

Abstract
Salmonella enterica causes systemic diseases (typhoid and paratyphoid fever), nontyphoidal septicemia (NTS), and gastroenteritis in humans and other animals worldwide. An important but underrecognized emerging infectious disease problem in sub-Saharan Africa is NTS in children and immunocompromised adults. A current goal is to identify Salmonella mutants that are not pathogenic in the absence of key components of the immune system such as might be found in immunocompromised hosts. Such attenuated strains have the potential to be used as live vaccines. We have used transposon-directed insertion site sequencing (TraDIS) to screen mutants of Salmonella enterica serovar Typhimurium for their ability to infect and grow in the tissues of wild-type and immunodeficient mice. This was to identify bacterial genes that might be deleted for the development of live attenuated vaccines that would be safer to use in situations and/or geographical areas where immunodeficiencies are prevalent. The relative fitness of each of 9,356 transposon mutants, representing mutations in 3,139 different genes, was determined in gp91−/− phox mice. Mutations in certain genes led to reduced fitness in both wild-type and mutant mice. To validate these results, these genes were mutated by allelic replacement, and resultant mutants were retested for fitness in the mice. A defined deletion mutant of cysE was attenuated in C57BL/6 wild-type mice and immunodeficient gp91−/− phox mice and was effective as a live vaccine in wild-type mice.
INTRODUCTION
Salmonella enterica causes systemic diseases (typhoid and paratyphoid fever), nontyphoidal septicemia, and gastroenteritis in humans and other animals worldwide. Salmonella enterica serovar Typhi and S. enterica serovar Paratyphi are restricted to humans and cause typhoid and paratyphoid fever, respectively. These are important febrile illnesses often seen in crowded and impoverished populations with inadequate sanitation that are exposed to unsafe water and food and in travelers visiting countries where these diseases are endemic (1). In 2010, typhoid and paratyphoid fevers together were estimated to account for 190,200 deaths (2). Mortality rates in untreated typhoid fever can be 10 to 15%. In some cases patients recover but remain carriers of the bacteria for many years. Invasive nontyphoidal salmonellae (iNTS) are estimated to cause over 2.1 million illnesses and 416,000 deaths in Africa annually despite antimicrobial treatment. Vaccine development for iNTS disease is a high priority (3). iNTS disease is linked with conditions which impair the immune system, such as a lack of antibodies in young children and multiple comorbidities, such as advanced HIV infection, malaria, hemolysis, sickle cell disease (SCD), malnutrition, and cytokine pathway deficiencies (4, 5).
Current measures for controlling Salmonella infections are far from ideal. The emergence of new multidrug-resistant strains has reduced the usefulness of most antimicrobials (6). Prevention by implementation of hygiene measures alone is proving insufficient. Currently licensed vaccines are far from optimal, and no effective vaccine or delivery system effective against NTS is available.
Live attenuated S. enterica vaccines are more effective in many different infection systems than nonliving ones due to their ability to generate protective T-helper 1 type immunity in addition to antibody responses (7). New live attenuated vaccines against Salmonella currently are being developed and tested (8). However, concerns remain about the use of live vaccines in general when these are targeted to areas where the diseases are endemic with a high incidence of conditions that impair the immune system, and there is reluctance to use live vaccines in animals bred for food production due to fears of side effects subsequent to the unlikely, but possible, spread to humans. Reversion to virulence would pose a serious threat to individuals with an impaired immune system who may inadvertently receive the live vaccine. Immunosuppression can be transient, latent, or undiagnosed, possibilities that are likely to be more frequent in children or in adults from countries where sophisticated diagnostic facilities are not always available.
We and others have found that some live attenuated Salmonella vaccines, such as aromatic-dependent (aro) mutants, htrA mutants, and aroA htrA double mutants, can cause lethal infections in immunodeficient mice such as those lacking in the production of reactive oxygen species (ROS), interleukin-12 (IL-12), or gamma interferon (IFN-γ) and in mice lacking T cells (9, 10). Therefore, bacterial mutants of these genes could be dangerous if used in patients with similar immune defects. We also have shown that other vaccines, specifically Salmonella pathogenicity island-2 (SPI-2) mutants of S. enterica, similar to the ones currently being tested in humans, regain virulence in ROS-deficient animals (11) and could prove dangerous in individuals with reduced efficiency of the oxygen-dependent antimicrobial mechanisms of phagocytes (e.g., chronic granulomatous disease [CGD] or malaria patients) (12, 13). These immunodeficiencies may very well be latent in the individual at the time of vaccination.
A current goal is to identify mutations that would allow the generation of live attenuated salmonellae that would not significantly regain virulence in the absence of key components of the immune system. The feasibility of this approach is supported by the evidence that some Salmonella mutants can retain this attenuation in situations of serious immunodeficiency in animal models of systemic infection. For example, SPI-2 mutants are attenuated in gene-targeted mice lacking IFN-γ, despite these strains being virulent in ROS-deficient animals (14).
Recent advances in high-throughput techniques to assess simultaneously the genotypes and relative fitness of individuals in complex pools of bacterial transposon (Tn) mutants allow us to utilize a genome-wide screen for genes necessary for bacterial fitness in the face of particular immunological challenges. Transposon-directed insertion site sequencing (TraDIS) is one such technique. TraDIS exploits very-high-throughput Illumina sequencing to obtain sequence reads directly from the region of DNA flanking each Tn insert in pools of mutants (15). This allows the location of the transposons to be precisely determined in a massively parallel manner. The number of reads obtained for each Tn allows the relative abundance of each mutant to be assessed, and a quantitative measure of fitness can be obtained by comparing data obtained before and after a selection step.
