

Abstract
Pain is a complex disorder with neurochemical and psychological components contributing to the severity, the persistence, and the difficulty in adequately treating the condition. Opioid and cannabinoids are two classes of analgesics that have been used to treat pain for centuries and are arguably the oldest of “pharmacological” interventions used by man. Unfortunately, they also produce several adverse side effects that can complicate pain management. Opioids and cannabinoids act at G protein-coupled receptors (GPCRs), and much of their effects are mediated by the mu-opioid receptor (MOR) and cannabinoid CB1 receptor (CB1R), respectively. These receptors couple to intracellular second messengers and regulatory proteins to impart their biological effects. In this chapter, we review the role of the intracellular regulatory proteins, β-arrestins, in modulating MOR and CB1R and how they influence the analgesic and side-effect profiles of opioid and cannabinoid drugs in vivo. This review of the literature suggests that the development of opioid and cannabinoid agonists that bias MOR and CB1R toward G protein signaling cascades and away from β-arrestin interactions may provide a novel mechanism by which to produce analgesia with less severe adverse effects.
Keywords: Opioid, Cannabinoid, Analgesia, Pain, Mu-opioid receptor, Cannabinoid CB1 receptor, Arrestin, Tolerance, Antinociception
1 Introduction
Pain is a complex disorder with neurochemical and psychological components contributing to the severity, the persistence, and the difficulty in adequately treating the condition. Although there are several different types of pharmaceutical drugs approved for the treatment of moderate to severe pain, it has been well documented that patients suffering from protracted persistent pain, especially those with cancer or neuropathic pain, often do not receive adequate relief from currently available analgesics (Brennan et al. 2007). Opioid and cannabinoids are two classes of analgesics that have been used to treat pain for centuries and are arguably the oldest of “pharmacological” interventions used by man. Unfortunately, they also produce several adverse side effects that can complicate pain management. Therefore, there remains a significant need to develop therapeutics with improved analgesic efficacy and reduced adverse effects. Since their discovery in the early 1990s, β-arrestins have proven to be important regulators of G protein-coupled receptors (GPCRs). Opioid and cannabinoids act at G protein-coupled receptors (GPCRs), and much of their effects are mediated by the mu-opioid receptor (MOR) and cannabinoid CB1 receptor (CB1R), respectively. These receptors couple to intracellular second messengers and regulatory proteins to impart their biological effects and β-arrestins may represent a means to fine-tune analgesic responses mediated by these receptors. In this chapter, we review studies that explore how β-arrestins impact opioid and cannabinoid drug responsiveness at the mu-opioid receptor (MOR) and cannabinoid CB1 receptor (CB1R) in vitro and in vivo with regard to how they influence the degree of analgesia and the side-effect profile of analgesic drugs.
2 Opioid and Cannabinoid Receptor Pharmacology
Opioids and cannabinoids produce their pharmacological effects through activation of GPCRs. There are four distinct genes coding for opioid receptors: the mu-, kappa-, and delta-opioid receptors (MOR, KOR, and DOR, respectively) and the opioid-like receptor1 [ORL-1 or the nociceptin receptor (NOP)] (Cox 2012; Pasternak 2013). The generation of genetic knockout mice has demonstrated that the majority of clinically used opioids including morphine produce their pharmacological effects primarily by activating the MOR (Matthes et al. 1996; Sora et al. 1997; Roy et al. 1998; Kieffer 1999; Kieffer and Gaveriaux-Ruff 2002). The MOR is widely distributed and expressed in neurons in the brain, spinal cord, and the periphery (Gutstein and Akil 2001). Two major types of cannabinoid receptors have been identified: cannabinoid subtype 1 (CB1R) and cannabinoid subtype 2 (CB2R). While there is evidence demonstrating a modulation of pain responses by actions at CB2 receptors (Jaggar et al. 1998; Malan et al. 2001; Sokal et al. 2003; Elmes et al. 2004; Hohmann et al. 2004; Ibrahim et al. 2006; LaBuda et al. 2005), CB1 receptors (CB1R) in the central nervous system play the most pronounced role in mediating the analgesic, motor, and psychoactive effects of cannabinoids (Zimmer et al. 1999; Kelly and Chapman 2001; Hohmann et al. 2005; Pertwee 2005; Suplita et al. 2006; Dziaduleqicz et al. 2007). CB1R are widely expressed in the central and peripheral nervous systems (SvÍzenská et al. 2008).
The clinically observed effects produced by opioid and cannabinoid analgesics can be determined by how effectively the MOR and CB1R signal at the cellular level. There is a rich literature describing MOR and CB1R signaling pathways that lead to antinociceptive responses (for reviews, see Williams et al. 2013; Raehal et al. 2011; Smith et al. 2010) (Fig. 1). Upon activation, both receptors couple predominantly to Gαi/o proteins (Law et al. 2000; Howlett et al. 2002). In descending pain processing pathways, MOR and CB1R coupling to inhibitory heterotrimeric G proteins leads to a decrease in calcium influx resulting in decreased GABA transmission. The decrease in GABA release promotes disinhibition of the OFF nociceptive neurons and direct inhibition of ON cells, resulting in antinociception (for reviews, see Fields 2004; Rea et al. 2007; Palazzo et al. 2010; Fig. 1). In addition to inhibiting calcium flux, G proteins can modulate the activity of several different second messengers and cellular effectors, which may generate both short-term and long-term changes at the molecular and cellular levels resulting in diverse biological effects, including alternate paths to antinociception as well as neuroadaptations such as physical dependence.
Fig. 1.
