

Abstract
Aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor regulating the expression of genes involved in xenobiotic response. Recent studies have suggested that AhR plays essential roles not only in xenobiotic detoxification but also energy metabolism. Thus, in this study, we studied the roles of AhR in lipid metabolism. Under high fat diet (HFD) challenge, liver-specific AhR knock-out (AhR LKO) mice exhibited severe steatosis, inflammation, and injury in the liver. Gene expression analysis and biochemical study revealed that de novo lipogenesis activity was significantly increased in AhR LKO mice. In contrast, induction of suppressor of cytokine signal 3 (Socs3) expression by HFD was attenuated in the livers of AhR LKO mice. Rescue of the Socs3 gene in the liver of AhR LKO mice cancelled the HFD-induced hepatic lipotoxicities. Promoter analysis established Socs3 as novel transcriptional target of AhR. These results indicated that AhR plays a protective role against HFD-induced hepatic steatosis and the subsequent lipotoxicity effects, such as inflammation, and that the mechanism of protection involves the direct transcriptional regulation of Socs3 expression by AhR.
Keywords: inflammation, lipid metabolism, liver, liver injury, liver metabolism, aryl hydrocarbon receptor (AhR), suppressor of cytokine signal 3 (Socs3)
Introduction
The aryl hydrocarbon receptor (AhR)2 is a ligand-activated transcription factor that possesses a basic helix-loop-helix/Per-Arnt-Sim domain and mediates a variety of toxic and biological effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds (1–3). Upon binding to ligands, AhR forms a heterodimer with the AhR nuclear translocator (Arnt) in the nucleus. Then, this complex transactivates the target genes such as xenobiotic-metabolizing enzymes by binding to xenobiotic response element (XRE) sequences in their promoter region (4–6). In addition to direct transcriptional regulation, AhR controls gene expressions thorough interactions with other transcription factors such as nuclear factor κ-light chain enhancer, estrogen receptor, and E2F (7–9).
Recent studies using Ahr knock-out (KO) mice have suggested that AhR is required for not only xenobiotic responses but also for several endobiotic responses, including the development of tissues and reproduction (10–12). These functional roles of AhR in endobiotic responses are supported by the identification of endogenous ligands or activators (13–22).
One possible function of AhR is the regulation of energy metabolism for the following reasons. First, AhR is highly expressed in metabolically active tissues, including the liver, adipose tissue, and macrophages. Second, the expressions of AhR and its target gene are increased in obese mice (23, 24). Third, global AhR KO mice exhibit spontaneous lipid accumulation and fibrosis in the liver (11). Moreover, a comprehensive analysis of gene expression in mice treated with low-dose 2,3,7,8-tetrachlorodibenzo-p-dioxin revealed that hepatic AhR is associated with lipid, glucose, and cholesterol metabolism (25). Finally, identified endogenous ligands or activators, such as arachidonic acid metabolites, modified LDL, and glucose, are known to be involved in energy and lipid metabolism (14, 21, 22).
In this study, to better understand the role of AhR in lipid metabolism in the liver, mice with specific deletions of Ahr in the liver (AhR LKO) were subjected to a high fat diet (HFD) challenge. The results revealed that AhR plays protective roles against HFD-induced hepatic steatosis and the subsequent lipotoxicity through the direct transcriptional regulation of Socs3 expression.
Experimental Procedures
Animals, Diet, Drug Treatment, and Histology
AhR LKO mice were generated as described previously (26). Briefly, AhRflox/flox mice were crossed to C57BL/6J mice carrying the Cre recombinase gene driven by the albumin promoter (The Jackson Laboratory, Bar Harbor, ME). Mice homozygous for the floxed allele and hemizygous for the Cre transgene (AhR LKO) were obtained by crossing AhRflox/−/CreAlb mice to AhRflox/flox mice. Deletion of the Ahr gene was observed specifically in hepatocytes but not in non-parenchymal cells and other tissues (26). Littermates that were negative for the Cre transgenes (AhRflox/flox) were used as experimental controls. Mice were maintained at 23 ± 1 °C with 50 ± 10% relative humidity under a standard 12-h light/dark cycle with free access to water and food. Composition of a high fat diet (CLEA Japan Inc., Tokyo, Japan) is summarized in supplemental Table S1. Chow diet was obtained from Oriental Yeast Co., Ltd. (Tokyo, Japan). Composition of micronutrients in chow diet and high fat diet was matched (5% minerals and 1.2–1.5% vitamins). When necessary, mice received 3-methylcholanthrene (3MC) (20 mg/kg) (Wako Pure Chemical Industries, Ltd., Osaka, Japan) by oral gavage for 3 consecutive days (27). For histology, tissues were fixed in 4% formaldehyde, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E). For immunohistochemical analysis, liver paraffin-embedded sections were deparaffinized, rehydrated, and pretreated with 100 μg/ml proteinase K in 0.05 m Tris-HCl (pH 7.5) for 10 min at room temperature for antigen retrieval. Sections were blocked using 10% rat serum with 1% BSA in phosphate-buffered saline (PBS) for 60 min, followed by incubation with anti-F4/80 antibody (1:500; ab6640, Abcam Cambridge, MA) overnight at 4 °C. After washing, endogenous peroxidase activity was quenched using 0.3% H2O2 in PBS for 10 min, and sections were incubated with anti-rat biotinylated secondary antibody, streptavidin-horseradish peroxidase (Peroxidase Vectastain ABC Kit, Vector Laboratories), according to the manufacturer's instructions. Sections were then developed using the 3,3′-diaminobenzidine substrate (ImmPACT DAB, Vector Laboratories, Burlingame, CA) and counterstained with hematoxylin. To evaluate the pathological results, the stained slides (at least 15 slides per sample) were reviewed by multiple pathologists in a blinded manner. The experimental protocol was approved by the ethics review committee for animal experimentation at Nihon University.
Metabolic Studies
Energy expenditure (VO2, VCO2, and RQ) was measured by an indirect calorimeter (Muromachi, Tokyo, Japan). Food intake was continuously recorded on a computer and analyzed using FDMWIN software (Melquest, Toyama, Japan). Glucose tolerance test and insulin tolerance test were performed by intraperitoneal injection of glucose solution (2 g/kg body weight) or insulin (0.5 units/kg body weight; Lilly), respectively. Glucose levels were monitored before and after injection using blood glucose strips (Arkray, Kyoto, Japan).
Measurement of Tissue Lipid Content
To measure the liver and skeletal muscle triglyceride content, lipids were extracted from the tissues by the method of Carson (28). To determine liver cholesterol and phospholipid contents, lipids were extracted by the method of Folch et al. (29). Tissue contents of triglyceride, cholesterol, and phospholipid were measured using a commercially available reagent (Wako). Diglyceride content was measured by diacylglycerol ELISA kit (Cloud-Clone Corp, Houston, TX).
Biochemical Analysis of Blood
Serum levels of non-esterified fatty acid (Wako), adiponectin (Otsuka Pharmaceutical Co., Ltd., Tokyo, Japan), insulin, leptin (Morinaga Institute of Biological Science, Inc., Kanagawa, Japan), alanine aminotransferase (ALT) (Wako), and aspartate aminotransferase (AST) (Wako) were determined using a commercial assay kit according to the manufacturer's instructions.