In the current study, we have used a stringent mouse model of immunodeficiency to perform a genome-wide screen with the potential to identify genes that can disable reversion to virulence in immunodeficient hosts and that therefore could be included in new generations of live vaccines against invasive salmonelloses.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
All wild-type (WT) strains, defined mutants, and plasmids are summarized in Table 1. The preparation of electrocompetent Escherichia coli and S. Typhimurium cells and transformations was performed as previously described (21). Bacteria were grown on Luria-Bertani (LB) medium. Media were supplemented with the appropriate antibiotic for selection (ampicillin, 100 μg/ml; kanamycin, 50 μg/ml; chloramphenicol, 10 μg/ml; tetracycline, 12.5 μg/ml).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or descriptiona | Source and/or reference |
|---|---|---|
| S. Typhimurium | ||
| SL1344 | Virulent in mice with an i.v. LD50 of <20 CFU for innately susceptible mice | 16 |
| SL1344 Mu Tn library | Random Tn mutants; Knr | 17 |
| SL1344 Tn5 Tn library | Random Tn mutants; Knr | 17 |
| SL1344 aroC | ΔaroC, Cmr | This study |
| SL1344 aroC aroD htrA | ΔaroC, Cmr; ΔaroD, Knr; ΔhtrA, Tcr | This study |
| SL1344 aroC ssaV | ΔaroC, Cmr; ΔssaV, Knr | This study |
| SL1344 cydC | ΔcydC, Cmr | This study |
| SL1344 cydD | ΔcydD, Cmr | This study |
| SL1344 cydC cydD | ΔcydC ΔcydD, Cmr | This study |
| SL1344 cysE | ΔcysE, Cmr | This study |
| SL1344 dksA | ΔdksA, Cmr | This study |
| SL1344 ftsK | ΔftsK, Cmr | This study |
| SL1344 miaA | ΔmiaA, Cmr | This study |
| SL1344 nuoK | ΔnuoK, Cmr | This study |
| SL1344 nuo operon | Δnuo operon, Cmr | This study |
| SL1344 ptsI | ΔptsI, Cmr | This study |
| SL1344 recD | ΔrecD, Cmr | This study |
| SL1344 secG | ΔsecG, Cmr | This study |
| SL1344 seqA | ΔseqA, Cmr | This study |
| SL1344 sucA | ΔsucA, Cmr | This study |
| SL1344 suc operon | Δsuc operon, Cmr | This study |
| SL1334 thdF | ΔthdF, Cmr | This study |
| SL1344 tol operon | Δtol operon, Cmr | This study |
| SL1344 tol pal operon | Δtol pal operon, Cmr | This study |
| SL1344 yqiC | ΔyqiC, Cmr | This study |
| E. coli | ||
| DH5α | Subcloning efficiency DH5α competent cells; F− Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17(rK− mK+) phoA supE44 thi-1 gyrA96 relA1 λ− | Thermo Fisher Scientific |
| Plasmids | ||
| pACYC177 | Apr, Knr | New England Biolabs, 18 |
| pACYC184 | Cmr, Tcr | New England Biolabs, 18 |
| pBR322 | Apr, Tcr | New England Biolabs, 19 |
| pBADλRED | Apr | 20 |
LD50, 50% lethal infectious dose.
Recombinant DNA techniques.
Standard methods were used for molecular cloning (22). Chromosomal and plasmid DNA purification and routine DNA modifications, including restriction endonuclease digestion of DNA, modifications of DNA, and ligations, were carried out using commercial kits and supplies according to the manufacturers' instructions (Qiagen, Thermo Fisher Scientific, and New England BioLabs). DNA concentration and purity were measured using a NanoDrop ND-1000 spectrophotometer. PCR primers (see Table S1 in the supplemental material) were designed using Primer3 (http://frodo.wi.mit.edu/) and purchased from Sigma (Sigma-Genosys, United Kingdom). PCRs were performed in 25-μl reaction volumes in 0.2 ml Eppendorf tubes in a PerkinElmer Gene Amp 2400 thermal cycler. Reaction mixtures contained 200 μM deoxynucleoside triphosphates (dNTPs), 2 mM Mg2+, 0.01 volume of Proof Start DNA polymerase (2.5 U μl−1; Qiagen), 0.1 volume of polymerase buffer (10×), 1 μM forward and reverse primers, and template DNA (∼50 ng plasmid DNA or ∼100 ng chromosomal DNA). Thermal cycler conditions were 94°C for 10 min and then 35 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min, followed by a final extension at 72°C for 10 min.
Generation of defined mutants of S. Typhimurium.
Mutants were generated using a modification of the ET cloning procedure (23, 24) as previously described (20). PCR was used to amplify the chloramphenicol resistance cassette from pACYC184 (18), the kanamycin resistance cassette from pACYC177 (18), or the tetracycline resistance cassette from pBR322 (19) with 5′ and 3′ 60-bp homology arms complementary to the flanking regions of the gene to be deleted. Approximately 1 μg of linear PCR product was used for integration into the chromosome using a modification of the Lambda Red method (25), as previously detailed (26). Transformants were selected by plating onto medium containing chloramphenicol, kanamycin, or tetracycline as appropriate. Screening for the loss of the pBADλred helper plasmid was as previously described (27), using MAST ID Intralactam circles (MAST Diagnostics, Bootle Merseyside, United Kingdom) to screen for the absence of beta-lactamase in bacterial colonies. (Further details of the construction of mutants are provided in the supplemental material).
Mouse infections.
All animals were handled in strict accordance with good animal practice as defined by the relevant international (Directive of the European Parliament and of the Council on the Protection of Animals Used for Scientific Purposes, Brussels 543/5) and local (Department of Veterinary Medicine, University of Cambridge) animal welfare guidelines. All animal work was approved by the ethical review committee of the University of Cambridge and was licensed by the United Kingdom Government Home Office under the Animals (Scientific Procedures) Act 1986. Sex- and aged-matched 9- to 12-week-old C57BL/6 wild-type mice (Harlan Olac Ltd.) and gp91−/− phox mice (bred at the Wellcome Trust Sanger Institute) were infected by intravenous (i.v.) injection of bacterial suspensions in a volume of 0.2 ml. Statically grown cultures of wild-type S. Typhimurium SL1344 and defined mutants were grown overnight at 37°C from single colonies in 10 ml LB broth and then diluted in phosphate-buffered saline (PBS) to the appropriate concentration for inoculation. Bacterial cultures for the pools of Tn5 and Mu Tn mutants (TraDIS pools) were grown as described previously (14). Inocula were enumerated by plating dilutions onto LB agar plates. Mice were killed by cervical dislocation, and the livers and spleens were aseptically removed and homogenized in sterile water using a Colworth Stomacher 80. The resulting homogenate was diluted in a 10-fold series in PBS, and LB agar pour plates were used to enumerate viable bacteria.
