

Abstract
Mouse gene inactivation has shown that the transcription factor Sox11 is required for mouse palatogenesis. However, whether Sox11 is primarily involved in the regulation of palatogenesis still remains elusive. In this study, we explored the role of Sox11 in palatogenesis by analyzing the developmental mechanism in cleft palate formation in mutants deficient in Sox11. Sox11 is expressed both in the developing palatal shelf and in the surrounding structures, including the mandible. We found that cleft palate occurs only in the mutant in which Sox11 is directly deleted. As in the wild type, the palatal shelves in the Sox11 mutant undergo outgrowth in a downward direction and exhibit potential for fusion and elevation. However, mutant palatal shelves encounter clefting, which is associated with a malpositioned tongue that results in physical obstruction of palatal shelf elevation at embryonic day 14.5 (E14.5). We found that loss of Sox11 led to reduced cell proliferation in the developing mandibular mesenchyme via Cyclin D1, leading to mandibular hypoplasia, which blocks tongue descent. Extensive analyses of gene expression in Sox11 deficiency identified FGF9 as a potential candidate target of Sox11 in the modulation of cell proliferation both in the mandible and the palatal shelf between E12.5 and E13.5. Finally we show, using in vitro assays, that Sox11 directly regulates the expression of Fgf9 and that application of FGF9 protein to Sox11-deficient palatal shelves restores the rate of BrdU incorporation. Taken together, the palate defects presented in the Sox11 loss mutant mimic the clefting in the Pierre Robin sequence in humans.
Keywords: cell proliferation, craniofacial development, FGF, gene regulation, mouse, micrognathia, Pierre Robin sequence, cleft palate, Sox11
Introduction
Cleft palate and cleft lip are among the most common congenital birth defects in humans, with a prevalence ranging between 1/700 and 1/1000 (1). In the mouse, palatogenesis is initiated at embryonic day 11.5 (E11.5)3 when the maxillary prominences outgrow as bilateral protrusions in the oral cavity to form paired palatal shelves (2). As development progresses, the palatal shelves undergo extensive growth vertically along both sides of the developing tongue until E14.0, when the palatal shelves rapidly elevate into the horizontal position above the tongue. Horizontal growth continues until the bilateral shelves achieve midline contact to form a transient midline epithelial seam at E14.5. The degradation of this seam completes the formation of the secondary palate around E15.0. Factors that interfere with growth, elevation, or fusion can result in the formation of cleft palate (2–4).
In addition to the cleft palate caused by intrinsic factors, clefting of the secondary palate occurs as a consequence of defects in other craniofacial structures. Clinically, it is referred to as the Pierre Robin sequence (PRS), which is comprised of mandibular hypoplasia (micrognathia or retrognathia), displaced tongue position, and cleft secondary palate (5). Previous studies from a few mutant mouse strains, including deficiencies in Hoxa2, Snail1/2, Prdm16, Alk2, Sox9, and Erk2, have shown that the cleft secondary palate resulting from the failed palatal elevation is a consequence of the physical obstruction by a malpositioned tongue (6–12), partially mimicking the clefting of PRS in humans (2, 13, 14). In many of the murine mutant models of PRS-like clefting, the expression of the gene of interest is detected in the palatal shelves themselves in addition to the relevant structure, like the mandible (2). However, the regulatory mechanism of how the loss of the gene of interest results in primary defects and influences the development of the Pierre Robin sequence-like palate clefting still remains unclear.
Sox11, together with Sox4 and Sox12, makes up the Sox C subgroup of a family of transcription factors with a Sry-related HMG box (Sox) DNA binding domain (15, 16). The mouse and human genomes contain 20 orthologous pairs of Sox genes, and these 20 genes are divided into eight subgroups on the bases of sequence similarity and genomic organization (17). SoxC genes are all single-exon genes and encode a protein containing two highly identical functional domains: a Sox DNA-binding domain (84% identity) in the N-terminal half of the protein and a transactivation domain (66% identity) at the C terminus (18, 19). SoxC genes have been implicated in the widespread and highly overlapping expression of the SoxC genes in the developing mouse embryo (19–22). In orofacial development, Sox4 and Sox12 share similar expression domains, with Sox11 being expressed in the developing palatal shelves and tooth germ epithelium (19). These features suggest that the three SoxC proteins could exhibit similar functional properties. However, although SOX4 maps to human chromosome 6p23, a region implicated by several CLP linkage studies (23–26), SOX12 (20p12) and SOX11 (2p25) in humans map to a region not implicated in cleft lip/cleft palate by linkage studies. Sox4-null mutant mice die in mid-gestation because of severe cardiac outflow tract defects (27). Sox12-null mice have no obvious malformations and undergo a normal lifespan and fertility (22). Sox11-null mutants exhibit neonatal lethality, with cleft palate in 100% of the mutant mice and CLP in 70% of the mutant mice (21). The mutant pups also usually have cardiac defects, pulmonary hypoplasia, asplenia, eyes open at birth, omphalocele, and other defects (21). Thus, the Sox11 mutant provides a candidate model for evaluating the etiology of SoxC genes associated with human cleft palate. However, little is known about the intrinsic role of Sox11 in palatogenesis. In this study, using a mutant deficient in mouse Sox11, we explore the potential role of Sox 11 in the regulation of palatal growth by analyzing the cellular and molecular mechanisms by which the loss of Sox11 results in the formation of cleft secondary palate. We uncover that the failure of tongue descent in the Sox11 loss mutant physically hampers elevation of the palatal shelves that otherwise are capable of elevation and fusion, resulting in the cleft secondary palate. We also reveal that Sox11 is necessary for growth of the mandible and the palatal shelves, in which Sox11 acts upstream of Fgf9 in regulating cell proliferation.
Experimental Procedures
Animals
Generation of the Sox11fx/fx mice has been described previously (28). Mouse strains for Wnt1-Cre, Dermo1-Cre, Osr2-IRESCre, K14-Cre, Foxg1-Cre, Nestin-Cre, and EIIa-Cre were purchased from The Jackson Laboratory. The morning that a virginal plug was detected was considered E0.5. The transgenic mice were maintained in a C57/B6 background in a standard animal vivarium. All of the animal experimental protocols in this study were approved by The Committee of Laboratory Animals (HNU-M2010-0701), Hangzhou Normal University, Hangzhou, China.
Histology, Immunohistochemistry, and in Situ Hybridization
Embryos were collected at the desired developmental stages and fixed in 4% paraformaldehyde overnight at 4 °C. For sectioning of in situ hybridization, embryos were dehydrated through graded alcohols, embedded in paraffin, and sectioned at 12 μm. In situ hybridization was carried out as described previously, with digoxigenin-labeled antisense RNA probes (29). To conduct immunohistochemical staining, samples were fixed for 1 h in 4% paraformaldehyde in PBS, transferred to 30% sucrose in PBS, and stored at 4 °C overnight. Frozen sections (12 μm) were rinsed in PBS and blocked in 0.3% Triton X-100 and 2% BSA in PBS for 1 h at room temperature. Sections were washed three times in PBST (0.1% Triton X-100/PBS), blocked in 5% BSA for 30 min, and incubated with primary antibodies diluted with 5% BSA at 4 °C overnight in a humid chamber. Sections were subsequently washed in PBST, three times for 10 min each. Secondary antibodies (1:1000) diluted in 5% BSA were applied for 30 min in the dark. 3.3′-diaminobenzidine substrate kit was used for peroxidase (Vector Laboratories). The peroxidase reaction was stopped by rinsing with PBS followed by 2% paraformaldehyde fixation. Primary antibodies used in this study were commercially purchased from Abcam: Cyclin D1 (ab134175), CyclinD2 (ab3085), Ki67 (ab15580), Fgfr9 (ab71395), Fgfr1 (ab636010), and Fgfr2 (ab10648). For histology, serial sections of 7 μm were stained with hematoxylin and eosin. The tongue heights were measured on the basis of serial coronal sections of embryonic heads between E13.5 and E16.5 as described previously (8). Frontal sections through palatal shelves were selected from the anterior, middle, and posterior regions, with the middle region consisting of sections through the maxillary first molar tooth buds.
Cell Proliferation and TUNEL Assays
Cell proliferation rates were measured by BrdU labeling, and apoptosis was determined using the TUNEL assay. Briefly, timed pregnant mice were injected intraperitoneally with BrdU solution (5 mg/100 g of body weight) (29) from a BrdU labeling and detection kit (Roche) 20 min prior to being sacrificed. Embryonic heads (n = 3 for both the wild type and mutant) were fixed in 10% neutral formalin and then processed for paraffin sectioning at 5 μm for immunohistochemical staining according to the instructions of the manufacturer. BrdU-positive cells were counted (≥20 consecutive fields at ×40 magnification from three samples for both the wild type and mutants) and calculated as the percentage of labeled cells among total nuclear stained cells within a defined arbitrary area. Statistical significance was calculated using Student's t test. Apoptosis was assayed by TUNEL staining using the in situ cell death assay kit according to the instructions of the manufacturer (Roche).
