

Abstract
Motivation: The Cancer Genome Atlas (TCGA) RNA-Sequencing data are used widely for research. TCGA provides ‘Level 3’ data, which have been processed using a pipeline specific to that resource. However, we have found using experimentally derived data that this pipeline produces gene-expression values that vary considerably across biological replicates. In addition, some RNA-Sequencing analysis tools require integer-based read counts, which are not provided with the Level 3 data. As an alternative, we have reprocessed the data for 9264 tumor and 741 normal samples across 24 cancer types using the Rsubread package. We have also collated corresponding clinical data for these samples. We provide these data as a community resource.
Results: We compared TCGA samples processed using either pipeline and found that the Rsubread pipeline produced fewer zero-expression genes and more consistent expression levels across replicate samples than the TCGA pipeline. Additionally, we used a genomic-signature approach to estimate HER2 (ERBB2) activation status for 662 breast-tumor samples and found that the Rsubread data resulted in stronger predictions of HER2 pathway activity. Finally, we used data from both pipelines to classify 575 lung cancer samples based on histological type. This analysis identified various non-coding RNA that may influence lung-cancer histology.
Availability and implementation: The RNA-Sequencing and clinical data can be downloaded from Gene Expression Omnibus (accession number GSE62944). Scripts and code that were used to process and analyze the data are available from https://github.com/srp33/TCGA_RNASeq_Clinical.
Contact: stephen_piccolo@byu.edu or andreab@genetics.utah.edu
Supplementary information: Supplementary material is available at Bioinformatics online.
1 Introduction
The Cancer Genome Atlas Research Network has profiled thousands of human tumors to discover various types of molecular-level aberrations that occur within tumors. Researchers have used these data to derive new insights about tumorigenesis and to validate and inform experimental findings (The Cancer Genome Atlas Research Network et al., 2013). To facilitate such analyses, The Cancer Genome Atlas (TCGA) provides ‘Level 3’ RNA-Sequencing (RNA-Seq) data, which have been aligned to the reference genome using MapSplice (Wang et al., 2010), quantified at the gene and transcript levels using RSEM (Li and Dewey, 2011) and standardized using upper-quartile normalization (Bullard et al., 2010; Li and Dewey, 2011; Wang et al., 2010). However, the use of these data comes with some caveats. First, some analytic tools designed specifically for RNA-Seq data—for example, DESeq2 (Love et al., 2014)—require the user to input integer-based read counts, yet Level 3 read counts are represented as non-integer numbers. Second, the upper-quartile normalization method scales gene counts by the upper-quartile value of the non-zero distribution; however, when a sample has a relatively high number of zero counts or genes with extremely high read counts, the value distributions may vary considerably across samples (Dillies et al., 2013). Third, when researchers seek to compare the TCGA Level 3 data against clinical covariates and outcomes, additional processing steps are necessary to match RNA-Seq identifiers to the clinical data. Users without computational training may face difficulty performing these steps, and scientists may duplicate each other’s efforts.
The TCGA consortium also provides the RNA-Seq data in raw form. Thus it is possible for researchers to reprocess the data using alternative computational pipelines. We obtained raw sequencing data for 9264 tumor samples and 741 normal samples across 24 cancer types (Table 1) and reprocessed the data using the Subread algorithm (Liao et al., 2014), which shows high concordance with other existing methods regarding assignment of reads to genes but takes a relatively short time for processing (SEQC/MAQC-III Consortium, 2014). RNA transcripts often span multiple exon-exon junctions, making it challenging for aligners to map reads that are smaller than the transcript length. Rsubread’s ‘vote-and-seed’ read-mapping technique addresses this problem by breaking the reads into relatively small segments, mapping the segments to the reference genome and identifying locations where adjacent segments map to different exons. This approach has been shown to be more accurate in mapping junction reads than other aligners, including MapSplice (Liao et al., 2013). The Rsubread package, which implements the Subread algorithm, is convenient for this task because: (i) it can be applied to both single- and paired-end reads; (ii) it is considerably faster and requires less computer memory than many other methods and (iii) it requires no external software packages for processing, whereas many other packages require a series of steps that span multiple packages.
Table 1.