TraDIS analysis of S. Typhimurium mutant pools.
The Illumina sequence reads from the input (bacteria inoculated into mice) and output (bacteria recovered from infected livers and spleens) pools (ENA no. ERA201074) were generated using single-end short-read sequencing with Tn-specific primers and by massively parallel sequencing of Tn-flanking regions in the input and output mutant pools. Genomic DNA was prepared and 2 μg fragmented by Covaris to an average size of ∼300 bp. Following end repair and A tailing, a modified Illumina adapter, synthesized and annealed by Integrated DNA Technologies (IDT) using oligonucleotides SplA5_top and SplA5_bottom (see Table S1 in the supplemental material), was ligated to the fragments for 40 min at 20°C. Ligated fragments were cleaned using Ampure XP beads (Beckman Coulter) with a bead-to-sample ratio of 0.8 to 1. PCR enrichment of fragments containing the Tn was performed using a primer homologous to each end of the Tn (Mu_AG_5′PCR, Mu_AG_3′PCR, Tn5_AG_5′PCR, or Tn5_AG_3′PCR; see Table S1) in conjunction with an adapter-specific primer containing an index tag (SplAP5.x; see Table S1). PCR products were cleaned using Ampure XP beads (Beckman Coulter), quantified by quantitative PCR (qPCR), and then pooled. The resultant products were sequenced on a HiSeq2500 (Illumina) using a specially modified recipe to overcome difficulties generated by the monotemplate Tn sequence. Briefly, a Tn-specific sequencing primer (Mu_AG_5′seq, Mu_AG_3′seq, Tn5_AG_5′seq, or Tn5_AG_3′seq; see Table S1) anneals to the Tn 10 bases away from the genomic DNA junction. Sequencing takes place with no imaging for the first 10 cycles followed by imaging for the next 40 cycles. This gives a 40-bp genomic DNA read. The template is denatured, the same sequencing primer is reannealed, and 10 cycles of sequencing takes place to give a 10-bp Tn read.
Sequence analysis.
Illumina reads generated from the genomic DNA of the input and output pools containing Tn5 and Mu Tn insertions were stripped of the 5′ Tn tags using Cutadapt version 1.8.1 (28), and the remainder of each sequence read was mapped to the S. Typhimurium SL1344 chromosome and plasmid sequences (GenBank accession numbers FQ312003, HE654724, HE654725, and HE654726) using BWA mem version 0.7.12-r1039 (29). The raw input and output read counts were loaded into R, version 3.0.2. A step was performed to eliminate nonspecific background reads by filtering out the insertions with a total read count across all samples of <100 reads. DESeq2 version 1.4.5 (30) was used to compare the reads mapped to specific insertion positions in the input pools and the corresponding insertion positions in the output pools. Following normalization to account for differential sequence coverage between samples, dispersions in the replicate samples were estimated, and a negative binomial distribution model implemented in DESeq2 was used to test the hypothesis that fitness score was equal to zero under the assumption that a particular mutant was present at equivalent levels of sequence coverage in the input and output pools. We obtained P values for all mutants, and the P values then were adjusted for multiple testing using the Benjamini and Hochberg false discovery rate (31).
A fitness score for each Tn mutant was calculated as a log2 transformation of the ratio of normalized output-input read counts. Hence, the calculated fitness scores (log2-fold change) represent the difference in abundance of each mutant in the output pool relative to the input. We defined significantly attenuated mutants as those with a negative log2-fold change and an adjusted P value (Benjamini-Hochberg) of ≤0.05.
For comparative purposes, the sequence data from our previous study investigating the survival of the same Tn mutants in BALB/c mice (32) were analyzed using the same methods as those described above, and the corresponding mutants in the two data sets (i.e., those with reads mapping to the same genomic locus in the same orientation) were identified.
RESULTS AND DISCUSSION
S. Typhimurium TraDIS infections in C57BL/6 wild-type and gp91−/− phox mice.
Gp91 is one of the subunits of the NADPH oxidase essential for ROS production by phagocytes. ROS deficiency becomes apparent from the very early stages of an S. enterica infection, leading to a reduction in initial killing of the bacteria and accelerated growth in the first few days of infection. Therefore, a short course of infection (48 h) was required to investigate the behavior of S. Typhimurium mutants in the gp91−/− phox mice.
We screened 21 pools of 480 Tn mutants of S. Typhimurium in each pool, covering ∼10,000 Tn5 and Mu Tn mutants, at a dose of ∼5 × 104 log10 CFU (the mean input dose in each of the 21 pools was 4.65 ± 0.04 log10 CFU; errors are given as standard deviations) in duplicate 8-week-old C57BL/6 wild-type (WT) or gp91−/− phox mice. The dose and pool size were dictated by the need to allow the progression of the infection until the 48-h time point without it reaching lethal bacterial numbers. We administered pools of S. Typhimurium Tn mutants via the parenteral i.v. route in order to relate our outputs to those of genes and mechanisms of immunity involved in the control of the systemic phase of an invasive infection. The control of growth at this systemic level (i.e., restraint of bacterial growth and dissemination) is an absolute requirement for survival in S. enterica typhoidal and septicemic infections in several target hosts (32).
Mice were killed at 48 h postinfection (p.i.) when the mean bacterial load per liver was, on average, 2.90 log10 CFU greater in the gp91−/− phox mice than in C57BL/6 WT mice (gp91−/− phox mice, 8.51 ± 0.33 log10 CFU; C57BL/6 WT mice, 5.62 ± 0.20 log10 CFU), and the mean bacterial load per spleen was, on average, 2.71 log10 CFU greater in gp91−/− phox mice than in C57BL/6 WT mice (gp91−/− phox mice, 8.42 ± 0.27 log10 CFU; C57BL/6 WT mice, 5.72 ± 0.20 log10 CFU).
