

Abstract
Lipophilic bisphosphonium salts are among the most promising antiprotozoal leads currently under investigation. As part of their preclinical evaluation we here report on their mode of action against African trypanosomes, the etiological agents of sleeping sickness. The bisphosphonium compounds CD38 and AHI-9 exhibited rapid inhibition of Trypanosoma brucei growth, apparently the result of cell cycle arrest that blocked the replication of mitochondrial DNA, contained in the kinetoplast, thereby preventing the initiation of S-phase. Incubation with either compound led to a rapid reduction in mitochondrial membrane potential, and ATP levels decreased by approximately 50% within 1 h. Between 4 and 8 h, cellular calcium levels increased, consistent with release from the depolarized mitochondria. Within the mitochondria, the Succinate Dehydrogenase complex (SDH) was investigated as a target for bisphosphonium salts, but while its subunit 1 (SDH1) was present at low levels in the bloodstream form trypanosomes, the assembled complex was hardly detectable. RNAi knockdown of the SDH1 subunit produced no growth phenotype, either in bloodstream or in the procyclic (insect) forms and we conclude that in trypanosomes SDH is not the target for bisphosphonium salts. Instead, the compounds inhibited ATP production in intact mitochondria, as well as the purified F1 ATPase, to a level that was similar to 1 mM azide. Co-incubation with azide and bisphosphonium compounds did not inhibit ATPase activity more than either product alone. The results show that, in T. brucei, bisphosphonium compounds do not principally act on succinate dehydrogenase but on the mitochondrial FoF1 ATPase.
Keywords: Trypanosoma brucei, Mitochondrion, FoF1 ATPase, Succinate dehydrogenase, Phosphonium salt, SDH complex
Graphical abstract
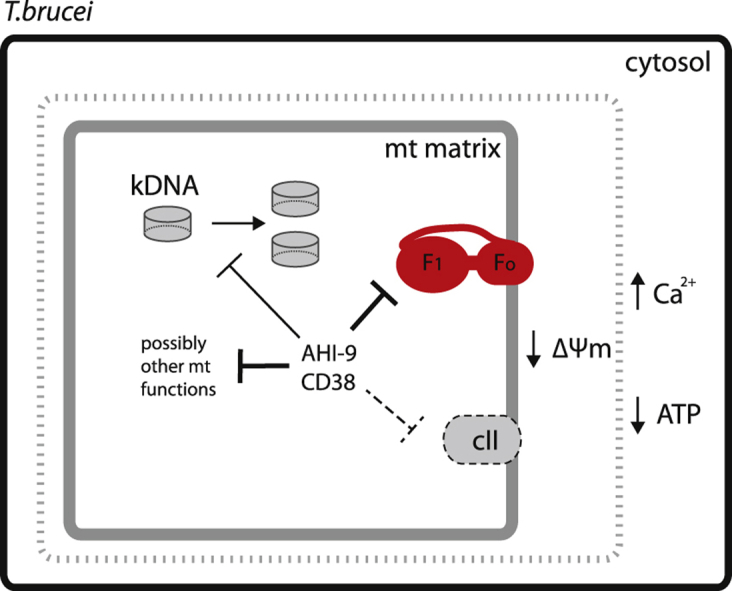
Highlights
-
•
Bisphosphonium salts display highly promising antiprotozoal activity.
-
•
It has been reported that, in Leishmania, they act on the mitochondrial SDH complex.
-
•
We show that in Trypanosoma brucei SDH is not essential and not the drug target.
-
•
Instead, we present strong evidence that the F1F0 ATPase is the target.
1. Introduction
African trypanosomes are responsible for a spectrum of important human and veterinary diseases south of the Sahara, transmitted by various tsetse fly species. Trypanosoma brucei gambiense causes a chronic but fatal human African trypanosomiasis (HAT, or sleeping sickness) in Western and Central Africa, whereas T. b. rhodesiense causes a much more acute illness in Eastern and Southern Africa (Brun et al., 2010); both forms of the disease are considered almost invariably fatal if left untreated. Whereas transmission of T. b. gambiense appears to be almost exclusively anthroponotic, T. b. rhodesiense is a zoonotic parasite, with many wild and domestic animals, particularly cattle, acting as reservoirs (Welburn et al., 2001). In addition, Trypanosoma congolense, T. b. brucei, Trypanosoma evansi and Trypanosoma vivax cause animal African trypanosomiasis (AAT), inflicting a terrible burden on agriculture in the tsetse belt and, for the latter two species, also in areas far beyond the testse habitat including the Indian subcontinent and South America (Desquesnes et al., 2013, Osório et al., 2008, Swallow, 1999).
While vector control in conjunction with extensive surveillance can have highly significant local impact on the disease burden, there is no realistic prospect of a vaccine (La Greca and Magez, 2011) and chemotherapy is practically the only option in most areas (Delespaux and de Koning, 2007, Jannin and Cattand, 2004). However, few drugs currently exist for either HAT or AAT, and these are old, suffer from severe side-effects and/or resistance, and are usually only effective against certain (sub) species or stages of the disease (Brun et al., 2010, Delespaux and de Koning, 2007). New therapeutic agents are therefore urgently necessary, preferably active against all African trypanosome species, and against both stages of the human disease. One promising lead is the class of benzyltriphenylphosphonium compounds that displays highly potent activity against rodent models of T. b. rhodesiense (Kinnamon et al., 1979), Trypanosoma cruzi (Kinnamon et al., 1977) and Leishmania donovani (Hanson et al., 1977) infections. Moreover, a large series of bisphosphonium salts were shown to possess strong anti-leishmanial (Luque-Ortega et al., 2010) and trypanocidal (Dardonville et al., 2015, Taladriz et al., 2012) activity in vitro. Many of these compounds showed high selectivity against the kinetoplastid parasites relative to human cell lines and, importantly, displayed no cross-resistance with existing trypanocides such as diamidines and melaminophenyl arsenicals (Taladriz et al., 2012).
The triphenylphosphonium (TPP) moiety has been used extensively as a vehicle to deliver drugs to mitochondrial targets (Cairns et al., 2015, Cortes et al., 2015, Smith et al., 2011). Among many applications, TPP has been used to deliver functional probes (Cairns et al., 2015), antioxidants (Kelso et al., 2001, Teixeira et al., 2012), anti-cancer drugs (Jara et al., 2014), and even liposomes (Benein et al., 2015) to mitochondria. TPP-linked natural compounds botulin and betulinic acid showed in vitro and in vivo activity against Schistosoma mansoni (Spivak et al., 2014). Crucially, TPP-mediated mitochondrial targeting appears to be generally safe, as a double-blind, placebo controlled study with human volunteers found no evidence of side-effects of the TPP-coupled antioxidant MitoQ over a 12-month period (Snow et al., 2010).
The mitochondrial accumulation of TPP-coupled drugs is driven by the strong inside-negative potential across the inner membrane of the mitochondrion. TPP, being a lipophilic cation with a highly dispersed charge is believed to diffuse freely across the inner membrane and be highly concentrated in the mitochondrial matrix, to an extent determined by the mitochondrial membrane potential Ψm and the plasma membrane potential Vm, as described by the Nernst equation (Cairns et al., 2015). Consistent with the extensive TPP literature, Luque-Ortega et al. showed that the anti-leishmanial TPP analogues targeted the parasite's mitochondrion, and proposed their principal action to be the inhibition of the succinate dehydrogenase complex (complex II) (Luque-Ortega et al., 2010). However, similar compounds also had strong activity against bloodstream form (BF) T. brucei (Dardonville et al., 2015, Taladriz et al., 2012), which has a much less elaborate mitochondrial metabolism, lacking for instance much of the Krebs cycle, and the cytochrome-dependent respiratory chain (Tielens and van Hellemond, 2009). Transfer of electrons to oxygen is instead mediated by a plant-like alternative oxidase (Chaudhuri et al., 1998) and the mitochondrial membrane potential is maintained by the FoF1 ATPase pumping protons from the mitochondrial matrix (Nolan and Voorheis, 1992, Schnaufer et al., 2005). We thus investigated whether the succinate dehydrogenase complex is expressed in BF T. brucei, and whether this might be the primary target for the trypanocidal activity of triphenylphosphonium salts and their analogues. We selected two compounds with strong trypanocidal activity (Taladriz et al., 2012) to represent the main two classes of aliphatic and aromatic bisphosphonium salts (Fig. 1).
Fig. 1.

Benzophenone-derived bisphosphonium salts used in this study.
2. Materials and methods
2.1. Effect of bisphosphonium compounds on growth of BF T. brucei
The effects of AHI-9 and CD38 on growth of BF Trypanosoma brucei brucei strain Lister 427 were investigated by incubating cultures under standard conditions (HMI-9/10% FBS; 37 °C, 5% CO2) in the presence or absence of 0.1 μM, 0.3 μM or 1 μM test compound for up to 72 h. Samples were taken in triplicate at the following times (h) after initiation of the experiment: 0, 4, 8, 12, 24, 28, 32, 36, 48, 52, 56, 60, 72. Cell density in the samples was determined using a haemocytometer. Compounds CD38 and AHI-9 were synthesized as reported previously (Luque-Ortega et al., 2010, Taladriz et al., 2012).