In Vitro Organ Culture and Protein Bead Implantation
For in vitro palatal fusion assays, secondary palatal shelves were dissected in cold PBS from E13.5 embryos. The bilateral palatal shelves were placed close to each other with the dorsal side upward on a Nucleopor filter membrane in TROWELL organ culture in a chemical defined medium as described previously (29). Forty-eight hours after culture, explants were collected and fixed in 4% paraformaldehyde for histological staining as described above.
For in vitro palatal shelf elevation assays, roller cultures were performed essentially as described previously (8). The embryonic head with either the mandible or tongue removed was placed into a 20-ml glass culture tube containing 2 ml of medium. The culture tube was secured in a vertical position on a rotary apparatus rotating at a speed of 4 rpm in an incubator at 37 °C and 5% CO2. Samples were harvested after 24 h of culture, and they were then processed for histological examination.
For bead implantation experiments, Affi-Gel blue-agarose beads (Bio-Rad) were soaked in proteins as described previously (29). The protein concentrations used were consistent throughout all experiments. The beads were washed in PBS and then incubated for 40 min at 37 °C in 100 μg/ml FGF9 (R&D Systems). Control beads were soaked with similar concentrations of BSA under the same conditions.
Expression Vectors and Promoter-Luciferase Constructs
The Sox11 expression vector was constructed by subcloning a Sox11 cDNA fragment containing the full-length protein-coding region into the pcDNA3-TOPO (Invitrogen) expression vector. To generate the Fgf9-luciferase reporter plasmid, an 1868-bp fragment (−1857 to +11 bp) containing the Sox11 putative binding site was amplified from C57/B6L mouse genomic DNA and then subcloned into the pGL2-Basic vector (Promega) at the HindIII site in the multiple cloning region. The following primers were used for cloning the Fgf9 promoter: 5′-AAG CTT AAA TTC ATT GCT TCC TCC GTC T-3′ (forward) and 5′-AAG CTT CCA TAG CAA CGT AAG TGC CTG-3′ (reverse). For the construction of the pGL2-Fgf9-Mut luciferase plasmid, the Sox11 binding site (AACAAAG mutated to ATAGAAT) was mutated from the pGL2-Fgf9 luciferase construct using QuikChange XL mutagenesis (Stratagene) following the instructions of the manufacturer.
Cell Culture, Transfection, and Luciferase Reporter Assays
293T cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. For luciferase reporter assays, cells were plated in 24-well tissue culture plates (Corning) and co-transfected with 0.1 μg of a luciferase reporter vector, 0.1 μg of the pRL-Renilla luciferase expression vector (Promega), and 0.1 μg of the Sox11 expression vector. Transfections were performed using Lipofectamine 2000 (Invitrogen) reagent in accordance with the instructions of the manufacturer. Luciferase activity was measured 48 h after transfection using the Dual-Luciferase assay kit (Promega) with a TD-20/20 luminometer (Turner Designs). Reporter expression was normalized to co-transfected Renilla luciferase activity, and the normalized value (RLUpGL2/RLUpRL-TK) was used for statistical analysis. Transfection data were summarized from six repeated experiments.
ChIP Assay
293T cells were transiently transfected with pFLAG-Sox11-expressing vectors using Lipofectamine 2000 (Invitrogen). The ChIP assay was realized with the Imprint chromatin immunoprecipitation kit (Sigma-Aldrich). For each ChIP reaction, 250,000 cells were used. The size of the sonicated DNA fragments used for immunoprecipitation was between 400 and 800 bp in length. Purified chromatin was immunoprecipitated using an anti-FLAG M2 antibody (Sigma). Eluted DNA fragments were purified to serve as templates for the PCR amplification. The input fraction corresponded to 2% of the chromatin solution before immunoprecipitation. IgG control served as the negative control to the FLAG antibody.
The primers used to amplify the area containing thepredicted Sox11 binding site were 5′-CTT CCC TCT CGC ACG CCG TC-3′ (forward) and 5′-GCG TCC CCA ACA CAA GCA CT-3′ (reverse), resulting in a 173-bp fragment (P2). The primers used as a negative control were 5′-CGA GGCT CCG GGA CGC GGT CC-3′ (forward) and 5′-ACG TTG GCG GCG GCG ACA AA-3′ (reverse), resulting in a 195-bp fragment (P1). Amplification was carried out using the following conditions: one cycle at 95 °C for 2 min, followed by 37 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s and one final incubation at 72 °C for 5 min.
Microarray and Quantitative Real-time PCR
For both microarray and real-time RT-PCR, secondary palatal shelves were dissected from E13.5 and E14.5 embryos for RNA extraction using an RNA isolation kit (Ambion, RNAqueous-4RNA). The following primer pairs were used for quantitative RT-PCR: Fgfr1, 5′-GGT GAA CGG GAG TAA GAT CGG-3′ (forward) and 5′-CCC CGC ATC CTC AAA GGA G-3′ (reverse); Fgfr2, 5′-CCT CGA TGT CGT TGA ACG GTC-3′ (forward) and 5′-CAG CAT CCA TCT CCG TCA CA-3′ (reverse); Fgf9, 5′-ATG GCT CCC TTA GGT GAA GTT-3′ (forward) and 5′-TCC GCC TGA GAA TCC CCT TT-3′ (reverse); Sox11, 5′-CGA GCC TGT ACG ACG AAG TG-3′ (forward) and 5′-AAG CTC AGG TCG AAC ATG AGG-3′ (reverse); Sox4, 5′-GAC CTG CTC GAC CTG AAC C-3′ (forward) and 5′-ACT CCA GCC AAT CTC CCG A-3′ (reverse); and Sox12, 5′-GGA GAC GGT GGT ATC TGG G-3′ (forward) and 5′-ATC ATC TCG GTA ACC TCG GGG-3′ (reverse).
Statistical Analysis
Data were analyzed for statistical significance by Student-Newman-Keuls test using SYSTAT software (SYSTAT Inc.), with values of p < 0.05 considered to be significant. All experiments were independently repeated at least three times.
Results
Sox11 Is Expressed in Developing Palatal Shelves and the Mandible
Expression of Sox11 in the mouse palatal shelf has been reported previously (19). To decipher whether Sox11 is involved in the regulation of palatal development, we performed in situ hybridization on sections of embryonic head using a non-radioactively labeled Sox11 riboprobe to characterize the localization of Sox11 mRNA in the palatal epithelium and mesenchyme during palatogenesis. At E11.5, when the palatal shelves were initiated as outgrowths of maxillary processes, Sox11 transcripts were detected in the shelves (Fig. 1A). A similar expression pattern was maintained during the vertical growth period from E12.5 and E13.5, with a high level of transcripts in both the mesenchyme and the epithelium, including the medial epithelial edge (MEE) of the palatal shelves (Fig. 1, B and C). The expression of Sox11 persisted in both the mesenchyme and the epithelium in E14.5, when the two bilateral palatal shelves met in the midline (Fig. 1D). Expression was down-regulated in the completely formed secondary palate at E15.5 (Fig. 1E). These data showed that Sox11 was dynamically expressed during the development of palatal shelves, where extensive epithelial and mesenchymal interactions occur.
FIGURE 1.
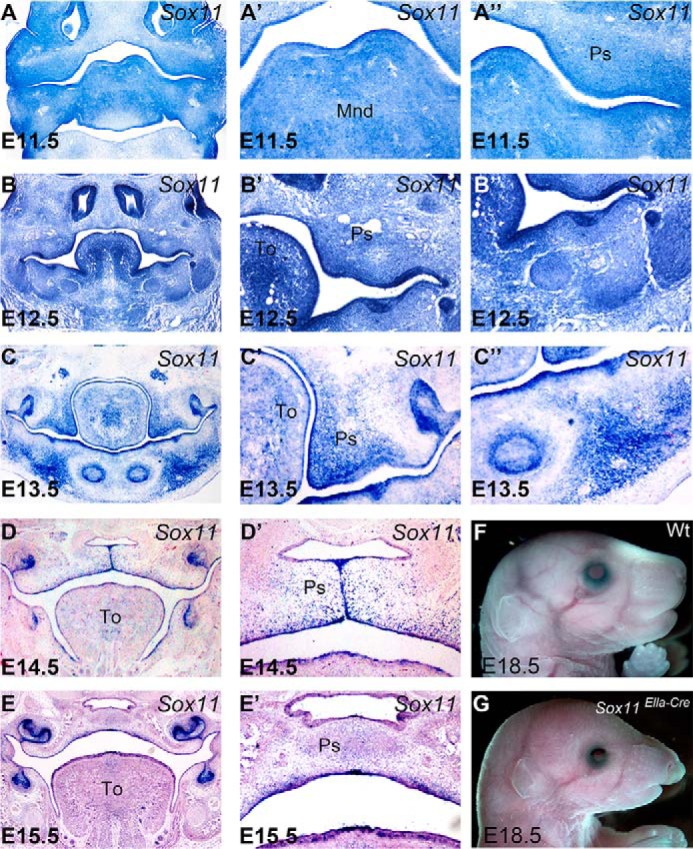
Sox11 expression in the developing palatal shelf and mandible. A–E', in situ hybridization on sections show the localization of mouse Sox11 mRNA in the developing palatal shelf between E11.5 and E15.5 in wild-type mice. In the developing palatal shelf, expression of Sox11 in both the mesenchyme and the epithelium is maintained throughout in the outgrowing palatal shelves (A, A', B, B', C, and C') until the two palatal shelves meet in the midline (D). The expression is down-regulated at E15.5, when the bilateral palatal shelves are completely merged to form the complete palate (E). All panels shown are from the middle palate region. Note that the Sox11 transcripts were detected in both mesenchyme and epithelium in the mandible at both E11.5 and E12.5 (A, A”, B, and B”) but is specifically detected in the mesenchyme, where the Meckel cartilage and mandibular bone arise at E13.5 (C and C”). F and G, a Sox11EIIa-Cre mouse displays severe craniofacial defects, including mandible hypoplasia (G), compared with the wild type (F). To, tongue; Mnd, mandible; Ps, palatal shelf.