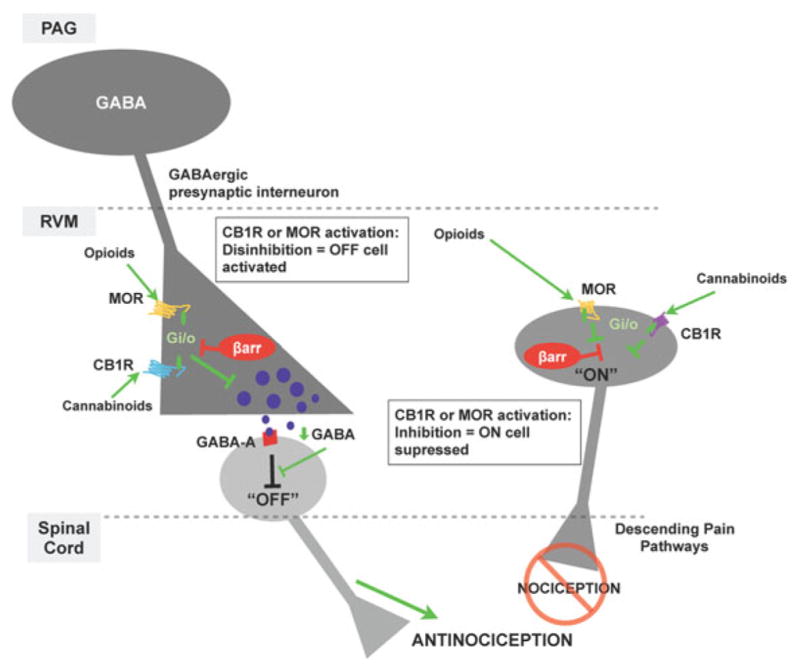
β-arrestin modulation of antinociceptive responses mediated by MOR or CB1R expressed in descending nociceptive processing pathways. Activation of cannabinoid 1 receptors (CB1) or mu-opioid receptors (MOR) on GABAergic interneurons in the rostroventral medulla (RVM) decreases GABA release via mechanisms downstream of coupling to inhibitory Gαi/o proteins (activation of GIRKs, inhibition of voltage-dependent calcium channels, and inhibition of adenylyl cyclase pathways, among others). Normal GABA tone suppress “OFF” cells in the RVM by acting at GABA-A receptors, which raise action potentials. When GABA levels decrease, the tonic inhibition of “OFF” cells is relieved (i.e., disinhibition) and “OFF” cells signal to suppress pain perception in the spinal cord (descending pain perception regulation). In addition, activation of MOR or CB1Rs expressed on GABAergic “ON” cells in the rostral ventromedial medulla inhibits firing of these cells. Collectively, disinhibition of “OFF” cells and direct inhibition of “ON” cells produce analgesia; an effect can be measured using a thermal nociception tests. Since β-arrestins can inhibit the G protein signaling mechanisms utilized by CB1R and MOR, they can ultimately decrease antinociception as demonstrated in this simplified model of antinociception regulation (Fields 2004; Rea et al. 2007; Palazzo et al. 2010)
Receptor signaling is determined not only by the activation of G protein-mediated signaling cascades, but also by several regulatory mechanisms including receptor desensitization, internalization, resensitization, and downregulation. Further, the signaling pathways and regulatory events can differ at a given receptor dependent on its cellular context. In other words, a receptor may signal via different G proteins when expressed in different neurons or may signal independently of G proteins altogether. Receptor regulation is essential as it aids in controlling the extent and duration of receptor signaling by preventing receptor overstimulation, promoting signal termination, and regulating cell surface expression of receptors. Although the signaling activity of MOR and CB1R can be regulated by several means, in vitro and in vivo studies have collectively shown that β-arrestins can substantially influence how the MOR and CB1 receptors respond to agonists.
3 β-Arrestin-Mediated MOR and CB1R Regulatory Mechanisms
β-Arrestins belong to a family of four arrestin proteins (Gurevich and Gurevich 2003). Arrestins 1 and 4 are almost exclusively expressed in rod and cone cells in the visual system (Shinohara et al. 1987; Yamaki et al. 1987; Murakami et al. 1993; Craft et al. 1994) and are therefore referred to as “visual” arrestins. Arrestin 2 and arrestin 3 were first discovered for their ability to regulate the β2-adrenergic receptor (Lohse et al. 1990) and, therefore, are also referred to as β-arrestin1 and β-arrestin2, respectively. Both β-arrestins are highly expressed in tissues throughout the central nervous system and periphery (Lohse et al. 1990; Attramadal et al. 1992; Gurevich and Benovic 2000; Gurevich et al. 2002; Gainetdinov et al. 2004; Bychkov et al. 2012) and have been shown to regulate the activity of MOR and CB1 receptors.
β-Arrestins play a multifaceted role in regulating how GPCRs respond to agonist stimulation. One of the many ways in which β-arrestins regulate MOR and CB1R signaling is by promoting receptor desensitization. Following GPCR phosphorylation by GPCR kinases, β-arrestins bind to the phosphorylated MOR, which prevents further interactions between the receptor and G proteins even in the continued presence of agonist resulting in diminished G protein-mediated signaling (Zhang et al. 1998; Whistler and von Zastrow 1998; Kovoor et al. 1997, 1998; Bohn et al. 2000; Lowe et al. 2002; Eisinger et al. 2002; Celver et al. 2001, 2004; Qiu et al. 2003; Bailey et al. 2004, 2009; Koch et al. 2004; Dang et al. 2009, 2011). Studies of the CB1R also reveal an important role for β-arrestins in desensitizing the agonist-stimulated, phosphorylated CB1R (Sim et al. 1996; Jin et al. 1999; Daigle et al 2008; Nguyen et al. 2012).
In addition to disrupting G protein signaling cascades, β-arrestins can play a role in determining the fate of MOR and CB1 receptors, from the initiation of clathrin-dependent endocytosis (Jin et al. 1999; Whistler et al. 1999; Celver et al. 2004; Koch et al. 2004; Haberstock-Debic et al. 2005; Walwyn et al. 2006, 2007; Daigle et al. 2008; Groer et al. 2011; Patierno et al. 2011; Ahn et al. 2013) (see Chap. 9) to the recruitment of ubiquitin (E3) ligases involved in lysosomal-mediated receptor degradation (Groer et al. 2011; Henry et al. 2012; Malik et al. 2012) (see Chap. 10). The temporal and spatial scaffolding that β-arrestins impart in determining receptor interactions with signaling and regulatory elements can drive specific receptor signaling pathways as well as determine whether the receptor is resensitized or degraded. Studies of the MOR suggest that β-arrestin1 may play a role in ubiquitinating MOR and facilitating its dephosphorylation and potentially resensitization, while β-arrestin2 is prominently involved in desensitizing the receptor (Kovoor et al. 1997, 1998; Celver et al. 2001; Bohn et al. 2000; Dang et al. 2011; Groer et al. 2011). CB1 receptors have been shown to require β-arrestin2 for CP55940-induced internalization in HEK 293 cells, and the allosteric modulator, ORG27569, was shown to direct CB1 receptors to activating ERK cascades in a β-arrestin1-dependent manner (Ahn et al. 2013). It is becoming increasingly evident that there are several means by which β-arrestins can impact how the MOR and CB1R respond to agonists, which may have great bearing on how β-arrestins can mediate overall responsiveness to analgesic drugs.