The input and output pool genomic DNA enriched for transposon junctions was sequenced using the Illumina platform as described in Materials and Methods. A total of 9,356 mutants (4,975 Mu and 4,381 Tn5) were unambiguously mapped at the level of the single nucleotide to the SL1344 genome (i.e., >94% of the Tn mutants screened were mapped). The reason for the lower number of mapped mutants than the number of mutants in the original input pools could be due to mutants not surviving the initial growth in the input pool and/or the presence of duplicate mutants in the library. A total of 3,139 different genes were disrupted by the 9,356 Tn insertions.
The sites of insertion of all transposons in the S. Typhimurium TraDIS mutant libraries, the identities of the genes that are disrupted, and the fitness scores of the mutants are listed in Table S2 in the supplemental material. The fitness scores of the mutants obtained during infection of C57BL/6 mice showed a high proportion of seemingly attenuated mutants (see Fig. S1a in the supplemental material). Additionally, the output read counts from the two replicate C57BL/6 mice did not show a good correspondence (see Fig. S1b), and the fitness scores did not correlate well with those obtained from BALB/c mice (see Fig. S1c) in our previous study (32) (Fig. 1). From these observations, we concluded that the C57BL/6 mice had exhibited stochastic loss of mutants for reasons unrelated to their genotype (i.e., random dropout). In contrast, only a small proportion of mutants were attenuated in the gp91−/− phox mice, with good correspondence between the replicates and with the data obtained from BALB/c mice (see Fig. S2). The random dropout was not identified in the BALB/c mice, probably because of the increased bacterial load used as the inoculum in this model (∼1 × 106 CFU in the BALB/c mice versus ∼5 × 104 CFU in the C57BL/6 mice), or in the gp91−/− phox mice due to the absence of bacterial killing in this model. Thus, the attenuated mutants in the C57BL/6 data set are likely to include false negatives due to the random dropout of mutants in this model (i.e., mutants that have been assigned as attenuated but that could be colonization proficient). This prevented an in-depth comparison of the different genes that contributed to fitness in the wild-type C57BL/6 and immunocompromised mice. However, the data from the screen in the gp91−/− phox mice were robust and provided us with solid foundations to select representative genes from the attenuated mutant lists and test defined allelic replacement mutants of these genes in wild-type and immunocompromised mice.
FIG 1.

Relative fitness scores (log2-fold change) plotted against normalized mean read counts. Fitness scores represent the log2-fold change in the sequence read counts of output pools compared with those of the input pools. Output pools were obtained from the livers of gp91−/− phox, C57BL/6, and BALB/c mice (32). Significantly attenuated mutants, represented as red closed circles, are a population of mutants showing negative fitness scores and an adjusted P value (Benjamini-Hochberg) of ≤0.05. Mutants with fitness scores which were not significantly different from zero are shown as black circles. The wide range of fitness scores exhibited in the C57B/L6 mice is indicative of large-scale stochastic loss (i.e., random dropout) of mutants during those infections.
Analysis of defined allelic replacement S. Typhimurium mutants in C57BL/6 WT and gp91−/− phox mice.
An ideal live attenuated vaccine would need to colonize but be attenuated in an immunocompetent host and to remain attenuated under conditions where the immune system may be impaired. Candidate genes for the generation of such vaccines will be among those that lead to attenuated mutant phenotypes in spleens and livers of both C57BL/6 WT and gp91−/− phox mice. We selected 14 genes that fit this pattern, cydC, cydD, cysE, dksA, ftsK, miaA, nuoK, ptsI, recD, secG, seqA, sucA, thdF, and yqiC, and deleted them by allelic replacement in S. Typhimurium. We also generated a double mutant knocking out both cydC and cydD, a mutant containing a deletion of the entire suc operon, a mutant containing a deletion of the entire nuo operon, a mutant containing a deletion of the tol operon (since many different tol mutants were attenuated and tol genes previously have been shown to have a role in virulence/fitness in mice [33]), and a mutant containing a deletion of the tol pal operon. All of these were tested individually for attenuation in C57BL/6 WT mice compared to levels in the SL1344 wild type, administered by i.v. injection, and at the input doses detailed in Table 2. We also included in the experiment controls mutants lacking aroC, aroC aroD htrA, and aroC ssaV, which have established roles in virulence and have been or are being used as vaccine candidates (34–36). Each gene, or genes, was inactivated separately by λRed recombinase-mediated integration of linear PCR amplicons by homologous recombination. Mice were killed at 7 days p.i. or earlier if clinical signs became apparent (Table 2). Defined mutants of cydC, cydD, cydC cydD, recD, seqA, and the suc operons were not attenuated in the C57BL/6 mice, and mice had to be killed at 3 days p.i., along with the mice infected with wild-type SL1344. Mice infected with defined ftsK, nuoK, and nuo operon mutants were killed on day 4 p.i., as the mice displayed clinical signs. Mice infected with a dksA mutant were killed on day 5 p.i. Mice infected with sucA and thdF mutants displayed clinical signs at 6 days p.i. and were killed, although interestingly viable bacterial counts in the livers and spleens were not high enough to be consistent with the observed signs of infection, indicating that the clinical signs are due to focal infection at other body sites. Mice infected with cysE, miaA, ptsI, secG, tol operon, tol pal operon, and yqiC mutants, as well as the controls infected with aroC, aroC aroD htrA, and aroC ssaV mutants, did not display any visible clinical signs on day 7 p.i.
TABLE 2.