In addition, we noted that in situ hybridization simultaneously displayed the expression of Sox11 in the other orofacial structures, including the mandible (Fig. 1, A”–C”), which shares a common origin with the palatal shelf (2, 13). The Sox11 transcript was detected both in the epithelium and in the mesenchyme throughout E11.5, E12.5, and E13.5, suggesting that the expression of Sox11 is not in a palatal shelf-specific manner and that Sox11 is also implicated in the development of these structures.
Loss of Sox11 Resulted in Cleft Palate Formation
A previous gene knockout study has shown that inactivation of Sox11 in mice resulted in cleft palate and cleft palate with cleft lip (21). To differentiate between the roles of Sox11 in the palatal epithelium versus the palatal mesenchyme, we generated the tissue-specific Sox11-null mutants using several tissue-specific Cre mouse lines, including Wnt1-Cre, Dermo1-Cre, and Osr2-IRES-Cre for the mesenchyme and K14-Cre, Foxg1-Cre, and Nestin-Cre for the epithelium. The conditional knockout mouse line (Sox11fx/fx) was crossed with tissue-specific Cre lines to inactivate Sox11 in either the mesenchyme or the epithelium. However, none of these tissue-specific Sox11 deletion mutants led to the formation of cleft palate when neonatal mice were assessed (data not shown). We attributed this to functional redundancy because of the highly overlapping expression of Sox11 with other Sox C genes in the palatal shelves (19). We then deleted Sox11 in both the epithelium and the mesenchyme by crossing EIIa-Cre mice with Sox11CKO mice to generate Sox11EIIa-Cre mice. The deletion of Sox11 was confirmed by both in situ hybridization and quantitative real-time PCR (supplemental Fig. 1). Interestingly, we found up-regulated expression of Sox4 and Sox12 in Sox11EIIa-Cre (supplemental Fig. 1).
Prenatally collected Sox11EIIa-Cre mutant mice exhibited craniofacial malformations, including mandibular hypoplasia (Fig. 1, F and G) and cleft palate with 100% penetrance and cleft lip in 70% of the mutants, consistent with results from the Sox11−/− mutants described previously (21). Scanning electron micrographs showed complete clefting of the secondary palate in Sox11EIIa-Cre mutant embryos (data not shown). Histological analysis during palatogenesis showed that the initial downward growth of the palatal shelves occurred normally in Sox11EIIa-Cre mice at E11.5, E12.5, and E13.5, during which the palatal shelves of Sox11EIIa-Cre mice were indistinguishable in size and shape from those of the wild type (Fig. 2, A–L). At E14.5, the palatal shelves in the wild-type embryos had already elevated to the horizontal position and begun to fuse at the midline (Fig. 2, M–O), and the bilateral palatal shelves had merged to form the complete secondary palate at E15.5 (Fig. 2, S–U). However, the bilateral palatal shelves of Sox11EIIa-Cre mutant embryos failed to elevate and remained at the vertical position during these stages (Fig. 2, P—R and V–X). It was also evident that the palatal shelves became regressed at the proximal portion in Sox11EIIa-Cre mutants, resulting in the reduction in the size of the palatal shelves at E15.5 (Fig. 2, V–X). Occasional elevation of one or both sides of the palatal shelves occurred in later stages in the Sox11EIIa-Cre mutant (supplemental Fig. 2), suggesting the possibility that Sox11 is not an intrinsic determinant for the elevation of the palatal shelves. Instead, the loss of Sox11 caused the retardation of the palatal shelf formation by delaying its elevation on time.
FIGURE 2.
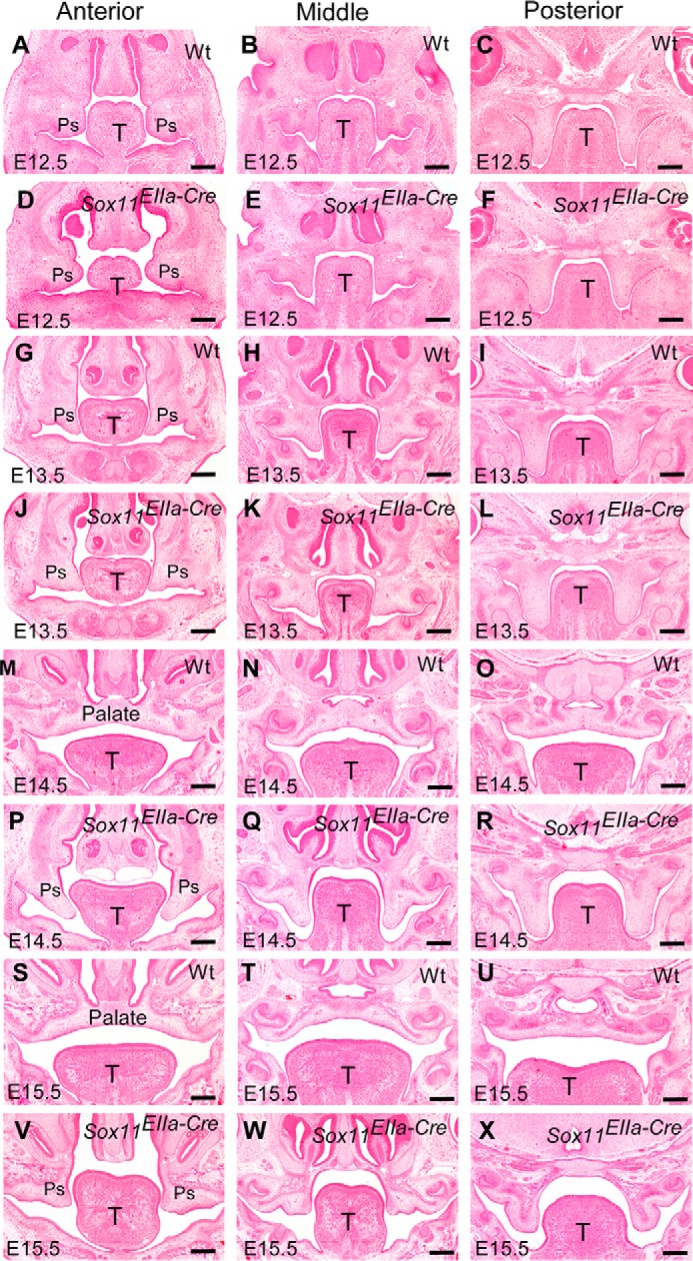
Sox11EIIa-Cre mutants display cleft palate with retardation to palatal shelf elevation. A–X, H&E staining on sections of embryonic heads shows the histology of developing palatal shelves. A–L, downward outgrowth of the palatal shelves shares similar features in Sox11EIIa-Cre mutants and their wild-type littermates at E12.5 (A–C versus D–F) and E13.5 (G–I versus J–L). Palatal shelves elevate to the horizontal position above the tongue dorsum and contact each other in the midline at E14.5 in the wild type (M–O) but remain in a downward position in the Sox11EIIa-Cre mutant (P–R). In comparison with a completely formed secondary palate in the wild type (S–U), Sox11EIIa-Cre mice still exhibit cleft palate with shelves in a downward position at E15.5 (V–X). T, tongue; Ps, palatal shelf. Scale bars = 200 μm.