Cancer types and total number of samples
| Cancer name | Abbreviated cancer name | Samples included |
|---|---|---|
| Adrenocortical carcinoma | ACC | 79 |
| Bladder urothelial carcinoma | BLCA | 414 |
| Breast invasive carcinoma | BRCA | 1119 |
| Cervical squamous cell carcinoma and endocervical adenocarconoma | CESC | 306 |
| Colon adenocarcinoma | COAD | 483 |
| Lymphoid neoplasm diffuse large B-cell lymphoma | DLBC | 48 |
| Glioblastoma multiforme | GBM | 170 |
| Head and neck squamous cell carcinoma | HNSC | 504 |
| Kidney chromophobe | KICH | 66 |
| Kidney renal clear cell carcinoma | KIRC | 542 |
| Kidney renal papillary cell carcinoma | KIRP | 291 |
| Acute myeloid leukemia | LAML | 178 |
| Brain lower grade glioma | LGG | 532 |
| Liver hepatocellular carcinoma | LIHC | 374 |
| Lung adenocarcinoma | LUAD | 541 |
| Lung squamous cell carcinoma | LUSC | 502 |
| Ovarian serous cystadenocarcinoma | OV | 430 |
| Prostate adenocarcinoma | PRAD | 502 |
| Rectum adenocarcinoma | READ | 167 |
| Skin cutaneous melanoma | SKCM | 472 |
| Stomach adenocarcinoma | STAD | 420 |
| Thyroid carcinoma | THCA | 513 |
| Uterine corpus endometrial carcinoma | UCEC | 554 |
| Uterine carcinoma | UCS | 57 |
A total of 9264 tumor samples across 24 cancer types are included in the database.
We used the featureCounts function within the Rsubread package to summarize the data to integer-based, gene-level read counts, and we calculated two types of normalized value: fragments per kilobase of exon per million reads mapped (FPKM) and transcripts per million (TPM) (Li and Dewey, 2011; Mortazavi et al., 2008; Wagner et al., 2012). In this pipeline, the FPKM and TPM values are calculated using the total number of mapped reads and the total number of non-overlapping exonic basepairs. Both FPKM and TPM methods account for the length of genomic features. FPKM corrects for the number of reads that have been sequenced, and TPM accounts for the average number of mapped bases per read. FPKM values are used widely, whereas TPM values have been shown to meet the invariant average criterion and thus may be more comparable across samples (Wagner et al., 2012). Importantly, FPKM and TPM are calculated using only data from an individual RNA-Seq sample; thus adding new samples to the dataset will not require changes to the existing expression values; such an approach is crucial for precision-medicine applications and for integrating data across technology platforms (Piccolo et al., 2012, 2013). Furthermore, because we have provided raw counts, it is possible for others to normalize the data using other methods with relative ease. We have made these data publicly available along with all clinical variables provided by TCGA for these samples. We have also aligned the RNA-Seq sample identifiers with the clinical identifiers.
2 Methods
2.1 HER2 gene-expression profiling data
Before analyzing TCGA data, we generated an experimental dataset that represented the effects of HER2 (ERBB2) overexpression in breast cancer cells. Using human mammary epithelial cells (HMECs), we produced five replicates, in which the HER2 protein had been experimentally activated, and 12 control green fluorescent protein (GFP) replicates. We used recombinant adenovirus to overexpress HER2 (Vector Biolabs) and GFP in the HMECs. The HMECs were grown in serum-free WIT-P media (Stemgent) and were starved of growth factors for 36 h prior to infection. HER2-expressing or GFP-expressing adenovirus (MOI 500) were added to HMEC cells in conditioned media and incubated with the cells for 18 h. Cells were washed with phosphate buffered saline, scraped into RNAlater (Ambion), and RNA was extracted from pelleted cells using an RNeasy kit (Qiagen) with DNase. To ensure that components were being expressed, we created lysates of HER2-adenovirus-vector and GFP-adenovirus-vector infected HMEC cells and analyzed these lysates for expression of HER2-pathway protein components by sodium dodecyl sulphate–polyacrylamide gel electrophoresis/Western blot. HER2 overexpression and activity was confirmed by Western blotting for HER2 and for activated HER2 (phospho-Tyr1173-HER2, Supplementary Fig. S1). cDNA libraries were prepared from the extracted RNA using the Illumina Stranded TruSeq protocol and then sequenced with the Illumina HiSeq 2000 sequencing platform with six samples per lane. Single-end reads of 101 base pairs were generated. This dataset is available on Gene Expression Omnibus via accession number GSE62820.