2.2. Determination of trypanocidal action using the Alamar blue assay
Fifty percent effective concentrations (EC50) were determined using the fluorescence viability indicator dye Alamar Blue (resazurin sodium salt, Sigma), exactly as described (Gould et al., 2013, Gould et al., 2008). Serial doubling dilutions of test compounds were prepared over two rows of a 96-well plate, leaving the last well without added drug as a control, before the addition of an equal volume of cell suspension to each well. Final cell density in each well was 105 BF T. brucei per mL. The plates were incubated for 48 h (37 °C, 5% CO2) before the addition of 20 μL resazurin solution (125 μg/mL in PBS pH7.4) per well and incubation for a further 24 h. Fluorescence was determined using a FLUOstar Optima (BMG Labtech, Durham, NC, USA) with excitation wavelength set at 544 nm and emission at 620 nm. Data were plotted to a sigmoid curve with variable slope using Prism 5.0 (GraphPad, San Diego, CA).
2.3. Assessing cell cycle progression in T. brucei
2.3.1. DAPI staining
Nuclei and kinetoplasts were visualized using the fluorescent dye 4,6-diamidino-2-phenylindole (DAPI) on BF trypanosomes after fixation. 50 μL of cells at ∼5 × 105 cells/mL were spread onto a glass microscope slide, left to air dry and fixed in methanol overnight at −20 °C. The slides were rehydrated with 1 mL of PBS for 10 min, which was allowed to evaporate (but not to completely dry). 50 μL of PBS containing 1 μg/mL DAPI and 1% of 1,4-diazabicyclo [2.2.2] octane (DAPCO) was added to the slides and spread by coverslip. Slides were observed under UV light on a Zeiss Axioplan microscope using a Hamamatso digital camera and Openlab software. 500 cells were recorded for each sample, and scored for DNA configuration into the following groups: 1N1K, 1N2K, 2N2K (Early) and 2N2K (Late) (N, nucleus; K, kinetoplast; ‘Early’ no ingression furrow; ‘Late’, cell division started with clear ingression furrow). The effect of test compounds on DNA configuration was determined at 0, 8, 12, 16 and 24 h; untreated cultures served as control.
2.3.2. Flow cytometry
DNA content of BF trypanosomes was measured by flow cytometry using the fluorescent dye propidium iodide (PI), as described (Hammarton et al., 2003, Ibrahim et al., 2011). Briefly, 1 mL of cell suspension at 106 cells/mL was transferred at each time point into microfuge tubes and centrifuged at 2500 rpm for 10 min at 4 °C, then resuspended and fixed in 1 mL of 70% methanol and 30% PBS and left at 4 °C overnight. The samples were washed twice with 1 mL of PBS and subsequently resuspended in 1 mL PBS containing propidium iodide and RNase A (both at 10 μg/mL), and incubated at 37 °C for 45 min while protected from light. Samples were analysed by Becton Dickinson FACSCalibur using the FL2-Area detector and CellQuest software. ModFit LT software was used to quantify the flow cytometry results.
2.4. Measuring intracellular calcium in BF T. brucei
Intracellular calcium levels were measured using the Screen Quest™ Fluo-8 Calcium Kit (ABD Bioquest, Sunnyvale, CA). T. b. brucei of strain Lister 427 were grown in HMI-9/FCS in vented flasks incubated (37 °C/5% CO2 for 48 h) and harvested by centrifugation (2500 × g, 10 min at 4 °C), resuspended to a final density of 4 × 106 cells/mL and divided into aliquots for determination at 0, 4, 8, 12 h of exposure to test compounds (in HMI-9/FCS, 37 °C/5% CO2). At an appropriate interval before the start of the measurement, aliquots of this culture were transferred to centrifuge tubes, centrifuged (10 min, 2500 g) and resuspended in the Fluo-8 dye-loading solution at exactly 4 × 106 cells/mL and further incubated at 37 °C for 30 min. The cells were then washed twice with assay buffer to remove any extracellular dye-loading solution. 90 μL of cell suspension was added to each well of a black-bottomed 96-well plate, and subsequently 10 μL of test compound at 10 × concentration was added (except for the 0 min recording); 10 μM calcium ionophore A23187 (Sigma) was used as a positive control and 10 μL buffer was added to a well that served as drug-free control. The plate was then incubated in a FLUOstar OPTIMA fluorimeter at 37 °C, and the fluorescence was observed for 10 min using cycles of 4 s.
2.5. Mitochondrial membrane potential of BF T. brucei
2.5.1. Analysis of drug-treated cells
The mitochondrial membrane potential of treated and untreated cells was assessed by using Tetramethylrhodamine ethyl ester (TMRE) (Denninger et al., 2007, Figarella et al., 2006). The cell density was adjusted to 106 cells/mL with and without test compounds for the start of the experiment. 1 mL of sample was transferred at each time point into a microfuge tube and centrifuged at 2500 rpm for 10 min at 4 °C, and was then resuspended in 1 mL PBS containing 25 nM of TMRE, and cells were incubated at 37 °C for 30 min. Valinomycin (100 nM) and troglitazone (10 μM) were used as controls, as they are known to induce mitochondrial membrane depolarization and hyperpolarization, respectively (Denninger et al., 2007). All samples were analysed by flow cytometry using a FL2-heigth detector and CellQuest software. Mitochondrial membrane potential was expressed as the percentage of cells displaying fluorescence higher than 100 artificial units, which was calibrated at exactly 50% for the no-drug control sample of the zero incubation time point, as described (Ibrahim et al., 2011).
2.5.2. Analysis of RNAi cell lines
Cells in the exponential growth phase were stained with 60 nM of TMRE for 30 min at 37 °C. Cells were pelleted (1300 × g, 10 min, RT), resuspended in 2 mL of PBS (pH 7.4) and immediately analysed by flow cytometry (BD FACS Canto II Instrument). For each sample, 10,000 events were collected. Treatment with the protonophore FCCP (20 μM) was used as a control for mitochondrial membrane depolarization. Data were evaluated using BD FACSDiva (BD Company) software.
2.6. Plasmid construction, transfection, cell growth and RNAi induction
To create the SDH1 (Tb927.8.6580) RNAi construct, a 485 bp fragment was PCR amplified from T. brucei strain 427 genomic DNA with the following oligonucleotides: FW: GAT GGA TCC CTC TGG GCT TCG TGC CGC AA, REV: GGA AAG CTT TGC CAC GAC AAC AGC CGT CC utilizing the respective BamHI and HindIII restriction sites inherent in the primers (underlined). The digested amplicon was then cloned into the p2T7-177 plasmid (Wickstead et al., 2002). This plasmid was used for transfection of PF T. brucei Lister 427 29.13 and BF Lister 427 single marker (SM) T. brucei cells that are transgenic for the T7 RNA polymerase and the tetracyclin (tet) repressor (Wirtz et al., 1999). PF 29.13 cells were grown in vitro at 27 °C in SDM79 medium containing hemin (7.5 mg/mL), hygromycin (25 μg/mL), G-418 (10 μg/mL) and 10% foetal bovine serum. BF SM cells were grown in vitro at 37 °C in HMI-9 medium containing G-418 (2.5 μg/mL) and 10% foetal bovine serum. After the transfection, both cell lines were selected using phleomycin (2.5 μg/mL). The inducible expression of double-stranded RNA was triggered by the addition of 1 μg/mL tetracycline to the medium. Growth curves were generated by measuring the cell density of tet-treated and untreated cultures using the Z2 cell counter (Beckman Coulter Inc.). Throughout the experiment, PF and BF cultures were split daily to ensure they continuously maintained an exponential growth phase of 106 - 107 cells/mL and 105–106 cells/mL, respectively.
2.7. Electrophoresis and western blot analysis
Crude mitochondrial fraction was isolated as described previously (Koreny et al., 2012, Subrtova et al., 2015) and the mitochondria were lysed using digitonin at detergent:protein ratio of 4 mg:1 mg. High resolution clear native (hrCN) PAGE analysis was performed by fractionating 50 μg of mitochondrial lysate on a 3–12% clear native (CN) PAGE gel followed by a protein transfer onto a nitrocellulose membrane. Protein samples from mitochondrial lysates were also separated on SDS PAGE gels and blotted onto a PVDF membrane. Both membranes were incubated with a specific SDH1 peptide antibody (1:1000) (Koreny et al., 2012), followed by an incubation with a secondary HRP-conjugated anti-rabbit antibody (1:2000, BioRad). The specific reaction was visualized using the Clarity™ Western ECl Substrate (Biorad) on a ChemiDoc instrument (BioRad).
2.8. Succinate:ubiquinone reductase (SQR) activity assay
The specific SQR activity was measured in a crude mitochondrial preparation in 1 mL of SDH buffer (25 mM K2HPO4, pH 7.2, 5 mM MgCl2, 20 mM sodium succinate) containing specific inhibitors of respiratory complexes III and IV (0.0002% antimycin, 2 mM KCN) and DCIP (2,6-dichlorophenolindophenol (Sigma)), which acts as the electron acceptor. The reaction was started by adding 65 coenzyme Q2. The unit of succinate–dehydrogenase activity (U) is defined as an amount of enzyme in 1 mg of kinetoplastid proteins, which causes the conversion of 1 nmol of DCIP in 1 min, as monitored at 600 nm (Birch-Machin and Turnbull, 2001).