The Sox11EIIa-Cre Mutant Does Not Disrupt the Fusion Mechanism of Palatal Shelves
Given the normal downward outgrowth of palatal shelves and the retained elevation potential in the palatal shelf of Sox11EIIa-Cre, the formation of cleft secondary palate upon loss of Sox11 may be attributed to mechanisms involving fusion (2). To test whether the fusion competency of palatal shelves was altered in Sox11EIIa-Cre, we took the in vitro organ culture in which the bilateral palatal shelves were closely placed on Nucleopore filters for 48 h. Histological examination of cultured explants showed that complete fusion occurred in two contacted palatal shelves in Sox11EIIa-Cre (n = 3). It was evident that the epithelial seam disappeared (Fig. 3B) in a way comparable with that which occurred in the wild type (n = 3) (Fig. 3A). These in vitro data suggest that fusion would occur in the event that the two bilateral palatal shelves contacted each other in Sox11EIIa-Cre. We further examined the expression of molecular markers critical for fusion fate of the palatal shelf in Sox11EIIa-Cre in comparison with the wild type. In wild-type embryos, MMP13 (matrix metalloproteinase 13) is exclusively expressed in the MEE at E14.0, when palatal shelves are ready for fusion on the horizontal position until the MEE is degenerated (Fig. 3C) (30, 31). The proper expression of Jagged 2 in the epithelium is critical for the proper development of the palatal shelf (Fig. 3E), and inactivation of Jagged 2 resulted in the premature and ectopic fusion of the palatal shelf and oral epithelium (32, 33). The TGF-β3 expression domain in the horizontal palatal shelf in wild-type mice was mainly located in the MEE, with equal extension to the oral and nasal surfaces at E14 (Fig. 3G). TGF-β3 signaling has been demonstrated to be essential for palate fusion (3). Similar to the wild-type controls, the transcript of MMP13 was located in distal medial epithelium of the vertical palatal shelf in Sox11EIIa-Cre at E14 (Fig. 3D). Jagged 2 was also expressed in palatal and oral epithelium in Sox11EIIa-Cre, comparable with the wild type at E13.5 (Fig. 3F). The TGF-β3 mRNA was localized to the distal epithelium of the vertical palatal shelf at E14 in the Sox11EIIa-Cre mutant (Fig. 3H). Thus, the comparable expression patterns of marker molecules suggest that the fusion fate of MEE in the Sox11 mutant is not changed despite the retarded elevation at E14. This conclusion was further supported by the positive TUNEL staining in the medial portion of the downward palatal shelf at E14.5 in the Sox11EIIa-Cre mutant (Fig. 3J), when apoptosis occurred in the midline seam in the wild-type control (Fig. 3I). Taken together, these data suggest that the altered fusion program of the palatal shelf is not the reason for cleft palate in Sox11EIIa-Cre mice.
FIGURE 3.
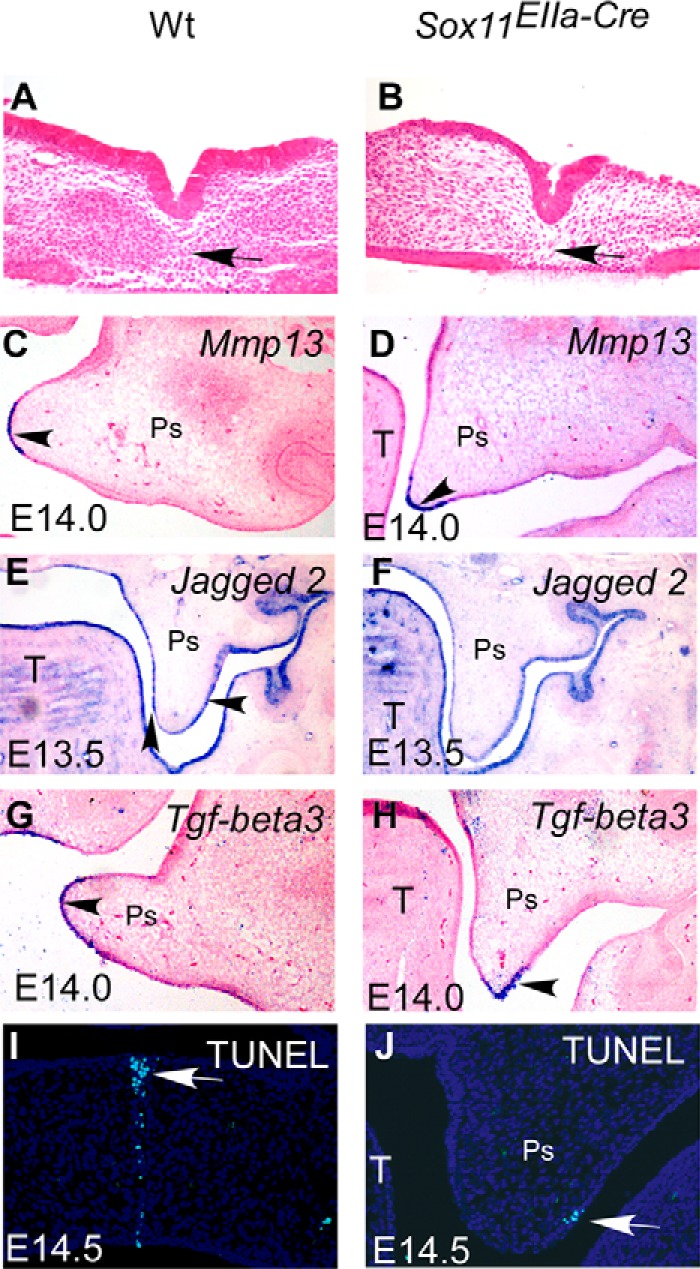
Fusion of palatal shelves is not impaired in Sox11EIIa-Cre mice. A and B, histological sections show that the palatal shelves from E13.5 Sox11EIIa-Cre embryos (B), like those in the wild-type control (A), fused with complete degradation of the midline seam in contact in an in vitro organ. Arrows indicate the merged cells in the midline seam. C–H, in situ hybridization shows that the expression of Mmp13 (C and D), Jagged-2 (E and F), and Tgf-β3 (G and H) is comparable in wild-type and Sox11EIIa-Cre mice. I and J, TUNEL assay shows cell apoptosis. Cell death is evident in the midline epithelial seam in the wild type (I) and in distal epithelial cells of the downward palatal shelf in the Sox11EIIa-Cre mutant (J). Ps, palatal shelf; T, tongue.
Malformed Tongue Position in Sox11EIIa-Cre Caused Physical Retardation of Palatal Elevation
On the other hand, Sox11EIIa-Cre mutant mice also showed a marked reduction in Meckel cartilage size at E13.5 (Fig. 4B) and in mandibular length and displaced tongue position during palatal elevation by E14.5 along the anterior-posterior axis, as exhibited by abnormal height in comparison with the wild-type littermates (Fig. 4, A–C), suggesting that the displaced tongue position was attributable to physical obstruction causing the retardation of palatal shelf elevation (2).
FIGURE 4.

Displaced tongue position obstructs palatal elevation in Sox11EIIa-Cre. A, measurement of tongue height along the anterior-posterior axis in Sox11EIIa-Cre and wild-type controls. Note that tongue height at E13.5 is comparable in Sox11EIIa-Cre and wild-type controls but remained significantly increased at E14.5, E15.5, and E16.5 in Sox11EIIa-Cre. *, p = 0.05 (n = 5); **, p < 0.01 (n = 5). A, anterior; M, middle; P, posterior. Data are represented as the mean ± S.D. B, skeletal preparations show the shorter mandibles in Sox11EIIa-Cre compared with wild-type mice. Scale bars = 1 mm. C, relative measurement of mandibular length. **, p < 0.01 (n = 5). Data are represented as the mean ± S.D. D, histological staining on sections of embryonic head explants by in vitro rolling culture. The elevation of palatal shelves is achieved either by removal of the tongue or removal of the mandible. Arrows point to the comparable horizontal positions in Sox11EIIa-Cre and wild-type controls. Ps, palatal shelf.
To determine whether the displaced tongue position retarded palate elevation in Sox11EIIa-Cre mice, we tested whether the palatal shelves possessed the intrinsic ability of elevation by in vitro organ culture on a rotary apparatus. E13.5 embryonic heads with their mandible or tongue removed were placed in a bottle subjected to roller culture. Samples were then collected and processed for histological examinations. As shown in Fig. 4D, the palatal shelves in samples of Sox11EIIa-Cre mutant (n = 6/6), similar to their wild-type littermates (n = 6/6), were able to elevate after 24 h in rolling culture. These observations indicate that the loss of transcription factor Sox11 does not intrinsically alter the morphogenetic potential for elevation but rather delay it. It also suggests that the palatal shelves in the Sox11 loss mutant still possess intrinsic competency for elevation.
Sox11 Is Required for Development of the Mandible
The aforementioned results, together with the expression of the Sox11 transcript in mandibular tissue, prompted us to examine the potential mechanism by which ablation of Sox11 led to growth defects of the mandibular primordia. To understand the cellular mechanism forming a hypoplastic mandible in the absence of Sox11, we assessed the regulation of cell proliferation in the mandibular mesenchyme by immunohistochemistry using antibodies against Ki67 and CyclinD1. A decrease in Cyclin D1 expression was demonstrated in the mandibular mesenchyme at E11.5 through E13.5 in Sox11EIIa-Cre (Fig. 5, A–F), suggesting that cell proliferation was affected through regulation of a type-D Cyclin. Coincidently, the mandible displayed a remarkable decrease in Ki67 expression in mesenchymal cells in and around the Meckel cartilage at E13.5 (Fig. 5, G–I). Taken together, these data provide evidence that the Sox11 deletion resulted in compromised mandibular growth through regulation of mesenchymal cell proliferation. Consistent with this suggestion, we detected that the expression of Runx-2, an early marker for differentiation of osteoblasts, was compromised in Sox11EIIa-Cre at E11.5 and E13.5 (Fig. 5, J–M).
