

Abstract
Aberrant accumulation of protein aggregates is a pathological hallmark of many neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). Although a buildup of protein aggregates frequently leads to cell death, whether it is the key pathogenic factor in driving neurodegenerative disease remains controversial. HDAC6, a cytosolic ubiquitin-binding deacetylase, has emerged as an important regulator of ubiquitin-dependent QC autophagy, a lysosome-dependent degradative system responsible for the disposal of misfolded protein aggregates and damaged organelles. Here, we show that in cell models, HDAC6 plays a protective role against multiple disease-associated and aggregation-prone cytosolic proteins by facilitating their degradation. We further show that HDAC6 is required for efficient localization of lysosomes to protein aggregates, indicating that lysosome targeting to autophagic substrates is regulated. Supporting a critical role of HDAC6 in protein aggregate disposal in vivo, genetic ablation of HDAC6 in a transgenic SOD1G93A mouse, a model of ALS, leads to dramatic accumulation of ubiquitinated SOD1G93A protein aggregates. Surprisingly, despite a robust buildup of SOD1G93A aggregates, deletion of HDAC6 only moderately modified the motor phenotypes. These findings indicate that SOD1G93A aggregation is not the only determining factor to drive neurodegeneration in ALS, and that HDAC6 likely modulates neurodegeneration through additional mechanisms beyond protein aggregate clearance.
Keywords: ALS, SOD1 mutant, HDAC6, aggresome, aggregates, autophagy, lysosome
Introduction
Neurodegenerative diseases have become a major public health issue in the aging populations [1-4]. Clinical pathogenic features of neurodegenerative diseases generally reflect the unique neuronal cell type affected. For example, the loss of dopaminergic neurons in Parkinson's disease leads to tremor [5] and the death of motor neurons in amyotrophic lateral sclerosis (ALS) causes paralysis [3]. In spite of diverse symptoms, these diseases share a common pathological feature: the formation of inclusion bodies made of misfolded protein aggregates in affected neurons [6]. Supporting a relevant role of protein aggregation in neurodegeneration, most genetic mutations found in familial cases of neurodegenerative disease produce mutant proteins that are prone to misfolding and aggregation [6]. The prominent examples include SOD1G93A for ALS and expansion of polyglutamine tracts in huntingtin (Huntington's disease), androgen receptors (spinal and bulbar muscular atrophy) and ataxin-1 (autosomal dominant cerebellar ataxia) [1-4].
Misfolded protein-derived aggregates can form in the cytoplasm or nucleus. Once aggregates accumulate, an elaborate cellular quality control (QC) system is activated to clear these toxic entities [7,8]. Generally, protein aggregates cannot be properly unfolded for proteasome-mediated degradation. In cell models, protein aggregates are frequently concentrated to the microtubule organizing center (MTOC) where they are further processed by autophagy [9]. This process leads to formation of a peri-nuclear inclusion body, termed the aggresome, which shares some features with inclusion bodies, such as Lewy bodies, found in neurodegenerative diseases [9,10]. Accumulating evidence suggests that the aggresome, rather than being a mere junkyard for protein aggregates, is actively processed and cleared through the coordinated actions of the proteasome and autophagy [11].
In the cellular QC system that targets protein aggregates and damaged organelles for autophagic degradation, the ubiquitin-binding cytoplasmic deacetylase HDAC6 has emerged as an important player [12-15]. HDAC6 is a component of the aggresome and Lewy bodies in Parkinson's disease patients [6,10,15]. Through its ubiquitin-binding domain (BUZ), HDAC6 binds and facilitates the transport of ubiquitinated misfolded proteins via microtubules to form the aggresome [10], and subsequently, coordinates the clearance of aggresomes by autophagy [11,12,14,16,17]. In this context, HDAC6 is not required for autophagy activation per se; rather, it stimulates autophagosome-lysosome fusion via an actin cytoskeleton dependent mechanism [11,12]. HDAC6- and ubiquitin-dependent QC autophagy is also involved the clearance of damaged mitochondria tagged by parkin, a process implicated in early onset Parkinson's disease (AR-JP) [18].
Collectively, these findings indicate that HDAC6 plays a central role in the clearance of cytosolic protein aggregates and, consequently, neurodegeneration induced by toxic protein aggregates. In this report, we show that genetic inactivation of HDAC6 indeed significantly inhibits the clearance of multiple forms of cytosolic aggregates in a cell model and SOD1G93A mutant protein in a mouse ALS model. Cell-based assays reveal that HDAC6 is also required for efficient targeting of lysosomes to ubiquitinated protein aggregates, uncovering a novel regulation of autophagy-dependent inclusion body clearance. Despite a large increase in ubiquitinated SOD1G93A protein aggregates upon HDAC6 knockout, surprisingly, the motor phenotypes of SOD1G93A mice are only moderately affected by HDAC6 deletion. These results indicate that SOD1G93A aggregation is not the only toxic factor in driving ALS pathogenesis.