2.9. F1-ATPase purification
Mitochondrial vesicles were purified as described previously (Acestor et al., 2011). Afterwards they were resuspended in buffer A (0.25 M sucrose, 50 mM Tris–HCl pH 8, 1 mM DTT) and the suspension was sonicated on power 8.5 for 3 × 40 s, with an incubation on ice of 1 min between sonications to prevent heating the sample. The samples were further sonicated for 10 15-s bursts, again left on ice for 1 min between the sonications (Sonicator 3000, Misonix, 19 mm probe, 250 W). The submitochondrial particles were then pelleted overnight in a SW60Ti rotor at 52,000 g at 4 °C.
The SMP pellet was resuspended in buffer B (0.25 M sucrose, 50 mM Tris HCl pH 8.0, 1 mM DTT, 4 mM EDTA, 2 mM ADP). Chloroform, saturated with 2 M Tris–HCl pH 8.5, was added to the pellet and then vigorously shaken for 20 s followed by immediate centrifugation for 5 min at 8500 rpm at room temperature. The top (light brown) layer was taken, leaving the middle (dark brown) and the bottom (chloroform containing) layers behind. The F1 depleted SMPs and residual chloroform were removed by centrifugation in a SW28 rotor at 27,000 rpm for 30 min at room temperature, using chloroform resistant polyallomer tubes. The F1-ATPase containing supernatant was carefully removed to avoid any chloroform contamination, blown with nitrogen for 15 min to remove all traces of chloroform. The F1-ATPase was filtered, concentrated down to 750 μl and loaded on a Superdex 200 10/300 GL. The F1-ATPase was purified on a FPLC using a specific F1-column buffer (20 mM Tris pH 8.5, 200 mM NaCl, 1 mM DTT, 4 mM EDTA, 1 mM ADP, 0.002% PMSF).
2.10. ATPase activity measurements
The amount of ATP hydrolysis in a crude mitochondrial preparation or by affinity purified F1 ATPase was measured by ATP regenerating assay (Pullman et al., 1960) that couples the hydrolysis of ATP with the oxidation of NADH. Crude mitochondrial lysates (15 μg) or purified F1 ATPase (0.7 μg) were incubated with different concentrations (5–200 μM) of CD38 and AHI-9 for 5 min after which the mix was transferred to a 1-mL UV cuvette containing an ATPase buffer (50 mM Tris, pH 8, 50 mM KCl, 2 mM MgSO4, 200 μM NADH, 1 mM phosphoenol pyruvate, 5 μL of lactate dehydrogenase (Sigma) and 9 μL of pyruvate kinase (Sigma)). The reaction was then initiated by adding 2 mM MgATP (from 100× solution of 400 mM Tris, 200 mM MgSO4, 200 mM ATP) and the change of absorbance at 340 nm was measured for 5 min. Azide (1 mM) was used as a known inhibitor of F1 ATPase activity (Bowler et al., 2006). Specific ATPase activity of mitochondrial lysate or of purified F1 ATPase was calculated from the amount of NADH oxidized in 1 min, and normalised to the amount of the protein in the sample.
2.11. ATP production
ATP production in digitonin-extracted mitochondria was measured following a protocol described previously (Allemann and Schneider, 2000). Briefly, crude mitochondrial fractions from the RNAi knockdown cell lines were obtained by digitonin extraction (Tan et al., 2002). ATP production in these samples was induced by the addition of 67 μM ADP and 5 mM of succinate. The mitochondrial preparations were preincubated for 10 min on ice with the inhibitors malonate (6.7 mM) and atractyloside (33 μg/mL). The concentration of ATP was determined by a luminometer (Orion II, Berthold detection systems) using the CLS I ATP bioluminescence assay kit (Roche Applied Science).
2.12. Surface plasmon resonance (SPR)
SPR experiments were performed at 25 °C with a Biacore X100 apparatus (GE Healthcare, Biacore AB, Uppsala, Sweden) in MES buffer (10 mM 2-(N-morpholino)ethanesulfonic acid, 1 mM EDTA, 100 mM NaCl, 0.005% surfactant P20, pH 6.25). The 5′-biotin labelled DNA hairpins 5′-CGAATTCGTCTCCGAATTCG-3′ and 5′-CGCGCGCGTTTTCGCGCGCG-3′ (the loop is underlined) were purchased from Sigma–Aldrich with HPLC purification. The SPR measurements were carried out as described (Rios Martinez et al., 2014).
3. Results
3.1. Actions of bisphosphonium compounds on T. brucei growth and cell cycle
We investigated the actions of two bisphosphonium compounds on bloodstream form (BF) T. b. brucei. The compounds CD38 and AHI-9 were selected as (i) displaying sub-micromolar activity against the parasite, (ii) possessing >300-fold in vitro selectivity with a human cell line and (iii) representing the two main classes of phosphonium trypanocides, possessing either linear saturated aliphatic substituents (n-pentyl; CD38) or aromatic substituents (phenyl and 3-methylphenyl; AHI-9) on the phosphonium moiety (Taladriz et al., 2012).
Both compounds were confirmed to be active against T. brucei, with sub-micromolar activity against BF cells and about an order of magnitude less activity against the procyclic forms (PF) (Table 1). At concentrations up to the EC50 values the compounds caused delayed growth rather than cell death and even at concentrations several fold above EC50 (1 μM) the initial effect appeared to be immediate (CD38) or slightly delayed (AHI9) growth arrest, followed by cell death 16–24 h later (Fig. 2). We thus investigated whether the compounds induced cell cycle arrest in a specific phase of the cycle, using flow cytometry to measure the DNA content in single cells of the population. As expected, we found that untreated control cells were less prone to enter S-phase when the culture entered the late log-phase of growth, leading to a small increase in G1-phase cells after 16 h; the proportion of G1-phase cells had initially decreased as the culture progressed from stationary to early log phase; the proportion of cells in S-phase steadily declined during late log phase (Fig. 3A). Similar effects, but much more pronounced and at much lower cell densities, were observed in the cultures treated with CD38 or AHI-9: both displayed a steep decline in S-phase cells after 8 h, accompanied by a significant increase in G1-phase cells (Fig. 3A, B), although the cultures were in virtual growth arrest and at comparatively low density (Fig. 3C). We thus conclude that bisphosphonium compounds prevent trypanosomes from entering the cell cycle and initiating DNA synthesis.
Table 1.
EC50 values for CD38 and AHI-9 compounds against BF427 and PF427 T. brucei cells.
| EC50 CD38 [μM] | EC50 AHI-9 [μM] | |
|---|---|---|
| T. brucei BF427 | 0.52 ± 0.038 | 0.14 ± 0.016 |
| T. brucei PF427 | 3.54 ± 0.14 | 1.19 ± 0.08 |
| P value | 0.00003 | 0.00004 |
The values represent the average and SEM of three independent determinations. The P value given is of an unpaired Student's t-test comparing the EC50 values for BF and PF trypanosomes.
Fig. 2.

Effect of different concentrations of two bisphosphonium compounds on the growth of BF T. brucei brucei over 72 h. Cell cultures were seeded at 105 cells/mL. Microscopic cell counts were performed in triplicate using a haemocytometer at various time points as indicated. The results shown are the average of three independent determinations; error bars depict standard errors.
Fig. 3.

Cell cycle analysis of BF T. b. brucei. (A) Control culture grown under standard conditions (blue closed symbols) or in the presence of 0.7 μM CD38 analysed for DNA content (red open symbols). Percentages of cells in each cell cycle stage were determined after staining of the permeabilised cells with propidium iodide, followed by flow cytometry. The data was further analysed by using the ModFit LT software to accurately separate and quantity the different peaks. (B) Identical culture grown in parallel but in the presence of 0.7 μM AHI-9 (green open symbols). The same control data as in panel A is shown for easy comparison. Panel C shows the cell densities of the cultures in panels A and B during this experiment, measured by using microscopic cell counts, performed in triplicate, using a haemocytometer. The results shown in panel A–C are the average of three independent experiments. Error bars depict standard errors, when not shown fall into the symbol. Statistical significance was determined using Student's unpaired T-test against the corresponding control group (*P < 0.05, **P < 0.01, ***P < 0.001). D. Percentage of the T. brucei population in each stage of the cell division cycle, with and without incubation with 0.7 μM CD38 over 24 h, as determined by fluorescence microscopy after staining with DAPI. For each culture, about 500 cells were counted and scored in terms of nuclei and kinetoplasts. N = nuclei, K = kinetoplasts, E = early stage of division, L = late stage of division (furrow ingression). Significant differences from the drug free control were calculated by using the Chi-squared test. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
CD38 also caused a highly significant decrease in cells in the first stages of trypanosomal cell division, i.e. with 1 nucleus and 2 kinetoplasts (1N2K), or with 2 of each (2N2K) but no cleavage furrow initiated (2N2K-Early), after 8 h of treatment; the percentage of cells in all stages of division (IN2K, 2N2K) continued to decline sharply with the exposure time (Fig. 3D), as no new cells entered the cell division cycle. This experiment showed that bisphosphonium-treated cells were unable to replicate kinetoplast DNA and/or initiate kinetoplast division. It was not possible to reliably distinguish between the two possibilities, although we did not observe increased DAPI fluorescence associated with the kinetoplast (i.e. kDNA content) after CD38 treatment (not shown); however, it is uncertain whether this would have been sufficiently unambiguous. However, it has previously been reported that CD38 does not bind directly to a poly(dA.dT)2 DNA polymer (Dardonville et al., 2006), and we here follow that up with SPR measurements of CD38 and AHI-9 binding to two other DNA. Neither compound showed measurable binding to the CGAATTCG oligonucleotide, and only CD38 interacted, weakly, with the CGCGCGCG sequence, with a KD of 7.0 × 10−6 M (Supplementary Fig. S1). This seems inconsistent with a direct action on kinetoplast replication by kDNA binding.