FIGURE 5.

Cell proliferation in the mandibular mesenchyme is reduced in Sox11EIIa-Cre. A–H', immunohistochemistry shows a decrease in Cyclin D1 expression during mandible development in the mutant mandible at E11.5 (B versus A), E12.5 (D versus C), and E13.5 (F versus E). A significant reduction in Ki67 is evident in the mesenchyme (mc) in and surrounding Meckel cartilage at E13.5 (H versus G). G' and H' are enlarged pictures of the boxed areas in G and H, respectively. I, relative proliferation as number of Ki67-positive cells in the boxed area in the wild type (G) versus Sox11EIIa-Cre (H). Arrows indicate the developing mandibular bone. Data are mean ± S.D. of three independent samples. J—M, in situ hybridization showing the expression of Runx2 in the mandibular mesenchyme at E11.5 (J and K) and the mesenchyme surrounding Meckel cartilage at E13.5 (L and M).
Sox11 Is Required for Proper Palatal Shelf Growth
Because Sox11 is expressed in the developing palatal shelves, to investigate the intrinsic role of Sox11, we also examined whether there were alterations in cell proliferation and cell survival during palate development in Sox11 mutant mice. No differences in cell apoptosis were found in the palatal shelves Sox11EIIa-Cre mutant embryos at E12.5 and at E13.5 (data not shown). Cell proliferation in E12.5 palatal shelves in Sox11EIIa-Cre was also comparable with that in wild-type tissue by BrdU incorporation assay (data not shown). However, at E13.5, we detected a 25% reduction (p < 0.01) in the palatal mesenchyme and a 19% reduction (p < 0.01) in palatal epithelial cells in Sox11EIIa-Cre mutant embryos (Fig. 6, B–C) in comparison with their wild-type littermates (Fig. 6, A, A', and C). Similarly, proliferation of both epithelial and mesenchymal cells in the proximal bend region of the palatal shelf was also significantly reduced in Sox11EIIa-Cre mutant embryos (Fig. 6, B, B”, and D) in comparison with the wild-type littermates (Fig. 6, A, A”, and D).
FIGURE 6.
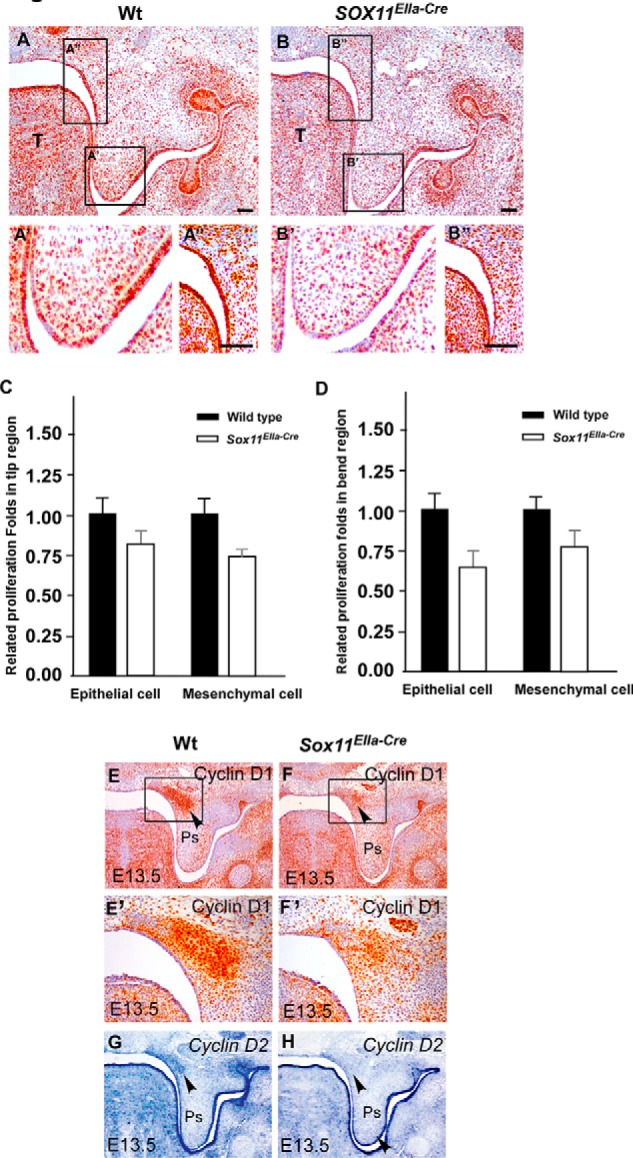
Cell proliferation of the palatal shelf is reduced in Sox11EIIa-Cre mice. A and B, immunohistochemistry shows the BrdU incorporation in E13.5 palatal shelves. In comparison with the wild type (A), the BrdU-labeled cells in the Sox11EIIa-Cre mutant were significantly reduced in both the epithelium and the mesenchyme (B). A', A”, B', and B”, large magnification of the boxed regions represent the distal palatal region (A' and B') and bend region (A” and B”) in A and B, respectively. Scale bars = 100 μm. C, statistical data show the relative proliferation folds in the distal palatal shelf of the wild type (A') and Sox11EIIa-Cre mutant (B'). D, statistical analysis shows the relative proliferation folds in the bend region in wild-type control (A”, n = 7) and Sox11EIIa-Cre mutant mice (B”, n = 7). E and F, immunostaining shows a decrease in cyclin D1 expression in the proximal region of the palatal shelf in the Sox11EIIa-Cre mutant (F, arrowhead) in comparison with the wild type (E, arrowhead). E' and F' are enlarged pictures of the boxed areas in E and F, respectively. G and H, in situ hybridization exhibits reduced expression of cyclin D2 in the proximal region of the palatal shelf in Sox11EIIa-Cre mice (F, arrowhead) compared with the wild-type mice (G, arrowhead). T, tongue; Ps, palatal shelf.
To evaluate whether Sox11 may regulate d-type Cyclin-modulated cell proliferation in palatal shelves, we examined the expression of cyclin D1 and D2 in the palatal shelves of Sox11EIIa-Cre embryos by in situ hybridization and immunohistochemistry. Down-regulated expression of cyclin D1 protein (Fig. 6, E and F) and cyclin D2 mRNA in mesenchymal cells (Fig. 6, G and H) was exhibited. These data are consistent with our data from BrdU incorporation assays (Fig. 6, A–D).
Fgf9 Is the Downstream Target of Sox11 in Palatal and Mandibular Growth
To explore the molecular mechanism by which Sox11 deletion results in cleft palate formation, we analyzed gene expression profiles by microarray using RNA extracted from E13.5 and E14.5 palatal shelf tissues from Sox11EIIa-Cre and wild-type control mice (n = 2/genotype) to identify the potential downstream genes of Sox11. In parallel comparisons, we uncovered dozens of probe sets representing transcripts that were differentially expressed (≥1.5-fold, <5% false discovery rate (Fig. 7A). Among these genes, Fgf9 is the one differentially expressed (Fig. 7A) and implicated in palatogenesis (34, 35). We confirmed the down-regulation of Fgf9 by quantitative real-time PCR using mRNA extracted from palatal shelve tissues at E13.5 and E14.5 (Fig. 7B) and in situ hybridization (Fig. 7, C and C' versus D and D'). Interestingly, we also found that expression of Fgf9 was markedly reduced in the mesenchymal site where the mandibular bone arises in E13.5 Sox11EIIa-Cre compared with the wild type (Fig. 7, C” versus D”), suggesting that Fgf9 expression is a downstream target of Sox11 in the mandible. Moreover, we detected a decrease in the expression of the Fgf9 receptor Fgfr2 (36, 37) in the mandibular bone-forming site and the palatal shelf (Fig. 7, E–F”) but not Fgfr1 (Fig. 7, G and H). These data suggest that Fgf9 and Fgfr2 are potential downstream genes of Sox11 in the developing mandible and palatal shelf.
FIGURE 7.