Quantification of Serum and Liver FGF21 Levels
Serum or tissue extracts prepared in PBS were processed for quantification of FGF21 levels by using the mouse/rat FGF21 ELISA kit (R&D Systems) according to the manufacturer's instructions.
Gene Expression Analysis
Total RNA was extracted by use of RNAiso Plus (Takara Bio Inc., Kusatsu, Japan) according to the manufacturer's instructions. The cDNA was synthesized from 1.0 μg of total RNA by reverse transcriptase (Wako). Aliquots of cDNA were amplified on Stratagene Mx3000P real time PCR system (Agilent Technologies, Santa Clara, CA) using SYBR Green PCR reagents (Promega, Madison, WI). The mRNA expression levels were normalized against 36b4 and gapdh expression and are presented as relative expression levels.
Western Blot Analysis
Liver tissues and cells were lysed in commercial lysis buffer (Cell Signaling Technology Inc., Danvers, MA) containing 1 μm dithiothreitol and a protease inhibitor mixture. Protein concentrations were measured by the method of Bradford (30). The proteins were resolved on SDS-PAGE, transferred onto membranes, and probed with the antibodies against SOCS3, pSTAT3 (Tyr-705), STAT3, or β-actin (Cell Signaling Technology, catalog numbers 9145, 2923, 4970, and 9132, respectively). Immunoreactive proteins were visualized with electrochemiluminescence Western blotting detection reagents from Thermo Fisher Scientific Inc. (Waltham, MA).
De Novo Lipogenesis Activity
De novo lipogenesis activity was determined by the incorporation of [1-14C]acetate into the lipid (31, 32). In brief, mice fed a HFD for 10 weeks were anesthetized, and a liver slice was taken from each mouse. Liver slices (20∼25 mg) were incubated with Dulbecco's modified Eagle's medium containing 0.5 mm sodium acetate and 74 KBq/ml [1-14C]sodium acetate (1.868 GBq/mmol; PerkinElmer Life Sciences) for 90 min at 37 °C. After incubation, liver slices were heated with ethanolic KOH for 1 h at 70 °C. Nonsaponified lipids were removed with petroleum ether. The aqueous solutions were acidified, and lipids were extracted with petroleum ether. Radioactivity was measured by a liquid scintillation counter (Hitachi Aloka Medical, Ltd., Tokyo, Japan).
Thiobarbituric Acid-reactive Substances
The level of thiobarbituric acid-reactive substances (TBARS) in the liver tissue was measured using a TBARS assay kit (Cayman Chemical Co., Ann Arbor, MI).
Cell Culture
HepG2 cells (the European Collection of Cell Culture) and RAW264.7 cells (RIKEN Cell Bank, Ibaraki, Japan) were maintained in minimum Eagle's medium supplemented with 10% fetal bovine serum (FBS) or Dulbecco's modified Eagle's medium with 10% FBS, respectively.
Knockdown and Overexpression of Socs3
To prepare mouse Socs3 knockdown cells, RAW264.7 cells were transfected with either control stealth interfering RNA (siRNA) or stealth siRNA targeted for mouse Socs3 (Thermo Fisher Scientific) by using an X-tremeGENE siRNA transfection reagent (Roche Diagnostics, Tokyo, Japan) according to the manufacturer's instructions. To prepare mouse Socs3-overexpressing cells, mouse Socs3 cDNA was subcloned into CMV-HA vector (Takara Bio). RAW264.7 cells were transfected with CMV-HA vector or CMV-HA mouse SOCS3 expression vector by using FuGENE HD (Promega).
Electrophoretic Mobility Shift Assay (EMSA)
Mouse AhR and Arnt proteins were prepared using TnT in vitro transcription and translation system (Promega). The binding reactions were performed as described previously (33). Protein-DNA complexes were resolved by electrophoresis through 5% polyacrylamide gel in 0.5× tris/borate/EDTA at 4 °C. For oligonucleotide competition experiments, unlabeled oligonucleotides were added to the reaction mixture in a 100-fold molar excess to the radiolabeled probe. EMSA probe sequences are labeled in Fig. 9C.
FIGURE 9.

Socs3 is a novel transcriptional target of AhR. A, qRT-PCR analysis of Socs3 expression in the livers of AhRflox/flox mice and AhR LKO mice treated with an AhR agonist, 3MC (20 mg/kg), or corn oil for 3 consecutive days under a regular diet (n = 3–4). *, p < 0.05 relative to same genotype treated with corn oil (vehicle). +, p < 0.05 relative to AhRflox/flox mice treated with 3MC. B, qRT-PCR analysis of SOCS3, SOCS1, and CYP1A1 expression in HepG2 cells treated with 3MC (3 μm) for the indicated period of time (n = 3). *, p < 0.05 relative to 0 h (untreated cells). C, partial DNA sequences of the mouse and human Socs3 gene promoters. The putative Socs3/XRE sequences are underlined, and the mutated nucleotides are in capital letters. D, specific binding of the in vitro-synthesized AhR/aryl hydrocarbon receptor nuclear translocator heterodimer to the Socs3/XRE and the Cyp1a1/XRE in EMSA. In the competition lanes, unlabeled probes were present in 100-fold molar excess relative to the radiolabeled probe. E, luciferase activity in DMSO-treated or 3MC (3 μm)-treated HepG2 cells transfected with the reporter plasmids containing the mouse or human Socs3 promoter and its mutant variants in the presence of AhR, Arnt, or pcDNA 3.1 empty vector. The value of pcDNA3.1-transfected cells treated with DMSO was normalized to 1. The data represent the average and standard deviation from three independent experiments. *, p < 0.05 relative to DMSO-treated cells transfected with same plasmids. +, p < 0.05 relative to wild type (WT) reporter gene-transfected cells treated with same compound (DMSO or 3MC). F, ChIP analysis of the interaction between AhR and the region containing the Socs3/XRE in mice treated with corn oil (vehicle) or 3MC for 3 consecutive days (left) and HepG2 cells treated with 3MC (3 μm) for 16 h (right). C, AhRflox/flox mice; KO, AhR LKO mice. G, representative Western blot of total STAT3 and phosphorylated STAT3 in HepG2 cells exposed to IL-6 (10 ng/ml) for the indicated period of time after treatment with DMSO or 3MC (3 μm) for 16 h.