Materials and Methods
Cell lines and plasmids
Control and HDAC6 knockdown 293T and A549 cells were prepared as described previously [10]. GFP-tagged CFTRΔF508, AR-112Q, Htt-81Q, SOD1-G93A and ataxin1-82Q expression plasmids were prepared by Qiagen Maxi-prep kit and transfected using Fugene 6 as following manufacturer's protocols. Wild type and HDAC6 KO MEFs reconstituted with various HDAC6 constructs were prepared as described previously [19]. Lipofectamine LTX (Invitrogen) was used for transfection into MEFs according to manufacturer's protocol.
Antibodies and reagents
The following antibodies were used: HDAC6 (H-300; Santa Cruz), acetyl-α-tubulin (Sigma), ubiquitin (Biomol), LAMP-1 (Hybridoma Bank, Iowa), GFP (Roche), active-caspase 3 (Millipore), human SOD1 (Millipore), and actin (Sigma).
Animal care and husbandry
Mice were cared for by the animal facility at Duke University with a regular maintenance program. HDAC6+/- female mice (129/Black Swiss or B6SJL genetic background) were mated to SOD1G93A [B6SJL-Tg(SOD1-G93A)1Gur/J, Jackson Laboratory] to generate litters. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Animal Research Committee of Duke University (Permit Number: A321-09-11). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Cytotoxicity assay using anti-active caspase 3 antibody
Control and HDAC6 knockdown A549 cells were cultured on glass coverslips, followed by transient transfection of GFP-tagged CFTRAF508, AR-112Q, Htt-81Q, SOD1-G93A or ataxin1-82Q expression plasmids with or without shRNA-resistant HDAC6 expression plasmid. Cells were fixed and stained with anti-GFP (green) and anti-active caspase 3 antibody (red). Count active caspase 3 positive cells.
Immunofluorescence microscopy
Immunostaining was performed as described previously [20,21]. Cells were cultured on glass coverslips, followed by treatment with MG132 at 2.5 μM for 24 h. Cells were washed with PBS, incubated with growth media for 18 hr to clear soluble ubiquitinated proteins, and then processed for immunostaining using anti-ubiquitin and anti-LAMP1 antibodies. Images were acquired by a spinning-disk confocal microscope (Leica DMI6000C).
Filter-Trap assay
Filter-trap assay for aggregates was performed as described [22]. The total protein load was normalized to the volume of the soluble fraction.
Immunohistochemical analysis for mouse spinal cord sections
Mice were transcardially perfused with 4% paraformaldehyde in phosphate buffer (pH 7.4). Lumbar spinal cords were dissected out and embedded in O.C.T. compound. Frozen lumbar spinal cord tissue sections (thickness: 20μm) were subjected to immunostaining with anti-ubiquitin antibody followed by FITC-conjugated secondary antibody and DAPI staining.
Rotarod test
Mice were tested for their motor coordination using Rota-rod apparatus (Ugo Basile, Comerio, VA , Italy) with an accelerating speed protocol from 4 to 40 rpm for 5min. The longest latency to fall from 3 trials was recorded; 180 seconds was chosen as the arbitrary cutoff time for disease onset [9].
Stride length measurement test
The stride test was performed as previously published [5]. Mice had their hindpaws dipped in nontoxic ink and were trained to walk across a 1 m white board for recording footprints. Stride lengths were measured in millimeters.
Paw grip endurance test
Forelimb grip strength was tested with a grip meter analysis for mouse (Bioseb) as previously described [7]. The average of 3 trials was presented.
Statistical analysis
Two-tailed Student's t-test was conducted for statistical analysis of quantitative data.
Results
HDAC6-deficient cells are more sensitive to cytosolic protein aggregate-induced toxicity
We have previously shown that HDAC6 promotes the processing and clearance of cytosolic protein aggregates induced by proteasome inhibition [12]. To determine if HDAC6 plays a general protective role against cytoplasmic protein aggregates, we expressed several disease-associated, aggregation-prone proteins in control and HDAC6-knockdown 293T cells. Cystic fibrosis transmembrane conductance regulator ΔF508 mutant (CFTR-ΔF508-GFP), androgen receptor 112Q (AR-112Q-GFP) mutants, huntingtin-81Q (Htt-81Q-GFP), and superoxide dismutase 1 G93A mutant (SOD1-G93A-GFP) formed cytosolic aggregates, while ataxin-1 82Q-GFP produced nuclear aggregates (data not shown). To determine the amount of aggregates, 1% SDS-insoluble fractions (Marked as I) were prepared and subjected to Western blotting with a GFP antibody (Fig. 1Ai) and quantified (Fig. 1Aii). As shown in Fig. 1, HDAC6 knockdown increased the accumulation of cytoplasmic type of aggregates (CFTR-ΔF508 and AR-112Q, marked by brackets) but had little effect on nuclear ataxin-1 82Q aggregates in the SDS-insoluble fraction (Fig. 1Ai right panel). These results support the notion that HDAC6 selectively regulates the clearance of cytosolic protein aggregates, consistent with its subcellular localization [21].