CFU in the organs of C57BL/6 WT mice infected with defined mutants of S. Typhimurium, TraDIS fitness scores, and the number of mutants in each gene identified by TraDIS
| Mutation | Inoculum (log10 CFU) | Organ | Counts in organs (mean log10 CFU ± SD) | Day p.i. | TraDIS fitness score |
|---|---|---|---|---|---|
| cydC | 5.60 | Liver | 8.19 (0.05) | 3 | −3.3; 1 mutant |
| Spleen | 8.03 (0.19) | −2.8; 1 mutant | |||
| cydD | 5.51 | Liver | 8.40 (0.11) | 3 | −2.6 (upper), −3.8 (lower); 3 mutants |
| Spleen | 8.34 (0.12) | −2.2 (upper), −4.1 (lower); 3 mutants | |||
| cydC cydD | 5.54 | Liver | 7.86 (0.28) | 3 | Not available |
| Spleen | 8.00 (0.36) | Not available | |||
| recD | 5.70 | Liver | 8.47 (0.21) | 3 | −4.9; 1 mutant |
| Spleen | 8.07 (0.33) | −3.2; 1 mutant | |||
| seqA | 5.65 | Liver | 8.37 (0.09) | 3 | −5.5; 1 mutant |
| Spleen | 8.18 (0.07) | −2.6; 1 mutant | |||
| suc operon | 5.49 | Liver | 8.54 (0.01) | 3 | −0.8 (upper), −5.3 (lower); 4 mutants |
| Spleen | 8.30 (0.04) | −0.3 (upper), −5.9 (lower); 4 mutants | |||
| SL1344 WT | 3.28 | Liver | 5.47 (0.23) | 3 | Not available |
| Spleen | 5.72 (0.16) | Not available | |||
| ftsK | 5.64 | Liver | 7.61 (0.21) | 4 | −5.1; 1 mutant |
| Spleen | 7.14 (0.22) | −5.8; 1 mutant | |||
| nuoK | 5.72 | Liver | 8.35 (0.33) | 4 | −3.3 (upper), −4.8 (lower); 2 mutants |
| Spleen | 8.05 (0.06) | −2.8 (upper), −4.4 (lower); 2 mutants | |||
| nuo operon | 5.73 | Liver | 8.35 (0.13) | 4 | −0.3 (upper), −4.4 (lower); 18 mutants |
| Spleen | 8.10 (0.13) | −1.7 (upper), −6.6 (lower); 18 mutants | |||
| dksA | 5.69 | Liver | 8.49 | 5 | −4.3; 1 mutant |
| Spleen | 8.19 | −3.9; 1 mutant | |||
| sucA | 5.71 | Liver | 5.99 (0.61) | 6 | −5.4; 1 mutant |
| Spleen | 6.71 (0.27) | −5.9; 1 mutant | |||
| thdF | 5.48 | Liver | 7.00 (1.98) | 6 | −2.4 (upper), −6.4 (lower); 2 mutants |
| Spleen | 7.07 (1.57) | −2.7 (upper), −3.8 (lower); 2 mutants | |||
| aroC | 5.32 | Liver | 5.30 (0.16) | 7 | −4.0 (upper), −4.2 (lower); 2 mutants |
| Spleen | 5.89 (0.08) | −3.2 (upper), −3.9 (lower); 2 mutants | |||
| aroC aroD htrA | 5.36 | Liver | 2.63 (0.08) | 7 | Not available |
| Spleen | 3.46 (0.11) | Not available | |||
| aroC ssaV | 5.51 | Liver | 5.02 (0.18) | 7 | Not available |
| Spleen | 5.64 (0.05) | Not available | |||
| cysE | 5.51 | Liver | 5.61 (0.11) | 7 | −2.8; 1 mutant |
| Spleen | 5.88 (0.15) | −4.1; 1 mutant | |||
| miaA | 5.53 | Liver | 4.53 (0.19) | 7 | −5.0 (upper), −7.2 (lower); 4 mutants |
| Spleen | 4.68 (0.10) | −4.4 (upper), −5.8 (lower); 4 mutants | |||
| ptsI | 5.57 | Liver | 6.99 (0.53) | 7 | −1.5; 1 mutant |
| Spleen | 6.95 (0.44) | −2.1; 1 mutant | |||
| secG | 5.65 | Liver | 6.67 (1.09) | 7 | −5.2; 1 mutant |
| Spleen | 5.95 (0.48) | −5.7; 1 mutant | |||
| tol operon | 5.74 | Liver | 3.27 (0.20) | 7 | −4.4 (upper), −5.4 (lower); 6 mutants |
| Spleen | 3.93 (0.09) | −4.5 (upper), −9.0 (lower); 6 mutants | |||
| tol pal operon | 5.54 | Liver | 3.42 (0.10) | 7 | Not available |
| Spleen | 4.07 (0.16) | Not available | |||
| yqiC | 5.65 | Liver | 6.53 (0.72) | 7 | −6.0; 1 mutant |
| Spleen | 6.39 (0.21) | −5.9; 1 mutant |
Given their attenuation in WT mice, cysE, miaA, ptsI, secG, and yqiC mutants, along with the tol operon and tol pal operon mutants, were used to infect gp91−/− phox mice. We also included aroC, aroC aroD, and ssaV mutants as controls. Mice were killed at 7 days p.i. or earlier if clinical signs became apparent (Table 3). Mice infected with defined ptsI, ssaV, tol operon, and tol pal operon mutants were killed on day 3 p.i., when they displayed clinical signs. Mice infected with aroC, miaA, and secG mutants were killed on day 4 p.i. Mice infected with a yqiC mutant were killed on day 5 p.i. when they displayed clinical signs. Mice infected with an aroC aroD or cysE mutant were killed on day 7 p.i., which was the predefined endpoint of the experiment. The cysE and the aroC aroD mutants had higher bacterial counts in the organs of the gp91−/− phox mice than in the C57BL/6 WT mice. In order to determine how long the gp91−/− phox mice could survive infection with a cysE mutant, we performed a small pilot study, infecting gp91−/− phox mice with 5.72 log10 CFU of the SL1344 cysE mutant and then allowing the infection to proceed until the mice displayed clinical symptoms. The mice had to be killed on day 8 p.i., when the bacterial loads in the livers were 8.33 ± 0.11 log10 CFU and the bacterial loads in the spleens were 7.81 ± 0.05 log10 CFU. We next proceeded to compare the cysE mutant to an aroC mutant in gp91−/− phox mice (Fig. 2). At three different input doses, the time until the appearance of clinical signs was consistently delayed for the cysE mutant compared to that for the aroC mutant.
TABLE 3.
