

Abstract
Considerable epidemiological and laboratory data have suggested that caffeine, a nonselective adenosine receptor antagonist, may protect against the underlying neurodegeneration of Parkinson’s disease (PD). Although both caffeine and more specific antagonists of the A2A subtype of adenosine receptor (A2AR) have been found to confer protection in animal models of PD, the dependence of caffeine’s neuroprotective effects on the A2AR is not known. To definitively determine its A2AR dependence, the effect of caffeine on MPTP neurotoxicity was compared in wild-type (WT) and A2AR gene global knockout (A2A KO) mice, as well as in CNS cell type-specific (conditional) A2AR knockout (cKO) mice that lack the receptor either in postnatal forebrain neurons or in astrocytes. In WT and in heterozygous A2AR KO mice caffeine pretreatment (25 mg/kg ip) significantly attenuated MPTP-induced depletion of striatal dopamine. By contrast in homozygous A2AR global KO mice caffeine had no effect on MPTP toxicity. In forebrain neuron A2AR cKO mice, caffeine lost its locomotor stimulant effect, whereas its neuroprotective effect was mostly preserved. In astrocytic A2AR cKO mice, both caffeine’s locomotor stimulant and protective properties were undiminished. Taken together, these results indicate that neuroprotection by caffeine in the MPTP model of PD relies on the A2AR, although the specific cellular localization of these receptors remains to be determined.
Keywords: Adenosine A2A receptors, MPTP, Caffeine, Neuroprotection, Parkinson’s disease
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). The neuropathological signs of PD occur long before any substantive clinical symptoms appear and it is estimated that at the time of symptom onset there may be 60–80% loss of striatal dopamine (Bernheimer et al., 1973). Like idiopathic PD, parkinsonism induced by acute exposure to the dopaminergic neuron protoxin 1-methyl-4-phenyl-1,2,3,6 tetra-hydropyridine (MPTP) results from degeneration of nigrostriatal dopaminergic neurons and associated loss of striatal dopamine (Langston et al., 1984; Ricaurte et al., 1987). Most of the biochemical, pathological and clinical features that occur following a substantial lesion from MPTP treatment in animal models resemble symptoms observed after losing 80% of total striatal dopamine (Langston et al., 1984; Kopin and Markey, 1988; Jackson-Lewis et al., 2007). Despite some limitations (e.g., the lack of Lewy body-like inclusions or reliable behavioral deficits characteristic of PD), acute MPTP intoxication remains one of the best-characterized animal models of PD and recapitulates neurochemical and anatomical features of the disease (Dawson et al., 2002; Jackson-Lewis et al., 2007).
Caffeine is the most consumed psychoactive drug in the world and, like classical psychostimulants, produces behavioral effects such as increased motor activation, arousal, and reinforcement. Multiple epidemiological studies have also shown that people who consume more caffeinated beverages are substantially less likely to develop PD (Ross et al., 2000, Ascherio et al., 2001, 2004). Although caffeinated coffee, tea and soda comprise many chemical constituents, the finding that regular but not decaffeinated coffee consumption is predictive of reduced PD risk (Ascherio et al., 2001; Palacios et al., 2012) implicates caffeine as the basis of the inverse association. However, epidemiological studies do not directly address causality and it remains unknown whether caffeine protects against the neurodegeneration underlying PD.
Laboratory studies have supported a true neuroprotective effect of caffeine in PD by demonstrating its biological plausibility in multiple animal models of the disease. When caffeine is co-administered with MPTP to mice at doses comparable with those of typical human exposure, it dose-dependently attenuates the loss of striatal dopamine triggered by MPTP using different exposure paradigms and in several mouse strains (Chen et al., 2001; Xu et al., 2002). It is shown that caffeine can be given up to two hours before or after MPTP and still confer protection against MPTP-induced dopamine loss. Moreover, caffeine’s metabolites, paraxanthine and theophylline, also provide protection against MPTP neurotoxicity (Xu et al., 2010). Other preclinical studies have shown that caffeine protects against dopaminergic neuron degeneration, dopamine loss and/or associated behavioral changes induced by 6-hydroxydopamine (6-OHDA) in rats (Joghataie et al., 2004; Aguiar et al., 2006; Kelsey et al., 2009) and by the pesticide combination of paraquat and manebin mice (Kachroo et al., 2010). Interestingly, the neuroprotective effect of caffeine could be dissociated from its psychomotor stimulant properties (Xu et al., 2002; Yu et al., 2008); whereas the latter showed tolerance to repeated administration, under the same conditions caffeine’s neuroprotective action persisted unabated (Xu et al., 2002).
Although the convergence of caffeine’s clinical correlations and protective effects in animal models supports a true beneficial effect in reducing PD risk, the mechanisms underlying neuroprotection by caffeine remain a matter of debate. Pharmacological studies indicate that its CNS effects are mediated primarily by its antagonistic actions at the A1 and A2A subtypes of adenosine receptors (Fredholm and Persson, 1982; Nehlig et al., 1992; Fredholm and Lindström, 1999; Fisone et al., 2004; Ferré et al., 2008). Interestingly, A2AR blockade, but not A1R blockade mimics caffeine’s protective effects in several experimental models of PD (Schwarzschild et al.,2006). In rodents, selective A2AR antagonists attenuate the loss of dopaminergic neurons in the SNpc or dopamine depletion in the striatum induced by either systemic administration of MPTP or acute infusion of 6-OHDA in the medial forebrain bundle (Chen et al., 2001; Ikeda et al., 2002: Pierri et al., 2005). A critical role of the A2AR in the MPTP model of PD was confirmed by the phenotype of global A2AR KO mice, which show preserved striatal dopamine content after acute MPTP multiple dose administration (Chen et al., 2001).