Fig. 1. HDAC6-knockdown cells are sensitive to misfolded proteins associated with neurodegeneration.
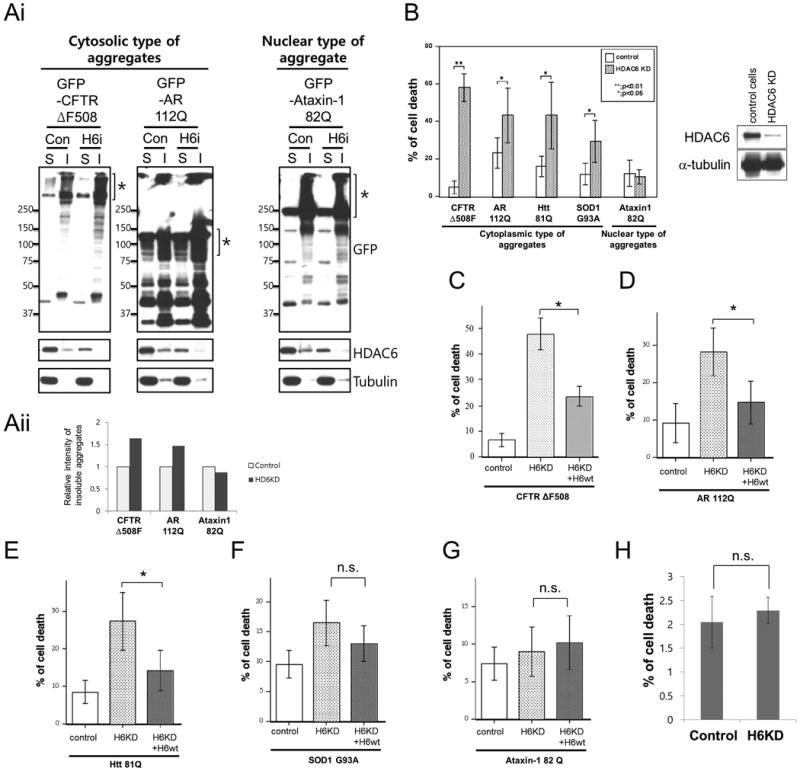
(Ai) Control and HDAC6-knockdown 293T cells were transfected with GFP-fused cytosolic (GFP-CFTR ΔF508 and GFP-AR-112Q) or nuclear aggregates (GFP-Ataxin-1 82Q) and fractionated into SDS soluble (S) and SDS insoluble fractions (I). Each fractionated sample was subjected for immunoblot analysis using anti-GFP, anti-HDAC6 and anti-tubulin antibodies. (Aii) Western blotting images were analyzed by BIO-1D™ software and relative band intensity of SDS insoluble aggregates were presented in bottom panel graph. (B) Control and HDAC6-knockdown A549 cells were transfected with cytosolic (CFTR ΔF508, AR112Q, Htt 81Q, SOD1G93A) or nuclear protein aggregates (ataxin-1) expression plasmids. Cells were immunostained with anti-active-caspase-3 antibody to check apoptotic cells and the average of apoptotic cells percent from three independent experiments are presented with standard deviation as error bar (t-test, *: p<0.05 and **: p<0.01). HDAC6 knockdown level was checked by Western blot using anti-HDAC6 and anti-tubulin antibodies (right panel). (C-G) shRNA-resistant HDAC6 was co-transfected with CFTRΔF508 (C), AR 112Q (D), Htt 81Q (E), SOD1G93A (F) or Ataxin-1 82Q (G) expression plasmids in HDAC6 knockdowned A549 cells. Apoptotic cells were stained with anti-active-caspase 3 antibody and the average of apoptotic cells percentage from three independent experiments are presented with standard deviation as error bar (t-test, *: p<0.05 and **: p<0.01). (H) Control and HDAC6 knockdown A549 cells were subjected to cytotoxicity assay using CytoTox-ONEtm. Basal cytotoxicity were measured by % of LDH release and presented as % of cell death. Averages from 3 independent experiments are presented with standard deviation as error bar (n.s.: not significant).