3.2. The mitochondrion as a target for the trypanocidal activity of bisphosphonium salts
It has been reported that CD38 elicits anti-leishmanial activity by inhibiting the succinate dehydrogenase complex (also known as complex II) in the L. donovani mitochondrion (Luque-Ortega et al., 2010), and a mitochondrial target would be consistent with the observed phenotype of being unable to initiate kinetoplast division (above).
Incubation of BF T. brucei with CD38 or AHI-9 for 4 h, at concentrations close to their respective EC50 values, caused a dose-dependent reduction of the mitochondrial membrane potential Ψm (Fig. 4A). At 0.7 μM bisphosphonium compound, small effects were observed after just 30 min (P < 0.05 for CD38) and much more dramatic reductions were seen after 4 h of exposure (P < 0.001 for both compounds) until the mitochondrial membranes appeared to be almost fully depolarized (Fig. 4B). This was accompanied by a similar drop in cellular ATP levels, which was highly significantly different from control (P < 0.01) and was statistically identical to treatment with the F1Fo ATPase inhibitor oligomycin after 1 h. This level of cellular ATP remained essentially constant for at least 12 h of treatment with either of the test compounds (Fig. 4C). The decrease of cellular ATP is in agreement with the observed redirection of glycolysis to 1 molecule of glycerol and 1 molecule of pyruvate due to Ψm dissipation (see the discussion). Another cellular effect was that after four hours of incubation with CD38 or AHI-9 the level of free intracellular Ca2+ started to increase (Fig. 4D), which was consistent with the depolarized mitochondria being unable to maintain their capacity as calcium reservoir (Huang et al., 2013).
Fig. 4.

Effects of bisphosphonium compounds on mitochondrial functionality in live BF trypanosomes. (A) Dose-dependency of effect of CD38 and AHI-9 on mitochondrial membrane potential Ψm after four hours of exposure, measured by flow cytometry using the indicator fluorophore TMRE. Data are the triplicate and SEM of three independent experiments. Statistical significance was determined using an unpaired Student's t-test: **, P < 0.01; ***, P < 0.001. (B) Effects of 0.7 μM CD38 and AHI-9 on Ψm over a period of 8 h, using 0.1 μM valinomycin and 10 μM troglitazone as controls for membrane depolarisation and hyperpolarisation, respectively. Data are the triplicate and SEM of three independent experiments. Statistically significant differences were observed for CD38 (P < 0.05, 30 min; P < 0.01, 4 h; P < 0.001, 8 and 12 h) and for AHI-9 (P < 0.001 for 4, 8 and 12 h). (C) The effects of 0.7 μM CD38 or AHI-9 on the total cellular ATP content. Oligomycin (2 μg/mL) was used as positive control and was significantly different from untreated controls at all time points (P < 0.01) whereas the bisphosphonium compounds only induced significantly reduced ATP levels at 60 min (P < 0.01). All data are the average and SEM of 3–4 independent experiments. (D) Assessment of intracellular calcium levels using the Fluo-8 indicator dye. Data are presented as percentage of the fluorescence level at time = 0 h for the untreated control cells. Fluorescence was significantly increased after treatment with 0.7 μM CD38 (P < 0.05, 8 h; P < 0.01, 12 h) or AHI-9 ((P < 0.05, 8 and 12 h). Average and SEM of three independent experiments.
3.3. Assessment of succinate dehydrogenase as a potential target for bisphosphonium compounds in T. brucei
Because the target of bisphosphonium salts in L. donovani cells was suggested to be the succinate dehydrogenase (SDH) complex (Luque-Ortega et al., 2010), we explored, in both PF and BF T. brucei, its steady state protein level, the assembly of the complex, and whether its depletion elicits a reduced growth rate. First, we determined the steady state levels of a specific subunit of the SDH complex, SDH1, which contains the cofactor FAD and is responsible for the electron entry into the complex. When mitochondrial lysate was fractionated on a TGX stain-free precast gel followed by western blot analysis using anti-SDH1 antibody, the correct size band for SDH1 (66 kDa) was detected. The highest level of expression is seen in PF cells and much lower amounts of SDH1 subunit are detected in BF cells (Fig. 5A, upper panel). Equal loading of protein samples was verified by UV detection of proteins (Fig. 5A, lower panel). Independently-generated mitochondrial samples were analysed repeatedly with similar results, confirming the relatively low intensity of the SDH1 signal in BF cells compared to PF, and clearly show a much lower steady-state SDH1 protein level in the former life cycle stage.
Fig. 5.

Abundance and essentiality of SDH complex in PF and BF cells. A. Steady state abundance of SDH1 subunit in PF and BF cells. Mitochondria were isolated from equal numbers of cells from T. brucei PF and BF. For each sample 50 μg of protein was loaded onto a BioRad TGX stain-free precast gel. After transfer to a PVDF membrane, the blot was probed with a rabbit antibody raised against a peptide of T. brucei SDH1. The 50 kDa unknown protein visualized using UV light served as a loading control. B. Relative levels of assembled SDH complex in PF and BF cells. For each sample 50 μg of mitochondrial protein was loaded onto a high resolution clear native (hrCN) PAGE, which was blotted and probed with the anti-SDH1 peptide antibody. C. Growth curve of the PF SDH1 RNAi cell line. PF 29.13 cells were transfected with a linearized p2T7-177 vector containing a 485bp fragment of the SDH1 cds. The resulting clonal transfectants were then analysed for a growth phenotype when RNAi was induced with 1 μg/mL tetracycline. These growth curves are representative of several clones. NON, non-induced cells; IND, induced cells. D. Growth curve of BF SDH1 RNAi. BF T. brucei SM cells were transfected with the same p2T7-177 plasmid and the growth curve analysis was performed as in C. E. Western blot analysis of PF SDH1 RNAi cells. Whole cell lysates made from equal amounts of cells after the indicated of tetracycline induction, and loaded onto a 12% SDS PAGE gel. After transfer, the PVDF membrane was probed with the anti-SDH1 antibody. To confirm that the upper of the two bands was specific for SDH1, the tetracycline was washed out (-tet) and the reappearance of the upper band was observed within 48 h (-tet, day 5). F. Western blot analysis of BF SDH1 RNAi cells. Mitochondrial lysate from noninduced (N) cells and cells induced by tetracycline (+tet) for 1 and 3 days were analysed. G. Mitochondrial membrane potential in PF T. brucei expressing RNAi against SDH1. Units indicated are arbitrary units of TMRE-associated fluorescence. NON, non-induced RNAi cells; Day 1, Day 3, Day 5, signifies the time in days after RNAi induction. The protonophore FCCP was included as a positive control to indicate the level of fluorescence in cells with depolarised mitochondrial membranes; fluorescence in FCCP-treated cells was highly significantly different from all other test groups. ***, P < 0.001, unpaired Student's t-test, n = 5.
To gain better insight about the significance of the function of the SDH complex in PF and BF cells, the level of assembled SDH complex was analysed by high resolution clear native (hrCNE) PAGE. The expected band of ∼500 kDa (Acestor et al., 2011) is prominent in the PF sample, but only a very faint band is detected in BF mitochondrial lysate (Fig. 5B), which correlates well with the relative steady state levels of SDH1 in the respective life stages of T. brucei. Interestingly, two bands were detected in the PF sample, suggesting that the complex may form higher molecular weight aggregates, or associate with another macromolecular structure, as suggested by Kovarova et al. for the mammalian complex II (Kovarova et al., 2013).
The activity of the SDH complex can be followed spectrophotometrically as a change in the absorbance of DCIP, with electrons flowing from succinate to DCIP via ubiquinone Q2. Each cell line was also incubated with the specific SDH inhibitor malonate to verify that only this specific activity was detected. The succinate-ubiquinone reductase (SQR) activity measured in BF cells corresponded to only 7% of the activity of SQR measured in PF cells (Table 2). These data closely correspond with the relative steady state protein levels of SDH complex (see above), and confirm that the SDH complex is much more abundant and is thus likely to play a more important role in the mitochondria of PF than BF cells.
Table 2.
SQR activity in intact PF427 and BF427 Trypanosoma brucei.
| Cells | SDH specific activity (Ua/mg) ± SEMb | % of PF activity | % of effect of malonate |
|---|---|---|---|
| PF 427 | 1.2 ± 0.011 | 100 | 100 |
| PF 427 + mal | 0.21 ± 0.006 | 17 | |
| BF 427 | 0.084 ± 0.007 | 7 | 100 |
| BF 427 + mal | 0.0381 ± 0.002 | 45 |
SQR activity in PF and BF T. brucei cells was measured spectrophotometrically as described in the methods section, in the presence or absence of 1 mM malonate (mal).