Chromatin Immunoprecipitation (ChIP) Assay
The ChIP assay was performed essentially as described elsewhere (27, 33). In brief, 12-week-old AhRflox/flox and AhR LKO male mice were treated with corn oil or 3MC (20 mg/kg) by oral gavage for 3 consecutive days. Mice were sacrificed 20 h after administration, and liver tissues were harvested. Liver tissues were homogenized and then cross-linked with formaldehyde. HepG2 cells were treated with dimethyl sulfoxide (DMSO) or 3MC (3 μm) for 16 h, cross-linked in formaldehyde, and lysed. The obtained liver and cell extracts were subjected to immunoprecipitation with anti-AhR antibody (SA210, Enzo Life Sciences, Farmingdale, NY). Parallel samples were incubated with normal IgG (sc-2027, Santa Cruz Biotechnology, Inc. Dallas, TX) as a negative control. The following PCR primers were used: mSOCS3 +20, 5-TGCTCTTACGACCGCTGTCTCT-3′; mSOCS3 +210, 5′-AGATGTTGGCAGCCGTGAAGTCTAC-3′; mSOCS3 −1906, 5′-ATTGTAATTTACAAAGCACTTTC-3′; mSOCS3 −1773, 5′-AAATGGATTCTCCCTTTCTCTG-3′; mCYP1A1 −866, 5′-GTTTCAAAATAATACATTCAGATCTT-3′; mCYP1A1 −745, 5′-AACAGGTAAAAGACTGATGGACAGGC-3′; hSOCS3 −74, 5′-GCCGGCCGCGCAGTTCCAGGAA-3′; hSOCS3 +137, 5′-ACCAACCGGGAGGGGACCAGGAGA-3′; hCYP1A1 −425, 5′-TTCTCACGCGAGCCGGGACTCAGTAA-3′; hCYP1A1 −237, 5′-AAGAGAGGGTACGGGAAGCTCCAT-3′; hCYP1A1 −9215, 5′-TCAGCTGACTGTCTTGGACTTGGT-3′; and hCYP1A1 −9094, 5′-ATTGAATTTAAGCGTGGCAGGCCC-3′.
Reporter Gene Assay
The 5′-regulatory region (−2905 to +926 bp) of the mouse Socs3 gene was amplified by PCR using mouse genomic DNA as the PCR template and the following oligonucleotides: mSOCS3 −2905 (5′-GACGTTCCTAAAAGCATGCATGCAT-3′) and mSOCS3 +926 (5′-GGATCTGCGCGGCGGTGGCTG-3′). The human SOCS3 promoter (−1399 bp to +848 bp) was amplified by PCR using human genomic DNA and the following oligonucleotides: hSOCS3 −1399 (5′-AGTGGGCCGAGGCTGGGTAG-3′) and hSOCS3 +848 (5′-GGCGCACGGAGCAGCGTGGATCTG-3′). The PCR-amplified fragments were cloned into the pGL3-basic vector (Promega). The site-directed mutagenesis was performed by the PCR overextension method and confirmed by DNA sequencing (34). HepG2 cells were transfected with the plasmids by using a FuGENE HD (Promega) (9, 33). After 24 h of incubation, the transfection medium was replaced with fresh medium containing 3MC or DMSO. The cells lysates were extracted 16 h later and assayed with a dual-luciferase reporter assay system (Promega). The pRL-tk vector (Promega) was used as a normalization control to correct for variable transfection efficiencies. All transfections were performed in triplicate.
Statistical Analysis
When applicable, the results are represented as the means ± S.D. Statistical analysis was performed by Student's t test or one-way analysis of variance with Tukey's post hoc test. A value of p < 0.05 was accepted as statistically significant.
Results
Deletion of the Ahr Gene in the Liver Accelerates High Fat Diet-induced Hepatic Steatosis
To understand the roles of AhR in lipid metabolism in the liver, AhRflox/flox mice and liver-specific AhR knock-out (AhR LKO) mice were subjected to HFD challenge for 12 weeks. As shown in Fig. 1, A–C, there were no differences in body mass, the tissue weights of the liver, the white adipose tissue and the kidney, or the state of adiposity between the two genotypes (Fig. 1, A–C). HFD feeding induced ectopic fat accumulation in the liver and the skeletal muscle (Fig. 1D). In particular, the level of hepatic triglyceride in AhR LKO mice was far greater than that in AhRflox/flox mice under the HFD condition (Fig. 1D). In contrast, this genotype-dependent increase of fat accumulation was not observed in the skeletal muscle (Fig. 1D). Analysis of the liver histology revealed that HFD feeding induced severe microvascular steatosis in AhR LKO mice (Fig. 1E). In the liver, the level of diglyceride in AhR LKO mice was significantly higher than that in AhRflox/flox mice under the HFD condition (Fig. 1F). In contrast, there was no significant difference in the level of cholesterol and phospholipid between the genotypes in the liver (Fig. 1F).
FIGURE 1.

Deletion of the Ahr gene in the liver accelerates HFD-induced hepatic steatosis. AhRflox/flox mice and AhR LKO mice were fed a chow diet or HFD for 12 weeks before being analyzed (n = 5–6). A, body weight. B, tissue weight of the liver, the epididymal white adipose tissue (WAT), and the kidney (n = 5–6). C, representative hematoxylin and eosin (H&E) staining of epididymal white adipose tissue. Magnification, ×40. Scale bar, 200 μm. D, triglyceride content in the liver and skeletal muscle. E, representative H&E staining of a liver section around the central vein. Magnification, ×100. Scale bar, 50 μm. F, content of diglyceride, phospholipid, and cholesterol in the liver (n = 5–6). G, FGF21 content in the liver (n = 5–6). H, food intake (kcal per day). I, top panel, representative daily changes of oxygen consumption (VO2), carbon dioxide production (VCO2), and RQ. Bottom panel, daily average of VO2, VCO2, and RQ (n = 5–6). #, p < 0.05 relative to AhRflox/flox mice fed a chow diet. *, p < 0.05 relative to AhRflox/flox mice fed a HFD.
The determination of circulating biochemical parameters showed that there were no significant differences in the level of nonesterified fatty acid, insulin, glucose, FGF21, or adipocytokines such as adiponectin and leptin between the genotypes (Table 1). Also, the level of hepatic FGF21, food intake, O2 consumption, CO2 production, and RQ value in the AhR LKO mice was similar to that of AhRflox/flox mice (Fig. 1, G–I).
TABLE 1.
Serum biochemical markers of AhRflox/flox mice and AhR LKO mice fed an HFD (n = 6)
| AhRflox/flox | AhR LKO | |
|---|---|---|
| Nonesterified fatty acid (mEq/liter) | 0.87 ± 0.21 | 0.79 ± 0.41 |
| Glucose (mg/dl) | 216 ± 45.3 | 187 ± 32.3 |
| Insulin (ng/ml) | 0.64 ± 0.19 | 0.62 ± 0.13 |
| FGF21 (pg/ml) | 796.5 ± 105.3 | 874.8 ± 302 |
| Adiponectin (μg/ml) | 34.9 ± 3.9 | 31.5 ± 3.9 |
| Leptin (ng/ml) | 2.46 ± 0.48 | 2.18 ± 0.31 |
Hepatic steatosis is linked with altered insulin signaling. Thus, to examine whether there is a difference in insulin sensitivity between the genotypes, the mice were subjected to a glucose tolerance test and insulin tolerance test. Under chow diet conditions, AhR LKO mice exhibited improved whole-body insulin sensitivity (Fig. 2, A and B) slightly but significantly (Fig. 2, A and B). However, HFD feeding aggravated whole-body insulin sensitivity in both genotypes, and the differences of scores observed under chow condition were diminished (Fig. 2, A and B).
FIGURE 2.

HFD feeding attenuates the effects of deletion of the hepatic Ahr gene on insulin sensitivity. AhRflox/flox mice and AhR LKO mice were fed a chow diet or HFD for 12 weeks before being analyzed (n = 6). A, glucose tolerance test. B, insulin tolerance test. The area under the curve (AUC) was calculated for respective group. #, p < 0.05 relative to AhRflox/flox mice fed a chow diet. *, p < 0.05 relative to AhRflox/flox mice on the same diet.