HDAC6-deficient A549 cells are more susceptible to cell death induced by proteasome inhibition [10]. If HDAC6 is selectively required for cytosolic protein aggregate clearance, it is expected that HDAC6-deficient cells would be more sensitive to toxicity caused by cytosolic aggregates than nuclear aggregates. As shown in Fig. 1B, apoptosis was significantly increased in HDAC6-knockdown A549 cells expressing cytosolic aggregation-prone proteins, CFTR-ΔF508, AR-112Q, Htt-81Q and SOD1-G93A. In contrast, apoptosis induced by the nuclear aggregation-prone ataxin-1 82Q was comparable between control and HDAC6 knockdown A549 cells (Fig. 1B). Expression of a shRNA-resistant HDAC6 in HDAC6 knockdown A549 significantly reduced the cytotoxicity associated with CFTR-AF508, AR-112Q and Htt-81Q, and a more moderate effect on SOD1-G93A (Fig. 1C-F). As expected, HDAC6 re-expression had no effect on ataxin-1 82Q toxicity cells (Fig. 1G). Of note, HDAC6 knockdown itself did not cause cell death (Fig. 1H), as it was previously reported [10]. These data indicate that HDAC6 plays a cytoprotective role against toxicity associated with cytosolic protein aggregates.
HDAC6 is required for the proper targeting of lysosomes to ubiquitinated protein aggregates
Autophagy is the primary cytoplasmic degradation machinery responsible for the disposal of protein aggregates [23]. It involves the formation of double membrane-vesicles that sequester cytosolic constituents to form autophagosome, which then fuse to lysosomes where the contents are degraded. We found that the majority of large ubiquitinated protein aggregates (∼60%) in wild type MEFs were encircled by lysosomes in the perinuclear region (Fig. 2A and 2B). In contrast, lysosomes in HDAC6 KO MEF were dispersed to the cell periphery and not concentrated to protein aggregates. This finding indicates that targeting of lysosomes to protein aggregates is defective in HDAC6 KO MEFs (Fig. 2A and 2B). Interestingly, Iwata et al. also showed HDAC6 knockdown led to periplasmic dispersion of lysosomes [16]. Importantly, failure to target lysosomes to protein aggregates was efficiently reversed by the reintroduction of wild type, but not catalytic inactive (CD) or ubiquitin binding-deficient (ABUZ) mutant, HDAC6 (Fig. 2A and 2B). Taken together, these results indicate that HDAC6 is required for targeting lysosomes to protein aggregates, which could enable more efficient autophagy-dependent aggregate degradation.
Fig. 2. HDAC6 is required for efficient targeting of lysosomes to aggregates.
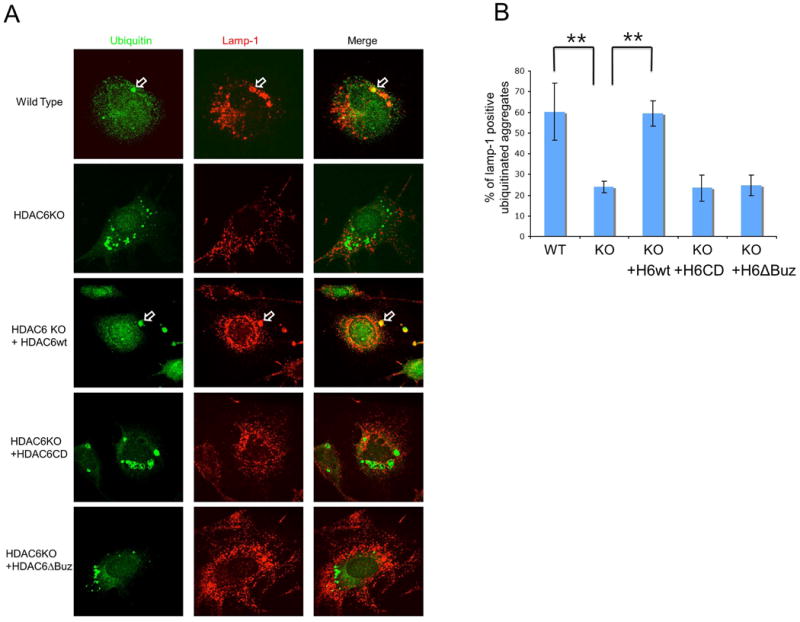
(A) Wild type, HDAC6 KO and HDAC6 KO MEFs that stably express human HDAC6WT, HDAC6CD or HDAC6ΔBuz were treated with MG132 (2.5 μM for 24 hours) and changed with normal growth media for 18 hours to allow the degradation of non-aggregated protein. Cells were immunostained with anti-LAMP-1 antibody for lysosomes and anti-ubiquitin antibody for aggregates. Open arrows indicated ubiquitin-positive aggregates that contain LAMP-1. (B) Quantification of lysosomes targeted to ubiquitin-positive aggregates. The graph represents the percent of LAMP-1-positive aggregates. The mean of LAMP-1-positive aggregates from 3 independent experiments (more than 10 images per each experiment) are presented with standard deviation for error bar. (t-test, **p<0.01)