Sox11 acts upstream of Fgf9 expression in the palatal shelf and mandible. A, heatmap of representative microarray probe sets representing transcripts that were differentially expressed (≥1.5-fold, <5% false discovery rate). Mut, Sox11EIIa-Cre. B, RT-PCR using RNA extracted from E13.5 and E14.5 palatal shelves. The Sox11 transcript is completely deleted in Sox11EIIa-Cre. The expression levels of Fgf9 and Fgfr2 but not Fgfr1 are reduced in the Sox11EIIa-Cre mutant in comparison with that in the wild type. Data are represented as the mean ± S.D. and are representatives of at least three independent experiments. C–H, in situ hybridization shows decreased expression of Fgf9 (D) and Fgfr2 (F), but not Fgfr1 (H versus G), in E13.5 Sox11EIIa-Cre palatal shelves and the mandible (D) compared with the wild type (C). Boxed regions specify reduced expression of Fgf9 in the epithelium (D' versus C') and Fgfr2 in the mesenchyme of the bend region of the palatal shelf (F' versus E'). Note the markedly reduced expression of mesenchymal Fgf9 (D” versus C”) and Fgfr2 (F” versus E”) in the E13.5 mandible. Arrows indicate the palatal shelf epithelium in C', the mandibular bone-forming site in C”, the mesenchyme in the bend region of the palatal shelf in E', and the mandibular bone-forming site in E”.
To further determine whether the transcription factor Sox11 regulates developmental processes by directly binding to Fgf9, we tested the potential binding of Sox11 to the regulatory region of Fgf9 by in vitro ChIP assays. Sox11 protein binds to the consensus sequence (5′-(A/T)(A/T)CAA(A/T)G-3′) in the minor groove of DNA and can function either as a transcriptional activator or repressor (38, 39). By using rVista software to analyze conserved regions within the Fgf9 promoter, we identified the potential Sox11 binding site, which was located in the −1671 to −1664 bp region upstream of the transcription start site (Fig. 8A). We utilized the oligo pairs (see “Experimental Procedures”) that amplify either the potential binding site of Sox11 in the regulatory regions of Fgf9 (Fig. 8A, P2) or non-binding site oligo pairs as a negative control (Fig. 8A, P1). PCR showed that, after immunoprecipitation of linked chromatin, there was a specific enrichment of FLAG-tagged Sox11 to a DNA fragment that corresponded to a potential site with antibodies against FLAG compared with IgG controls (Fig. 8B). Luciferase reporter assays were further performed to test the functional significance of the Sox11 binding region (Fig. 8, A and C). The reporter was significantly activated by overexpression of CMV-Sox11. In contrast, these luciferase activities were dramatically diminished when the Sox11 binding site was deleted in the Fgf9-promoter vector (Fig. 8C). In summary, we found that transcription factor Sox11 activates Fgf9 expression by directly binding to this promoter region of Fgf9.
FIGURE 8.

Sox11 directly regulates Fgf9 expression. A, schematic of the mouse Fgf9 gene from the upstream promoter region of the transcription start site (TSS, angled arrow) to the downstream encoding region (Fgf9). A putative Sox11 binding position (A/T)(A/T)CAA(A/T)G-3′) is found in the promoter region. Primer pairs were designed to amplify a 197-bp promoter region containing a Sox11 binding site (P2) and promoter region containing no Sox11 binding site (P1). B, ChIP assays of Sox11 protein binding to endogenous Fgf9. Chromatin extracts from 293T cells transiently expressing FLAG-tagged Sox11 protein were precipitated in the presence of nonspecific mouse IgG or antibody against FLAG. PCR products using primer pairs amplifying P2 and control P1 from the immunoprecipitated chromatin and input sample were resolved by agarose gel electrophoresis. C, Fgf9-luciferase reporter containing a Sox11 binding site and expression vector of Sox11 (CMV-Sox11) were transiently transfected into 293T cells. Reporter expression was normalized to co-transfected Renilla luciferase activity, and the normalized value (RLUpGL2/RLUpRL-TK) was used for statistical analysis. **, p < 0.01. Data are represented as the mean ± S.D. and are representatives of at least three independent experiments. D and E, Fgf9 protein restores cell proliferation of the palatal shelf in Sox11EIIa-Cre. D, immunostaining shows the BrdU-labeled cells in the palatal shelves in organ culture (arrows). BrdU incorporation was reduced in Sox11EIIa-Cre in comparison with the wild type. However, the number of BrdU-positive cells was increased when Fgf9 protein beads were implanted in Sox11EIIa-Cre. E, statistical analysis of BrdU labeling in palatal shelves implanted with either Fgf9 beads or BSA control. **, p < 0.01 (n = 5).
Fgf9 Maintains the Cell Proliferation of the Palatal Shelf in the Absence of Sox11
To investigate whether Fgf9 maintains cell proliferation downstream of Sox11, we detected cell proliferation in the palatal shelves in organ culture by implanted Fgf9 protein-saturated beads. BrdU was applied for 30 min at the end of the culture period. Immunostaining showed that the ratio of BrdU incorporation was significantly increased by implanted Fgf9 beads but not by BSA control beads (Fig. 8, D and E), indicating that Fgf9 protein treatment restored the cell proliferation of Sox11EIIa-Cre palatal shelves.
Discussion
The formation of cleft palate with or without cleft lip resulting from the loss of mouse Sox11 has been known for a decade (21). However, the question of whether or how Sox11 is involved in the regulation of mammalian palatogenesis has not been addressed. In this study, we found that the cleft secondary palate in the Sox11 loss mutant is mainly due to a failed palatal shelf elevation. Interestingly, loss of Sox11 influences the proliferation of the mandibular mesenchymal cells, resulting in a small mandible (hypoplasia). As a result of the reduced mandible size in the Sox11 loss mutant, the tongue failed to move downward mechanically, preventing palatal shelf elevation from a vertical to a horizontal position, causing cleft secondary palate. We thus provide evidence that extrinsic influence is the major factor in clefting of the secondary palate of the Sox11 mutant, resembling the human PRS. Similar murine models have been revealed in the Prdm16 loss-of-function mutant and loss of mesenchymal signaling for Erk2 or Fgfr (12, 14, 40).
Morphogenetic events in murine palatogenesis, particular in the elevation of the palatal shelves from a vertical to a horizontal position, depends on both intrinsic factors from the shelf itself and the extrinsic forces coinciding with the downward movement of the tongue (2, 13, 41). Proper cell proliferation is essential for the proper growth of craniofacial structures. In Sox11EIIa-Cre mice, the mandible is evidently narrow and small, and the tongue remains heightened above the palatal shelves on both sides. On the other hand, the mutant palatal shelves without the tongue are able to elevate in an in vitro organ culture. These results, together with data of the enriched Sox11 transcript in the mandibular mesenchyme and the proportionally small Meckel cartilage and mandibular bone in the Sox11 mutant, favor a non-intrinsic palate role for Sox11 during palatal shelf elevation. Formation of Meckel cartilage in mouse development occurs at approximately E13.5 and is closely correlated with mandibular outgrowth (42) 1 day before palatal elevation. In the Sox11EIIa-Cre mutant, the impaired growth of Meckel cartilage may influence mandible size and cause clefting in the secondary palate (11, 40).
In addition, we also show that expression of Sox11 in the palatal shelf is required for the maintenance of proper palatal growth. Sox11 may regulate cell proliferation in the proximal bending region on the nasal side of the palatal shelf through d-cyclins, such as cyclin D1 and cyclin D2, suggesting a regulatory role for Sox11 in the maintenance of cell proliferation in the palatal mesenchyme. This indication is consistent with the fact that the oronasal patterning is highly regulated through epithelium-mesenchyme interactions in the developing palatal shelf (43), in which the interaction between Fgf7 and Shh in the nasal region of the palatal mesenchyme could be one of the candidate pathways to determine the oronasal patterning of the palatal shelf in mice (43).
The expression pattern of Sox11 in the mandibular tissues is consistent with a function in the regulation of cell proliferation. Sox-C genes encode regulatory transcription factors critical for the cell cycle both in vitro and in vivo (18, 44–46). SOX4 overexpression was shown to promote cell cycle arrest and apoptosis of HCT116 colon carcinoma cells (47). Morpholino-targeted disruption of Sox11 in zebrafish also identified that Sox11 plays a crucial role in enhancing osteoblast differentiation by facilitating the proliferation of mesenchymal and osteoblast progenitor cells (45). BrdU incorporation assays have shown a significant reduction in cell proliferation in the neural tube, the branchial arches, and somites of Sox4/Sox11 double mutant mice (48). Highly overlapping transcript locations of Sox11 and Sox12 in the mouse palatal shelf have been described previously (19). Our data reveal that the expression of Sox4 and Sox11 partially overlaps in the epithelium and mesenchyme of the developing palatal shelf (supplemental Fig. 1A). It is thus reasonable to predict the possibility that the function of Sox11 in mouse palatogenesis may be compensated for by Sox4 and Sox12. The functional redundancy of SoxC genes has been described in the development of the CNS (18, 19). None of the single Sox C gene deletions in mice exhibited any CNS defects (21, 22), indicating the functional compensation for the loss of any individual SoxC gene (18) by other SoxC genes. Recent compound mutations of Sox C genes have also clearly shown that the more the SoxC genes were deleted, the more severe were the defects that resulted. For instance, Sox4/Sox11 double-null mice die at early organogenesis at E10.5, indicating that three SoxC genes are redundantly required for successful organogenesis (48). However, this seems not to be the case in the developing palatal shelf, where deletion of Sox11 results in clefting in palate and lip even though the expression of Sox4 and Sox12 is up-regulated prior to the palatal shelf elevation in the Sox11EIIa-Cre mutant. Nevertheless, the question of whether Sox11 function can be replaced by the other SoxC genes still remains to be addressed by genetic replacement of Sox11 with other SoxC genes. Our data reveal the regulatory effect of the transcription factor Sox11 on the cell proliferation of both mesenchymal and epithelial cells.