To gain insight into the mechanism by which the deficiency of AhR promotes HFD-induced hepatic steatosis, we examined the expression of genes related to lipid metabolism by qRT-PCR analysis. Under the HFD conditions, AhR LKO mice showed an increased expression of lipogenesis-related genes such as sterol-response element-binding protein-1c (Srebp-1c) and its target genes, including steraroyl coenzyme decarboxylase 1 (Scd1), acetyl-CoA carboxylase 1 (Acc1), and glycerol-3-phosphate acyltransferase 1 (Gpat1) (Fig. 3A). Also, gene expression level of adipocyte differentiation-related protein (Adrp), a lipid droplet coat protein, and hormone-sensitive lipase (Hsl) in AhR LKO mice was higher than that in AhRflox/floxmice (Fig. 3B). In contrast, no significant difference was seen in the expression level of genes involved in fatty acid uptake, β-oxidation, and gluconeogenesis (Fig. 3, C–E).
FIGURE 3.

Deletion of the Ahr gene in the liver increases expression of lipogenesis-related genes under the HFD condition. AhRflox/flox mice and AhR LKO mice were fed a chow diet or HFD for 12 weeks before being analyzed (n = 5–6). Gene expression in the mouse liver was analyzed by qRT-PCR. A, expression of genes involved in lipogenesis. B, expression of genes related to lipid droplet or lipolysis. C, expression of genes involved in fatty acid uptake. D, expression of genes involved in β-oxidation. E, expression of genes involved in gluconeogenesis. #, p < 0.05 relative to AhRflox/flox mice fed a chow diet. *, p < 0.05 relative to AhRflox/flox mice fed a HFD.
Deficiency of the Ahr Gene in the Liver Promotes HFD-induced Hepatic Inflammation
The results described above suggest that the increase of hepatic steatosis in AhR LKO mice is due to abnormal hepatic function rather than a dysfunction of other tissues or deterioration of insulin sensitivity. Hepatic steatosis is often accompanied by inflammation. Thus, in the next set of experiments, we analyzed the state of inflammation and liver damage in AhR LKO mice fed a HFD. Under the HFD conditions, a markedly higher infiltration of inflammatory cells was observed in the livers of AhR LKO mice as compared with that in the livers of AhRflox/flox mice (Fig. 4, A and B). The elevation of hepatic inflammation status was further confirmed by gene expression analysis for pro-inflammatory genes (Fig. 4C). Moreover, HFD feeding increased the activity of both serum ALT and AST, indicators of liver damage, in AhR LKO mice but not in AhRflox/flox mice (Fig. 4D). These data indicate that the deletion of hepatic AhR accelerates the onset of HFD-induced inflammation and damage.
FIGURE 4.

Deletion of the Ahr gene in the liver accelerates HFD-induced inflammation. AhRflox/flox mice and AhR LKO mice were fed a chow diet or HFD for 12 weeks before being analyzed (n = 5–6). A, left panel, representative H&E staining of a liver section around the central vein with arrows indicating inflammatory cell infiltration. Magnification, ×100 (top and middle) and ×200 (bottom). Scale bar, 50 μm (top and middle) and 100 μm (bottom). Right panel, the colony number of inflammatory cells per high power field. B, representative immunohistochemistry to detect F4/80 in the liver of mice fed a HFD (magnification, ×200. Scale bar, 50 μm). C, qRT-PCR analysis of inflammatory-related gene expression. D, activity of ALT and AST in serum. #, p < 0.05 relative to AhRflox/flox mice fed a chow diet. *, p < 0.05 relative to AhRflox/flox mice fed a HFD. N.D., not detected.
Deletion of the Ahr Gene Attenuates the HFD-dependent Induction of Socs3 Expression
In the progression of inflammation, the JAK/STAT pathway plays critical roles in the transduction of the cytokine-dependent signal. This pathway is negatively and tightly regulated by the suppressor of cytokine signal (SOCS) family (35). Among SOCS family members, SOCS3 acts as a potent negative regulator of the pro-inflammatory-induced STAT3 signaling pathway. Interestingly, HFD feeding increases the expression of Socs3 in the liver (Fig. 5A). However, the degree of HFD-induced Socs3 expression in AhR LKO mice was far lower than that in AhRflox/flox mice (Fig. 5A). Western blot analysis of liver proteins confirmed the lower SOCS3 expression in AhR LKO mice fed a HFD (Fig. 5B). Also, functional loss of SOCS3 in the AhR LKO mouse liver was demonstrated by the increase of the phosphorylated form of STAT3 (Fig. 5B).
FIGURE 5.
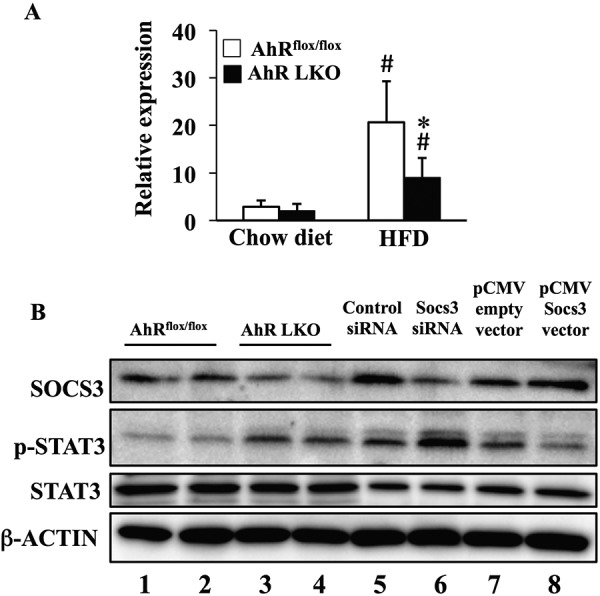
Deletion of the Ahr gene in the liver attenuates the HFD-dependent induction of Socs3 expression. AhRflox/flox mice and AhR LKO mice were fed a chow diet or HFD for 12 weeks before being analyzed (n = 5). A, qRT-PCR analysis of Socs3 gene expression in the liver. B, representative Western blot of SOCS3, total STAT3, and phosphorylated STAT3 in the liver extract of mice fed a HFD. β-Actin was used as a loading control. Lanes 1 and 2 were run using the liver extract prepared from two distinct AhRflox/flox mice, and lanes 3 and 4 were run using the liver extract prepared from two distinct AhR LKO mice. Lane 6 was run using a cell extract prepared from Socs3 siRNA-transfected RAW264.7 cells as negative control. Lane 8 was run using a cell extract prepared from Socs3 expression vector-transfected RAW264.7 cells as positive control. Lanes 5 and 7 were transfection control for Socs3 siRNA-transfected cells (lane 6) and Socs3 expression vector-transfected cells (lane 8), respectively. #, p < 0.05 relative to AhRflox/flox mice fed a chow diet. *, p < 0.05 relative to AhRflox/flox mice fed a HFD.