Genetic ablation of HDAC6 leads to prominent accumulation of ubiquitinated SOD1G93A protein aggregates in a SOD1G93A ALS mouse model
ALS patients accumulate a characteristic protein inclusion body, termed Lewy body-like Hyaline inclusions [24]. The expression of ALS-associated SOD1-G93A mutant in cultured cells can also form aggresome-like inclusion bodies [25]. We next investigated whether genetic ablation of HDAC6 would affect protein aggregation and neurodegeneration in a SOD1G93A transgenic ALS mouse model. We bred HDAC6 heterozygote female mice (129/Black Swiss mixed background) with SOD1G93A males (B6SJL background) to produce HDAC6WT, HDAC6KO, HDAC6WT/SOD1G93A and HDAC6KO/SOD1G93A male mice for analyses. We first used immuno-staining to assess the amount of ubiquitinated protein aggregates in the spinal cords from HDAC6WT, HDAC6KO, HDAC6WT/SOD1G93A, and HDAC6KO/SOD1G93A mice at 21 weeks old. As expected, a few ubiquitin-positive inclusion body-like structures were detected in the cross sections of the lumbar spinal cord from HDAC6WT/SOD1G93A mice (Fig 3A, arrowheads, bottom-left panel). No such a signal was detected in WT control mice (Fig 3A, top-left panel). In stark contrast, dramatically more ubiquitinated aggregates were detected in spinal cord sections from HDAC6KO/SOD1G93A double mutant mice (Fig. 3A, bottom-right panel). Of note, similar to what we have previously reported [12], modest ubiquitinated protein aggregates were detected in the lumbar spinal cord of HDAC6KO mice (Fig. 3A. top-right panel). To further confirm the elevated accumulation of SOD1G93A aggregates in HDAC6KO/SOD1G93A mice, we measured the amount of ubiquitinated and SOD1G93A protein aggregates in lysates prepared from lumbar spinal cords by the filter-trap assay. Consistent with immunostaining results, protein aggregates, as detected by anti-ubiquitin and anti-hSOD1 antibodies, were greatly increased in spinal cords from HDAC6KO/SOD1G93A double mutant mice compare to those from SOD1G93A single transgenic mice (Fig. 3B). The same spinal cord lysates were subjected to Western analysis and showed that soluble SOD1G93A proteins were not affected by HDAC6 deficiency (Fig. 3C). Taken together, these data demonstrate that HDAC6 deficiency causes a significant accumulation of SOD1G93A protein aggregates in SOD1G93A transgenic mice.
Fig. 3. HDAC6 deficiency results in accumulation of SOD1 G93A mutant protein aggregates in the spinal cord.
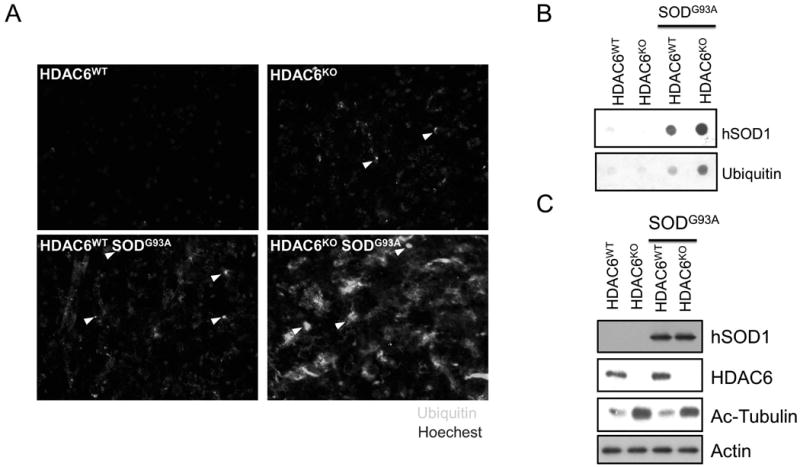
(A) 129/Black Swiss/B6SJL background mice for each genotype were sacrificed at 20 weeks of age, and dissected spinal cord were subjected to the immunostaining with anti-ubiquitin antibody for ubiquitinated protein aggregates. Arrowheads indicate ubiquitinated protein aggregates. (B) Spinal cord lysates (100 ug) from indicated genotypes at 20 weeks old were subjected to filter trap assay to measure protein aggregates, using anti-ubiquitin (bottom panel) and anti-human SOD1 antibody (top panel). (C) The same spinal cord lysates were subjected to immunoblotting using anti-human SOD1, anti-HDAC6, anti-acetyl tubulin and anti-actin antibodies.