In order to determine whether protection by caffeine in fact requires the A2AR we investigated the effect of A2AR depletion on the ability of caffeine to protect against MPTP toxicity in A2AR KO and littermate control mice. In addition to a global (constitutive) A2AR KO line we employed conditional (Cre-loxP system) KO mice with cell-specific disruption of the A2AR gene either in astrocytes or (cortical and striatal) neurons based on transgenic cre expression driven by GFAP (Glial fibrillary acidic protein) or CamKIIα promoters, respectively. Our findings demonstrate an A2AR-dependent mechanism by which caffeine protects against MPTP neurotoxicity. They also show that astrocytic A2ARs are not required and forebrain neuronal A2ARs cannot fully account for caffeine’s neuroprotective effect in this model of PD.
Experimental procedures
Transgenic animals
Breeding and characterization of global A2AR knockout mice (A2A KO), CaMKIIα gene promoter-driven forebrain neuron A2AR cKO mice (CaMKIIα-A2AR KO) as well as the GFAP gene promoter-driven astrocyte A2AR cKO (gfap-A2AR KO) mice have been described previously (Chen et al., 1999, 2002; Bastia et al., 2005; Yu et al., 2008; Matos et al., 2015). Briefly, chimeric A2AR KO mice (F0) that were derived from 129-Steel embryonic stem cells were bred to C57BL/6 mice, resulting in mice of mixed C57BL/6 x 129-Steel background. The mixed line was then repeatedly backcrossed to pure C57BL/6 mice over 10 generations, yielding an A2AR KO line congenic for the C57BL/6 background. A2AR KO, heterozygous and wild-type (WT) littermates (male, 4–14 months old) were used in the global KO study. Both fb-A2AR KO and astro-A2AR KO mice were generated using a Cre/loxP strategy. Cre recombinase gene expression was controlled by either the forebrain (cortical and striatal) neuron-specific CaMKII-α promoter (Bastia et al., 2005) or astrocyte-specific GFAP gene promoter, as previously described elsewhere (Bajenaru et al., 2002). Both transgenic cre mice and “floxed” A2AR gene (A2Aflox/flox) mice were backcrossed for 10–12 generations to C57Bl/6 mice (Charles River; Wilmington, MA). Homozygous floxed (A2Aflox/flox) mice were crossed with cre(+) mice, and female cre(+) (A2Aflox/+) offspring were then crossed with A2Aflox/+ males. Their cre(+) A2Aflox/flox and cre(−) A2Aflox/flox offspring (male and females, 2–9 months old) were used in this study. Mice were housed five in each cage in temperature- and humidity-controlled rooms with a 12-h dark:light cycle and had free access to food and water. All experiments were conducted in accordance with Massachusetts General Hospital and NIH Guidelines on the ethical use of animals.
Drug treatment
In all experiments involving MPTP, a single injection of caffeine (25 mg/kg dissolved in saline) or saline was administered 10 min before a single intraperitoneal injection of MPTP (35mg/kg, HCl salt of MPTP dissolved in saline; corresponding to a MPTP•HCl dose of 29 mg/kg.
Locomotor activity
Horizontal locomotor activity was assessed in standard polypropylene cages (15 × 25 cm) placed into frames equipped to generate and detect five evenly spaced, parallel infrared light beams across the width of each cage just above its floor (San Diego Instruments, San Diego). Mice were habituated for more than 12 hours overnight in the testing cages that were placed in a behavioral suite. Basal spontaneous locomotion was recorded for at least 180 min. Then locomotor behavior was monitored during the light phase for another 240 min after caffeine (25 mg/kg or saline) ip injection. Locomotion, scored as the number of adjacent photobeam breaks (ambulation), was determined as described before (Xu et al., 2002).
Measurement of dopamine
One week after MPTP treatment, mice were killed by rapid cervical dislocation. The striatum was dissected out from the right cerebral hemisphere, frozen on dry ice and stored at −80°C until use. Each striatum was weighed, homogenized with 150 mM phosphoric acid and 0.2 mM EDTA and centrifuged at 12,000 g for 15 min at 4°C. Supernatants were analyzed for dopamine content using standard reverse-phase HPLC with electrochemical detection, according to our previously published protocol (Chen et al., 2001; Xu, et al., 2006). The dopamine content was calculated as picomoles per milligram of tissue, and these values are presented within the figures as percentage of change from respective saline–saline-treated controls.
Statistics
All values are expressed as mean ± SEM. Differences among means in dopamine content after MPTP treatment as well as peak locomotion were analyzed using two-way ANOVA with treatment (saline or caffeine) and genotype as independent factors. When ANOVA showed significant differences, pair-wise comparisons between means were tested by Fisher’s Least Significant Difference (LSD) post hoc testing. In all analyses, the null hypothesis was rejected at the 0.05 level. The analysis was generated using GraphPad.
Results
1. Caffeine’s protection against MPTP-induced dopamine depletion is lost in A2AR global KO mice
We first evaluated the effect of global genetic deletion of A2AR on caffeine’s neuroprotection against MPTP-induced dopamine depletion. MPTP (35 mg/kg ip single injection) significantly depleted striatal dopamine content measured one week later in WT, heterozygous and global A2AR KO mice. As reported previously (Xu et al. 2006), caffeine (25 mg/kg single ip injection 10 minutes before MPTP) significantly attenuated MPTP-induced dopamine depletion in WT mice. Similarly, caffeine also significantly attenuated MPTP-induced dopamine loss in heterozygous mice. However, caffeine pretreatment did not attenuated MPTP-induced dopamine loss in global A2AR KO mice (Figure 1). Although the global A2AR KO can have protective phenotype in multi-dose MPTP toxicity paradigms (Chen et al, 2001; Yu et al, 2008) no appreciable effect of the KO itself was observed here using a single MPTP injection paradigm (69% vs 75% loss of striatal dopamine induced by MPTP in A2AR KO vs WT mice, respectively; p>0.05).
