCNQ2 channel gating. Proc Natl Acad Sci USA 110, 20093–20098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. Bimodal regulation of an Elk subfamily K+ channel by phosphatidylinositol 4,5-bisphosphate. J Gen Physiol 146, 357–374 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian J. S., Kagan A. & McDonald T. V. Molecular analysis of PIP2 regulation of HERG and IKr. Am J Physiol Heart Circ Physiol 287, H2154–2163 (2004). [DOI] [PubMed] [Google Scholar]
- Delmas P. & Brown D. A. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6, 850–862 (2005). [DOI] [PubMed] [Google Scholar]
- Kwon Y., Hofmann T. & Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Molecular Cell 25, 491–503 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M. et al. Selective phosphorylation modulates the PIP2 sensitivity of the CaM-SK channel complex. Nat Chem Biol 10, 753–759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady J. D. et al. Interplay between PIP3 and calmodulin regulation of olfactory cyclic nucleotide-gated channels. Proc Natl Acad Sci USA 103, 15635–15640 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Garduno A. M. et al. KV10.1 K+-channel plasma membrane discrete domain partitioning and its functional correlation in neurons. Biochimica Et Biophysica Acta-Biomembranes 1838, 921–931 (2014). [DOI] [PubMed] [Google Scholar]
- Hammond G. R. et al. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337, 727–730 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T. et al. A rapid method for determining dynamic binding capacity of resins for the purification of proteins. Protein Expr Purif 60, 147–150 (2008). [DOI] [PubMed] [Google Scholar]
- Gustina A. S. & Trudeau M. C. HERG potassium channel regulation by the N-terminal eag domain. Cell Signal 24, 1592–1598 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartchow C. A. et al. Biosensor-based small molecule fragment screening with biolayer interferometry. J Comput Aided Mol Des 25, 669–676 (2011). [DOI] [PubMed] [Google Scholar]
- Hou S. W., Xu R., Heinemann S. H. & Hoshi T. Reciprocal regulation of the Ca2+ and H+ sensitivity in the SLO1 BK channel conferred by the RCK1 domain. Nat Struct Mol Biol 15, 403–410 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre C. et al. Port-a-Patch and Patchliner: High Fidelity Electrophysiology for Secondary Screening and Safety Pharmacology. Comb Chem High T Scr 12, 24–37 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.