HDAC6 moderately affects the pathogenesis of ALS in SOD1G93A model
We next investigated whether HDAC6 also modifies the motor phenotypes induced by SOD1G93A. As shown in Fig. 4A, HDAC6KO mice did not show significant difference from WT mice in the accelerating rotarod test, indicating that HDAC6 inactivation by itself does not lead to a motor phenotype. We then evaluated mice from breeding HDAC6KO female (129/Black Swiss) with SOD1G93A male (B6SJL). The analysis of HDAC6KO/SOD1G93A mice showed a motor phenotype (defined as when mice could not stay on the rod for 180 seconds) 2 weeks earlier than SOD1G93A single transgenic mice did although the difference did not reach statistical significance (Fig. 4B). HB-9 positive motor neurons in the spinal cords of HDAC6KO/SOD1G93A were reduced compared to HDAC6WT/SOD1G93A at 20 weeks old mice (Fig. 4C). Thus, despite the further accumulation of protein aggregates and some reduction in motor neurons (Fig. 3), the deletion of HDAC6 only moderately accelerates motor phenotypes caused by SOD1G93A.
Fig. 4. HDAC6 deficiency moderately accelerates the pathogenesis of ALS disease in mouse model.
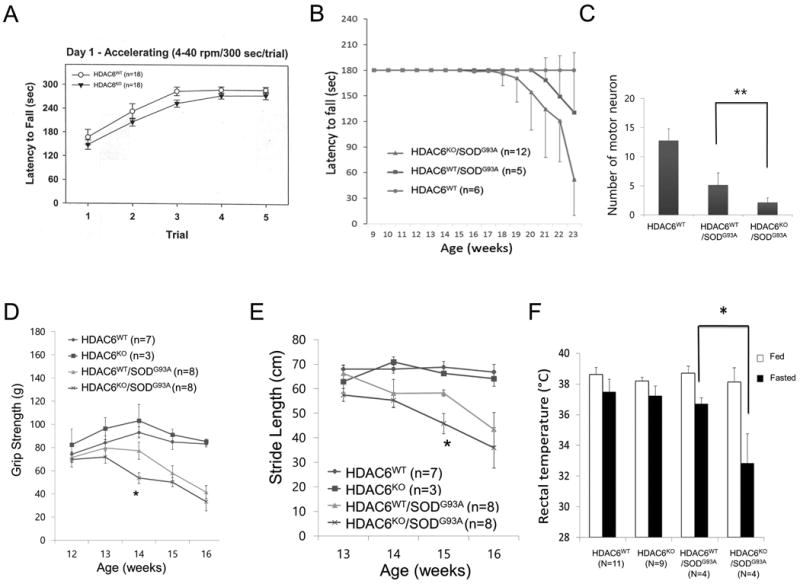
(A) Wild type and HDAC6 KO mice with 129/Black Swiss with double genetic background were subjected to accelerating rotarod test (4-40rpm/300 sec) at 6 month old. Average of latency to fall are presented with standard error as error bar. (B) 129/Black Swiss/B6SJL triple genetic background mice with indicated genotypes were subjected to accelerating rotarod test (4-40rpm/180 sec) at every week from 9 weeks to 23 weeks old. Latency to fall was recorded as highest score of three trials and presented as average of group. Cut-off is 180 seconds. Average of latency to fall were presented with standard deviation as error bar. (C) The mouse spinal cord sections are stained with anti-HB-9 antibody to visualize motor neuron for counting at 20 weeks old. Quantification of motor neuron in spinal cords from indicated genotype mouse are presented with standard deviation for error bar. (t-test, **: p<0.01) (D) B6SJL single genetic background mouse with indicated genotypes were subjected to a paw grip endurance test to measure muscle strength. Grip strength was monitored every week. The averages are presented with standard deviation as error bar. (t-test; *: p<0.05) (E) The same cohorts were subjected to stride length measurement tests to assess motor function. Stride length was measured every week. The averages are presented with standard deviation as error bar. (t-test; *: p<0.05) (F) Body temperature of indicated genotypes (12 weeks old) was measured under fed and 24 hours fasted condition, using rectal temperature probe. The averages are presented with standard deviation as error bar. (t-test; *: p<0.05)
To minimize the effect of different genetic backgrounds that may generate variation, HDAC6KO mice were backcrossed into B6SJL for 4 generations and mated with SOD1G93A (B6SJL) male mice. In B6SJL single background littermates, overall disease onset was shortened (at ∼14 weeks old) in contrast to 129/Black Swiss/B6SJL triple background mice described in the previous section (about 18 weeks old). In the new genetic background, we performed paw grip endurance (PaGE) and stride length measurement (SLM) tests. In 14 week-old mice, we observed a statistically significant decrease in PaGE performance in SOD1G93A/HDAC6KO mice compared to SOD1G93A/HDAC6WT mice (Fig. 4D). In the SLM test, SOD1G93A/HDAC6KO mice showed significantly shorter stride length than SOD1G93A/HDAC6WT at 15 weeks old age (Fig. 4E). Taken together, the HDAC6KO/SOD1G93A cohort showed moderate acceleration of motor phenotypes compared with the HDAC6WT/SODG93A cohorts. Thus, in both genetic backgrounds, the effect of HDAC6 deletion on motor function of SOD1G93A mice was modest.