Figure 1. Caffeine attenuated MPTP-induced striatal dopamine loss in WT and HZ, but not global A2AR KO male mice.
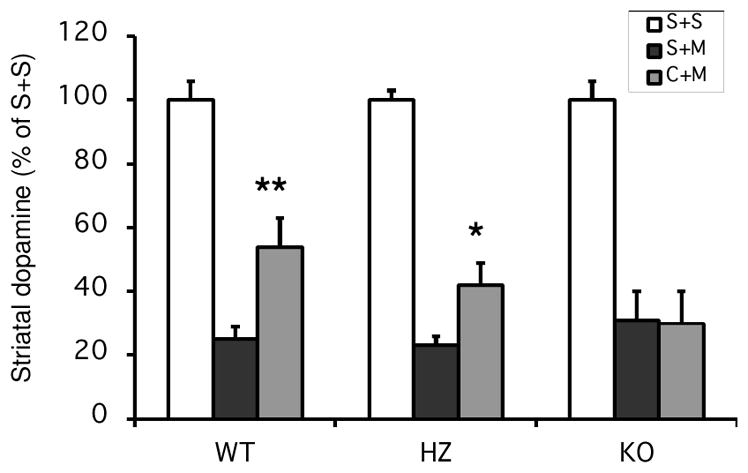
Saline or Caffeine (25 mg/kg ip) were administered 10min before saline or MPTP (35 mg/kg ip single injection). Dopamine content was determined one week after drug treatments. S, saline; M, MPTP; C, Caffeine. N=4 for saline treatments and N=5–14 for MPTP treatments. Bars represent striatal dopamine levels (mean ± SEM) calculated as percentage of their respective control (i.e., saline + saline treatment group). Statistically significant differences among the means of dopamine content in MPTP treated mice were determined by a two-way analysis of variance followed by Fisher’s Least Significant Difference test. There is a significant difference in caffeine versus saline treated groups (F[1,53]=7.11, p<0.05). *p<0.05 and **p<0.01 compared with respective S+M group.
2. Caffeine’s protection against MPTP-induced dopamine loss persists, although its motor stimulant effect is lost in forebrain neuronal A2A KO mice
We next examined the contribution of the A2AR in forebrain neurons to the neuroprotection by caffeine in MPTP-induced neurotoxicity. MPTP induced significant and similar dopamine loss in fb-A2AR KO mice and their non-transgenic littermate controls. In contrast to our findings with the global A2AR KO, caffeine pretreatment significantly attenuated MPTP-induced dopamine depletion in both fb-A2AR KO and control mice (Figure 2. The amount of residual dopamine with caffeine pretreatment was ~180% greater than without caffeine in control mice, whereas it was only ~64% greater in fb-A2AR KO mice (p=0.052, t-test; comparing the ‘Caffeine + MPTP’ mice without and with the cre transgene as shown in Figure 2). These data would suggest that caffeine’s attenuation of MPTP-induced striatal dopamine loss is at least partially independent of forebrain neuronal A2AR, though it may contribute partially to caffeine’s protective effect. In contrast to what was found for caffeine’s neuroprotective effect, caffeine stimulated locomotor activities in fb-A2AR WT but not fb-A2AR KO mice (Figures 3).
Figure 2. Caffeine’s attenuation of MPTP-induced striatal dopamine loss is at least partially independent of forebrain neuronal A2ARs.
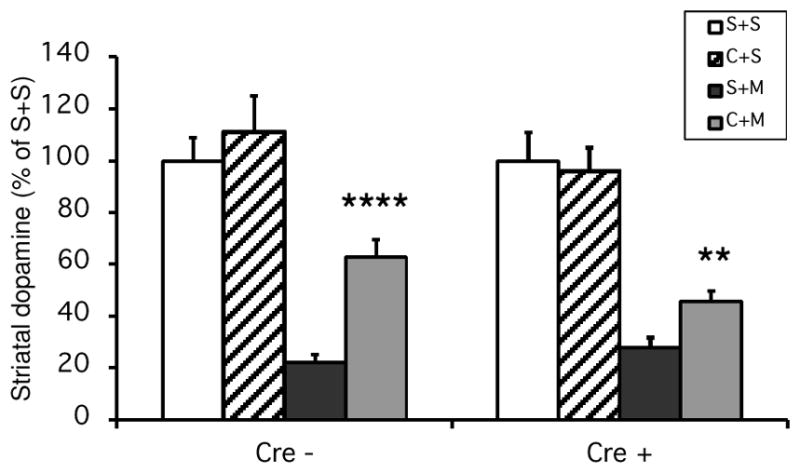
Saline or Caffeine (25 mg/kg ip) was administered 10 min before saline or MPTP (35 mg/kg ip single injection). Striatal dopamine was determined one week after drug treatments. N=3–7 for saline treatments and N=11–22 for MPTP treatments. Bars represent striatal dopamine levels (mean ± SEM) calculated as percentage of their respective control (i.e., saline + saline treatment group). Statistically significant differences among the means of dopamine content in MPTP treated mice were determined by a two-way analysis of variance followed by Fisher’s Least Significant Difference test. There is a significant difference in caffeine versus saline treated groups (F[1,60]=42.28, p<0.0001). **p<0.01, ****p<0.0001 compared with respective S+M group.
Figure 3. Dependence of caffeine-induced locomotion on neuronal A2ARs.