CFU in the organs of gp91−/− phox mice infected with defined mutants of S. Typhimurium, TraDIS fitness scores, and the number of mutants in each gene identified by TraDIS
| Mutation | Inoculum (log10 CFU) | Organ | Counts in organs (mean log10 CFU ± SD) | Day p.i. | TraDIS fitness score |
|---|---|---|---|---|---|
| ptsI | 5.60 | Liver | 7 | 3 | −4.5; 1 mutant |
| Spleen | 8.40 (0.06) | −4.0; 1 mutant | |||
| ssaV | 5.69 | Liver | 9.01 (0.10) | 3 | −2.9 (upper), −5.3 (lower); 4 mutants |
| Spleen | 8.63 (0.11) | −3.1 (upper), −5.6 (lower); 4 mutants | |||
| tol operon | 5.48 | Liver | 8.30 (0.80) | 3 | −5.8 (upper), −7.6 (lower); 7 mutants |
| Spleen | 8.00 (1.00) | −5.8 (upper), −6.8 (lower); 7 mutants | |||
| tol pal operon | 5.45 | Liver | 8.28 (0.29) | 3 | Not available |
| Spleen | 7.53 (0.44) | Not available | |||
| aroC | 5.56 | Liver | 7.83 (0.32) | 4 | −5.6 (upper), −6.0 (lower); 2 mutants |
| Spleen | 7.59 (0.48) | −5.3 (upper), −6.5 (lower); 2 mutants | |||
| miaA | 5.60 | Liver | 9.86 (0.04) | 4 | −4.4 (upper), −6.1 (lower); 4 mutants |
| Spleen | 9.27 (0.32) | −5.8 (upper), −7.7 (lower); 4 mutants | |||
| secG | 5.34 | Liver | 8.85 (0.38) | 4 | −6.9; 1 mutant |
| Spleen | 8.64 (0.14) | −7.3; 1 mutant | |||
| yqiC | 5.51 | Liver | 9.19 (0.21) | 5 | −0.3; 1 mutant |
| Spleen | 8.05 (0.13) | −7.7; 1 mutant | |||
| aroC aroD | 5.76 | Liver | 8.06 (0.14) | 7 | Not available |
| Spleen | 7.75 (0.08) | Not available | |||
| cysE | 5.63 | Liver | 7.89 (0.07) | 7 | −5.2; 1 mutant |
| Spleen | 7.55 (0.22) | −5.9; 1 mutant |
FIG 2.
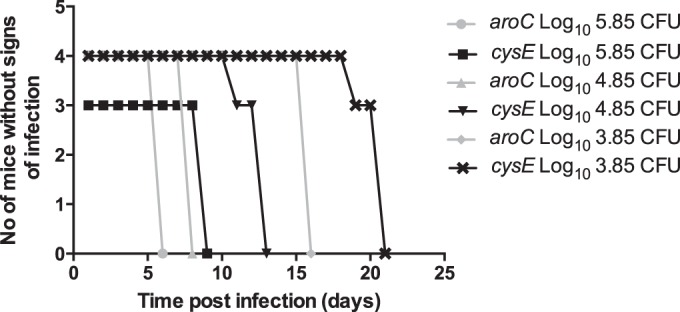
Time to presentation of clinical signs in gp91−/− phox mice infected with various doses of the SL1344 cysE or SL1344 aroC mutant. gp91−/− phox mice were infected i.v. with 5.85 log10 CFU, 4.85 log10 CFU, or 3.85 log10 CFU of either the SL1344 aroC (gray symbols) or SL1344 cysE (black symbols) mutant, and the time to clinical signs was monitored.
As a prerequisite to a vaccination study, we determined the time it took for the SL1344 cysE mutant to be cleared from the liver and spleen of WT C57BL/6 mice (Fig. 3). We found that by day 84 p.i., mice infected i.v. had cleared all of the SL1344 cysE mutant from the liver and spleen, while in the orally infected group it took until day 91 p.i. for the organs to be clear of the SL1344 cysE mutant.
FIG 3.
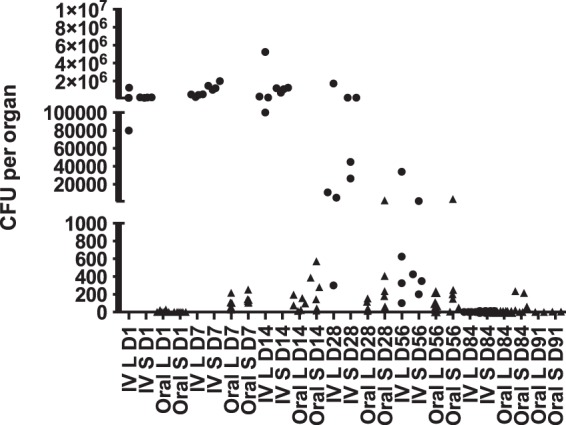
WT C57BL/6 mice were infected i.v. (circles) or orally (triangles) with 5.80 log10 CFU or 8.68 log10 CFU, respectively, of the SL1344 cysE mutant, and bacterial counts in the livers (L) and spleens (S) were monitored at various times postinfection until the bacteria had cleared from the organs.
We next vaccinated WT C57BL/6 mice, either i.v. or orally, with the SL1344 cysE mutant and then challenged the mice, 91 days postimmunization, orally with virulent SL1344 and monitored the bacterial counts in the livers and spleens at various times postinfection (Fig. 4). The majority of the vaccinated mice were protected from challenge with the virulent SL1344. However, during the challenge experiment it was necessary to kill six mice (five from the orally immunized group and one from the i.v. immunized group) because they were displaying signs of infection. Thus, 97% of the SL1344 cysE mutant i.v. vaccinated C57BL/6 mice and 86% of the SL1344 cysE mutant orally vaccinated C57BL/6 mice were protected against challenge with virulent SL1344.
FIG 4.