Recently, metabolic dysregulation has been reported in ALS mouse model where starvation could magnify the thermogenic defect in ALS mutant mice [2]. We therefore investigated if HDAC6 status affects the thermogenic phenotype in SOD1G93A mice. We recorded rectal temperature of 12 week-old mice that did not yet show motor phenotypes. As shown in Fig. 4F (white column), no significant difference in rectal temperature was detected among genotypes under fed conditions. After 24 hours fasting, rectal temperature trended lower in all genotypes. Importantly, HDAC6KO/SOD1G93A mice showed a significant and prominent decrease of rectal temperature to about 32.5°C (Fig. 4F, black column). This result indicates that the genetic deletion of HDAC6 modifies the thermogenic phenotype of SOD1G93A transgenic mouse model.
Discussion
In this report, we determine the contribution of disease-associated protein aggregates in the development of neurodegenerative disease. Through the characterization of HDAC6-deficient cells, we show that HDAC6 plays a cytoprotective role against toxic effects of cytosolic but not nuclear aggregate-prone proteins. These findings are consistent with the proposed role of HDAC6 in promoting ubiquitin-dependent QC autophagy that degrades protein aggregates. However, the genetic analysis of SOD1G93A mice has revealed that a marked increase in SOD1G93A aggregates caused by HDAC6 ablation did not lead to significant aggravation of motor phenotypes. This unexpected finding has uncovered a more complex picture of protein aggregation in neurodegenerative disease.
There are several possible mechanisms to explain the apparent uncoupling of SOD1G93A aggregation from the motor phenotypes. First, it is possible that SOD1G93A aggregation plays only a minor role in driving motor dysfunction under normal laboratory conditions. Accordingly, SOD1G93A toxicity would be less sensitive to the status of HDAC6. It should be noted that in Drosophila and C. elegans, loss of HDAC6 significantly enhanced the toxicity induced by transgenic α–synuclein, an aggregation-prone protein associated with Parkinson's disease [26,27]. We did also observe fewer motor neurons in SOD1G93A; HDAC6KO double mutant mice than in the SOD1G93A transgenic mice (Fig. 4C). Thermogenesis under starvation is also significantly reduced in HDAC6KO/SOD1G93A mice compared to the single transgenic mice (Fig. 4F). Collectively, these findings clearly indicate a functional interaction between HDAC6 deletion and SOD1G93A. However, these differences did not translate into robust motor phenotypes. Whether this differential effect of HDAC6 reflects the intrinsic difference between SOD1-G93A and α–synuclein aggregates or a more complex biology of neurodegeneration in mice remains to be determined. Second, HDAC6 has activities independent of autophagy. Pharmacological and genetic inhibition of HDAC6 increases the acetylation of α-tubulin at the lysine 40, which has been shown to promote intracellular trafficking and axonal transport of mitochondria and organelles [28,29]. As defects in axonal transport have been documented in neurodegenerative diseases [30], an increased axonal transport could potentially improve neuronal function [31]. Acetylation of a-tubulin also promotes anti-inflammatory responses [4]. Given the prominent role of neuroinflamation in the progression of ALS [32], inhibition of HDAC6 could potentially suppress neuroinflamation. These beneficial effects caused by HDAC6 ablation could potentially alleviate the toxicity associated with SOD1 aggregates, thereby explaining the relatively mild effect of HDAC6 on modifying motor phenotypes induced by SOD1G93A. It is conceivable that HDAC6-selective inhibitors that promote tubulin acetylation, but do not interfere with protein aggregate clearance, could have desirable pharmacological effects on ALS or related neurodegenerative disease.
Acknowledgments
This work is supported by research fund of 2013 Chungnam National University, 2013R1A1A2008576 (NRF of Korea) to J.Y. Lee, NIH grant R01NS077284 to H.Z. and 2R01-NS054022 and AR055613 (NIH) to T.P.Y.