Ambulation was scored as the number of adjacent photobeam breaks (mean ± SEM, N=8 for each group). Mice were habituated overnight and basal spontaneous locomotion was recorded for at least 180 min. Ambulation was recorded for another 240 min after caffeine (25 mg/kg ip) or saline injection. A. Locomotion after saline injection is similar in WT and forebrain neuron A2AR cKO (CaMKII α -cre, A2Aflox/flox) mice. B. Caffeine-stimulated locomotion is significantly reduced in forebrain neuron A2AR cKO mice compared to that of WT mice. C. Bars represent peak locomotion (mean ± SEM) 180 minutes after caffeine or saline injection. Statistically significant differences among the peak locomotions were determined by a two-way analysis of variance followed by Fisher’s Least Significant Difference test. There is a significant difference in Cre versus No Cre mice (F[1,28]=6.77, p<0.05). There is also a significant difference in caffeine versus saline treated groups (F[1,28]=18.46, p<0.001). ***p<0.001 compared with caffeine treated group No Cre mice.
3. Caffeine’s neuroprotective and motor stimulant effects are unaltered in astrocyte-specific A2AR KO mice
Finally, we investigated the roles of A2AR in astrocytes in caffeine’s neuroprotection and motor stimulant effect. MPTP induced significant striatal dopamine loss in gfap-A2AR KO and their WT littermates. Caffeine pre-treatment significantly and similarly attenuated MPTP-induced dopamine loss in both gfap-A2AR WT and gfap-A2AR KO mice (Figure 4). We also studied caffeine’s motor stimulant effect in these gfap-A2AR KO and their WT littermates. In contrast to what was found in fb-A2AR KO mice, caffeine treatment stimulated locomotion in both gfap-A2AR KO mice to the same extent as in their matched controls (Figure 5).
Figure 4. Caffeine attenuated MPTP-induced striatal dopamine loss in both WT and astrocyte-directed A2AR cKO mice.

Saline or Caffeine (25 mg/kg ip) was administered 10 min before saline or MPTP (35 mg/kg ip single injection). Striatal dopamine was determined one week after drug treatments. N=3–6 for saline treatments and N=9–20 for MPTP treatments. Bars represent striatal dopamine levels (mean ± SEM) calculated as percentage of their respective control (i.e., saline + saline treatment group). Statistically significant differences among the means of dopamine content in MPTP treated mice were determined by a two-way analysis of variance followed by Fisher’s Least Significant Difference test. There is a significant difference in caffeine versus saline treated groups (F[1,51]=17.69, p=0.0001). *p<0.05, ***p<0.001 compared with respective S+M group.
Figure 5. Lack of dependence of caffeine-induced locomotion on astrocytic A2ARs.

Ambulation was scored as the number of adjacent photobeam breaks (mean ± SEM, N=8 for each group). Mice were habituated overnight and basal spontaneous locomotion was recorded for at least 180 min. Ambulation was recorded for another 240 min after caffeine (25 mg/kg ip) or saline injection. A. Locomotion is similar after saline injection in WT and astrocyte-directed A2AR cKO (gfap-cre, A2Aflox/flox) mice. B. Caffeine also stimulated similar locomotion in WT and astrocyte-directed A2AR cKO mice. Bars represent peak locomotion (mean ± SEM) 180 minutes after caffeine or saline injection. There is no statistical difference in locomotion after either saline or caffeine injection between Cre and No Cre mice.
Discussion
The main finding of our study is the demonstration that caffeine’s neuroprotection depends on adenosine A2AR. Although the use of a global A2AR KO helps establish an essential role of this receptor in caffeine’s neuroprotective effect, constitutive KO methodology has limitations such as its inability to distinguish between a role for the receptor during development or adulthood (Bockamp et al., 2002). Our results also suggest that caffeine’s protective effect is mostly dependent on A2AR other than those located on forebrain (striatal and cortical) neurons and on astrocytes.
Adenosine receptor antagonism has been shown to be the main mechanism of action responsible for the CNS effects of caffeine. Several studies have indicated that caffeine exerts its psychostimulant effects acting as a nonselective adenosine A1R and A2AR receptor antagonist (Fredholm and Persson, 1982; Nehlig et al., 1992; Fredholm and Lindström, 1999; Fisone et al., 2004; Ferré, 2008). Interestingly, the expression of these two adenosine receptors in the brain display very different patterns: whereas A1R are present throughout the brain, the expression of the A2AR is largely restricted to the striatum. Although brain A2AR were initially thought to be exclusively located in this region, several studies provided evidence also for the presence of A2AR in the hippocampus and neo-cortex (Rebola et al., 2005; reviewed in Cunha et al., 2001) at a lower density (Lopes et al., 2004).
Adenosine A2AR in the striatum act on the striatopallidal pathway to control locomotor activity, whereas the sparser extra-striatal A2AR may serve other functions such as facilitation of neurotransmitter release and modulation of neurodegeneration. In addition, compared to the predominant postsynaptic role, there may be a modulation of the pre-synaptic terminals by facilitating the evoked release of neurotransmitters (reviewed in Cunha et al., 2001). Moreover, several studies have focused on the importance of forebrain A2AR for PD neuroprotection (Carta et al., 2009) and possible extra-neuronal protective effects of A2AR antagonist (Yu et al., 2008).
Previous reports have suggested that adenosine may contribute to the pathological changes of Parkinson’s disease by triggering the activation of surrounding glial cells (Hirsch et al. 1999). A2AR activation is in fact partially responsible for cerebral inflammation and excitotoxicity (Popoli et al. 1995; Okusa et al. 1999; Sullivan et al. 1999) and A2ARs located on glial cells might play a role in neuroprotection mediated by A2AR antagonists against acute (Yu et al., 2008) and chronic MPTP-induced striatal dopamine depletion (Sonsalla et al., 2012) MPTP-induced striatal dopamine depletion.
Whereas several studies have reported a clear role of A1R in the behavioral effects of acutely administered caffeine with involvement of A2AR preferentially under conditions of chronic caffeine treatment (Karcz-Kubicha et al., 2003; Antoniou et al., 2005; Orrúet al., 2013), we have confirmed (Figure 3) that caffeine’s acute locomotor stimulant effect also requires the A2AR, specifically those expressed in forebrain (e.g., striatal) neurons (Yu et al, 2008).