4 β-Arrestin Regulation of MOR- and CB1R-Mediated Antinociception In Vivo
Due to the lack of selective inhibitors of arrestins at the turn of the century, the role of β-arrestins in opioid-induced analgesia was initially determined by assessing pain responses following morphine treatment of genetically modified mice lacking either β-arrestin1 or β-arrestin2 (Bohn et al. 1999, 2000, 2002). When administered a single dose of morphine, β-arrestin1-KO mice respond normally compared to wild-type (WT) mice (Bohn et al. 2004); however, differences become readily apparent when β-arrestin2-KO mice are treated with morphine. β-Arrestin2-KO mice display enhanced and prolonged morphine-induced antinociception in paradigms that evaluate supra-spinal (hot plate) and spinal (tail flick) antinociceptive responses to a noxious thermal stimulus (Bohn et al. 1999, 2002, 2004; Raehal and Bohn 2011), suggesting that in the absence of the desensitizing effect of the β-arrestin2, MOR responsiveness is enhanced.
Similar responses to morphine observed in β-arrestin2-KO mice have been reported in normal mice and rats in which small interfering RNA (siRNA) or antisense oligonucleotides are used to knockdown β-arrestin2 in pain processing regions. siRNA inhibition of β-arrestin2 in the periaqueductal gray of mice enhances acute morphine-induced antinociception and delays the development of antinociceptive tolerance in the hot plate test (Li et al 2009). Morphine-induced antinociceptive tolerance in the tail flick test has also been shown to be significantly reduced in rats in which β-arrestin2 expression is knocked down in the spinal cord (Przewlocka et al. 2002; Yang et al. 2011). Moreover, the antinociceptive effects of morphine in the hot plate test are absent in rats overexpressing β-arrestin2 in the periaqueductal gray (Jiang et al. 2006). In contrast, siRNA knockdown of β-arrestin1 in rat periaqueductal gray had no effect on morphine-induced antinociception or the development of antinociceptive tolerance in the hot plate test (Li et al. 2009). This finding is consistent with cell culture studies wherein the morphine-bound MOR preferentially interacts with β-arrestin2 (Bohn et al. 2004; Groer et al. 2011).
Interestingly, not all MOR agonists produce enhanced antinociception in β-arrestin2-KO mice. Methadone, fentanyl, and etorphine produce a similar degree of antinociception as in their WT littermates in the hot plate test (Bohn et al. 2004; Raehal et al. 2011). In cell culture studies, these agonists promote both β-arrestin1 and β-arrestin2 interactions with the MOR, whereas morphine weakly and selectively recruits β-arrestin2 (Zhang et al. 2008; Whistler et al 1999; Bohn et al. 2004; Groer et al. 2007; McPherson et al 2010; Molinari et al. 2010). Therefore, morphine is a unique MOR agonist that selectively elicits β-arrestin2 regulation of the MOR; in its absence, β-arrestin1 may functionally substitute for the loss of β-arrestin2 in response to other MOR agonists.
Studies investigating the role of β-arrestins in CB1R-mediated analgesia and side effects have primarily been performed using β-arrestin2-KO mice. Upon acute treatment with the cannabinoid receptor agonists Δ9-tetrahydrocannabinoid (THC) or CP55940, only THC produces an enhanced and prolonged response in the β-arrestin2-KO mice compared to vehicle controls as assessed by the hot plate (Breivogel et al. 2008) and tail flick (Breivogel et al. 2008; Nguyen et al 2012) tests. Similar to CP55940, other cannabinoid agonists including methanandamide and JWH-015 produce the same degree of antinociception in both WT and KO mice in the tail flick test (Breivogel et al. 2008). While it appears that the degree of MOR phosphorylation may be related to the propensity of an agonist to substitute β-arrestin1 for β-arrestin2 in desensitizing the MOR, the same correlations have yet to be made for the CBR1. However, a recent report demonstrates that the two β-arrestins may have distinct roles in regulating the CB1R in an agonist-dependent manner, with β-arrestin1 mediating signaling events and β-arrestin2 determining internalization profiles in cell lines (Ahn et al. 2013).
5 β-Arrestin Regulation of Basal Nociceptive Resposes
An investigation of nociceptive behaviors in drug-naïve animals revealed that β-arrestin2-KO mice display longer basal warm water tail flick response latencies (Bohn et al. 2002; Breivogel et al. 2008; Lam et al. 2011; Nguyen et al. 2012), which can be blocked by the opioid antagonists naltrexone or naloxone (Bohn et al. 2002; Lam et al. 2011), but not the kappa- and delta-selective antagonists, norbinaltorphamine and naltrindole, respectively. This suggests that the prolonged response latencies are due to increased basal MOR activity or that the MOR is more sensitive to the presence of endogenous opioid peptides such as enkephalins and endomorphins. Alternatively, the deletion of β-arrestin2 may enhance the sensitivity of pro-nociceptive GPCRs. There is some evidence to support this idea as the transient receptor potential vanilloid 1 (TRPV1) channel, the transducer of thermal and chemical pain transmission, was found to be desensitized by β-arrestin2 (Por et al. 2012, 2013). Neurokinin receptors can also be regulated by β-arrestins (Schmidlin et al. 2003); however, no neurokinin receptor-mediated phenotypes have been reported in the β-arrestin2-KO mice. Further, while β-arrestin2-KO mice display prolonged basal antinociceptive response in assays with thermal endpoints such as the tail flick and Hargreaves tests (Bohn et al. 2002; Lam et al 2011), their basal responses in tests of mechanical stimulation using von Frey filaments are similar to their WT littermates (Lam et al. 2011), suggesting that β-arrestin2 may contribute to basal nociception thresholds in only certain types of pain pathways.
6 β-Arrestin Regulation of MOR and CB1R in Neurons
The manner in which β-arrestin2 regulates the MOR in distinct neuronal populations found in brain regions involved in modulating pain have been evaluated using β-arrestin2-KO mice. In periaqueductal gray and brain stem from KO mice, the MOR-selective agonist, DAMGO (D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin), produces a greater degree of receptor coupling to G protein compared to WT controls, suggesting that in the absence of the desensitizing β-arrestin2, the capacity for MOR to couple to G protein signaling cascades is enhanced (Bohn et al. 1999, 2000). Likewise, in the absence of β-arrestin2, DAMGO and morphine produce less inhibition of voltage-gated calcium channels in dorsal root ganglion neurons, which are involved in transmitting nociceptive information (Walwyn et al. 2007). Further, under basal conditions, constitutive coupling to these channels is also enhanced (Walwyn et al. 2007).