2.2 TCGA data acquisition
We downloaded TCGA Level 3 data via the Synapse portal for 12 cancer types (https://www.synapse.org/#!Synapse:syn1695324). This included 3468 samples that had been preprocessed using TCGA’s standard pipeline.
To reprocess TCGA data with Rsubread, we downloaded FASTQ formatted files for all available TCGA tumor samples via the National Cancer Institute’s Cancer Genomics Hub (Wilks et al., 2014). This included a total of 9264 tumor samples across 24 cancer types (Table 1). Some patient samples were sequenced multiple times; in these cases, we included each replicate.
We downloaded TCGA clinical data in ‘Biotab’ format on May 20, 2015 from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm) and extracted all reported clinical variables from the nationwidechildrens.org_clinical_patient_[cancerTypeAbbreviatedInLowerCase].txt files. In these files, 12-character patient identifiers were used, whereas the RNA-Seq sample identifiers were longer. To make it easier to integrate these two data sources, we converted the short IDs to full IDs by matching the ‘bcr_patient_barcode’ values in the clinical files. For patients who had multiple RNA-Seq replicates, we provide multiple columns in the clinical data file. We set values as ‘NA’ when no information was reported in the clinical files for a given patient. If there were multiple sequences available for a tumor sample, we duplicated the clinical variables available for that sample. In total, we included 548 clinical variables.
2.3 Data processing and normalization
For our HER2 expression-profiling data, we calculated gene-level values using the same steps that the TCGA consortium uses to produce ‘Level 3’ values. The reference data, Perl scripts and parameters used in this pipeline are described here: https://cghub.ucsc.edu/docs/tcga/UNC_mRNAseq_summary.pdf. In some cases, the software versions specified in the above document were unable to handle single-end reads. In these cases, we used the latest versions of these software tools that were able to handle single-end reads. Below we list these versions:
MapSplice v 12_07 (Wang et al., 2010)
RSEM v1.2.12 (Li and Dewey, 2011)
UBU v1.2 (https://github.com/mozack/ubu/)
Picard-tools v1.82 (http://picard.sourceforge.net)
BedTools v2.17.0 (Quinlan and Hall, 2010)
For our HER2 data and for the samples from TCGA, we used the Rsubread package (version v1.14.2; Liao et al., 2014) to align the reads and to produce gene-level summarized values. We used the UCSC hg19 reference for alignment and the corresponding gene annotation format file available from http://support.illumina.com/sequencing/sequencing_software/igenome.html. Within this pipeline, we obtained integer-based gene counts using the featureCounts function in the Rsubread package (Liao et al., 2014). We used the limma (version 3.20.9; Smyth, 2004) and edgeR (version v3.6.8; Nikolayeva and Robinson, 2014; Robinson et al., 2010) packages to calculate FPKM values (Li and Dewey, 2011) and a custom script to convert FPKM to TPM values (Li and Dewey, 2011; Wagner et al., 2012). We used R version 3.1.0 and Bioconductor version 2.14 (Gentleman et al., 2004; R Core Team, 2014; http://www.R-project.org/). When evaluating pre-normalized gene counts, we used the ‘expected_count’ column in the ‘.genes.results’ files generated by RSEM, and Rsubread’s raw, integer-based gene counts. All processed TCGA data can be accessed on Gene Expression Omnibus via accession number GSE62944. This includes integer-based gene counts and FPKM and TPM values as well as clinical data.
2.4 Statistical procedures
When comparing gene-expression values between groups in this study, we calculated the standardized mean difference using Hedges’ formula (Hedges, 1981, 1985). We used the coefficient of variation (CV) to assess variability. We used the Random Forests classification algorithm implemented in the caret package (Kuhn, 2008).
The data-processing pipelines and analysis scripts that we used for this manuscript are available from https://github.com/srp33/TCGA_RNASeq_Clinical.