Although the basis of caffeine’s neuroprotective properties appear to be in some ways distinct from those of its behavioral actions (Xu et al, 2002; Yu et al, 2008), we have now provided definitive evidence that caffeine’s ability to protect dopaminergic neurons can be entirely dependent on the A2AR, as previous suggested by other studies. For example, selective A2AR antagonists mimicked the protective effects of caffeine by reducing both MPTP (Chen et al., 2001) and 6-OHDA (Ikeda et al., 2002) induced neurotoxicity, mimicking the protective effects of caffeine. The findings suggest that caffeine’s protective effects, which can be mimicked by A2AR depletion (Chen et al. 1999; Li et al, 2009), are in fact mediated by the A2AR, as described in other neurotoxicity models like those for stroke (Fredholm et al. 1996) and traumatic brain injury (Li et al, 2008).
While our main results are generally in agreement with the findings of Chen et al. in 2001 and Yu et al. in 2008, the depletion of A2AR in the global A2AR KO mice in the current study was not sufficient to attenuate MPTP neurotoxicity in contrast to the earlier studies. This apparent discrepancy could be related to the different MPTP•HCl doses and dosing paradigms used across these experiments (a single injection of 35 mg/kg ip here compared to multiple injections of 20 mg/kg ip hours apart in the earlier studies). Differences in MPTP dose amounts and timing are known to induce different patterns of neuronal death. (e.g., apoptotic vs necrotic; Meredith et al., 2011), and thus potentially different dependencies on the A2AR. Interestingly, recent evidence that caffeine may produce some CNS effects through inverse agonism (rather than competitive antagonism of endogenous adenosine) at the A2AR (Fernández-Dueñas et al., 2014) suggests a possible explanation for how caffeine’s protective effect could be A2AR-dependent, without A2AR depletion having an effect of its own, in the MPTP paradigm employed in our study.
Selective deletion of A2AR in forebrain neurons showed that caffeine’s attenuation of MPTP-induced striatal dopamine loss is at least partially independent of forebrain neuronal A2ARs, although locomotion appeared fully dependent upon them. These results are in agreement with previous studies that suggested that selective A2AR antagonists (KW-6002) did not require forebrain neurons to protect against MPTP neurotoxicity (Yu et al., 2008). Extra-striatal A2AR, characterized in Bastia et al. 2005, have been shown to have a critical role in providing a prominent effect on psychomotor activity induced by amphetamine, cocaine and phencyclidine (Bastia et al., 2005; Shen et al., 2008) and a recent study has also considered this extra-striatal A2AR to have a fundamental role of in PD neuroprotection (Carta et al., 2009).
In our study, selective depletion of fb-A2AR did not abolish the effect of caffeine on MPTP-induced dopamine loss, suggesting that caffeine neuroprotection on acute MPTP toxicity does not fully depend on A2AR in forebrain neurons. On the other hand, we found out that caffeine’s locomotor activating properties are still entirely dependent upon the presence of fb-A2AR, suggesting that caffeine stimulates motor activity through A2AR in forebrain neurons, likely those in the striatum. In complete agreement with these results, the selective deletion of A2AR in forebrain neurons abolished motor effects of A2AR antagonist KW-6002 (Yu et al., 2008). They are, however, in contrast with a previous report on the selective deletion of A2AR from forebrain neurons preventing dopaminergic neuron loss and gliosis in the SNpc following multiple MPTP injections (Carta et al., 2009). These discrepancies might involve other paradigm variables, such as different MPTP treatment paradigms as above, or different readouts for dopaminergic toxicity (TH-positive SNpc cells vs striatal dopamine content).
Astrocytic A2ARs also warranted consideration as candidate mediators of neuroprotection by caffeine. Brain glial cells may express A2ARs (Fiebich et al. 1996; Saura et al. 2005), and A2ARs on astrocytes may contribute to excitotoxic neurodegeneration (Nishizaki, 2004; Matos et al, 2012). However, using astrocyte-directed conditional A2AR KO mice generated by a Cre-loxP system based on the specificity of GFAP gene promoter, we showed that the selective depletion of A2AR from astrocytes affected neither caffeine’s motor effect nor its protection against MPTP-induced neurotoxicity. Thus at least in the paradigms we employed, caffeine does not require astrocyte A2ARs to protect dopaminergic neurons or stimulate motor activity.
Other possible cellular sources of A2ARs should be considered in addition to striatal and cortical neurons and astrocytes. These include A2AR-expressing microglial cells (Fiebich et al., 1996; Hasko et al., 2005) and oligodendrocytes (Stevens et al., 2002). Caffeine or selective A2AR antagonists were shown to attenuate microglia recruitment to sites of injury and to reduce the production of pro-inflammatory cytokines (Brambilla et al., 2003; Brothers et al., 2010; Rebola et al., 2011). Microglial activation during inflammatory processes observed in the brains of older rats is partially reversed by chronic caffeine (Brothers et al., 2010). Furthermore, activation of A2ARs has been associated with release of brain-derived neurotrophic factor (BDNF) and proliferation of microglial cells, which are intrinsically related events of neuroinflammation. There is also evidence that non-CNS cells expressing the A2AR, such as bone marrow cells, may contribute to ischemic brain cell injury, as suggested by Yu et al, in 2004. In addition, it seems that these receptors in peripheral cells may be modulators of inflammatory cytokine production (Ran et al., 2015).