The locus coeruleus (LC) is another brain region that contributes to the descending pain suppression pathway. In these neurons, acute enkephalin treatment produces the same degree of MOR-mediated desensitization of inward rectifying potassium channels in β-arrestin2-KO as WT controls (Dang et al. 2009). However, inhibition of ERK1/2 in β-arrestin2-KO mice alleviates desensitization, indicating that MOR desensitization in LC neurons is not solely dependent upon β-arrestin2 (Dang et al. 2009). Investigations of MOR resensitization profiles in LC neurons using the same system revealed that disruption of β-arrestin2-mediated receptor endocytosis enhances MOR resensitization rates (Dang et al. 2011). Further, WT but not βarrestin2-KO neurons treated chronically with morphine showed reduced rates of MOR resensitization, suggesting that β-arrestin2 determines MOR recycling profiles when morphine is used (Dang et al. 2011). Similarly, chronic morphine also impairs MOR recycling rates in WT but not KO. LC neurons isolated WT and β-arrestin2-KO mice (Quillinan et al 2011). However, this effect appears to be morphine dependent, as chronic methadone impairs MOR resensitization to the same extent in both genotypes (Quillinan et al. 2011).
Although the loss of β-arrestin2 has an overall effect of enhancing the antinociceptive efficacy of morphine, the manner by which β-arrestin2 regulates MOR to produce this response can vary depending on neuronal type as well as the agonist used. In the intact animal, it may also depend upon the pain state induced. While β-arrestin2 may play a substantial role in regulating MOR in vivo, it is not an exclusive modulator and likely part of a chain of events that ultimately determine receptor responsiveness. For example, treatment with the JNK inhibitors SP6 or BI78D3 or the PKC inhibitor bisindolylmaleimide VIII prior to morphine reverses the enhanced antinociceptive effect observed in β-arrestin2-KO mice to WT control levels in the tail flick test (Mittal et al. 2012). These studies suggest that activated JNK and/or PKC contribute to morphine-induced spinal antinociception. However, inhibition of JNK has no effect on fentanyl-induced antinociception in either genotype, indicating that the effect is ligand dependent (Mittal et al. 2012). In addition, elevated basal antinociceptive responses observed in β-arrestin2-KO mice are unaffected by JNK inhibition (Mittal et al. 2012). The lack of effect observed in the absence of drug with fentanyl may be explained by β-arrestin1 compensation in the β-arrestin2-KO mice or could depend on other regulatory proteins.
When examined for CB1R responsiveness, WT and β-arrestin2-KO mice were found to have similar levels of CB1R expression in the brain, although β-arrestin2-KO mice displayed greater antinociceptive responses to Δ9-THC (Breivogel et al. 2008; Nguyen et al. 2012). When examined in spinal cord and across brain regions, the deletion of β-arrestin2 did not significantly impact upon CP55940-stimulated G protein coupling. However, after chronic treatment with Δ9-THC, spinal cords from WT mice display significant decreases in CP55940-stimulated coupling to G protein and a downregulation of CB1R binding while these adaptations are not observed in β-arrestin2-KO mice (Nguyen et al. 2012).
7 β-Arrestin Mediation of Adverse Side Effects
While cannabinoids and opioids are effective pain relievers, they also produce a number of adverse side effects. Therefore, in addition to understanding how MOR and CB1R promote analgesia, there has also been significant interest in understanding the mechanisms that contribute to the side effects resulting from their activation. In the absence of β-arrestin2 morphine produces enhanced and prolonged analgesia, yet significantly less antinociceptive tolerance, physical dependence, constipation, and respiratory suppression (Bohn et al 1999, 2000, 2002; Raehal et al. 2005, 2011). The reduction in antinociceptive tolerance that develops following chronic morphine treatment appears to be due to a loss of β-arrestin2 desensitization of the MOR (Bohn et al. 2000). Following repeated Δ9-THC administration, β-arrestin2-KO mice develop antinociceptive tolerance in the tail flick assay, yet to a lesser extent than that observed in the WT mice (Nguyen et al. 2012). This attenuated tolerance was shown to correlate with decreased CB1R desensitization of G protein coupling in cerebellum, caudal periaqueductal gray, and spinal cord (Nguyen et al. 2012)
In addition to tolerance, morphine produces physical dependence, constipation, and respiratory suppression in mice and in humans. When tested in the β-arrestin2-KO mice, morphine induces less constipation, an effect that appears to be due to altered β-arrestin2 regulation of MOR at the level of the colon (Raehal et al. 2005). Studies of colon preparations derived from the β-arrestin2-KO mice assayed in organ baths to assess morphine’s inhibition of contractility suggest that morphine tolerance develops following repeated morphine treatment in the absence of β-arrestin2, while it does not develop in colon from WT mice (Kang et al. 2012).
At high doses, chronic morphine treatment produces physical dependence in the β-arrestin2-KO mice that is indistinguishable from the effects in the WT mice (Bohn et al. 2000; Raehal et al. 2011); however at lower doses of morphine infusion, β-arrestin2-KO mice are protected from the onset of dependence as evidenced by a decrease in the severity of the antagonist-precipitated withdrawal response (Raehal et al. 2011). It would seem that β-arrestin2 might facilitate the neuroadaptations that underlie the development of dependence but that it may not be exclusively required for the pathways to ensue.
Morphine-induced respiratory suppression is also decreased in the β-arrestin2-KO mice; however, like morphine-induced constipation and physical dependence, the mechanisms by which β-arrestin2 mediates these neuroadaptations remain unclear. If β-arrestin2 was primarily acting to desensitize MOR involved in mediating these side effects, one might predict that the severity of these side effects would be enhanced in β-arrestin2-KO mice. However, the side effects are less severe in the KO mice, suggesting that β-arrestin2 may be involved in mediating these responses in vivo. In cell culture studies β-arrestin2 has been shown to act as a scaffolding molecule to promote MOR signaling (Zhang et al. 2008). In neuronal preparations, β-arrestin2-mediated MOR signaling has been observed in dorsal root ganglion neurons from β-arrestin2-KO mice, wherein DAMGO and morphine are less efficacious in inhibiting voltage-gated calcium currents (Walwyn et al. 2007). Fentanyl-induced MOR activation of ERK1/2 has also been shown to utilize β-arrestin2 in primary striatal neurons (Macey et al. 2006). However, β-arrestin2-mediated MOR signaling has not yet clearly been demonstrated in tissues associated with the onset of the side effects.