Sox C transcription factors have been identified as the direct regulators of several genes critical for mouse organogenesis. In humans, Tcfap2a (TFAP2A) maps to human chromosome 6p24, a region repeatedly implicated in linkage studies of human CLP (24–26, 49). The Sox11 transcription factor has a binding site in the regulatory region of Tcfap2a, which drives the expression of Tcfap2 in cranial neural crest cells and their derivatives, such as the mesenchyme of the nasal prominences. Chimeric experiments where Tcfap2-null cells were mixed with wild-type cells resulted in mouse embryos with various isolated defects, including cleft palate, suggesting that the proper regulatory effect on Tcfap2 transcription is essential for human facial development (50, 51). SoxC genes bind directly to the promoter region to promote the expression of Tead2 (48). Teads encode transcription factors that are associated with Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) to function as co-transcription factors to regulate the cell proliferation and survival underlying the Hippo signaling pathway (52).
Our study reveals that Sox11 acts upstream of Fgf9 in mandibular mesenchymal cells as the mandibular bone is arising. This observation is further supported by biochemical evidence that Sox11 directly regulates the transcription of Fgf9, which acts in turn as a mitogen in the developing mandibular mesenchyme and palatal shelf (35). Additionally, we show that Sox11 expression in the epithelium and the mesenchyme of the developing palatal shelf is present until completion of palatal development (19), which also suggests that Sox11 is involved in the molecular control of palatal growth. Sox11 has been found to regulate Fgf9 in the epithelium of the palatal shelf and receptor Fgfr2 (36) in the nasal side mesenchyme of the palatal shelf. Fgfr-mediated control of cell proliferation has previously been recognized in the lift movement of palatal shelves (53). Ablation of Fgfr1 in the ectomesenchyme results in a cell proliferation delay and cleft palate by impeding palatal shelf elevation prior to shelf fusion (53). However, in contrast to the clefting secondary palate in our Sox11 loss mutation that highlights the extrinsic influence of a malpositioned tongue and micrognathia on the clefting the secondary palate, the cleft palate in loss of function in Fgfr1 is due to the disturbance of cell signaling to delay cell proliferation in both the mesenchyme and epithelium of developing palatal shelves, preventing the shelves from proper elevation and fusion (53). Moreover, Fgf9 associated with Pitx2 has been identified to mediate TGF-β signaling and regulate cell proliferation in the palatal mesenchyme during mouse palatogenesis (35). Thus, we suggest the existence of a Sox11/Fgf9/Fgfr2 regulatory pathway in controlling the growth of the mandible and palatal shelf, consistent with the critical role of Fgf9 in the regulation of growth (34).
In summary, although the palatal shelf in Sox11 mutant mice fails to elevate on time, it possesses the morphogenetic potential for elevation and fusion. Moreover, loss of Sox11 does not result in the ectopic, premature fusion between the downward palatal epithelium and other oral epithelium that occurred in Jag2−/−, Fgf10−/−, and Mn1−/− mutant mice (32, 33, 54), partially because of the normal Jagged-2 expression in the Sox11 mutant palatal shelf. The results that facial structural impairments, such as mandibular dysplasia (shorter in length) and tongue displacement, accompany cleft secondary palate in Sox11 mutant mice support that Sox11 deletion during palatogenesis is evidently associated with secondary effects in the Sox11EIIa-Cre mutants (21), mimicking the clefting in PRS in humans.
Author Contributions
H. H. and Z. Z. designed the research. X. Z. collected animal materials. H. H., X. Y., M. B., X. M., and H. C. performed the experiments. H. H. and X. Y. analyzed the data. L. G. and M. Q. helped perform the analysis with constructive discussions. Z. Z. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank all members of the Zhang laboratory at the Institute of Developmental and Regenerative Biology, Hangzhou Normal University for suggestions during the generation of these data.
This work was supported by Natural Science Foundation of China Grant 31071287 and Natural Science Foundation of Zhejiang Province Grant LZ12C12002 (to Z. Z) and Natural Science Foundation of ChinaGrant 31101046 and Natural Science Foundation of Zhejiang Province Grant LY15C070005 (to H. H.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figures 1 and 2.
- E11.5
- embryonic day 11.5
- PRS
- Pierre Robin sequence
- MEE
- medial epithelial edge.
References
- 1. Murray J. C. (2002) Gene/environment causes of cleft lip and/or palate. Clin. Genet. 61, 248–256 [DOI] [PubMed] [Google Scholar]
- 2. Bush J. O., and Jiang R. (2012) Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139, 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gritli-Linde A. (2007) Molecular control of secondary palate development. Dev. Biol. 301, 309–326 [DOI] [PubMed] [Google Scholar]
- 4. Juriloff D. M., and Harris M. J. (2008) Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. A Clin. Mol. Teratol. 82, 63–77 [DOI] [PubMed] [Google Scholar]
- 5. Tan T. Y., Kilpatrick N., and Farlie P. G. (2013) Developmental and genetic perspectives on Pierre Robin sequence. Am. J. Med. Genet. C Semin. Med. Genet. 163C, 295–305 [DOI] [PubMed] [Google Scholar]
- 6. Mori-Akiyama Y., Akiyama H., Rowitch D. H., and de Crombrugghe B. (2003) Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. U.S.A. 100, 9360–9365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang X., Goudy S. L., Ketova T., Litingtung Y., and Chiang C. (2008) Gli3-deficient mice exhibit cleft palate associated with abnormal tongue development. Dev. Dyn. 237, 3079–3087 [DOI] [PubMed] [Google Scholar]
- 8. Song Z., Liu C., Iwata J., Gu S., Suzuki A., Sun C., He W., Shu R., Li L., Chai Y., and Chen Y. (2013) Mice with Tak1 deficiency in neural crest lineage exhibit cleft palate associated with abnormal tongue development. J. Biol. Chem. 288, 10440–10450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gendron-Maguire M., Mallo M., Zhang M., and Gridley T. (1993) Hoxa-2 mutant mice exhibit homeotic transformation of skeletal elements derived from cranial neural crest. Cell 75, 1317–1331 [DOI] [PubMed] [Google Scholar]
- 10. Dudas M., Sridurongrit S., Nagy A., Okazaki K., and Kaartinen V. (2004) Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech. Dev. 121, 173–182 [DOI] [PubMed] [Google Scholar]
- 11. Murray S. A., Oram K. F., and Gridley T. (2007) Multiple functions of Snail family genes during palate development in mice. Development 134, 1789–1797 [DOI] [PubMed] [Google Scholar]
- 12. Parada C., Han D., Grimaldi A., Sarrión P., Park S. S., Pelikan R., Sanchez-Lara P. A., and Chai Y. (2015) Disruption of the ERK/MAPK pathway in neural crest cells as a potential cause of Pierre Robin sequence. Development 142, 3734–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parada C., and Chai Y. (2015) Mandible and tongue development. Curr. Top. Dev. Biol. 115, 31–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bjork B. C., Turbe-Doan A., Prysak M., Herron B. J., and Beier D. R. (2010) Prdm16 is required for normal palatogenesis in mice. Hum. Mol. Genet. 19, 774–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bowles J., Schepers G., and Koopman P. (2000) Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 227, 239–255 [DOI] [PubMed] [Google Scholar]
- 16. Chew L. J., and Gallo V. (2009) The Yin and Yang of Sox proteins: activation and repression in development and disease. J. Neurosci. Res. 87, 3277–3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schepers G. E., Teasdale R. D., and Koopman P. (2002) Twenty pairs of sox: extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev. Cell 3, 167–170 [DOI] [PubMed] [Google Scholar]
- 18. Penzo-Méndez A. I. (2010) Critical roles for SoxC transcription factors in development and cancer. Int. J. Biochem. Cell Biol. 42, 425–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dy P., Penzo-Méndez A., Wang H., Pedraza C. E., Macklin W. B., and Lefebvre V. (2008) The three SoxC proteins–Sox4, Sox11 and Sox12–exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res. 36, 3101–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hargrave M., Wright E., Kun J., Emery J., Cooper L., and Koopman P. (1997) Expression of the Sox11 gene in mouse embryos suggests roles in neuronal maturation and epithelio-mesenchymal induction. Dev. Dyn. 210, 79–86 [DOI] [PubMed] [Google Scholar]