Conclusion
The finding of the complete loss of neuroprotection by caffeine in global A2AR KO mice establishes the adenosine A2AR as a critical mediator of caffeine’s neuroprotective effects in the MPTP model of Parkinson’s disease. Our conditional KO data suggest that caffeine’s neuroprotection is at least partially independent of A2ARs in the forebrain neurons or astrocytes. The exact location of A2AR that is responsible for caffeine’s neuroprotection is still unknown. Understanding the neurobiological basis of caffeine’s putative benefit is of increasing translational significance not only in explaining the epidemiological between caffeine use and reduced risk of PD (Liu et al, 2012, Ascherio et al., 2001, 2003). The findings also support therapeutic development of caffeine and more specific adenosine A2AR antagonists as candidate disease-modifying agents, for example, with a long-term clinical trial of caffeine having been initiated in PD (http://clinicaltrials.gov/show/NCT01738178). Continued research is warranted to identify CNS A2ARs other than those residing on striatal and cortical neurons that may contribute to neurodegeneration.
Highlights.
Neuroprotection by caffeine requires the adenosine A2AR
Neuroprotection by caffeine depends in part on A2ARs other than those on forebrain neurons
Caffeine’s locomotor activating properties are dependent upon forebrain A2ARs
Caffeine does not require astrocyte A2ARs to protect dopaminergic neurons
Acknowledgments
Source of support: This work is supported by NIH grants 5R01ES010804 and 5K24NS60991 and DOD grant W81XWH-11-1-0150. The Parkinson’s Disease Foundation has provided funding supporting to Daniel Garbin Di Luca as a summer research fellow.
Footnotes
Disclosure of potential conflicts of interest:
None reported.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Kui Xu, Email: kuixu@hotmail.com.
Daniel Garbin Di Luca, Email: dilucadaniel@gmail.com.
Marco Orrú, Email: orrumarco@gmail.com.
Yuehang Xu, Email: xuy@helix.mgh.harvard.edu.
Jiang-Fan Chen, Email: chenjf@bu.edu.
Michael A. Schwarzschild, Email: michaels@helix.mgh.harvard.edu.
References
- Aguiar LM, Nobre HV, Jr, Macêdo DS, Oliveira AA, Freitas RM, Vasconcelos SM, Cunha GM, Sousa FC, Viana GS. Neuroprotective effects of caffeine in the model of 6-hydroxydopamine lesion in rats. Pharmacol Biochem Behav. 2006;84:415–419. doi: 10.1016/j.pbb.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Weisskopf MG, O’Reilly EJ, McCullough ML, Calle EE, Rodriguez C, Thun MJ. Coffee consumption, gender, and Parkinson’s disease mortality in the cancer prevention study II cohort: the modifying effects of estrogen. Am J Epidemiol. 2004;160:977–984. doi: 10.1093/aje/kwh312. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Zhang SM, Hernan MA, Kawachi I, Colditz GA, Speizer FE, Willett WC. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann Neurol. 2001;50:56–63. doi: 10.1002/ana.1052. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Schwarzschild MA, Zhang SM, Colditz GA, Speizer FE. Caffeine, postmenopausal estrogen, and risk of Parkinson’s disease. Neurology. 2003;60:790–795. doi: 10.1212/01.wnl.0000046523.05125.87. [DOI] [PubMed] [Google Scholar]
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastia E, Xu YH, Scibelli AC, Day YJ, Linden J, Chen J-F, Schwarzschild MA. A Crucial Role for Forebrain Adenosine A2A Receptors in Amphetamine Sensitization. Neuropsychopharmacology. 2005;30:891–900. doi: 10.1038/sj.npp.1300630. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Bockamp E, Maringer M, Spangenberg C, Fees S, Fraser S, Eshkind L, Oesch F, Zabel B. Of mice and models: improved animal models for biomedical research. Physiol Genomics. 2002;11:115–132. doi: 10.1152/physiolgenomics.00067.2002. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Cottini L, Fumagalli M, Ceruti S, Abbracchio MP. Blockade of A2A adenosine receptors prevents basic fibroblast growth factor-induced reactive astrogliosis in rat striatal primary astrocytes. Glia. 2003;43:190–194. doi: 10.1002/glia.10243. [DOI] [PubMed] [Google Scholar]
- Brothers HM, Marchalant Y, Wenk GL. Caffeine attenuates lipopolysaccharide-induced neuroinflammation. Neurosci Lett. 2010;480:97–100. doi: 10.1016/j.neulet.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta AR, Kachroo A, Schintu N, Xu K, Schwarzschild MA, Wardas J, Morelli M. Inactivation of neuronal forebrain A receptors protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurochem. 2009;111:1478–1489. doi: 10.1111/j.1471-4159.2009.06425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Moratalla R, Impagnatiello F, Grandy DK, Cuellar B, Rubinstein M, Beilstein MA, Hackett E, Fink JS, Low MJ, Ongini E, Schwarzschild MA. The role of the D(2) dopamine receptor (D(2)R) in A(2A) adenosine receptor (A(2A)R)-mediated behavioral and cellular responses as revealed by A(2A) and D(2) receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98:1970–1975. doi: 10.1073/pnas.98.4.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli N, Jr, Schwarzschild MA. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J Neurosci. 2001b;21:RC143. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- Dawson T, Mandir A, Lee M. Animal models of PD: pieces of the same puzzle? Neuron. 2002;35:219–222. doi: 10.1016/s0896-6273(02)00780-8. [DOI] [PubMed] [Google Scholar]
- Fernández-Dueñas V, Gómez-Soler M, López-Cano M, Taura JJ, Ledent C, Watanabe M, Jacobson KA, Vilardaga JP, Ciruela F. Uncovering caffeine’s adenosine A2A receptor inverse agonism in experimental parkinsonism. ACS Chem Biol. 2014 Nov 21;9(11):2496–501. doi: 10.1021/cb5005383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S. An update on the mechanisms of the psychostimulant effects of caffeine. J Neurochem. 2008;105:1067–1079. doi: 10.1111/j.1471-4159.2007.05196.x. [DOI] [PubMed] [Google Scholar]
- Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebicke-Haerter PJ. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia. 1996;18:152–160. doi: 10.1002/(SICI)1098-1136(199610)18:2<152::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Fisone G, Borgkvist A, Usiello A. Caffeine as a psychomotor stimulant: mechanism of action. Cell Mol Life Sci. 2004;61:857–872. doi: 10.1007/s00018-003-3269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Lindström K. Autoradiographic comparison of the potency of several structurally unrelated adenosine receptor antagonists at adenosine A1 and A(2A) receptors. Eur J Pharmacol. 1999;380:197–202. doi: 10.1016/s0014-2999(99)00533-6. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Adén U, Lindström K, Bona E, Hagberg H. Caffeine and ischemia--effects on immediate early genes and adenosine receptors. Adv Neurol. 1996;71:469–474. [PubMed] [Google Scholar]
- Fredholm BB, Persson CG. Xanthine derivatives as adenosine receptor antagonists. Eur J Pharmacol. 1982;81:673–676. doi: 10.1016/0014-2999(82)90359-4. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S, Damier P, Brugg B, Faucheux BA, Michel PP, Ruberg M, Muriel MP, Mouatt-Prigent A, Agid Y. Glial cell participation in the degeneration of dopaminergic neurons in Parkinson’s disease. Adv Neurol. 1999;80:9–18. [PubMed] [Google Scholar]
- Hasko G, Pacher P, Vizi ES, Illes P. Adenosine receptor signaling in the brain immune system. Trends Pharmacol Sci. 2005;26:511–516. doi: 10.1016/j.tips.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Kurokawa M, Aoyama S, Kuwana Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J Neurochem. 2002;80:262–270. doi: 10.1046/j.0022-3042.2001.00694.x. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc. 2007;2:141–151. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- Joghataie MT, Roghani M, Negahdar F, Hashemi L. Protective effect of caffeine against neurodegeneration in a model of Parkinson’s disease in rat: behavioral and histochemical evidence. Parkinsonism Relat Disord. 2004;10:465–468. doi: 10.1016/j.parkreldis.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Kachroo A, Prasad K, Irizarry MC, Richfield EK, Schwarzschild MA. Caffeine protects against combined paraquat and maneb-induced neurotoxicity of dopaminergic nigral neurons. Program #265.20.2007 Society for Neuroscience Annual Meeting; San Diego, CA. 2007.2007. [Google Scholar]
- Karcz-Kubicha M, Quarta D, Hope BT, Antoniou K, Müller CE, Morales M, Schindler CW, Goldberg SR, Ferré S. Enabling role of adenosine A1 receptors in adenosine A2A receptor-mediated striatal expression of c-fos. Eur J Neurosci. 2003;18:296–302. doi: 10.1046/j.1460-9568.2003.02747.x. [DOI] [PubMed] [Google Scholar]
- Kelsey JE, Langelier NA, Oriel BS, Reedy C. The effects of systemic, intrastriatal, and intrapallidal injections of caffeine and systemic injections of A2A and A1 antagonists on forepaw stepping in the unilateral 6-OHDA-lesioned rat. Psychopharmacology (Berl) 2009;201:529–539. doi: 10.1007/s00213-008-1319-0. [DOI] [PubMed] [Google Scholar]
- Kopin IJ, Markey SP. MPTP toxicity: implication for research in Parkinson’s disease. Annu Rev Neurosci. 1988;11:81–96. doi: 10.1146/annurev.ne.11.030188.000501. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Rebert CS, Irwin I. Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl 1-1,2,3,6, tetrahydropyridine (MPTP) in the squirrel monkey. Brain Res. 1984;292:390–394. doi: 10.1016/0006-8993(84)90777-7. [DOI] [PubMed] [Google Scholar]
- Li W, Dai S, An J, Li P, Chen X, Xiong R, Liu P, Wang H, Zhao Y, Zhu M, Liu X, Zhu P, Chen JF, Zhou Y. Chronic but not acute treatment with caffeine attenuates traumatic brain injury in the mouse cortical impact model. Neuroscience. 2008;151:1198–1207. doi: 10.1016/j.neuroscience.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Li W, Dai S, An J, Xiong R, Li P, Chen X, Zhao Y, Liu P, Wang H, Zhu P, Chen J, Zhou Y. Genetic inactivation of adenosine A2A receptors attenuates acute traumatic brain injury in the mouse cortical impact model. Exp Neurol. 2009;215:69–76. doi: 10.1016/j.expneurol.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Liu R, Guo X, Park Y, Huang X, Sinha R, Freedman ND, Hollenbeck AR, Blair A, Chen H. Caffeine intake, smoking, and risk of Parkinson disease in men and women. Am J Epidemiol. 2012;175:1200–1207. doi: 10.1093/aje/kwr451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LV, Halldner L, Rebola N, Johansson B, Ledent C, Chen JF, Fredholm BB, Cunha RA. Binding of the prototypical adenosine A2A receptor agonist, CGS 21680, to the cerebral cortex of adenosine A1 and A2A receptor knockout mice. Br J Pharmacol. 2004;141:1006–1014. doi: 10.1038/sj.bjp.0705692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos M, Augusto E, Santos-Rodrigues AD, Schwarzschild MA, Chen JF, Cunha RA, Agostinho P. Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia. 2012;60:702–716. doi: 10.1002/glia.22290. [DOI] [PubMed] [Google Scholar]