When treated with methadone or fentanyl, β-arrestin2-KO mice display the same degree of antinociceptive tolerance and physical dependence as WT controls (Raehal et al. 2011). Studies exploring the effects of other opioids on constipation and respiration have not been published, with the exception of loperamide. Loperamide (clinically used as the antidiarrheal Imodium®) is a peripherally restricted MOR agonist and does not delay colon transit times in the β-arrestin2-KO mice (Raehal et al. 2005). The β-arrestin-mediated effects on opioid constipation appear to be restricted to the periphery, as the genotype-dependent differences are not preserved when WT and β-arrestin2-KO are administered morphine by intracerebroventricular route (Bohn and Raehal 2006). Interestingly, loperamide, unlike morphine, leads to robust phosphorylation of the MOR in cell-based assays, which makes it more pharmacologically similar to methadone and fentanyl than morphine (unpublished observations). These findings suggest that the MOR in colon functions to delay transit via a β-arrestin2-dependent mechanism, although more studies must be undertaken to fully elucidate these mechanisms.
Cannabinoids also produce physical dependence as evidenced by signs of withdrawal upon cessation of drug taking, although to date, the role of β-arrestin2 has not been investigated in cannabinoid dependence. Cannabinoids also induce catalepsy and both genotypes display equivalent response profiles upon Δ9-THC in the catalepsy ring test. However, the β-arrestin2-KO mice develop tolerance to a greater extent in this assay following chronic Δ9-THC administration. It is not clear what mechanism underlies this behavioral adaption; however, the degree of CB1R desensitization of G protein coupling was greater in the cortex, globus pallidus, and substantia nigra of β-arrestin2-KO mice compared to the WT mice, suggesting that β-arrestin2-mediated desensitization of CB1R in these regions may play some role in mediating Δ9-THC-induced catalepsy (Nguyen et al. 2012).
In mice, cannabinoids as well as opioids induce hypothermia. Morphine induces a greater drop in body temperature over time in β-arrestin2-KO mice compared to WT mice. β-arrestin2-KO display greater hypothermia in response to Δ9-THC; however, tolerance to hypothermia develops in both genotypes (Breivogel et al. 2008; Nguyen et al. 2012). Interestingly, other agonists, including CP55940, methanandamide, and JWH-073, do not reveal a difference between genotypes in the hypothermia studies (Breivogel et al. 2008).
8 Therapeutic Potential of Biased Agonists in Pain Treatment
Biased agonists selectively engage one GPCR signaling pathway over another, such as coupling to a G protein over recruiting a β-arrestin (see Chap. 3). The pharmacological and genetic studies of rodents to date suggest that developing an opioid that does not recruit β-arrestin may represent a means to enhance antinociceptive efficacy while avoiding certain side effects. Presently, there have been a few reports of “biased” MOR agonists and their effects in vivo. One such compound herkinorin is a selective MOR agonist that does not recruit β-arrestin1 or β-arrestin2 in cell culture assays (Groer et al. 2007). In an inflammatory pain model in rat, herkinorin reduces formalin-induced flinching to the same degree as morphine when administered at the same dose (10 mg/kg, i.pl.), an effect that is reversed by the opioid antagonist naloxone (Lamb et al. 2012). Moreover, antinociceptive tolerance to herkinorin does not develop to repeated treatment over a 5-day period and it produces antinociception in morphine-tolerant rats (Lamb et al. 2012).
Another recently described compound, TRV130, has been reported to be a selective MOR agonist that produces robust G protein coupling but does not recruit β-arrestin2 (DeWire et al. 2013). In mice and rats, TRV130 is approximately five times more potent than morphine in tests (hot plate and tail flick) of thermal nociception. In a rat incisional pain model, TRV130 was as effective as morphine in treating tactile allodynia (DeWire et al. 2013). When given acutely, TRV130 also produces less constipation and respiratory suppression compared to mice treated with equi-efficacious doses of morphine (DeWire et al. 2013). The initial studies with these MOR “biased” agonists lend further support to the idea that developing an MOR agonist that does not engage β-arrestins but fully activates G protein signaling may provide a novel therapeutic avenue to improve pain treatment with opioids.
It also appears that biased agonism may represent a promising path for CB1R therapeutic development. The studies described herein indicate that β-arrestin2 regulates cannabinoid receptor-mediated thermal antinociception in a ligand- and tissue-dependent manner and developing cannabinoid agonists that do not promote receptor interactions with β-arrestins may improve the therapeutic profile of these types of analgesic drugs, while at the same time may promote desensitization for the catalepsy side effect. The continued development of biased agonists at each of these receptors and the study of their effect across species and physiologies will certainly broaden the degree of applicability of this merging concept.
9 Conclusions
It is apparent that β-arrestin-dependent regulation of MOR and CB1 receptors can profoundly impact how these receptors respond to their respective classes of analgesic drugs. Collectively, the studies discussed in this chapter show that β-arrestin regulation of MOR and CB1 receptors is complex, so that the nature of how β-arrestins affect these receptors is influenced by the agonist acting at the receptor and the cellular environment in which the receptor is expressed. Moreover, opioid and CB1 agonists can direct receptors toward interactions with particular β-arrestins, which can affect the overall cellular signaling profile and biological response that is observed. The in vivo studies also suggest that not engaging β-arrestin interactions with the MOR or CB1 receptors can produce analgesia with less severe side effects. While initial studies in which selective and “biased” ligands at the MOR show promise in improving the overall therapeutic profile of opioid analgesics, extensive preclinical and clinical studies will be required to ultimately determine if a “biased” MOR and/or CB1R strategy will lead to the development of an analgesic with a wider safety margin that will improve the clinical treatment of pain.