3 Results
3.1 Evaluation of biological replicates
Our initial goal was to generate a gene-expression signature representing HER2 activation and to use that signature to identify breast tumors in TCGA where the HER2 pathway was active. For consistency with TCGA, we initially processed the RNA-Seq signature data using the same pipeline used by the TCGA consortium (see Materials and Methods). However, upon examining these data, we observed inconsistencies across our biological replicates. For example, as illustrated in Figure 1, we found that some replicates exhibited considerably different patterns of expression for genes that showed the greatest differences in expression between HER2-active cells and GFP controls. Concerned that such inconsistencies could reduce the effectiveness of our signature-based predictions, we examined the data further and explored the Rsubread pipeline as an alternative.
Fig. 1.

Heat maps of normalized expression values for the 200 genes most differentially expressed between HER2-activated HMECs (n = 5) and GFP-treated controls (n = 12). Each column in the heat maps represents data for a given HMEC replicate. Each row represents data for a given gene
We hypothesized that the inconsistencies we observed in our biological replicates may have resulted from differences in the total number of mapped reads, from genes expressed at extremely high levels or from differences in the number of zero-count genes per sample. Others have described these factors as potential limitations of the upper-quartile normalization step used in the TCGA Level 3 processing pipeline (Dillies et al., 2013). Accordingly, we reprocessed the data using Rsubread and performed various analyses to understand the effects of these variables for data processed using either pipeline. In addition, we performed various analyses to compare the performance of the two datasets in various biomedical research contexts (Supplementary Table S1).
3.2 Raw gene count analysis
Initially, we compared raw (non-normalized) gene counts between the TCGA Level 3 and Rsubread processing pipelines for our HER2 (n = 5) and control (n = 12) replicates. The TCGA Level 3 pipeline produces expected counts as floating point (non-integer) numbers, whereas Rsubread produces integer-based gene counts, which represent the number of mapped reads per gene. For both pipelines, the HER2 gene counts were significantly overexpressed in HER2 activated cells relative to control samples (Supplementary Fig. S2). However, the difference in expression between HER2-activated cells and controls was greater for the Rsubread data (standardized mean difference for TCGA: 10.0; Rsubread: 23.8).
To explore these differences further, we compared the total number of mapped reads per sample between the two pipelines. For HER2-activated samples, the total number of mapped reads was much more variable for the TCGA Level 3 data than for the Rsubread data (Fig. 2). Two of the HER2-activated samples—the same samples (2 and 4) that showed visual differences in Figure 1—had a considerably smaller number of total mapped reads when the TCGA pipeline was used. Upon plotting the empirical cumulative distribution of the total mapped reads per sample (Fig. 3 and Supplementary Fig. S3), we observed that the same HER2-activated samples showed different overall expression patterns, due to a relatively high number of genes with zero read counts. These observations suggest that Rsubread is less sensitive to differences in library size and that it more consistently identifies genes expressed at extremely low levels.
Fig. 2.

Total mapped reads per sample for data processed using the TCGA Level 3 and Rsubread pipelines. For the TCGA Level 3 pipeline, the number of mapped reads varied widely for the HER2 samples, and samples 2 and 4 (see Fig. 1) had a considerably lower number of mapped reads. In contrast, the number of mapped reads for Rsubread was consistent across the samples
Fig. 3.