The unit (U) of SQR is defined as the amount of enzyme required for the conversion of 1 nmol of 2,6-dichlorophenolindophenol/min/mg of mitochondrial proteins.
Each measurement was performed in triplicate.
It has been shown that the SDH complex possesses an essential function in PF T. brucei (strain EATRO1125) when grown in the absence of glucose, while its function was unimportant for cells grown in the presence of glucose (Coustou et al., 2008). Nevertheless, both tested compounds exert a cytotoxic effect for PF 427 Lister strain T. brucei grown in the presence of glucose (Table 1). To address whether the SDH complex is essential for the viability of this specific strain of T. brucei, and under the specific conditions used in our study, a cell line expressing RNAi targeting the SDH1 subunit was generated. When PF SDH1 RNAi cells were grown in glucose-rich media, no significant difference in growth of non-induced and tetracycline-induced RNA cells was observed. (Fig. 5C) confirming that the SDH complex is not essential for PF cells under glucose-rich growth conditions. To test whether the SDH complex is important for the viability of BF T. brucei cells, the BF SDH RNAi cell line was examined upon induction with tetracycline. Importantly, there was no growth phenotype at all (Fig. 5D). To verify successful depletion of SDH1 protein in the PF and BF RNAi induced cell lines, a western blot analysis was performed with the specific SDH1 antibody. In PF RNAi tetracycline-induced cells a band of the appropriate size had disappeared after 1 day, but a band of somewhat lower apparent weight remained unchanged (Fig. 5E). In order to confirm that the upper band was specific for SDH1, the tetracycline was washed out from the media (Fig. 5E, -tet samples) which led to the reappearance of the upper band within 48 h. In BF SDH1 RNAi cells, a single distinct band of the expected size was observed in non-induced cells (N), but this band was undetectable after just 24 h of RNAi induction (Fig. 5F). Furthermore, the specific SQR activity of SDH complex in PF RNAi SDH1 cell line was decreased by 62% after 1 day of RNAi induction, and almost completely absent by day 3 (Table 3).
Table 3.
SQR activity in PF RNAi SDH1 noninduced (N) and tetracycline induced (+tet) cells in the presence of absence of malonate (mal).
| Cells | SDH specific activity (Ua/mg) ± SEMb | % of 29–13 activity | % of RNAi N activity |
|---|---|---|---|
| PF 29-13 | 0.658 ± 0.021 | 100 | |
| PF 29-13 + mal | 0.156 ± 0.004 | 24 | |
| PF RNAi Sdh1 N | 0.497 ± 0.005 | 100 | |
| PF RNAi Sdh1 N/+mal | 0.107 ± 0.008 | 22 | |
| PF RNAi Sdh1 1d +tet | 0.19 ± 0.0046 | 38 | |
| PF RNAi Sdh1 3d +tet | 0.08 ± 0.005 | 16 |
The unit (U) of SQR is defined as an amount of enzyme required for conversion of 1 nmol of 2,6-dichlorophenolindophenol/min/mg of mitochondrial proteins.
Each measurement was performed in triplicate.
The mitochondrial membrane potential in BF cells is maintained exclusively by the hydrolytic activity of the FoF1 ATPase complex (Schnaufer et al., 2005, Subrtova et al., 2015), while in PF cells the proton motive force is generated by the respiratory chain complexes (Gnipova et al., 2012, Horvath et al., 2005). In order to determine whether the depletion of SDH complex has any effect on the mitochondrial membrane potential in PF cells, the electrochemical gradient was measured using a flow cytometry analysis with the dye TMRE that accumulates only in charged mitochondria. The membrane potential remained unchanged after even 5 days of RNAi induction (Fig. 5G) indicating that the SDH complex does not significantly contribute to Ψm in PF T. brucei cells.
3.4. The effects of bisphosphonium compounds on mitochondrial ATPases
The reductions in Ψm and cellular ATP levels are the early effects seen after the treatment of T. brucei with bisphosphonium compounds (see above). However, we here show that, in T. brucei, the SDH complex is not essential and does not contribute to the membrane potential. It seems therefore unlikely that the principal trypanocidal activity of bisphosphonium salts would be on this complex and we thus explored other potential mitochondrial targets - in particular, the FoF1 ATPase complex that maintains Ψm in BF cells.
First, in vitro ATPase assays were performed using a purified mitochondrial fraction, which was lysed by digitonin. The prepared lysed mitochondrion-enriched fractions from PF cells were treated with various concentrations of CD38 (Fig. 6A) or AHI-9 (Fig. 6B). Since there are many ATPases in these samples, the fractions were also treated with the F1-ATPase inhibitor azide. Typically, 1 mM azide inhibits 30–40% of all measured ATP hydrolysis activity in such samples (Zikova et al., 2009). Increasing concentrations of either drug elicited a similar (CD38) or identical (AHI-9) level of inhibition as the azide control, albeit at much higher concentrations than their measured EC50 values. These inflated concentrations might be an artifact of the in vitro assay performed on lysed instead of intact and charged mitochondria, as the latter would be able to concentrate the drugs, driven by Ψm. Importantly, the same level of inhibition was observed when the crude mitochondria were treated with azide together with a bisphosphonium salt, indicating that the inhibitions were not additive and both inhibitors were therefore acting on the same target. Furthermore, the enzymatic activity was more strongly affected by AHI-9, which typically has a stronger effect on trypanosome cells than CD38.
Fig. 6.

ATPase activity of mitochondrion-enriched fractions of PF427 treated with CD38 (A) and AHI-9 (B). Approximately 15 μg of hypotonically isolated mitochondria were incubated with the indicated test compounds for 5 min and then transferred to ATPase assay buffer. Changes in NADH absorbance were monitored at 340 nm for 5 min. Specific ATPase activity from mitochondrial lysates was calculated from the amount of oxidized NADH per minute, and related to the amount of total protein. The specific ATPase activity in the non-induced sample was set at 100%. Assays were performed in triplicate. Error bars depict standard errors of the mean. Statistical significance was determined using Student's unpaired T-test.
To further evaluate the mechanism of action of bisphosphonium salts, we determined whether these compounds also affect the rate of ATP synthesis by FoF1-ATP synthase. This experimental set-up relies on a complete and functional oxidative phosphorylation pathway and is performed with intact, charged mitochondria isolated from PF cells (Allemann and Schneider, 2000). Malonate and atractyloside are used as positive controls, as the former inhibits the succinate dehydrogenase complex (i.e. the entry of the electrons), and the latter inhibits the ATP/ADP carrier (i.e. the supplier of the substrate). Not surprisingly, the results indicated that both CD38 and AHI-9 dose-dependently inhibited ATP production in intact T. brucei mitochondria, with concentrations similar to the measured EC50 values moderately inhibitory and higher concentrations inhibiting ATP production almost completely (Fig. 7). While this assay does not rule out succinate dehydrogenase as a potential target, the dose-dependency shows unambiguously that an active mitochondrial Ψm is important to these in vitro assays, as the test compounds were much more active than on mitochondrial lysates.
Fig. 7.

Effect of CD38 (A) and AHI-9 (B) treatment on ATP production in PF mitochondria. Crude mitochondrial preparations were obtained by digitonin extraction and ATP production in untreated and treated samples was measured using succinate as a substrate. Malonate, specific inhibitor of succinate dehydrogenase was used to inhibit ATP production by oxidative phosphorylation and atractyloside was used to inhibit import of ADP into mitochondria. ATP production in untreated mitochondria was set to 100%. The bars represent means expressed as percentages from three independent RNAi inductions. Error bars are SEM.
Since the effects on total mitochondrial ATP hydrolytic and ATP synthetic activities were indicative of specific inhibition of the FoF1 ATP synthase/ATPase, we repeated the assay on highly purified F1-ATPase, and observed a similar dose dependency of inhibition as above on the lysed mitochondrial fractions, i.e. less potent than in intact and charged mitochondria. In this experiment, azide inhibits almost 100% of the activity of the purified F1-ATPase, confirming the purity of the sample. The increasing concentrations of the bisphosphonium test compounds reduced the ATP hydrolysis activity in a linear manner. Once again, AHI-9 displayed a significantly higher level of inhibition than CD38. In fact, 200 μM AHI-9 inactivated F1-ATPase almost to the same extent as azide (Fig. 8). We conclude that inhibition of the F1-ATPase is at a minimum an important contributing factor in the mechanism of action of bisphosphonium compounds on T. brucei.
Fig. 8.

Activity of T. brucei purified F1-ATPase treated with CD38 (A) and AHI-9 (B). ATPase activity was measured as described in the legend to Fig. 6, in triplicate. Error bars depict standard errors of mean.