Contributor Information
Kirsten M. Raehal, Email: kirsten.raehal@pharma.com, The Scripps Research Institute, 130 Scripps Way #2A2, Jupiter, FL 33458, USA, Purdue Pharma, L.P., 6 Cedarbrook Dr, Cranbury Township, NJ 08512, USA
Laura M. Bohn, The Scripps Research Institute, 130 Scripps Way #2A2, Jupiter, FL 33458, USA
References
- Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1) J Biol Chem. 2013;288:9790–9800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, Lefkowitz RJ. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- Bailey CP, Kelly E, Henderson G. Protein kinase C activation enhances morphine-induced rapid desensitization of mu-opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol. 2004;66:1592–1598. doi: 10.1124/mol.104.004747. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Oldfield S, Llorent J, Caunt CH, Teschemacher AG, Roberts L, McArdle CA, Smith FL, Dewy WL, Kelly E, Henderson G. Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br J Pharmacol. 2009;158:157–164. doi: 10.1111/j.1476-5381.2009.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Raehal KM. Opioid receptor signaling: relevance for gastrointestinal therapy. Curr Opin Pharmacol. 2006;6:559–563. doi: 10.1016/j.coph.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Caron MJ. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J Neurosci. 2002;22:10494–10500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Caron MJ. G protein-coupled receptor kinase/beta-arrestin systems and drugs of abuse: psychostimulant and opiate studies in knockout mice. Neuromolecular Med. 2004;5:41–50. doi: 10.1385/NMM:5:1:041. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Lambert JM, Gerfin S, Huffman JW, Razdan RK. Sensitivity to Δ9-tetrahy-drocannabinol is selectively enhanced in beta-arrestin2−/− mice. Behav Pharmacol. 2008;19:298–307. doi: 10.1097/FBP.0b013e328308f1e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan F, Carr DB, Cousins M. Pain management: a fundamental human right. Anesth Analg. 2007;105:205–221. doi: 10.1213/01.ane.0000268145.52345.55. [DOI] [PubMed] [Google Scholar]
- Bychkov E, Zurkovsky L, Garret MB, Ahmed MR, Gurevich EV. Distinct cellular and subcellular distributions of G protein-coupled receptor kinase and arrestin isoforms in the striatum. PLoS One. 2012;7:e48912. doi: 10.1371/journal.pone.0048912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celver JP, Lowe J, Kovoor A, Gurevich VV, Chavkin C. Threonine 180 is required for G-protein-coupled receptor kinase 3- and beta-arrestin 2-mediated desensitization of the mu-opioid receptor in xenopus oocytes. J Biol Chem. 2001;276:4894–4900. doi: 10.1074/jbc.M007437200. [DOI] [PubMed] [Google Scholar]
- Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains of the mu-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- Cox BM. Recent developments in the study of opioid receptors. Mol Pharm. 2012;83:723–728. doi: 10.1124/mol.112.083279. [DOI] [PubMed] [Google Scholar]
- Craft CM, Whitmore DH, Wiechmann AF. Cone arrestin identified by targeting expression of a functional family. J Biol Chem. 1994;269:4613–4619. [PubMed] [Google Scholar]
- Daigle TL, Kearn CS, Mackie K. Rapid CB1 cannabinoid receptor desensitization defined the time course of ERK1/2 MAP kinase signaling. Neuropharmacology. 2008;54:36–44. doi: 10.1016/j.neuropharm.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Napier IA, Christie MJ. Two distinct mechanisms mediate acute μ-opioid receptor desensitization in native neurons. J Neurosci. 2009;29:3322–3327. doi: 10.1523/JNEUROSCI.4749-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Chieng B, Azriel Y, Christie MJ. Cellular morphine tolerance produced by βarresitn-2-dependent impairment of μ-opioid receptor resensitization. J Neurosci. 2011;31:7122–7130. doi: 10.1523/JNEUROSCI.5999-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- Dziaduleqicz EK, Bevan SJ, Brain CT, Coote PR, Culshaw AG, Davis AJ, Edwards LJ, Fisher AJ, Fox AJ, Gentry C, Vecchia L, Loong Y, Lyothier I, McNair KO, Farrell C, Peacoc M, Portmann R, Schopfer U, Yaqoob M, Zadrobilek J. Napthlalen-11-yl-(4-pentyloxy-napthalen-1-yl) methanone: a potent, orally bioavailable human CB1/CB2 dual agonist with antihyperalgesic properties and restricted central nervous system penetration. J Med Chem. 2007;50:3851–3856. doi: 10.1021/jm070317a. [DOI] [PubMed] [Google Scholar]
- Eisinger DA, Ammer H, Schulz R. Chronic morphine treatment inhibits opioid receptor desensitization and internalization. J Neurosci. 2002;22:10192–10200. doi: 10.1523/JNEUROSCI.22-23-10192.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmes SJR, Jhaveri MD, Smart D, Kendall DA, Chapman V. Cannabinoid CB2 receptor activation inhibits mechanically evoked responses of wide dynamic range dorsal horn neurons in naïve rats and in rat models of inflammatory and neuropathic pain. Eur J Neurosci. 2004;20:2311–2320. doi: 10.1111/j.1460-9568.2004.03690.x. [DOI] [PubMed] [Google Scholar]
- Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5:565–575. doi: 10.1038/nrn1431. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Groer CE, Tidgewll K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induced mu-opioid receptor/arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer CE, Schmid CL, Jaeger AM, Bohn LM. Agonist-directed interactions with specific beta-arrestins determines mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J Biol Chem. 2011;286:31731–31741. doi: 10.1074/jbc.M111.248310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 2000;315:422–437. doi: 10.1016/s0076-6879(00)15859-8. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The new face of active receptor bound arrestin attracts new partners. Structure. 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience. 2002;109:421–436. doi: 10.1016/s0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- Gutstein HB, Akil H. Opioid analgesics. In: Hardman JG, Limbird LE, Gilman AG, editors. Goodman and Gilman’s The pharmacological basis of therapeutics. 10. McGraw Hill; New York: 2001. pp. 569–620. [Google Scholar]