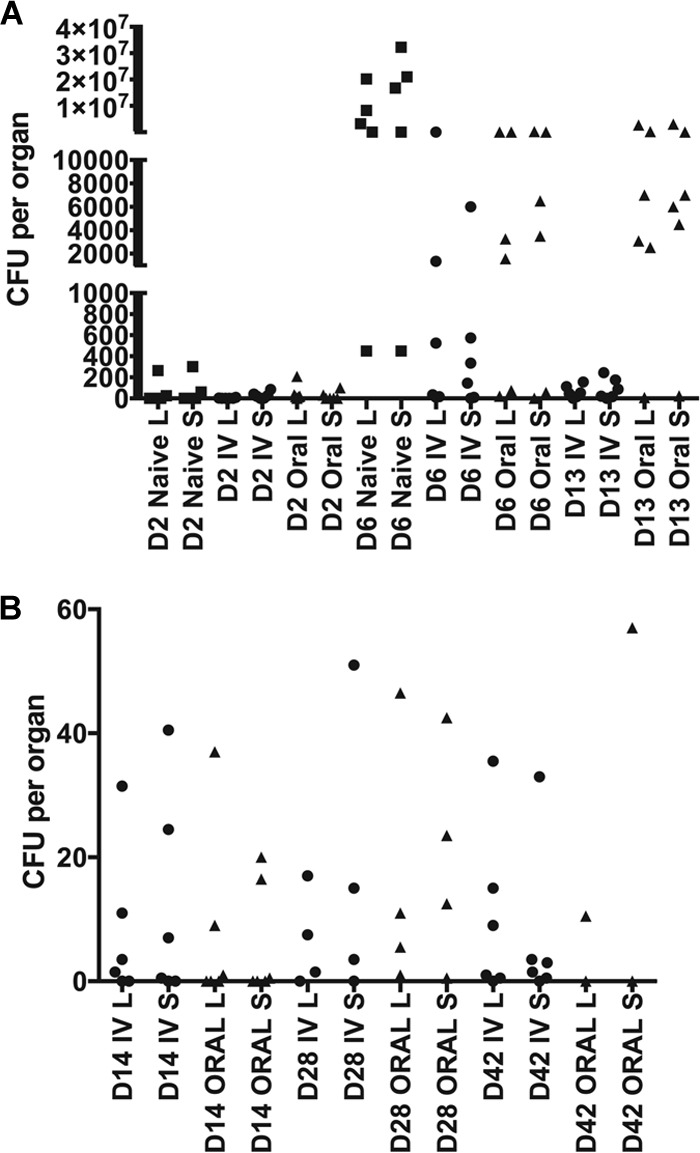
Vaccination study, immunizing C57BL/6 WT mice with the SL1344 cysE mutant, and subsequent challenge with WT SL1344. (A) WT C57BL/6 mice were vaccinated with the SL1344 cysE mutant administered at either 5.7 log10 CFU i.v. or 8.5 log10 CFU orally. At 91 days postimmunization, the vaccinated mice and an unvaccinated group of mice (naive control) were challenged orally with 8.8 log10 CFU of virulent SL1344. Bacterial counts in the livers (L) and spleens (S) were monitored at various times postchallenge (note that one mouse from the orally vaccinated group had to be killed on day 9 postchallenge, since it was displaying signs of infection). (B) WT C57BL/6 mice were vaccinated with the SL1344 cysE mutant administered at either 5.8 log10 CFU i.v. or 8.7 log10 CFU orally. At 91 days postimmunization, the vaccinated mice were challenged orally with 8.8 log10 CFU or virulent SL1344. Bacterial counts in the livers and spleens were monitored at various times postchallenge (note that one mouse from both the orally vaccinated and the i.v. vaccinated group had to be killed on day 9 postchallenge and one mouse each from the orally vaccinated group had to be killed on day 18, day 19, and day 34 postchallenge, since they were displaying signs of infection).
Conclusion.
Here, we have presented a report of the fitness of random Tn mutants of S. Typhimurium in vivo and described attenuated mutants based on the TraDIS analysis of pools of Tn mutants screened in C57BL/6 WT and gp91−/− phox mice. Our study provides proof-of-principle data on the feasibility of using TraDIS analysis in combination with infections of gene-targeted animals to allow the identification of bacterial mutants as live vaccine candidates that would be safe in situations where the immune system is impaired.
We show that through TraDIS analysis it is possible to identify novel mutations in S. Typhimurium genes that still allow colonization in WT mice but importantly also reduce bacterial growth rate in vivo in severely immunodeficient gp91−/− phox mice. It is worth noting that the variance of defined mutants from their TraDIS fitness scores (Tables 2 and 3) likely is due to differences in competition dynamics for a given mutant relative to those of coscreened mutant bacteria, as we previously demonstrated (32), and the differences in time points at which the bacteria were enumerated p.i. between the TraDIS screen and the defined mutant experiments. Thus, some of the attenuated mutants identified in the screen are virulent when tested individually, while some others retain the attenuation; therefore, the screen is useful but requires further evaluation of mutations individually.
Through the TraDIS screens and validation of defined mutants, we identified that the SL1344 cysE mutant was attenuated for growth, was cleared from WT C57BL/6 mice, and had reduced growth in gp91−/− phox mice, although these mice succumbed to the infection 8 days p.i. The SL1344 cysE mutant offered protection when given i.v. or orally as a live vaccine to C57BL/6 WT mice; however, a few of the vaccinated mice succumbed to the challenge with the virulent SL1344, indicating that cysE does not offer full protection to all vaccinated animals.
Our work did not identify a single-gene S. Typhimurium mutant that would colonize WT animals at levels sufficient to be predicted to be immunogenic and that also would be completely unable to cause disease in the severely immunodeficient animals used in this study. The attractive possibility that different combinations of mutations could yield this ideal vaccine candidate remains to be explored. However, even mutations that reduce growth in immunocompromised hosts would be advantageous and could be used to construct live strains in a way that would crucially enable more time for medical intervention should an immunocompromised vaccinee develop signs of infection. Furthermore, the animal model that we used was of extreme immunodeficiency, while it is likely that in the field one would encounter situations where macrophage function is only partially impaired. Therefore, it remains to be established whether some of the mutants identified from this study would be safer in models of partial immunodeficiency.