Rescue of Socs3 Expression in AhR LKO Mice Attenuates HFD-induced Steatosis and the Subsequent Lipotoxicity
To demonstrate the development of severe HFD-induced steatosis was, at least partly, due to a decrease in SOCS3 expression in AhR LKO mice, hepatic Socs3 expression was rescued in AhR LKO mice by breeding with AhRflox/flox mice carrying the transgene of Socs3 (S3 Tg mice) (Fig. 6A). The overexpression of SOCS3 in the liver of S3 Tg mice was validated by Western blotting (Fig. 6B). In addition, the reduction of the active form of STAT3 provided evidence of functional rescue of SOCS3 in S3 Tg mice and AhR LKO mice carrying transgene of Socs3 (AhR LKO/S3 Tg mice) (Fig. 6B). To evaluate the protective roles of SOCS3 for HFD-induced lipotoxicity, AhRflox/flox mice, AhR LKO mice, AhR LKO/S3 Tg mice, and S3 Tg mice were subjected to HFD challenge for 10 weeks. There was no difference in the body weight or tissue weight among the four groups under either a chow diet or HFD condition (Fig. 6D). Histological analysis revealed that the rescue of SOCS3 in AhR LKO mice improved the status of HFD-induced steatosis (Fig. 7A). Attenuation of HFD-induced steatosis by the rescue of SOCS3 expression was confirmed by the observation that the triglyceride level and the expression of lipogenic genes in the AhR LKO/S3 Tg mouse liver were lowered to levels similar to those in the AhRflox/flox mouse liver (Fig. 7, B and C). Physiological measure of lipogenesis revealed that de novo lipogenesis activity in the AhR LKO mouse liver was significantly higher than that in the AhRflox/flox mouse liver (Fig. 7D). However, the increase of lipogenesis activity was attenuated by the overexpression of SOCS3 in the liver (Fig. 7D). As shown in Fig. 4, the degree of the damage induced by HFD feeding in the AhR LKO mouse liver was more severe than that in the AhRflox/flox mouse liver. However, these lipotoxicities were relieved in the AhR LKO/S3 Tg mouse liver (Fig. 8, A and B). In addition, the effect of deletion of the hepatic Ahr gene and the rescue of the Socs3 gene on the level of oxidative stress markers such as TBARS was determined. As expected, HFD feeding increased the level of TBARS in all genotypes, and the degree of augmentation was more pronounced in AhR LKO mice (Fig. 8C). However, the level of TBARS in the AhR LKO/S3 Tg mouse liver was as low as that in the AhRflox/flox mouse liver under the HFD condition (Fig. 8C).
FIGURE 6.

Generation of AhR LKO mice expressing transgene of the Socs3. A, schematic representations of the transgene construct. The transgene is under the control of the mouse albumin promoter. B, representative Western blot of SOCS3, phosphorylated STAT3, total STAT3, and β-actin in the liver extract. Each lane was run using samples from two different male mice. AhRflox/flox mice, AhR LKO mice, AhR LKO/S3 Tg mice, and S3 Tg mice were fed a HFD. C, body weight of mice fed a chow diet or HFD for 10 weeks (n = 5–7). D, tissue weight of mice fed a chow diet or HFD for 10 weeks (n = 5–7).
FIGURE 7.

Rescue of Socs3 expression in AhR LKO mice attenuates HFD-induced steatosis. AhRflox/flox mice, AhR LKO mice, AhR LKO/S3 Tg mice, and S3 Tg mice were fed a chow or HFD for 10 weeks (n = 5–7). A, representative H&E staining of the liver section (magnification, ×100. Scale bar, 50 μm). B, triglyceride content in the liver. C, qRT-PCR analysis of the expression of lipogenic genes. D, de novo lipogenesis activity in the mouse liver of mice fed a HFD. #, p < 0.05 relative to AhRflox/flox mice fed a chow diet. *, p < 0.05 relative to AhRflox/flox mice fed a HFD (in B and D) or AhRflox/flox mice fed a same diet (C). +, p < 0.05 relative to AhR LKO mice fed a HFD.
FIGURE 8.

Rescue of Socs3 expression in AhR LKO mice attenuates HFD-induced lipotoxicity. AhRflox/flox mice, AhR LKO mice, AhR LKO/S3 Tg mice, and S3 Tg mice were fed a chow diet or HFD for 10 weeks (n = 5–7). A, activity of ALT and AST in serum. B, qRT-PCR analysis of the expression of inflammation-related genes (n = 5–7). C, TBARS level in the liver. #, p < 0.05 relative to AhRflox/flox mice fed a chow diet. *, p < 0.05 relative to AhRflox/flox mice fed a HFD. +, p < 0.05 relative to AhR LKO mice fed a HFD.
Socs3 Is a Novel Transcriptional Target of AhR
The lower level of induction of Socs3 expression in AhR LKO mice under the HFD condition suggested that the Socs3 gene is a transcriptional target of AhR (Fig. 5). Therefore, we examined whether the activation of AhR would increase the Socs3 expression. Activation of AhR by the treatment with 3MC, a ligand of AhR, increased Socs3 expression in the liver of AhRflox/flox mice. In the liver of the AhR LKO mice, the Socs3 expression was slightly induced by the AhR agonist treatment, but the level of expression was far less than that in the AhRflox/flox mouse liver (Fig. 9A). In addition, the expression of SOCS3 was induced by the activation of AhR in human hepatoma HepG2 cells in a time-dependent manner as well as a well known AhR target gene, CYP1A1 (Fig. 9B). In contrast, 3MC treatment had little effect on the expression of SOCS1 (Fig. 9B). Inspection of the 5′-regulatory sequences of the Socs3 gene revealed the presence of an XRE in the mouse and human genome at nucleotides +166 to +171 and +49 to +54, respectively (Fig. 9C). EMSA revealed the AhR/Arnt heterodimers bound to the DNA probes containing mouse Socs3/XRE and human SOCS3/XRE (Fig. 9D). The specificity of the complex formation was confirmed by competition experiments (Fig. 9D). To determine whether AhR can mediate the transactivation via Socs3/XRE, we cloned the mouse 2.9-kb and human 1.4-kb 5′-flanking region of the Socs3 gene. As shown in Fig. 9E, 3MC treatment increased both mouse and human SOCS3 promoter activity in the presence of AhR, although these inductions were significantly suppressed by the introduction of a mutation in the XRE (Fig. 9E). Also, the results of ChIP assay confirmed the ligand-dependent recruitment of the AhR to mouse and human SOCS3/XRE on the genome (Fig. 9F).
To understand the effects of Socs3 induction by AhR on cytokine signaling, HepG2 cells pre-treated with either DMSO or 3MC were exposed to IL-6. Phosphorylation of STAT3 was induced and became maximal at 15 min of the treatment with IL-6 in DMSO-treated cells. In contrast, 3MC-treated cells showed no substantial increase of phosphorylated STAT3 by IL-6 treatment (Fig. 9G).