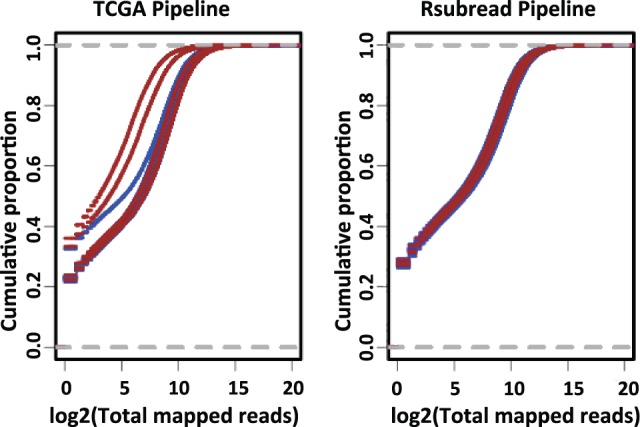
Empirical cumulative distribution of total mapped reads using raw gene counts. In all cases, the cumulative distributions were more consistent for Rsubread than for the TCGA pipeline produced gene counts data. The aberrantly expressed samples in the TCGA data are the same samples (GFP sample 4, HER2 samples 2 and 4) that showed visually different expression patterns in the heat maps (see Fig. 1). GFP samples (n = 12) are represented in blue and HER2 samples (n = 5) are represented in brown color
3.3 Normalized gene expression analysis
We observed similar findings for the normalized values produced using either pipeline. The empirical cumulative distribution of total normalized expression was more consistent for the Rsubread data (FPKM and TPM) than for the TCGA Level 3 data (Supplementary Fig. S4). HER2 gene-expression levels were less variable across the replicates for the Rsubread values than for the Level 3 data (CV for FPKM = 0.09; TPM = 0.06; Level 3 = 0.30). Differences in expression between HER2 activated cells and controls were also greater for the Rsubread data (standardized mean difference for FPKM = 66.9; TPM = 67.2; Level 3 = 25.8; see Supplementary Fig. S4). In addition, across all genes for the control and HER2-activated replicates, the coefficients of variation were smaller for the Rsubread processed data than for the TCGA Level 3 data (Supplementary Fig. S5). These observations remained consistent, even if we excluded the two HER2 replicates that showed different gene-count distributions in the TCGA Level 3 data (Supplementary Table S2).
We calculated the number of zero-expression genes per GFP sample using the genes that overlap between the TCGA Level 3 and Rsubread TPM data. The Level 3 data contained a higher number of zero-expressing genes per GFP replicate (Level 3 median: 4452; Rsubread TPM: 4174). For each gene that had at least one zero value across the replicates, we calculated the number of samples that had a zero value for a given gene. The average was 7.50 (out of 12) for TCGA Level 3 and 8.92 for Rsubread. Although the Level 3 samples had a higher overall number of zero values across all genes (Supplementary Fig. S6), these values were less consistent for a given gene. These findings suggest that the alignment, count estimation and/or upper-quartile normalization steps used in the Level 3 pipeline lead to variability across the replicates and that the Rsubread FPKM and TPM values are more consistent across replicates.
Having observed these patterns in our replicates, we processed 9264 RNA-Seq samples from TCGA using the Rsubread package. We performed various comparative analyses using the samples that overlapped with the Level 3 data that had been distributed via the Pan Cancer 12 project (The Cancer Genome Atlas Research Network et al., 2013). We limited our comparative analyses to the genes (n = 19 584) and samples (n = 3380) that overlapped between these datasets. Across all samples, the number of zero-count genes was significantly higher in the TCGA Level 3 data than in the Rsubread data, (t-test P value < 0.001; Level 3 median = 2742.5; Rsubread TPM = 1910.0; see Supplementary Fig. S7). In addition, we calculated Pearson’s correlation coefficients between replicates for the 13 patients that were common between TCGA PANCAN12 and our Rsubread TPM data (Supplementary Table S3 and Fig. S8). Across the replicates, the Pearson’s correlation coefficients were higher for the Rsubread processed replicates (median = 0.86) than for the TCGA Level 3 replicates (median = 0.79).
3.4. Downstream analyses
Next, we used a sparse binary factor regression method (West et al., 2001) to derive a gene-expression signature that would predict whether the HER2 pathway was active in a given TCGA breast-tumor sample. This technique results in a probabilistic estimate for each tumor sample that indicates whether the pathway is active. We applied this approach to data from both processing pipelines and compared the estimates of HER2 pathway activity between tumor samples that had been confirmed via immunohistochemistry to be HER2 positive (n = 149) or negative (n = 513). For both data-processing pipelines, the probabilistic estimates of HER2 pathway activity were significantly higher for HER2-positive versus HER2-negative samples (see Supplementary Fig. S9 and Table S4). However, the predictions for the Rsubread data were less variable than for the TCGA Level 3 data (see Supplementary Table S5), and the standardized mean difference between the groups was greater for the Rsubread data (TCGA Level 3: 0.44; Rsubread FPKM: 0.52; Rsubread TPM: 0.59). This finding was robust to the exclusion of HER2 samples 2 and 4 (Supplementary Table S2). Thus, using an empirical approach to estimate HER2 pathway activity, the Rsubread data resulted in more reliable and consistent conclusions when validated against traditional methods.