References
- 1.Sopher BL, Thomas PS, Jr, LaFevre-Bernt MA, Holm IE, Wilke SA, Ware CB, Jin LW, Libby RT, Ellerby LM, La Spada AR. Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron. 2004;41:687–99. doi: 10.1016/s0896-6273(04)00082-0. [DOI] [PubMed] [Google Scholar]
- 2.Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11159–64. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O'Halloran TV, Siddique T. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7142–7. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang B, Rao YH, Inoue M, Hao R, Lai CH, Chen D, McDonald SL, Choi MC, Wang Q, Shinohara ML, Yao TP. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nature communications. 2014;5:3479. doi: 10.1038/ncomms4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernagut PO, Diguet E, Labattu B, Tison F. A simple method to measure stride length as an index of nigrostriatal dysfunction in mice. Journal of neuroscience methods. 2002;113:123–30. doi: 10.1016/s0165-0270(01)00485-x. [DOI] [PubMed] [Google Scholar]
- 6.Miki Y, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K. Accumulation of histone deacetylase 6, an aggresome-related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology : official journal of the Japanese Society of Neuropathology. 2011;31:561–8. doi: 10.1111/j.1440-1789.2011.01200.x. [DOI] [PubMed] [Google Scholar]
- 7.Weydt P, Hong SY, Kliot M, Moller T. Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport. 2003;14:1051–4. doi: 10.1097/01.wnr.0000073685.00308.89. [DOI] [PubMed] [Google Scholar]
- 8.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 9.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends in cell biology. 2000;10:524–30. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 10.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 11.Hao R, Nanduri P, Rao Y, Panichelli RS, Ito A, Yoshida M, Yao TP. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Molecular cell. 2013;51:819–28. doi: 10.1016/j.molcel.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. The EMBO journal. 2010;29:969–80. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy. 2007;3:643–5. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- 14.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 15.Su M, Shi JJ, Yang YP, Li J, Zhang YL, Chen J, Hu LF, Liu CF. HDAC6 regulates aggresome-autophagy degradation pathway of alpha-synuclein in response to MPP+-induced stress. Journal of neurochemistry. 2011;117:112–20. doi: 10.1111/j.1471-4159.2011.07180.x. [DOI] [PubMed] [Google Scholar]
- 16.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. The Journal of biological chemistry. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 17.Gal J, Chen J, Barnett KR, Yang L, Brumley E, Zhu H. HDAC6 regulates mutant SOD1 aggregation through two SMIR motifs and tubulin acetylation. The Journal of biological chemistry. 2013;288:15035–45. doi: 10.1074/jbc.M112.431957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. The Journal of cell biology. 2010;189:671–9. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao YS, Hubbert CC, Lu J, Lee YS, Lee JY, Yao TP. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol Cell Biol. 2007;27:8637–47. doi: 10.1128/MCB.00393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JY, Kim H, Ryu CH, Kim JY, Choi BH, Lim Y, Huh PW, Kim YH, Lee KH, Jun TY, Rha HK, Kang JK, Choi CR. Merlin, a tumor suppressor, interacts with transactivation-responsive RNA-binding protein and inhibits its oncogenic activity. J Biol Chem. 2004;279:30265–73. doi: 10.1074/jbc.M312083200. [DOI] [PubMed] [Google Scholar]
- 21.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–8. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 22.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–58. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato S, Hayashi H, Nakashima K, Nanba E, Kato M, Hirano A, Nakano I, Asayama K, Ohama E. Pathological characterization of astrocytic hyaline inclusions in familial amyotrophic lateral sclerosis. Am J Pathol. 1997;151:611–20. [PMC free article] [PubMed] [Google Scholar]
- 25.Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12571–6. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Usenovic M, Knight AL, Ray A, Wong V, Brown KR, Caldwell GA, Caldwell KA, Stagljar I, Krainc D. Identification of novel ATP13A2 interactors and their role in alpha-synuclein misfolding and toxicity. Human molecular genetics. 2012;21:3785–94. doi: 10.1093/hmg/dds206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du G, Liu X, Chen X, Song M, Yan Y, Jiao R, Wang CC. Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Molecular biology of the cell. 2010;21:2128–37. doi: 10.1091/mbc.E10-03-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:3571–83. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Current biology : CB. 2006;16:2166–72. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 30.Perlson E, Maday S, Fu MM, Moughamian AJ, Holzbaur EL. Retrograde axonal transport: pathways to cell death? Trends in neurosciences. 2010;33:335–44. doi: 10.1016/j.tins.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taes I, Timmers M, Hersmus N, Bento-Abreu A, Van Den Bosch L, Van Damme P, Auwerx J, Robberecht W. Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Human molecular genetics. 2013;22:1783–90. doi: 10.1093/hmg/ddt028. [DOI] [PubMed] [Google Scholar]
- 32.Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science. 2006;312:1389–92. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]