Discussion
The results in this study revealed that deletion of the Ahr gene in the liver leads to the development of severe hepatic steatosis and the subsequent lipotoxicity effects, including liver inflammation and injury, under the HFD condition (Figs. 1, 3, and 4). Hepatic steatosis, termed as nonalcoholic fatty liver disease, appears to be associated with abnormal lipid metabolism-related diseases such as obesity and type 2 diabetes. In patients with nonalcoholic fatty liver disease, ∼60% of liver triglyceride content is derived from free fatty acid from adipose tissue, 26% from de novo lipogenesis, and 15% from diet (36). However, the results in this study showed that there were no substantial differences in the body or tissue weight, food intake, energy expenditure, levels of circulating fatty acids or adipocytokine, and whole-body insulin sensitivity between AhRflox/flox mice and AhR LKO mice under the HFD condition. Consequently, disorder of lipid metabolism in the liver was considered to be a main cause of exacerbated ectopic fat accumulation in the tissue. Indeed, a series of gene expressions related to lipogenesis and actual de novo lipogenesis activity were found to be up-regulated in the AhR LKO mouse liver (Figs. 3 and 7). In conjunction with the increased lipogenesis, one of the notable changes in the livers of AhR LKO mice was a lowered induction of Socs3 expression and subsequent lowered activation of STAT3 when the mice were challenged with HFD feeding (Fig. 5). SOCS3, a negative inflammatory factor, is induced by HFD in the liver, the skeletal muscle, and the adipose tissue (37–39) and inhibits inflammatory signal transduction via binding to tyrosine phosphorylation sites on cytokine receptors (40–42). Similar to AhR LKO mice, liver-specific Socs3 KO mice fed a HFD showed the severe hepatic steatosis associated with increased de novo lipogenesis and inflammation (40). The mechanism involves HFD-dependent increase of Srebp-1c expression (40). A recent study revealed that SOCS3 acts as an inhibitor of the JAK/STAT5a pathway and disturbs lipogenesis by decreasing Srebp-1c expression (43). Also, AhR modulated the basal expression of Socs3 in spleen and brain (44). These results suggest that the lipotoxicities observed following HFD feeding in AhR LKO mice were attributed to a lower level of Socs3 expression. In confirmation of this hypothesis, we demonstrated that the rescue of Socs3 in the AhR LKO mouse liver improved the degree of hepatic steatosis, inflammation, and reactive oxygen species production level similar to that in the AhRflox/flox mouse liver (Figs. 7 and 8). We also revealed that AhR directly regulates transcription of the Socs3 gene (Fig. 9, A–F). A recent study reported by Brant et al. (44) showed that AhR is involved in a SOCS3-mediated immune response. Consequently, we are led to conclude that AhR plays protective roles against HFD-induced hepatic steatosis and the subsequent lipotoxicity via direct transcriptional regulation of Socs3 expression in the liver.
The effects of the lower level of SOCS3 in the AhR LKO mouse liver were observed only under the condition of HFD feeding but not under chow diet feeding. Also, increased liver fat and inflammation in liver-specific Socs3 KO mice were HFD-dependent phenotypes (38). Therefore, the functional significance of Socs3 induction by AhR is associated with the pathogenesis induced by HFD. Hepatic steatosis induced by HFD feeding is accompanied by chronic inflammation. Inflammation has been implicated in the progression of steatosis and liver injury through production of proinflammatory cytokines such as TNFα and IL-1β (45, 46). These cytokines promote lipogenesis and triglyceride accumulation by regulating Srebp1c expression (47). In this study, we demonstrated that deletion of the hepatic Ahr gene increased inflammatory cell infiltration into the liver and the expression of pro-inflammatory cytokines (Fig. 4, A–C). Previous studies have similarly demonstrated that AhR inactivation induced inflammatory cell infiltration in the various tissues (48–50). These results suggest a critical role for AhR in the regulation of inflammatory responses. Similar to the phenotype of AhR LKO mice, deletion of the Socs3 gene increases infiltration of neutrophils and macrophages and heightens inflammation (40, 51–53). Given the facts that (i) expression of AhR and Socs3 is induced by HFD (Fig. 5), (ii) Socs3 is a direct transcriptional target of AhR (Fig. 9, A–F), (iii) the inflammation induced by HFD feeding is rescued by SOCS3 in the AhR LKO mouse liver (Fig. 7A and 8B), and (iv) pretreatment of HepG2 cells with AhR ligand weakened the IL-6-dependent phosphorylation of STAT3 (Fig. 9G), it would be appear that the functional significance of Socs3 induction by AhR is to suppress the progression of inflammation and the subsequent hepatic damages induced by HFD.
Hepatic steatosis is associated with insulin resistance. Previous studies have reported improved insulin sensitivity with Socs3 deletion (39–42). Under chow diet conditions, AhR LKO mice exhibited slight but significant improvement of whole-body insulin sensitivity (Fig. 2, A and B), as observed in Socs3 LKO mice (40). Therefore, the enhanced insulin actions may accelerate the early stage of lipogenesis in AhR LKO mice. However, HFD feeding markedly aggravated the insulin sensitivity in both genotypes, and as a result, the genotype-dependent differences in scores observed under chow diet conditions were diminished (Fig. 2, A and B). Thus, the HFD-dependent deterioration of hepatic steatosis in AhR LKO mice compared with AhRflox/flox mice is unlikely due to the alternation of insulin sensitivity between genotypes.
Previous studies have reported that pharmacological or transgenic activation of AhR induces hepatic steatosis (54–56). In contrast, global AhR KO mice develop spontaneous triglyceride accumulation and fibrosis in the liver (11). As shown in this study, deletion of the Ahr gene in the liver exacerbates hepatic steatosis induced by a HFD (Figs. 1, 3, and 4). These results indicate that hepatic AhR may play dual roles in the regulation of lipid metabolism. It is clear that overload of lipid in the liver is the first hit in the development of hepatic steatosis. However, the deposition of lipid in tissue is not sufficient in itself to induce hepatic damage. For example, although rescue of the Socs3 gene in AhR LKO mice improves the lipotoxicities to the level of those in AhRflox/flox mice under a HFD condition, AhR LKO mice overexpressing Socs3 still showed ectopic fat formation in the liver by HFD feeding (Fig. 7B). Thus, a second hit, such as inflammation and/or elevation of the reactive oxygen species level, is required to develop hepatic steatosis. The reports referred to above showed that pharmacologically activated or constitutively activated AhR triggers triglyceride accumulation in the liver, reflecting a role in the first hit of hepatic steatosis described above (54–56). In contrast, the results in this study demonstrated the increase of inflammation in AhR LKO mice under HFD feeding conditions. Therefore, AhR also plays the protective roles against the second hit in hepatic steatosis development. Because the mechanism by which AhR improves HFD-induced hepatic steatosis and the subsequent lipotoxicity involves direct transcriptional regulation of Socs3 gene in vivo, activation of AhR is required. This suggests that HFD feeding or the increase of ectopic fat may produce the AhR ligands or activators. Indeed, increased adiposity elevates the level of AhR and its target genes (23, 24). Although identification of the molecules acts as the ligands or the activator produced under HFD conditions has to be elucidated in future studies, recent studies have identified various low molecular compounds as AhR endogenous ligands or activators, which include glucose, sheared LDL, cAMP derivatives, and tryptophan derivatives (13–22). These compounds and the related compounds could be contained in HFD or be derived through the metabolic pathway. It is now well recognized that AhR is, directly or indirectly, associated with several pathophysiological processes, including atherosclerosis, inflammation, immunomodulation, and cancer. Consequently,identification of an AhR ligand and/or activator in HFD may be useful for the development of treatments for metabolic liver diseases.
Author Contributions
T.W. performed the experiments and wrote the manuscript. H. S., K. M., H. S., and Y. U. performed the experiments. S. S. designed and supervised the study and wrote the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Dr. Wen Xie for providing the albumin promoter plasmid.