As an additional test, we examined how well we could distinguish between lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) samples in TCGA. This classification is clinically important to guide personalized therapy based on the molecular subtypes (The Cancer Genome Atlas Research Network, 2012, 2014). We used the Random Forests classification algorithm (Breiman, 2001) to identify gene-expression patterns that differ between these cancer types, and we performed 10-fold cross-validation to estimate how accurately tumors of either cancer type could be identified. For this analysis, we used TCGA Level 3 data and Rsubread normalized (TPM) data for 575 tumor samples that overlapped between these datasets. We used receiver operating characteristic (ROC) curves to assess classification accuracy and the balance between sensitivity and specificity in making these predictions. With the area under ROC curves (AUC) as a comparison metric and a probability threshold of 0.5, both datasets resulted in highly accurate predictions of lung-cancer histological type (AUC = 0.999 for Rsubread; AUC = 0.985 for TCGA Level 3); however, the TCGA Level 3 data resulted in 28 (out of 575) incorrect predictions, whereas the Rsubread data resulted in only 9 incorrect predictions (Fig. 4).
Fig. 4.

Receiver operating characteristics (ROC) curves (in black) showing the balance between sensitivity and specificity in classifying TCGA lung adenocarcinoma (LUAD) and lung squamous carcinoma (LUSC) samples using TCGA Level 3 and Rsubread pipeline processed RNA-Seq data. The gray shaded areas denote the confidence intervals associated with the ROC curve. The Rsubread data resulted in more accurate predictions than the TCGA Level 3 data when all the genes for the respective pipelines were used
Using the TCGA Level 3 data, Cline et al. (2013) suggested that a subset of the LUSC samples were ‘discordant’ with the remaining LUSC samples and exhibited ‘LUAD-like’ properties. Our Random Forests predictions for the Level 3 data led to similar conclusions. In contrast, when we use the Rsubread data, the ‘LUSC Discordant’ samples are classified mostly as ‘LUSC’. One difference between the two datasets is that the TCGA Level 3 data contain values for 20 217 genes (after excluding genes that have zero variance across all samples), whereas the Rsubread data contain values for 22 833 genes. Accordingly, we repeated the Random Forests classification analysis and limited each dataset so that it included only the 19 453 genes that overlap between the two datasets. With this approach, both datasets resulted in virtually identical results: most ‘LUSC Discordant’ samples were classified as ‘LUAD’. We examined the genes present in the Rsubread data but not in the TCGA Level 3 data and found various genes that show strong and consistent expression similarity between ‘LUSC Discordant’ and LUSC samples (Supplementary Fig. S10). Expression patterns for these genes are consistent and strong enough that they alter the Random Forests classification results for the ‘LUSC Discordant’ samples. Although these samples do exhibit expression patterns characteristic of LUAD for many genes, this analysis indicates that these samples should not necessarily be classified as LUAD tumors. We observed this difference because the Rsubread data were processed using relatively recent gene definitions; thus researchers who work with these data may have a more complete picture of tumor biology.
4 Discussion
To our knowledge, this compendium of RNA-Seq tumor data is the largest compiled to date. It includes 9264 tumor samples and 741 normal samples across 24 cancer types. These data offer an alternative to the lone pipeline used by the TCGA consortium. In contrast to the TCGA data portal, which provides the RNA-Seq data in individual files for each sample, we have compiled the Rsubread tumor data into aggregate data files; thus it will be easier for researchers to analyze the data and compare across cancer types. We have matched these data to clinical variables to ease the process of examining relationships between these variables and gene-expression levels.