- Haberstock-Debic H, Kin KA, Yu YJ, von Zastrow Morphine promotes rapid, arrestin-dependent endocytosis of mu-opioid receptors in striatal neurons. J Neurosci. 2005;25:7847–7857. doi: 10.1523/JNEUROSCI.5045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry AG, Hislop JN, Grove J, Thorn K, Marsh M, von Zastrow M. Regulation of endocytic clathrin dynamics by cargo ubiquitination. Dev Cell. 2012;23:519–532. doi: 10.1016/j.devcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A. Selecitve activation of cannabinoid CB2 receptors suppresses hyperalgeisa evoked by intradermal capsaicin. J Pharmacol Exp Ther. 2004;308:446–453. doi: 10.1124/jpet.103.060079. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontinin A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, Porreca F, Buckley NE, Makriyannis A, Malan TP. CB2 cannabinoid receptor mediation of nociception. Pain. 2006;122:36–42. doi: 10.1016/j.pain.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Jaggar SI, Hasnie FS, Sellaturay S, Rice ASC. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 recpetor agonist palmitoylethanolamine in visceral and somatic inflammatory pain. Pain. 1998;76:189–199. doi: 10.1016/s0304-3959(98)00041-4. [DOI] [PubMed] [Google Scholar]
- Jiang B, Shi Y, Li H, Kang L, Ma L. Decreased morphine analgesia in rat overexpressing beta-arrestin 2 at periaqueductal gray. Neurosci Lett. 2006;400:150–153. doi: 10.1016/j.neulet.2006.02.071. [DOI] [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, Chavkin C, Mackie K. Distinct domains of the CB1 cannaniboid receptor mediate desensitizaiton and internalization. J Neurosci. 1999;15:3773–3780. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Maguma HT, Smith TH, Ross GR, Dewy WL, Akbarali HI. The role of β-arrestin2 in the mechanism of morphine tolerance in the mouse and guina pig gastrointestinal tract. J Pharmacol Exp Ther. 2012;340:567–576. doi: 10.1124/jpet.111.186320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S, Chapman V. Selective cannabinoid CB1 receptor activation inhibits spinal nociceptive transmission in vivo. J Neurophysiol. 2001;86:3061–3064. doi: 10.1152/jn.2001.86.6.3061. [DOI] [PubMed] [Google Scholar]
- Kieffer BL. Opioids: first lessons from knockout mice. Trends Pharmacol Sci. 1999;20:19–26. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Koch T, Brandenburg LO, Liang Y, Schulz S, Beyer A, Schröder H, Höllt V. Phospholipase D2 modulates agonist-induced mu-opioid receptor desensitization and resensitization. J Neurochem. 2004;88:680–688. doi: 10.1046/j.1471-4159.2003.02189.x. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Nappey V, Kieffer BL, Chavkin C. Mu and delta opioid receptors are differentially desensitized by the coexpression of beta-adrenergic receptor kinase 2 and beta-arresitn2 in xenopus oocytes. J Biol Chem. 1997;272:27605–27611. doi: 10.1074/jbc.272.44.27605. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Celver JP, Wu A, Chavkin C. Agonist induced homologous desensitization of mu-opioid receptors mediated by G protein-coupled receptor kinases is dependent on agonist efficacy. Mol Pharmacol. 1998;54:704–711. [PubMed] [Google Scholar]
- LaBuda CJ, Koblish M, Little PH. Cannabinoid CB2 receptor agonist activity in the hindpaw incision model of postoperative pain. Eur J Pharmacol. 2005;527:172–174. doi: 10.1016/j.ejphar.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Lam H, Maga M, Pradhan A, Evans CJ, Maidment NT, Hales TG, Walwyn W. Analgesic tone conferred by constitutively active mu opioid receptors in mice lacking β-arrestin 2. Mol Pain. 2011;7:24. doi: 10.1186/1744-8069-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb K, Tidgewell K, Simpson DS, Bohn LM, Prisinzano TE. Antinociceptive effects of herkinorin, a MOP receptor agonist derived from salvinorin A in the formalin test in rats: new concepts in mu opioid receptor pharmacology. Drug Alcohol Depend. 2012;121:181–188. doi: 10.1016/j.drugalcdep.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor. J Biol Chem. 2000;277:15729–15735. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu X, Liu C, Kang J, Yang J, Pei G, Wu C. Improvement of morphine-mediated analgesia by inhibition of beta-arrestin 2 expression in mice periaqueductal gray matter. Int J Mol Sci. 2009;10:954–963. doi: 10.3390/ijms10030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. Beta-arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Lowe JD, Celver JP, Gurevich VV, Chavkin C. mu-Opioid receptors desensitize less rapidly than delta-opioid receptors due to less efficient activation of arrestin. J Biol Chem. 2002;277:15729–15735. doi: 10.1074/jbc.M200612200. [DOI] [PubMed] [Google Scholar]
- Macey TA, Lowe JD, Chavkin C. Mu opioid receptor activation of ERK 1/2 is GRK3 and arrestin dependent in striatal neurons. J Biol Chem. 2006;281:34515–34524. doi: 10.1074/jbc.M604278200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malan TP, Ibrahim MM, Deng JF, Liu A, Mata HP, Vanderah T, Porreca R, Makriyannis A. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- Malik R, Soh UJ, Trejo J, Marchese A. Novel roles for the E3 ubiquitin ligase atrophin-interacting protein 4 and signal transduction adaptor molecule 1 in G protein-coupled receptor signaling. J Biol Chem. 2012;287:9013–9027. doi: 10.1074/jbc.M111.336792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, LeMeur M, Dollé P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Lorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey COP, Rosethorne EM, Charlton SJ, Henderson G, Kelly E. μ-opioid receptors: correlation of agonist efficacy for signaling with ability to activate internalization. Mol Pharmacol. 2010;78:756–766. doi: 10.1124/mol.110.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal N, Tan M, Egbuta O, Desai N, Crawford C, Xie CW, Evans C, Walwyn W. Evidence that behavioral phenotypes of morphine in β-arrs−/− mice are due to the unmaking of JNK signaling. Neuropscyhopharmacology. 2012;37:1953–1962. doi: 10.1038/npp.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari P, Vezzi V, Sbraccia M, Grò C, Riitano D, Ambrosio C, Casella I, Costa T. Morphine-like opiates selectively antagonize receptor-arrestin interactions. J Biol Chem. 2010;285:12522–12535. doi: 10.1074/jbc.M109.059410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami A, Yajima T, Sakuma H, McLaren MJ, Inana G. X-arrestin: a new retinal arrestin mapping to the X chromosome. FEBS Lett. 1993;334:203–209. doi: 10.1016/0014-5793(93)81712-9. [DOI] [PubMed] [Google Scholar]