- 21. Sock E., Rettig S. D., Enderich J., Bösl M. R., Tamm E. R., and Wegner M. (2004) Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol. Cell Biol. 24, 6635–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoser M., Potzner M. R., Koch J. M., Bösl M. R., Wegner M., and Sock E. (2008) Sox12 deletion in the mouse reveals nonreciprocal redundancy with the related Sox4 and Sox11 transcription factors. Mol. Cell Biol. 28, 4675–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marazita M. L., Murray J. C., Lidral A. C., Arcos-Burgos M., Cooper M. E., Goldstein T., Maher B. S., Daack-Hirsch S., Schultz R., Mansilla M. A., Field L. L., Liu Y. E., Prescott N., Malcolm S., Winter R., Ray A., Moreno L., Valencia C., Neiswanger K., Wyszynski D. F., Bailey-Wilson J. E., Albacha-Hejazi H., Beaty T. H., McIntosh I., Hetmanski J. B., Tunçbilek G., Edwards M., Harkin L., Scott R., and Roddick L. G. (2004) Meta-analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q21 and 2q32–35. Am. J. Hum. Genet. 75, 161–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moreno L. M., Arcos-Burgos M., Marazita M. L., Krahn K., Maher B. S., Cooper M. E., Valencia-Ramirez C. R., and Lidral A. C. (2004) Genetic analysis of candidate loci in non-syndromic cleft lip families from Antioquia-Colombia and Ohio. Am. J. Med. Genet. A 125A, 135–144 [DOI] [PubMed] [Google Scholar]
- 25. Prescott N. J., Lees M. M., Winter R. M., and Malcolm S. (2000) Identification of susceptibility loci for nonsyndromic cleft lip with or without cleft palate in a two stage genome scan of affected sib-pairs. Hum. Genet. 106, 345–350 [DOI] [PubMed] [Google Scholar]
- 26. Scapoli L., Pezzetti F., Carinci F., Martinelli M., Carinci P., and Tognon M. (1997) Evidence of linkage to 6p23 and genetic heterogeneity in nonsyndromic cleft lip with or without cleft palate. Genomics 43, 216–220 [DOI] [PubMed] [Google Scholar]
- 27. Schilham M. W., Oosterwegel M. A., Moerer P., Ya J., de Boer P. A., van de Wetering M., Verbeek S., Lamers W. H., Kruisbeek A. M., Cumano A., and Clevers H. (1996) Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature 380, 711–714 [DOI] [PubMed] [Google Scholar]
- 28. Jiang Y., Ding Q., Xie X., Libby R. T., Lefebvre V., and Gan L. (2013) Transcription factors SOX4 and SOX11 function redundantly to regulate the development of mouse retinal ganglion cells. J. Biol. Chem. 288, 18429–18438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Z., Song Y., Zhao X., Zhang X., Fermin C., and Chen Y. (2002) Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 129, 4135–4146 [DOI] [PubMed] [Google Scholar]
- 30. Jin J. Z., Li Q., Higashi Y., Darling D. S., and Ding J. (2008) Analysis of Zfhx1a mutant mice reveals palatal shelf contact-independent medial edge epithelial differentiation during palate fusion. Cell Tissue Res. 333, 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blavier L., Lazaryev A., Groffen J., Heisterkamp N., DeClerck Y. A., and Kaartinen V. (2001) TGF-β3-induced palatogenesis requires matrix metalloproteinases. Mol. Biol. Cell 12, 1457–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Casey L. M., Lan Y., Cho E. S., Maltby K. M., Gridley T., and Jiang R. (2006) Jag2-Notch1 signaling regulates oral epithelial differentiation and palate development. Dev. Dyn. 235, 1830–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alappat S. R., Zhang Z., Suzuki K., Zhang X., Liu H., Jiang R., Yamada G., and Chen Y. (2005) The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev. Biol. 277, 102–113 [DOI] [PubMed] [Google Scholar]
- 34. Colvin J. S., White A. C., Pratt S. J., and Ornitz D. M. (2001) Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development 128, 2095–2106 [DOI] [PubMed] [Google Scholar]
- 35. Iwata J., Tung L., Urata M., Hacia J. G., Pelikan R., Suzuki A., Ramenzoni L., Chaudhry O., Parada C., Sanchez-Lara P. A., and Chai Y. (2012) Fibroblast growth factor 9 (FGF9)-pituitary homeobox 2 (PITX2) pathway mediates transforming growth factor β (TGFβ) signaling to regulate cell proliferation in palatal mesenchyme during mouse palatogenesis. J. Biol. Chem. 287, 2353–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmahl J., Kim Y., Colvin J. S., Ornitz D. M., and Capel B. (2004) Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 131, 3627–3636 [DOI] [PubMed] [Google Scholar]
- 37. Cinaroglu A., Ozmen Y., Ozdemir A., Ozcan F., Ergorul C., Cayirlioglu P., Hicks D., and Bugra K. (2005) Expression and possible function of fibroblast growth factor 9 (FGF9) and its cognate receptors FGFR2 and FGFR3 in postnatal and adult retina. J. Neurosci. Res. 79, 329–339 [DOI] [PubMed] [Google Scholar]
- 38. Wegner M. (1999) From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res. 27, 1409–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harley V. R., Lovell-Badge R., and Goodfellow P. N. (1994) Definition of a consensus DNA binding site for SRY. Nucleic Acids Res. 22, 1500–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu K., Karuppaiah K., and Ornitz D. M. (2015) Mesenchymal fibroblast growth factor receptor signaling regulates palatal shelf elevation during secondary palate formation. Dev. Dyn. 244, 1427–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ferguson M. W. (1977) The mechanism of palatal shelf elevation and the pathogenesis of cleft palate. Virchows Arch. A Pathol. Anat. Histol. 375, 97–113 [DOI] [PubMed] [Google Scholar]
- 42. Ramaesh T., and Bard J. B. (2003) The growth and morphogenesis of the early mouse mandible: a quantitative analysis. J. Anat. 203, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Han J., Mayo J., Xu X., Li J., Bringas P. Jr., Maas R. L., Rubenstein J. L., and Chai Y. (2009) Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development 136, 4225–4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paul M. H., Harvey R. P., Wegner M., and Sock E. (2014) Cardiac outflow tract development relies on the complex function of Sox4 and Sox11 in multiple cell types. Cell. Mol. Life Sci. 71, 2931–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gadi J., Jung S. H., Lee M. J., Jami A., Ruthala K., Kim K. M., Cho N. H., Jung H. S., Kim C. H., and Lim S. K. (2013) The transcription factor protein Sox11 enhances early osteoblast differentiation by facilitating proliferation and the survival of mesenchymal and osteoblast progenitors. J. Biol. Chem. 288, 25400–25413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cizelsky W., Hempel A., Metzig M., Tao S., Hollemann T., Kühl M., and Kühl S. J. (2013) sox4 and sox11 function during Xenopus laevis eye development. PloS ONE 8, e69372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pan X., Zhao J., Zhang W. N., Li H. Y., Mu R., Zhou T., Zhang H. Y., Gong W. L., Yu M., Man J. H., Zhang P. J., Li A. L., and Zhang X. M. (2009) Induction of SOX4 by DNA damage is critical for p53 stabilization and function. Proc. Natl. Acad. Sci. U.S.A. 106, 3788–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bhattaram P., Penzo-Méndez A., Sock E., Colmenares C., Kaneko K. J., Vassilev A., Depamphilis M. L., Wegner M., and Lefebvre V. (2010) Organogenesis relies on SoxC transcription factors for the survival of neural and mesenchymal progenitors. Nat. Commun. 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schultz R. E., Cooper M. E., Daack-Hirsch S., Shi M., Nepomucena B., Graf K. A., O'Brien E. K., O'Brien S. E., Marazita M. L., and Murray J. C. (2004) Targeted scan of fifteen regions for nonsyndromic cleft lip and palate in Filipino families. Am. J. Med. Genet. A 125A, 17–22 [DOI] [PubMed] [Google Scholar]
- 50. Donner A. L., and Williams T. (2006) Frontal nasal prominence expression driven by Tcfap2a relies on a conserved binding site for STAT proteins. Dev. Dyn. 235, 1358–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nottoli T., Hagopian-Donaldson S., Zhang J., Perkins A., and Williams T. (1998) AP-2-null cells disrupt morphogenesis of the eye, face, and limbs in chimeric mice. Proc. Natl. Acad. Sci. U.S.A. 95, 13714–13719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Azzolin L., Panciera T., Soligo S., Enzo E., Bicciato S., Dupont S., Bresolin S., Frasson C., Basso G., Guzzardo V., Fassina A., Cordenonsi M., and Piccolo S. (2014) YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170 [DOI] [PubMed] [Google Scholar]
- 53. Wang C., Chang J. Y., Yang C., Huang Y., Liu J., You P., McKeehan W. L., Wang F., and Li X. (2013) Type 1 fibroblast growth factor receptor in cranial neural crest cell-derived mesenchyme is required for palatogenesis. J. Biol. Chem. 288, 22174–22183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu W., Lan Y., Pauws E., Meester-Smoor M. A., Stanier P., Zwarthoff E. C., and Jiang R. (2008) The Mn1 transcription factor acts upstream of Tbx22 and preferentially regulates posterior palate growth in mice. Development 135, 3959–3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.