This work was supported in part by a grant-in-aid for scientific research from Japan Society for the Promotion of Science, Ministry of Education, Culture, Sports, Science and Technology, Japan (to S. S. and T. W), and the “High-Tech Research Center” Project for Private Universities, and a matching fund subsidy from the Ministry of Education, Culture, Sports, Science and Technology 2007. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
- AhR
- aryl hydrocarbon receptor
- 3MC
- 3-methylcholanthrene
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- CYP
- cytochrome P450
- HFD
- high fat diet
- LKO
- liver-specific knockout
- qRT-PCR
- quantitative reverse transcription-polymerase chain reaction
- XRE
- xenobiotic response element
- m
- mouse
- h
- human
- TBARS
- thiobarbituric acid reactive substance
- Tg
- transgene
- RQ
- respiratory quotient.
References
- 1. Ema M., Sogawa K., Watanabe N., Chujoh Y., Matsushita N., Gotoh O., Funae Y., and Fujii-Kuriyama Y. (1992) cDNA cloning and structure of mouse putative Ah receptor. Biochem. Biophys. Res. Commun. 184, 246–253 [DOI] [PubMed] [Google Scholar]
- 2. Burbach K. M., Poland A., and Bradfield C. A. (1992) Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. U.S.A. 89, 8185–8189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poland A., and Knutson J. C. (1982) 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 22, 517–554 [DOI] [PubMed] [Google Scholar]
- 4. Reyes H., Reisz-Porszasz S., and Hankinson O. (1992) Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256, 1193–1195 [DOI] [PubMed] [Google Scholar]
- 5. Hankinson O. (1995) The arylhydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 35, 307–340 [DOI] [PubMed] [Google Scholar]
- 6. Schmidt J. V., and Bradfield C. A. (1996) Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 12, 55–89 [DOI] [PubMed] [Google Scholar]
- 7. Beischlag T. V., Luis Morales J., Hollingshead B. D., and Perdew G. H. (2008) The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 18, 207–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ohtake F., Takeyama K., Matsumoto T., Kitagawa H., Yamamoto Y., Nohara K., Tohyama C., Krust A., Mimura J., Chambon P., Yanagisawa J., Fujii-Kuriyama Y., and Kato S. (2003) Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 423, 545–550 [DOI] [PubMed] [Google Scholar]
- 9. Watabe Y., Nazuka N., Tezuka M., and Shimba S. (2010) Aryl hydrocarbon receptor functions as a potent coactivator of E2F1-dependent transcription activity. Biol. Pharm. Bull. 33, 389–397 [DOI] [PubMed] [Google Scholar]
- 10. Fernandez-Salguero P., Pineau T., Hilbert D. M., McPhail T., Lee S. S., Kimura S., Nebert D. W., Rudikoff S., Ward J. M., and Gonzalez F. J. (1995) Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726 [DOI] [PubMed] [Google Scholar]
- 11. Schmidt J. V., Su G. H., Reddy J. K., Simon M. C., and Bradfield C. A. (1996) Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. U.S.A. 93, 6731–6736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abbott B. D., Schmid J. E., Pitt J. A., Buckalew A. R., Wood C. R., Held G. A., and Diliberto J. J. (1999) Adverse reproductive outcomes in the transgenic Ah receptor-deficient mouse. Toxicol. Appl. Pharmacol. 155, 62–70 [DOI] [PubMed] [Google Scholar]
- 13. Sinal C. J., and Bend J. R. (1997) Aryl hydrocarbon receptor-dependent induction of cyp1a1 by bilirubin in mouse hepatoma hepa 1c1c7 cells. Mol. Pharmacol. 52, 590–599 [DOI] [PubMed] [Google Scholar]
- 14. Ciolino H. P., Daschner P. J., and Yeh G. C. (1999) Dietary flavonols quercetin and kaempferol are ligands of the aryl hydrocarbon receptor that affect CYP1A1 transcription differentially. Biochem. J. 340, 715–722 [PMC free article] [PubMed] [Google Scholar]
- 15. Seidel S. D., Winters G. M., Rogers W. J., Ziccardi M. H., Li V., Keser B., and Denison M. S. (2001) Activation of the Ah receptor signaling pathway by prostaglandins. J. Biochem. Mol. Toxicol. 15, 187–196 [DOI] [PubMed] [Google Scholar]
- 16. Adachi J., Mori Y., Matsui S., Takigami H., Fujino J., Kitagawa H., Miller C. A. 3rd, Kato T., Saeki K., and Matsuda T. (2001) Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J. Biol. Chem. 276, 31475–31478 [DOI] [PubMed] [Google Scholar]
- 17. Zhang S., Qin C., and Safe S. H. (2003) Flavonoids as aryl hydrocarbon receptor agonists/antagonists: effects of structure and cell context. Environ. Health Perspect. 111, 1877–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wincent E., Amini N., Luecke S., Glatt H., Bergman J., Crescenzi C., Rannug A., and Rannug U. (2009) The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem. 284, 2690–2696 [DOI] [PubMed] [Google Scholar]
- 19. Opitz C. A., Litzenburger U. M., Sahm F., Ott M., Tritschler I., Trump S., Schumacher T., Jestaedt L., Schrenk D., Weller M., Jugold M., Guillemin G. J., Miller C. L., Lutz C., Radlwimmer B., et al. (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 [DOI] [PubMed] [Google Scholar]
- 20. Oesch-Bartlomowicz B., Huelster A., Wiss O., Antoniou-Lipfert P., Dietrich C., Arand M., Weiss C., Bockamp E., and Oesch F. (2005) Aryl hydrocarbon receptor activation by cAMP vs. dioxin: divergent signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 102, 9218–9223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McMillan B. J., and Bradfield C. A. (2007) The aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 104, 1412–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dabir P., Marinic T. E., Krukovets I., and Stenina O. I. (2008) Aryl hydrocarbon receptor is activated by glucose and regulates the thrombospondin-1 gene promoter in endothelial cells. Circ. Res. 102, 1558–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. La Merrill M., Kuruvilla B. S., Pomp D., Birnbaum L. S., and Threadgill D. W. (2009) Dietary fat alters body composition, mammary development, and cytochrome p450 induction after maternal TCDD exposure in DBA/2J mice with low-responsive aryl hydrocarbon receptors. Environ. Health Perspect. 117, 1414–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu J., Kulkarni S. R., Li L., and Slitt A. L. (2012) UDP-glucuronosyltransferase expression in mouse liver is increased in obesity- and fasting-induced steatosis. Drug Metab. Dispos. 40, 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sato S., Shirakawa H., Tomita S., Ohsaki Y., Haketa K., Tooi O., Santo N., Tohkin M., Furukawa Y., Gonzalez F. J., and Komai M. (2008) Low-dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor-mediated pathway in mouse liver. Toxicol. Appl. Pharmacol. 229, 10–19 [DOI] [PubMed] [Google Scholar]
- 26. Walisser J. A., Glover E., Pande K., Liss A. L., and Bradfield C. A. (2005) Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. U.S.A. 102, 17858–17863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wada T., Sunaga H., Ohkawara R., and Shimba S. (2013) Aryl hydrocarbon receptor modulates NADPH oxidase activity via direct transcriptional regulation of p40phox expression. Mol. Pharmacol. 83, 1133–1140 [DOI] [PubMed] [Google Scholar]
- 28. Carson L. A. (1963) Determination of serum triglycerides. J. Atheroscler. Res. 3, 334–336 [DOI] [PubMed] [Google Scholar]