- Nguyen PT, Schmid CL, Raehal KM, Selley DE, Bohn LM, Sim-Selley LJ. β-arrestin2 regulated cannabinoid CB1 receptor signaling and adaptation in a central nervous system region-dependent manner. Biol Psychiatry. 2012;71:714–724. doi: 10.1016/j.biopsych.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo E, Livio L, de Novellis V, Rossi F, Maione S. The role of cannabinoid receptor in the descending modulation of pain. Pharmaceuticals. 2010;3:2661–2673. doi: 10.3390/ph3082661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW. Molecular biology of opioid analgesia. J Pain Symptom Manage. 2013;29:2–9. doi: 10.1016/j.jpainsymman.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Patierno S, Anselmi L, Jaramill I, Scott D, Garcia R, Sternini C. Morphine induces μ opioid receptor endocytosis in guinea pig enteric neurons following prolonged receptor activation. Gastroenterology. 2011;140:618–626. doi: 10.1053/j.gastro.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids. AAPS J. 2005;7:E625–654. doi: 10.1208/aapsj070364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Por ED, Bierbower SM, Berg KA, Gomez R, Akopian AN, Wetsel WC, Jeske NA. β-Arrestin-2 desensitizes the transient receptor potential vanilloid 1 (TRPV1) channel. J Biol Chem. 2012;287:37552–37563. doi: 10.1074/jbc.M112.391847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Por ED, Gomez R, Akopian AN, Jeske NA. Phosphorylation regulates TRPV1 association with β-arrestin-2. Biochem J. 2013;451:101–109. doi: 10.1042/BJ20121637. [DOI] [PubMed] [Google Scholar]
- Przewlocka B, Sieja A, Starowicz K, Maj M, Bilecki W, Przewlocki R. Knockdown of spinal opioid receptors by antisense targeting beta-arrestin reduces morphine tolerance and allodynia in rat. Neurosci Lett. 2002;325:107–110. doi: 10.1016/s0304-3940(02)00246-x. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Law PY, Loh HH. Mu-opioid receptor desensitization: role of receptor phosphorylation, internalization and representation. J Biol Chem. 2003;278:36733–36739. doi: 10.1074/jbc.M305857200. [DOI] [PubMed] [Google Scholar]
- Quillinan N, Lau EK, Virk M, von Zastrow M, Williams JT. Recovery from μ-opioid receptor desensitization after chronic treatment with morphine and methadone. J Neurosci. 2011;31:4434–4443. doi: 10.1523/JNEUROSCI.4874-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology. 2011;63:1001–1019. doi: 10.1016/j.neuropharm.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arresitn2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev. 2011;63(4):1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea K, Roche M, Finn DP. Supraspinal modulation of pain by cannabinoids: the role of GABA and glutamate. Br J Pharmacol. 2007;152:633–648. doi: 10.1038/sj.bjp.0707440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Liu HC, Loh HH. Mu-opioid receptor-knockout mice: the role of mu-opioid receptor in gastrointestinal transit. Brain Res. 1998;56:281–283. doi: 10.1016/s0169-328x(98)00051-5. [DOI] [PubMed] [Google Scholar]
- Schmidlin F, Roosterman D, Bunnett NW. The third intracellular loop and carboxyl tail of neurokinin 1 and 3 receptors determine interactions with beta-arrestins. Am J Physiol Cell Physiol. 2003;285:C945–C958. doi: 10.1152/ajpcell.00541.2002. [DOI] [PubMed] [Google Scholar]
- Shinohara T, Dietzschold B, Craft CM, Wistow G, Early JJ, Donoso LA, Horwitz J, Tao R. Primary and secondary structure of bovine retinal S antigen (48-kDa protein) Proc Natl Acad Sci USA. 1987;84:6975–6979. doi: 10.1073/pnas.84.20.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim LJ, Hampson RE, Deadwyler SA, Childers SR. Effects of chronic treatment with delta9-tetrahydrocannabinol on cannabinoid-stimulated [35S]GTPgammaS autoradiography in rat brain. 1996;16:8057–8066. doi: 10.1523/JNEUROSCI.16-24-08057.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TH, Sim-Selley LJ, Selley DE. Cannabinoid CB1 receptor-interacting proteins: novel targets for central nervous system drug discovery? Br J Pharmacol. 2010;(160):454–66. doi: 10.1111/j.1476-5381.2010.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal DM, Elmes SJR, Kendall DA, Chapman V. Intraplantar injection of anandamide inhibits mechanically-evoked responses of spinal neurons via activation of CB2 receptors in anaesthetized rats. Neuropharmacol. 2003;45:404–411. doi: 10.1016/s0028-3908(03)00195-3. [DOI] [PubMed] [Google Scholar]
- Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, Miner LL, Uhl GR. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci USA. 1997;94:1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suplita RL, 2nd, Gutierrez T, Fegley D, Piomelli D, Hohmann AG. Endocannabinoids at the spinal level regulate, but do not mediate, nonopioid stress-induced analgesia. Neuropharmacol. 2006;50:372–379. doi: 10.1016/j.neuropharm.2005.10.007. [DOI] [PubMed] [Google Scholar]
- SvÍzenská I, Dubový P, Sulcová A. Cannabinoid receptor 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol Biochem Behav. 2008;90:501–511. doi: 10.1016/j.pbb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Walwyn WM, Wei W, Xie CW, Chiu K, Kieffer BL, Evans CJ, Maidment NT. Mu opioid receptor-effector coupling and trafficking in dorsal root ganglia neurons. Neuroscience. 2006;142:493–503. doi: 10.1016/j.neuroscience.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Walwyn W, Evans CJ, Hales TG. Beat-arrestin2 and c-Src regulate the constitutive activity and recycling of mu opioid receptors in dorsal root ganglion neurons. J Neurosci. 2007;27:5092–5104. doi: 10.1523/JNEUROSCI.1157-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, von Zastrow M. Morphine-activated opioid receptor eludes desensitization by beta-arrestin. Proc Natl Acad Sci USA. 1998;95:9914–9919. doi: 10.1073/pnas.95.17.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ. Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65:223–254. doi: 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaki K, Takahashi Y, Sakuragi S, Matsubara K. Molecular cloning of the S-antigen cDNA from bovine retina. Biochem Biophys Res Comm. 1987;142:904–910. doi: 10.1016/0006-291x(87)91499-9. [DOI] [PubMed] [Google Scholar]
- Yang CH, Huang HW, Chen KH, Chen YS, Sheen-Chen SM, Lin CR. Antinociceptive potentiation and attenuation of tolerance by intrathecal β-arrestin 2 small interfering RNA in rats. Br J Anaesth. 2011;107:774–781. doi: 10.1093/bja/aer291. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Roler for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci USA. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhao H, Qui Y, Loh HH, Law PY. Src phosphorylation of μ-receptor is responsible for the receptor switching from an inhibitory to a stimulatory signal. J Biol Chem. 2008;284:1990–2000. doi: 10.1074/jbc.M807971200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenhan M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci USA. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]