- Matos M, Shen HY, Augusto E, Wang Y, Wei CJ, Wang YT, Agostinho P, Boison D, Cunha RA, Chen JF. Deletion of Adenosine A2A Receptors from Astrocytes Disrupts Glutamate Homeostasis Leading to Psychomotor and Cognitive Impairment: Relevance to Schizophrenia. Biol Psychiatry. 2015;78:763–774. doi: 10.1016/j.biopsych.2015.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith GE, Rademacher DJ. MPTP Mouse Models of Parkinson’s Disease: An Update. J Parkinsons Dis. 2011;1:19–33. doi: 10.3233/JPD-2011-11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehlig A, Daval JL, Debry G. Caffeine and the central nervous system: mechanisms of action, biochemical, metabolic and psychostimulant effects. Brain Res Brain Res Rev. 1992;17:139–170. doi: 10.1016/0165-0173(92)90012-b. [DOI] [PubMed] [Google Scholar]
- Nishizaki T. ATP- and adenosine-mediated signaling in the central nervous system: adenosine stimulates glutamate release from astrocytes via A2a adenosine receptors. J Pharmacol Sci. 2004;94:100–102. doi: 10.1254/jphs.94.100. [DOI] [PubMed] [Google Scholar]
- Okusa MD, Linden J, Macdonald T, Huang L. Selective A2A adenosine receptor activation reduces ischemia-reperfusion injury in rat kidney. Am J Physiol. 1999;277:404–412. doi: 10.1152/ajprenal.1999.277.3.F404. [DOI] [PubMed] [Google Scholar]
- Orrú M, Guitart X, Karcz-Kubicha M, Solinas M, Justinova Z, Barodia SK, Zanoveli J, Cortes A, Lluis C, Casado V, Moeller FG, Ferré S. Psychostimulant pharmacological profile of paraxanthine the main metabolite of caffeine in humans. Neuropharmacology. 2013;67:476–484. doi: 10.1016/j.neuropharm.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios N, Gao X, McCullough ML, Schwarzschild MA, Shah R, Gapstur S, Ascherio A. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov Disord. 2012;27:1276–1282. doi: 10.1002/mds.25076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierri M, Vaudano E, Sager T, Englund U. KW-6002 protects from MPTP induced dopaminergic toxicity in the mouse. Neuropharmacology. 2005;48:517–524. doi: 10.1016/j.neuropharm.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Popoli P, Betto P, Reggio R, Ricciarello G. Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol. 1995;287:215–217. doi: 10.1016/0014-2999(95)00679-6. [DOI] [PubMed] [Google Scholar]
- Ran H, Duan W, Gong Z, Xu S, Zhu H, Hou X, Jiang L, He Q, Zheng J. Critical contribution of adenosine A2A receptors in bone marrow derived cells to white matter lesions induced bychronic cerebral hypoperfusion. J Neuropathol Exp Neurol. 2015;74:305–318. doi: 10.1097/NEN.0000000000000174. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Irwin I, Forno LS, DeLanney LE, Langston E, Langston JW. Aging and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced degeneration of dopaminergic neurons in the substantia nigra. Brain Res. 1987;403:43–51. doi: 10.1016/0006-8993(87)90120-x. [DOI] [PubMed] [Google Scholar]
- Rebola N, Canas PM, Oliveira CR, Cunha RA. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience. 2005;132:893–903. doi: 10.1016/j.neuroscience.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Rebola N, Simões AP, Canas PM, Tomé AR, Andrade GM, Barry CE, Agostinho PM, Lynch MA, Cunha RA. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J Neurochem. 2011;117:100–111. doi: 10.1111/j.1471-4159.2011.07178.x. [DOI] [PubMed] [Google Scholar]
- Ross GW, Abbott RD, Petrovitch H, Morens DM, Grandinetti A, Tung KH, Tanner CM, Masaki KH, Blanchette PL, Curb JD, Popper JS, White LR. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA. 2000;283:2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- Saura J, Angulo E, Ejarque A, Casadó V, Tusell JM, Moratalla R, Chen JF, Schwarzschild MA, Lluis C, Franco R, Serratosa J. Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J Neurochem. 2005;95:919–929. doi: 10.1111/j.1471-4159.2005.03395.x. [DOI] [PubMed] [Google Scholar]
- Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Targeting adenosine A2A receptors in Parkinson’s disease. Trends Neurosci. 2006;29:647–654. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Wong LY, Harris SL, Richardson JR, Khobahy I, Li W, et al. Delayed caffeine treatment prevents nigral dopamine neuron loss in a progressive rat model of Parkinson’s disease. Exp Neurol. 2012;234:482–487. doi: 10.1016/j.expneurol.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron. 2002;36:855–868. doi: 10.1016/s0896-6273(02)01067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan GW, Linden J. Role of A2A adenosine receptors in inflammation. Drug Dev Res. 1998;45:103–112. [Google Scholar]
- Xu K, Xu Y, Brown-Jermyn D, Chen JF, Ascherio A, Dluzen DE, Schwarzschild MA. Estrogen prevents neuroprotection by caffeine in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J Neurosci. 2006;26:535–541. doi: 10.1523/JNEUROSCI.3008-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Xu Y-H, Chen J-F, Schwarzschild MA. Neuroprotection by caffeine: Time course and role of its metabolites in the MPTP model of Parkinson Disease. Neuroscience. 2010;167:475–481. doi: 10.1016/j.neuroscience.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Xu YH, Chen JF, Schwarzschild MA. Caffeine’s neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity shows no tolerance to chronic caffeine administration in mice. Neurosci Lett. 2002;322:13–16. doi: 10.1016/s0304-3940(02)00069-1. [DOI] [PubMed] [Google Scholar]
- Yu L, Huang Z, Mariani J, Wang Y, Moskowitz M, Chen JF. Selective inactivation or reconstitution of adenosine A2A receptors in bone marrow cells reveals their significant contribution to the development of ischemic brain injury. Nat Med. 2004;10:1081–1087. doi: 10.1038/nm1103. [DOI] [PubMed] [Google Scholar]
- Yu L, Shen HY, Coelho JE, Araújo IM, Huang QY, Day YJ, Rebola N, Canas PM, Rapp EK, Ferrara J, Taylor D, Müller CE, Linden J, Cunha RA, Chen JF. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol. 2008;63:338–346. doi: 10.1002/ana.21313. [DOI] [PubMed] [Google Scholar]