4. Discussion
The phosphonium salts were discovered as potent chemotherapeutics against Trypanosoma cruzi and T. b. rhodesiense in the late 1970s (Kinnamon et al., 1977, Kinnamon et al., 1979). Thirty years later benzophenone-derived bisphosphonium salts were synthesized showing much higher potency against Leishmania parasites. Some of these phosphonium derivatives act as lipophilic cations with enhanced hydrophobicity that allow them to cross biological membranes and accumulate in organelles with a strong electrochemical gradient, e.g. mitochondria. Interestingly, in mammalian cells, these lipophilic phosphonium ions are being developed to deliver antioxidants or anti-cancer drugs to mitochondria as they pass easily through the lipid bilayers. Because of their disperse charged the electrochemical gradient drives their accumulation into the mitochondrial matrix concentrating them as high as 100–500 fold (Murphy, 2008).
The nature of the trypanocidal activity of these compounds has not been determined yet, but in L. donovani some bisphosphonium compounds including CD38 were shown to have a negative impact on proper mitochondrial function (Luque-Ortega et al., 2010). Upon treatment with a low drug concentration (0.7 μM) the mitochondria were swollen, the mitochondrial Ψm was significantly decreased, the oxygen consumption rate, using succinate as a substrate, was affected, and the cellular ATP levels were reduced. These results would suggest that the drug was either affecting a component of the oxidative phosphorylation pathway, or was acting as a kind of ionophore or charge carrier, depolarizing the mitochondrial membrane. The study proposed that in L. donovani the bisphosphonium compounds act through inhibition of the respiratory complex II, succinate dehydrogenase (Luque-Ortega et al., 2010).
While the available data certainly suggest that CD38 might be able to inhibit succinate dehydrogenase in Leishmania, it seems unlikely that this would be the mode of action for bisphosphonium drugs in T. brucei. Since metabolic activities of BF mitochondria are largely repressed with the exception of lipid metabolism (Guler et al., 2008), ion homeostasis (Nolan and Voorheis, 1992), calcium signalling (Huang et al., 2013, Vercesi et al., 1992), FeS cluster assembly (Kovarova et al., 2014) and acetate production for de novo lipid biosynthesis (Mazet et al., 2013), it is generally assumed that succinate dehydrogenase complex is largely inactive and unimportant for these cells. Therefore, we decided to investigate how bisphosphonium salts interact with the unique metabolism of BF trypanosomes.
We found that adding CD38 and AHI-9, at concentrations close to their trypanocidal EC50 values, into the T. brucei culture prevents the initiation and replication of kDNA, reduces cellular ATP levels, and depolarizes the mitochondrial membrane potential within a few hours of treatment; this was immediately followed by increased cytosolic Ca2+ levels. All these phenotypic changes point also to a mitochondrial target, although not necessarily to succinate dehydrogenase. Succinate dehydrogenase does not directly contribute to the membrane potential by pumping protons, as it passes its electrons to ubiquinone. Moreover, we show that in BF it is not essential, as RNAi knockdown presented no growth phenotype, and indeed succinate dehydrogenase is barely present as an assembled complex in this life-cycle stage. Furthermore, the succinate dehydrogenase complex is also not essential for PF T. brucei grown under high glucose conditions (this work and (Coustou et al., 2008)) and does not contribute to the mitochondrial membrane potential – yet, both of the tested drugs have a strong effect against PF cells (EC50 values in single digit μM range) and decreased the mitochondrial Ψm. We thus conclude that, in trypanosomes, inhibition of succinate dehydrogenase is not mode of action of the bisphosphonium compounds.
Interestingly, the potent trypanocidal diamidine DB75 induces a similar phenotype in BF cells, as its treatment collapses the mitochondrial Ψm and inhibits glucose-dependent cellular respiration, apparently through inhibition of the hydrolytic function of FoF1 ATPase (Lanteri et al., 2008), which, in BF cells, employs its reverse function in order to maintain the mitochondrial Ψm (Schnaufer et al., 2005, Subrtova et al., 2015). These actions of DB75 certainly overlap with those of the specific FoF1 ATPase inhibitor oligomycin, which decreases mitochondrial membrane potential, inhibits oxygen consumption and reduces cellular ATP levels by 50% (Nolan and Voorheis, 1992). We observed similarly reduced cellular ATP levels upon treatment with the bisphosphonium compounds CD38 and AHI-9. The observed reduction in ATP is consistent with inhibition of the FoF1 complex, which is followed by immediate mitochondrial membrane depolarization, as in BF cells respiration through alternative oxidase is sensitive to Ψm dissipation (Miller and Klein, 1980). The membrane depolarization in turn results in the loss of the glycerol-3-phosphate shuttle and the subsequent redirection of glycolysis to 1 molecule of glycerol and 1 molecular of pyruvate. Thus, pyruvate production falls by 50% upon oligomycin treatment (Nolan and Voorheis, 1992) and only 1 molecule of ATP is made from 1 molecule of glucose. Similarly, we report an approximately 50% reduction in cellular ATP concentration upon treatment with bisphosphonium drugs.
Moreover, our data strongly suggest that bisphosphonium salts CD38 and AHI-9 inhibit the hydrolytic activity of the FoF1 ATPase in vitro. Inhibition assays, performed on mitochondrial lysates from PF cells and on highly purified F1-ATPase, showed highly significant inhibition of ATPase activity to similar levels as achieved with 1 mM azide. AHI-9, particularly, inhibited the isolated F1 ATPase almost completely. Furthermore, a combination of azide plus CD38 or AHI-9 did not result in additional inhibition of ATPase activity compared to either drug separately, suggesting that no other mitochondrial ATPases play a significant role in the observed phenotype.
Interestingly, all the biochemical analyses performed revealed a greater sensitivity to AHI-9 than to CD38, which correlates nicely with the almost 4-fold lower EC50 value for the former compound. It should be noted that the in vitro assay, which measured ATPase activity in lysates, required concentration of the compounds that are considerably higher than the EC50 values, as the lipophilic cations greatly concentrate in the mitochondrion (Murphy, 2008), because Ψm creates a negative charge in the matrix. This assumption is completely consistent with our results from the ATP production assay, which utilizes intact and charged mitochondria from PF cells. In this assay, ATP production was inhibited by the bisphosphonium compounds at the concentration similar to EC50 values.
The reduction in cellular ATP levels may be the immediate cause of the observed failure in initiating the S-phase of the cell cycle (DNA synthesis). Trypanosomatid kDNA is a highly organized structure composed of thousands of minicircles and tens of maxicircles (Liu et al., 2005). To ensure that daughter cells receive completely identical kDNA networks, the replication process is highly synchronized with nuclear replication followed by cell division (Jensen and Englund, 2012). kDNA replication relies on ATP and hundreds of mitochondrial proteins (Beck et al., 2013), which are imported to mitochondria via the Ψm-dependent TIM machinery (Williams et al., 2008). Thus it is plausible to speculate that a lack of ATP and decrease of Ψm will interfere with proper initiation of kDNA replication and result in cell cycle arrest. Similar results were observed upon RNAi knock-down of the ATP/ADP carrier in PF T. brucei cells, in which reduced ATP levels led to failure of the overall cell division machinery (Gnipova et al., 2015). In contrast, a direct inhibition by bisphosphonium salts of kDNA replication, segregation and maintenance as their primary mode of action is far less likely, as (i) these compounds have a poor affinity to DNA as shown previously (Dardonville et al., 2006) and in this study, (ii) CD38 and AHI-9 cause a decrease in Ψm in T. brucei cells that lack mitochondrial DNA (Gould, Schnaufer and De Koning, unpublished), and (iii) interference with kDNA replication, either by RNAi or drug treatment, usually leads to complete loss of kDNA (Bruhn et al., 2011, Hiltensperger et al., 2012, Tyc et al., 2015).
In conclusion, our data strongly suggest that bisphosphonium compounds CD38 and AHI-9 primarily act on trypanosomes through inhibition of the F1-ATPase although additional mitochondrial targets cannot be excluded.
Acknowledgements
The assistance of Dr Laura Lagartera with the SPR experiments (DNA binding) is gratefully acknowledged.
This work was funded by the Czech Republic Ministry of Education ERC CZ grant LL1205, and EMBO Installation grant 1965 (both to AZ); AAMA was funded by a studentship from the Government of Saudi Arabia (Ref. JFU008).
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ijpddr.2015.12.002.
Contributor Information
Alena Zíková, Email: azikova@paru.cas.cz.
Harry P. de Koning, Email: Harry.de-Koning@glasgow.ac.uk.
Authorship contributions
Participated in research design: Panicucci, Dardonville, Zíková, De Koning.
Conducted experiments: Alkhaldi, Martinek, Panicucci, Dardonville.
Contributed new reagents or analytic tools: Dardonville.
Performed data analysis: Alkhaldi, Martinek, Panicucci, Zíková, De Koning.