Different RNA-Seq processing pipelines differ considerably in accuracy for quantifying gene-level expression values (Fonseca et al., 2014). However, our goal was not to perform an exhaustive benchmark comparison across the many tools available for processing RNA-Seq data, although others have shown that Rsubread performs quite well in such benchmarks at quantifying gene-expression levels (SEQC/MAQC-III Consortium, 2014). Rather our goals were to provide a new community resource and to provide evidence that this alternative dataset is of high quality and performs better in various downstream analyses than the standard TCGA data. We have demonstrated that Rsubread produces more consistent values across biological replicates, and we have provided evidence that our data lead to more biologically relevant conclusions. Tens of thousands of hours of computational processing time were necessary to compile this dataset. Thus we also hope to prevent the need for other scientists to invest similar resources.
Our dataset will be most useful to researchers who wish to compare gene-level expression values across samples. Researchers who wish to work with transcript- or exon-level values or who wish to identify splice junctions may find the TCGA Level 3 data useful for this purpose. Various Web-based portals exist for visualizing and analyzing TCGA data. These include cBioPortal for Cancer Genomics (Cerami et al., 2012; Gao et al., 2013) and the UCSC Cancer Genomics Browser (Zhu et al., 2009). Our data could be incorporated into these portals as an additional option for users who wish to analyze raw gene counts or to use the FPKM and TPM values that we provide.
We plan to update the data as more cancer types and tumor samples become available. We used open-source software to align and normalize the data and have made our processing code publicly available. In addition, we used single-sample normalization techniques to process the data. Thus, one can add new samples as they become available without affecting the existing data. However, we emphasize that it may still be necessary for researchers to correct for inter–sample variation when comparing data across batches and cancer types.
Supplementary Material
Acknowledgements
We gratefully acknowledge the use of the Shared Computing Cluster at Boston University for computational processing.
Funding
M.R. was funded by a National Library of Medicine training fellowship (T15LM007124). This study was supported by National Institutes of Health grants U01CA164720 (A.H.B. and W.E.J.) and R01ES025002 (W.E.J.).
Conflict of Interest: none declared.
References
- Breiman L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- Bullard J.H., et al. (2010) Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics, 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov., 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline M.S., et al. (2013) Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci. Rep., 3, 2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillies M.A., et al. (2013) A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinform., 14, 671–683. [DOI] [PubMed] [Google Scholar]
- Fonseca NA., et al. (2014) RNA-Seq gene profiling - A systematic empirical comparison. PLoS ONE., 9, e107026 doi:10.1371/journal.pone.0107026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal., 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.C., et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol., 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges L.V. (1981) Distribution theory for Glass' estimator of effect size and related estimators. J. Edu. Stat., 6, 107–128. [Google Scholar]
- Hedges L.V.O.I. (1985) Statistical Methods for Meta-Analysis. 1–16, Academic Press, London. [Google Scholar]
- Kuhn R.M. (2008) Building predictive models in R using the caret package. J. Stat. Soft., 28, 1–26. [Google Scholar]
- Li B., Dewey C.N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., et al. (2013) The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res., 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., et al. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love M.I., et al. (2014) Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., et al. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods, 5, 621–628. [DOI] [PubMed] [Google Scholar]
- Nikolayeva O., Robinson M.D. (2014) edgeR for differential RNA-seq and ChIP-seq analysis: an application to stem cell biology. Methods Mol. Biol., 1150, 45–79. [DOI] [PubMed] [Google Scholar]
- Piccolo S.R., et al. (2012) A single-sample microarray normalization method to facilitate personalized-medicine workflows. Genomics, 100, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S.R., et al. (2013) Multiplatform single-sample estimates of transcriptional activation. Proc. Natl. Acad. Sci. USA., 110, 17778–17783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2014) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- SEQC/MAQC-III Consortium. (2014) A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol., 32, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G.K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol., 3, Article3. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature, 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network et al. (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet., 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature, 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner G.P., et al. (2012) Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theor. Biosci., 131, 281–285. [DOI] [PubMed] [Google Scholar]
- Wang K., et al. (2010) MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res., 38, e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M., et al. (2001) Predicting the clinical status of human breast cancer by using gene expression profiles. Proc. Natl. Acad. Sci. USA., 98, 11462–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks C., et al. (2014) The Cancer Genomics Hub (CGHub): overcoming cancer through the power of torrential data. Database, 2014, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., et al. (2009) The UCSC Cancer Genomics Browser. Nat. Methods, 6, 239–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.