- 29. Folch J., Lees M., and Sloane-Stanley G. H. (1957) A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 30. Bradford M. M. (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 31. Jamdar S. C. (1978) Glycerolipid biosynthesis in rat adipose tissue. Influence of adipose-cell size and site of adipose tissue on triacylglycerol formation in lean and obese rats. Biochem. J. 170, 153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harada N., Oda Z., Hara Y., Fujinami K., Okawa M., Ohbuchi K., Yonemoto M., Ikeda Y., Ohwaki K., Aragane K., Tamai Y., and Kusunoki J. (2007) Hepatic de novo lipogenesis is present in liver-specific ACC1-deficient mice. Mol. Cell. Biol. 27, 1881–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wada T., Kang H. S., Angers M., Gong H., Bhatia S., Khadem S., Ren S., Ellis E., Strom S. C., Jetten A. M., and Xie W. (2008) Identification of oxysterol 7α-hydroxylase (Cyp7b1) as a novel retinoid-related orphan receptor α (RORα) (NR1F1) target gene and a functional cross-talk between RORα and liver X receptor (NR1H3). Mol. Pharmacol. 73, 891–899 [DOI] [PubMed] [Google Scholar]
- 34. Xie W., Barwick J. L., Simon C. M., Pierce A. M., Safe S., Blumberg B., Guzelian P. S., and Evans R. M. (2000) Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 14, 3014–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexander W. S., and Hilton D. J. (2004) The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 22, 503–529 [DOI] [PubMed] [Google Scholar]
- 36. Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., and Parks E. J. (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emanuelli B., Peraldi P., Filloux C., Chavey C., Freidinger K., Hilton D. J., Hotamisligil G. S., and Van Obberghen E. (2001) SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-alpha in the adipose tissue of obese mice. J. Biol. Chem. 276, 47944–47949 [DOI] [PubMed] [Google Scholar]
- 38. Steinberg G. R., Smith A. C., Wormald S., Malenfant P., Collier C., and Dyck D. J. (2004) Endurance training partially reverses dietary-induced leptin resistance in rodent skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 286, E57–E63 [DOI] [PubMed] [Google Scholar]
- 39. Ueki K., Kondo T., and Kahn C. R. (2004) Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol. 24, 5434–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sachithanandan N., Fam B. C., Fynch S., Dzamko N., Watt M. J., Wormald S., Honeyman J., Galic S., Proietto J., Andrikopoulos S., Hevener A. L., Kay T. W., and Steinberg G. R. (2010) Liver-specific suppressor of cytokine signaling-3 deletion in mice enhances hepatic insulin sensitivity and lipogenesis resulting in fatty liver and obesity. Hepatology 52, 1632–1642 [DOI] [PubMed] [Google Scholar]
- 41. Palanivel R., Fullerton M. D., Galic S., Honeyman J., Hewitt K. A., Jorgensen S. B., and Steinberg G. R. (2012) Reduced Socs3 expression in adipose tissue protects female mice against obesity-induced insulin resistance. Diabetologia 55, 3083–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jorgensen S. B., O'Neill H. M., Sylow L., Honeyman J., Hewitt K. A., Palanivel R., Fullerton M. D., Öberg L., Balendran A., Galic S., van der Poel C., Trounce I. A., Lynch G. S., Schertzer J. D., and Steinberg G. R. (2013) Deletion of skeletal muscle SOCS3 prevents insulin resistance in obesity. Diabetes 62, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang Y. L., Zhao F., Luo C. C., Zhang X., Si Y., Sun Z., Zhang L., Li Q. Z., and Gao X. J. (2013) SOCS3-mediated blockade reveals major contribution of JAK2/STAT5 signaling pathway to lactation and proliferation of dairy cow mammary epithelial cells in vitro. Molecules 18, 12987–13002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brant F., Miranda A. S., Esper L., Rodrigues D. H., Kangussu L. M., Bonaventura D., Soriani F. M., Pinho V., Souza D. G., Rachid M. A., Weiss L. M., Tanowitz H. B., Teixeira M. M., Teixeira A. L., and Machado F. S. (2014) Role of the aryl hydrocarbon receptor in the immune response profile and development of pathology during Plasmodium berghei Anka infection. Infect. Immun. 82, 3127–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yin M., Wheeler M. D., Kono H., Bradford B. U., Gallucci R. M., Luster M. I., and Thurman R. G. (1999) Essential role of tumor necrosis factor a in alcohol-induced liver injury in mice. Gastroenterology 117, 942–952 [DOI] [PubMed] [Google Scholar]
- 46. Miura K., Kodama Y., Inokuchi S., Schnabl B., Aoyama T., Ohnishi H., Olefsky J. M., Brenner D. A., and Seki E. (2010) Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 139, 323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lawler J. F. Jr., Yin M., Diehl A. M., Roberts E., and Chatterjee S. (1998) Tumor necrosis factor-α stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J. Biol. Chem. 273, 5053–5059 [DOI] [PubMed] [Google Scholar]
- 48. Thatcher T. H., Maggirwar S. B., Baglole C. J., Lakatos H. F., Gasiewicz T. A., Phipps R. P., and Sime P. J. (2007) Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-κB component RelB. Am. J. Pathol. 170, 855–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Di Meglio P., Duarte J. H., Ahlfors H., Owens N. D., Li Y., Villanova F., Tosi I., Hirota K., Nestle F. O., Mrowietz U., Gilchrist M. J., and Stockinger B. (2014) Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 40, 989–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sekine H., Mimura J., Oshima M., Okawa H., Kanno J., Igarashi K., Gonzalez F. J., Ikuta T., Kawajiri K., and Fujii-Kuriyama Y. (2009) Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol. Cell. Biol. 29, 6391–6400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wong P. K., Egan P. J., Croker B. A., O'Donnell K., Sims N. A., Drake S., Kiu H., McManus E. J., Alexander W. S., Roberts A. W., and Wicks I. P. (2006) SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J. Clin. Invest. 116, 1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu B. M., Ishida Y., Robinson G. W., Pacher-Zavisin M., Yoshimura A., Murphy P. M., and Hennighausen L. (2008) SOCS3 negatively regulates the gp130-STAT3 pathway in mouse skin wound healing. J. Invest. Dermatol. 128, 1821–1829 [DOI] [PubMed] [Google Scholar]
- 53. Yan C., Ward P. A., Wang X., and Gao H. (2013) Myeloid depletion of SOCS3 enhances LPS-induced acute lung injury through CCAAT/enhancer binding protein δ pathway. FASEB J. 27, 2967–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee J. H., Wada T., Febbraio M., He J., Matsubara T., Lee M. J., Gonzalez F. J., and Xie W. (2010) A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139, 653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. He J., Hu B., Shi X., Weidert E. R., Lu P., Xu M., Huang M., Kelley E. E., and Xie W. (2013) Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell. Biol. 33, 2047–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu P., Yan J., Liu K., Garbacz W. G., Wang P., Xu M., Ma X., and Xie W. (2015) Activation of aryl hydrocarbon receptor dissociates fatty liver from insulin resistance by inducing fibroblast growth factor 21. Hepatology 61, 1908–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.