Wrote or contributed to the writing of the manuscript: Zíková, De Koning.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- Acestor N., Zikova A., Dalley R.A., Anupama A., Panigrahi A.K., Stuart K.D. Trypanosoma brucei mitochondrial respiratome: composition and organization in procyclic form. Mol. Cell Proteom. 2011;10 doi: 10.1074/mcp.M110.006908. M110 006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allemann N., Schneider A. ATP production in isolated mitochondria of procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 2000;111:87–94. doi: 10.1016/s0166-6851(00)00303-0. [DOI] [PubMed] [Google Scholar]
- Beck K., Acestor N., Schulfer A., Anupama A., Carnes J., Panigrahi A.K., Stuart K. Trypanosoma brucei Tb927.2.6100 is an essential protein associated with kinetoplast DNA. Eukaryot. Cell. 2013;12:970–978. doi: 10.1128/EC.00352-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benein P., Almuteri M.A., Mehanna A.S., D'Souza G.G. Synthesis of triphenylphosphonium phospholipid conjugates for the preparation of mitochondriotropic liposomes. Methods Mol. Biol. 2015;1265:51–57. doi: 10.1007/978-1-4939-2288-8_4. [DOI] [PubMed] [Google Scholar]
- Birch-Machin M.A., Turnbull D.M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- Bowler M.W., Montgomery M.G., Leslie A.G., Walker J.E. How azide inhibits ATP hydrolysis by the F-ATPases. Proc. Natl. Acad. Sci. U. S. A. 2006;103:8646–8649. doi: 10.1073/pnas.0602915103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhn D.F., Sammartino M.P., Klingbeil M.M. Three mitochondrial DNA polymerases are essential for kinetoplast DNA replication and survival of bloodstream form Trypanosoma brucei. Eukaryot. Cell. 2011;10:734–743. doi: 10.1128/EC.05008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun R., Blum J., Chappuis F., Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–159. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- Cairns A.G., McQuaker S.J., Murphy M.P., Hartley R.C. Targeting mitochondria with small molecules: the preparation of MitoB and MitoP as exomarkers of mitochondrial hydrogen peroxide. Methods Mol. Biol. 2015;1265:25–50. doi: 10.1007/978-1-4939-2288-8_3. [DOI] [PubMed] [Google Scholar]
- Chaudhuri M., Ajayi W., Hill G.C. Biochemical and molecular properties of the Trypanosoma brucei alternative oxidase. Mol. Biochem. Parasitol. 1998;95:53–68. doi: 10.1016/s0166-6851(98)00091-7. [DOI] [PubMed] [Google Scholar]
- Cortes L.A., Castro L., Pesce B., Maya J.D., Ferreira J., Castro-Castillo V., Parra E., Jara J.A., Lopez-Munoz R. Novel gallate triphenylphosphonium derivatives with potent antichagasic activity. PLoS One. 2015;10:e0136852. doi: 10.1371/journal.pone.0136852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coustou V., Biran M., Breton M., Guegan F., Riviere L., Plazolles N., Nolan D., Barrett M.P., Franconi J.M., Bringaud F. Glucose-induced remodeling of intermediary and energy metabolism in procyclic Trypanosoma brucei. J. Biol. Chem. 2008;283:16342–16354. doi: 10.1074/jbc.M709592200. [DOI] [PubMed] [Google Scholar]
- Dardonville C., Alkhaldi A.A., De Koning H.P. SAR studies of diphenyl cationic trypanocides: superior activity of phosphonium over ammonium salts. ACS Med. Chem. Lett. 2015;6:151–155. doi: 10.1021/ml500408d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardonville C., Barrett M.P., Brun R., Kaiser M., Tanious F., Wilson W.D. DNA binding affinity of bisguanidine and bis(2-aminoimidazoline) derivatives with in vivo antitrypanosomal activity. J. Med. Chem. 2006;49:3748–3752. doi: 10.1021/jm060295c. [DOI] [PubMed] [Google Scholar]
- Delespaux V., de Koning H.P. Drugs and drug resistance in African trypanosomiasis. Drug Resist. Updates. 2007;10:30–50. doi: 10.1016/j.drup.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Denninger V., Figarella K., Schonfeld C., Brems S., Busold C., Lang F., Hoheisel J., Duszenko M. Troglitazone induces differentiation in Trypanosoma brucei. Exp. Cell Res. 2007;313:1805–1819. doi: 10.1016/j.yexcr.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Desquesnes M., Dargantes A., Lai D.H., Lun Z.R., Holzmuller P., Jittapalapong S. Trypanosoma evansi and surra: a review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. Biomed. Res. Int. 2013:321237. doi: 10.1155/2013/321237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figarella K., Uzcategui N.L., Beck A., Schoenfeld C., Kubata B.K., Lang F., Duszenko M. Prostaglandin-induced programmed cell death in Trypanosoma brucei involves oxidative stress. Cell Death Differ. 2006;13:1802–1814. doi: 10.1038/sj.cdd.4401862. [DOI] [PubMed] [Google Scholar]
- Gnipova A., Panicucci B., Paris Z., Verner Z., Horvath A., Lukes J., Zikova A. Disparate phenotypic effects from the knockdown of various Trypanosoma brucei cytochrome c oxidase subunits. Mol. Biochem. Parasitol. 2012;184:90–98. doi: 10.1016/j.molbiopara.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Gnipova A., Subrtova K., Panicucci B., Horvath A., Lukes J., Zikova A. The ADP/ATP carrier and its relationship to OXPHOS in an ancestral protist Trypanosoma brucei. Eukaryot. Cell. 2015;14:297–310. doi: 10.1128/EC.00238-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould M.K., Bachmaier S., Ali J.A., Alsford S., Tagoe D.N., Munday J.C., Schnaufer A.C., Horn D., Boshart M., de Koning H.P. Cyclic AMP effectors in African trypanosomes revealed by genome-scale RNA interference library screening for resistance to the phosphodiesterase inhibitor CpdA. Antimicrob. Agents Chemother. 2013;57:4882–4893. doi: 10.1128/AAC.00508-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould M.K., Vu X.L., Seebeck T., de Koning H.P. Propidium iodide-based methods for monitoring drug action in the kinetoplastidae: comparison with the Alamar Blue assay. Anal. Biochem. 2008;382:87–93. doi: 10.1016/j.ab.2008.07.036. [DOI] [PubMed] [Google Scholar]
- Guler J.L., Kriegova E., Smith T.K., Lukes J., Englund P.T. Mitochondrial fatty acid synthesis is required for normal mitochondrial morphology and function in Trypanosoma brucei. Mol. Microbiol. 2008;67:1125–1142. doi: 10.1111/j.1365-2958.2008.06112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarton T.C., Mottram J.C., Doerig C. The cell cycle of parasitic protozoa: potential for chemotherapeutic exploitation. Prog. Cell Cycle Res. 2003;5:91–101. [PubMed] [Google Scholar]
- Hanson W.L., Chapman W.L., Jr., Kinnamon K.E. Testing of drugs for antileishmanial activity in golden hamsters infected with Leishmania donovani. Int. J. Parasitol. 1977;7:443–447. doi: 10.1016/0020-7519(77)90004-2. [DOI] [PubMed] [Google Scholar]
- Hiltensperger G., Jones N.G., Niedermeier S., Stich A., Kaiser M., Jung J., Puhl S., Damme A., Braunschweig H., Meinel L., Engstler M., Holzgrabe U. Synthesis and structure-activity relationships of new quinolone-type molecules against Trypanosoma brucei. J. Med. Chem. 2012;55:2538–2548. doi: 10.1021/jm101439s. [DOI] [PubMed] [Google Scholar]
- Horvath A., Horakova E., Dunajcikova P., Verner Z., Pravdova E., Slapetova I., Cuninkova L., Lukes J. Downregulation of the nuclear-encoded subunits of the complexes III and IV disrupts their respective complexes but not complex I in procyclic Trypanosoma brucei. Mol. Microbiol. 2005;58:116–130. doi: 10.1111/j.1365-2958.2005.04813.x. [DOI] [PubMed] [Google Scholar]
- Huang G., Vercesi A.E., Docampo R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat. Commun. 2013;4:2865. doi: 10.1038/ncomms3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim H.M., Al-Salabi M.I., El Sabbagh N., Quashie N.B., Alkhaldi A.A., Escale R., Smith T.K., Vial H.J., de Koning H.P. Symmetrical choline-derived dications display strong anti-kinetoplastid activity. J. Antimicrob. Chemother. 2011;66:111–125. doi: 10.1093/jac/dkq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannin J., Cattand P. Treatment and control of human African trypanosomiasis. Curr. Opin. Infect. Dis. 2004;17:565–571. doi: 10.1097/00001432-200412000-00009. [DOI] [PubMed] [Google Scholar]
- Jara J.A., Castro-Castillo V., Saavedra-Olavarria J., Peredo L., Pavanni M., Jana F., Letelier M.E., Parra E., Becker M.I., Morello A., Kemmerling U., Maya J.D., Ferreira J. Antiproliferative and uncoupling effects of delocalized, lipophilic, cationic gallic acid derivatives on cancer cell lines. Validation in vivo in singenic mice. J. Med. Chem. 2014;57:2440–2454. doi: 10.1021/jm500174v. [DOI] [PubMed] [Google Scholar]
- Jensen R.E., Englund P.T. Network news: the replication of kinetoplast DNA. Annu. Rev. Microbiol. 2012;66:473–491. doi: 10.1146/annurev-micro-092611-150057. [DOI] [PubMed] [Google Scholar]
- Kelso G.F., Porteous C.M., Coulter C.V., Hughes G., Porteous W.K., Ledgerwood E.C., Smith R.A., Murphy M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- Kinnamon K.E., Steck E.A., Hanson W.L., Chapman W.L., Jr. In search of anti-Trypanosoma cruzi drugs: new leads from a mouse model. J. Med. Chem. 1977;20:741–744. doi: 10.1021/jm00216a001. [DOI] [PubMed] [Google Scholar]
- Kinnamon K.E., Steck E.A., Rane D.S. A new chemical series active against African trypanosomes: benzyltriphenylphosphonium salts. J. Med. Chem. 1979;22:452–455. doi: 10.1021/jm00190a019. [DOI] [PubMed] [Google Scholar]
- Koreny L., Sobotka R., Kovarova J., Gnipova A., Flegontov P., Horvath A., Obornik M., Ayala F.J., Lukes J. Aerobic kinetoplastid flagellate Phytomonas does not require heme for viability. Proc. Natl. Acad. Sci. U. S. A. 2012;109:3808–3813. doi: 10.1073/pnas.1201089109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova J., Horakova E., Changmai P., Vancova M., Lukes J. Mitochondrial and nucleolar localization of cysteine desulfurase Nfs and the scaffold protein Isu in Trypanosoma brucei. Eukaryot. Cell. 2014;13:353–362. doi: 10.1128/EC.00235-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova N., Mracek T., Nuskova H., Holzerova E., Vrbacky M., Pecina P., Hejzlarova K., Kluckova K., Rohlena J., Neuzil J., Houstek J. High molecular weight forms of mammalian respiratory chain complex II. PLoS One. 2013;8:e71869. doi: 10.1371/journal.pone.0071869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Greca F., Magez S. Vaccination against trypanosomiasis: can it be done or is the trypanosome truly the ultimate immune destroyer and escape artist? Hum. Vaccines. 2011;7:1225–1233. doi: 10.4161/hv.7.11.18203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanteri C.A., Tidwell R.R., Meshnick S.R. The mitochondrion is a site of trypanocidal action of the aromatic diamidine DB75 in bloodstream forms of Trypanosoma brucei. Antimicrob. Agents Chemother. 2008;52:875–882. doi: 10.1128/AAC.00642-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Liu Y., Motyka S.A., Agbo E.E., Englund P.T. Fellowship of the rings: the replication of kinetoplast DNA. Trends Parasitol. 2005;21:363–369. doi: 10.1016/j.pt.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Luque-Ortega J.R., Reuther P., Rivas L., Dardonville C. New benzophenone-derived bisphosphonium salts as leishmanicidal leads targeting mitochondria through inhibition of respiratory complex II. J. Med. Chem. 2010;53:1788–1798. doi: 10.1021/jm901677h. [DOI] [PubMed] [Google Scholar]
- Mazet M., Morand P., Biran M., Bouyssou G., Courtois P., Daulouede S., Millerioux Y., Franconi J.M., Vincendeau P., Moreau P., Bringaud F. Revisiting the central metabolism of the bloodstream forms of Trypanosoma brucei: production of acetate in the mitochondrion is essential for parasite viability. PLoS Negl. Trop. Dis. 2013;7:e2587. doi: 10.1371/journal.pntd.0002587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P.G., Klein R.A. Effects of oligomycin on glucose utilization and calcium transport in African trypanosomes. J. General Microbiol. 1980;116:391–396. doi: 10.1099/00221287-116-2-391. [DOI] [PubMed] [Google Scholar]
- Murphy M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta. 2008;1777:1028–1031. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- Nolan D.P., Voorheis H.P. The mitochondrion in bloodstream forms of Trypanosoma brucei is energized by the electrogenic pumping of protons catalysed by the F1F0-ATPase. Eur. J. Biochem. 1992;209:207–216. doi: 10.1111/j.1432-1033.1992.tb17278.x. [DOI] [PubMed] [Google Scholar]
- Osório A.L., Madruga C.R., Desquesnes M., Soares C.O., Ribeiro L.R., Costa S.C. Trypanosoma (Duttonella) vivax: its biology, epidemiology, pathogenesis, and introduction in the New World – a review. Mem. Inst. Oswaldo Cruz. 2008;103:1–13. doi: 10.1590/s0074-02762008000100001. [DOI] [PubMed] [Google Scholar]
- Pullman M.E., Penefsky H.S., Datta A., Racker E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. I. Purification and properties of soluble dinitrophenol-stimulated adenosine triphosphatase. J. Biol. Chem. 1960;235:3322–3329. [PubMed] [Google Scholar]
- Rios Martinez C.H., Lagartera L., Kaiser M., Dardonville C. Antiprotozoal activity and DNA binding of N-substituted N-phenylbenzamide and 1,3-diphenylurea bisguanidines. Eur. J. Med. Chem. 2014;81:481–491. doi: 10.1016/j.ejmech.2014.04.083. [DOI] [PubMed] [Google Scholar]
- Schnaufer A., Clark-Walker G.D., Steinberg A.G., Stuart K. The F1-ATP synthase complex in bloodstream stage trypanosomes has an unusual and essential function. EMBO J. 2005;24:4029–4040. doi: 10.1038/sj.emboj.7600862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R.A., Hartley R.C., Murphy M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Signal. 2011;15:3021–3038. doi: 10.1089/ars.2011.3969. [DOI] [PubMed] [Google Scholar]
- Snow B.J., Rolfe F.L., Lockhart M.M., Frampton C.M., O'Sullivan J.D., Fung V., Smith R.A., Murphy M.P., Taylor K.M., Protect Study G. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- Spivak A.Y., Keiser J., Vargas M., Gubaidullin R.R., Nedopekina D.A., Shakurova E.R., Khalitova R.R., Odinokov V.N. Synthesis and activity of new triphenylphosphonium derivatives of betulin and betulinic acid against Schistosoma mansoni in vitro and in vivo. Bioorg. Med. Chem. 2014;22:6297–6304. doi: 10.1016/j.bmc.2014.07.014. [DOI] [PubMed] [Google Scholar]
- Subrtova K., Panicucci B., Zikova A. ATPaseTb2, a unique membrane-bound FoF1-ATPase component, is essential in bloodstream and dyskinetoplastic trypanosomes. PLoS Pathog. 2015;11:e1004660. doi: 10.1371/journal.ppat.1004660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swallow B.M. Food and Agriculture Organization of the United Nations; Rome: 1999. Impacts of Trypanosomiasis on African Agriculture. [Google Scholar]
- Taladriz A., Healy A., Flores Perez E.J., Herrero Garcia V., Rios Martinez C., Alkhaldi A.A., Eze A.A., Kaiser M., de Koning H.P., Chana A., Dardonville C. Synthesis and structure-activity analysis of new phosphonium salts with potent activity against African trypanosomes. J. Med. Chem. 2012;55:2606–2622. doi: 10.1021/jm2014259. [DOI] [PubMed] [Google Scholar]
- Tan T.H., Bochud-Allemann N., Horn E.K., Schneider A. Eukaryotic-type elongator tRNAMet of Trypanosoma brucei becomes formylated after import into mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2002;99:1152–1157. doi: 10.1073/pnas.022522299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira J., Soares P., Benfeito S., Gaspar A., Garrido J., Murphy M.P., Borges F. Rational discovery and development of a mitochondria-targeted antioxidant based on cinnamic acid scaffold. Free Radic. Res. 2012;46:600–611. doi: 10.3109/10715762.2012.662593. [DOI] [PubMed] [Google Scholar]
- Tielens A.G., van Hellemond J.J. Surprising variety in energy metabolism within Trypanosomatidae. Trends Parasitol. 2009;25:482–490. doi: 10.1016/j.pt.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Tyc J., Klingbeil M.M., Lukes J. Mitochondrial heat shock protein machinery hsp70/hsp40 is indispensable for proper mitochondrial DNA maintenance and replication. mBio. 2015;6 doi: 10.1128/mBio.02425-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercesi A.E., Docampo R., Moreno S.N. Energization-dependent Ca2+ accumulation in Trypanosoma brucei bloodstream and procyclic trypomastigotes mitochondria. Mol. Biochem. Parasitol. 1992;56:251–257. doi: 10.1016/0166-6851(92)90174-i. [DOI] [PubMed] [Google Scholar]
- Welburn S.C., Picozzi K., Fevre E.M., Coleman P.G., Odiit M., Carrington M., Maudlin I. Identification of human-infective trypanosomes in animal reservoir of sleeping sickness in Uganda by means of serum-resistance-associated (SRA) gene. Lancet. 2001;358:2017–2019. doi: 10.1016/s0140-6736(01)07096-9. [DOI] [PubMed] [Google Scholar]
- Wickstead B., Ersfeld K., Gull K. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol. Biochem. Parasitol. 2002;125:211–216. doi: 10.1016/s0166-6851(02)00238-4. [DOI] [PubMed] [Google Scholar]
- Williams S., Saha L., Singha U.K., Chaudhuri M. Trypanosoma brucei: differential requirement of membrane potential for import of proteins into mitochondria in two developmental stages. Exp. Parasitol. 2008;118:420–433. doi: 10.1016/j.exppara.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz E., Leal S., Ochatt C., Cross G.A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- Zikova A., Schnaufer A., Dalley R.A., Panigrahi A.K., Stuart K.D. The F(0)F(1)-ATP synthase complex contains novel subunits and is essential for procyclic Trypanosoma brucei. PLoS Pathog. 2009;5:e1000436. doi: 10.1371/journal.ppat.1000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.