Extending the approach reported here to other gene-targeted mouse strains has the potential in the future to generate a more comprehensive picture of the functional interactions between the bacterial and host genomes and to identify bacterial mutants as live vaccine candidates that would be safe across a wide range of immunodeficiencies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Medical Research Council (MRC) grant G1100102. Tn libraries were previously constructed with funding from the Biotechnology and Biological Sciences Research Council grant APG19115. O.O. was supported by a Newton Trust grant awarded to A.J.G. M.M. was supported by the Wellcome Trust, grant number WT098051.
We have no conflicting financial interests. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01423-15.
REFERENCES
- 1.Crump JA, Mintz ED. 2010. Global trends in typhoid and paratyphoid Fever. Clin Infect Dis 50:241–246. doi: 10.1086/649541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, et al. 2012. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ao TT, Feasey NA, Gordon MA, Keddy KH, Angulo FJ, Crump JA. 2015. Global burden of invasive nontyphoidal Salmonella disease 2010. Emerg Infect Dis 21:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon MA, Banda HT, Gondwe M, Gordon SB, Boeree MJ, Walsh AL, Corkill JE, Hart CA, Gilks CF, Molneux ME. 2002. Non-typhoidal salmonella bacteraemia among HIV-infected Malawian adults: high mortality and frequent recrudescence. AIDS 16:1633–1641. doi: 10.1097/00002030-200208160-00009. [DOI] [PubMed] [Google Scholar]
- 5.Gordon MA. 2008. Salmonella infections in immunocompromised adults. J Infect 56:413–422. doi: 10.1016/j.jinf.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Gordon MA, Graham SM, Walsh AL, Wilson L, Phiri A, Molyneux E, Zijlstra EE, Heyderman RS, Hart CA, Molyneux ME. 2008. Epidemics of invasive Salmonella enterica serovar enteritidis and S. enterica Serovar typhimurium infection associated with multidrug resistance among adults and children in Malawi. Clin Infect Dis 46:963–969. doi: 10.1086/529146. [DOI] [PubMed] [Google Scholar]
- 7.Harrison JA, Villarreal-Ramos B, Mastroeni P, Demarco de Hormaeche R, Hormaeche CE. 1997. Correlates of protection induced by live Aro-Salmonella typhimurium vaccines in the murine typhoid model. Immunology 90:618–625. doi: 10.1046/j.1365-2567.1997.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacLennan CA, Martin LB, Micoli F. 2014. Vaccines against invasive Salmonella disease: current status and future directions. Hum Vaccin Immunother 10:1478–1493. doi: 10.4161/hv.29054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mastroeni P, Harrison JA, Robinson JH, Clare S, Khan S, Maskell DJ, Dougan G, Hormaeche CE. 1998. Interleukin-12 is required for control of the growth of attenuated aromatic-compound-dependent salmonellae in BALB/c mice: role of gamma interferon and macrophage activation. Infect Immun 66:4767–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinha K, Mastroeniv P, Harrison J, de Hormaeche RD, Hormaeche CE. 1997. Salmonella typhimurium aroA, htrA, and aroD htrA mutants cause progressive infections in athymic (nu/nu) BALB/c mice. Infect Immun 65:1566–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vazquez-Torres A, Xu Y, Jones-Carson J, Holden DW, Lucia SM, Dinauer MC, Mastroeni P, Fang FC. 2000. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science 287:1655–1658. doi: 10.1126/science.287.5458.1655. [DOI] [PubMed] [Google Scholar]
- 12.Gilchrist JJ, MacLennan CA, Hill AV. 2015. Genetic susceptibility to invasive Salmonella disease. Nat Rev Immunol 15:452–463. doi: 10.1038/nri3858. [DOI] [PubMed] [Google Scholar]
- 13.Mastroeni P, Ugrinovic S, Chandra A, MacLennan C, Doffinger R, Kumararatne D. 2003. Resistance and susceptibility to Salmonella infections: lessons from mice and patients with immunodeficiencies. Rev Med Microbiol 14:53–62. doi: 10.1097/00013542-200304000-00002. [DOI] [Google Scholar]
- 14.Khan SA, Stratford R, Wu T, Mckelvie N, Bellaby T, Hindle Z, Sinha KA, Eltze S, Mastroeni P, Pickard D, Dougan G, Chatfield SN, Brennan FR. 2003. Salmonella typhi and S. typhimurium derivatives harbouring deletions in aromatic biosynthesis and Salmonella pathogenicity island-2 (SPI-2) genes as vaccines and vectors. Vaccine 21:538–548. doi: 10.1016/S0264-410X(02)00410-3. [DOI] [PubMed] [Google Scholar]
- 15.Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G, Wain J, Parkhill J, Turner AK. 2009. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res 19:2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoiseth SK, Stocker BA. 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 21:238–239. [DOI] [PubMed] [Google Scholar]
- 17.Chaudhuri RR, Peters SE, Pleasance SJ, Northern H, Willers C, Paterson GK, Cone DB, Allen AG, Owen PJ, Shalom G, Stekel DJ, Charles IG, Maskell DJ. 2009. Comprehensive identification of Salmonella enterica serovar typhimurium genes required for infection of BALB/c mice. PLoS Pathog 5(7):e1000529. doi: 10.1371/journal.ppat.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang CY, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134:1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW. 1977. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene 2:95–113. [PubMed] [Google Scholar]
- 20.Mo E, Peters SE, Willers C, Maskell DJ, Charles IG. 2006. Single, double and triple mutants of Salmonella enterica serovar Typhimurium degP (htrA), degQ (hhoA), and degS (hhoB) have diverse phenotypes on exposure to elevated temperature and their growth in vivo is attenuated to different extents. Microb Pathog 41:174–182. doi: 10.1016/j.micpath.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Dower WJ, Miller JF, Ragsdale CW. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16:6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Russell D. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 23.Muyrers JPP, Zhang Y, Testa G, Stewart AF. 1999. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res 27:1555–1557. doi: 10.1093/nar/27.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grant AJ, Restif O, McKinley TJ, Sheppard M, Maskell DJ, Mastroeni P. 2008. Modelling within-host spatiotemporal dynamics of invasive bacterial disease. PLoS Biol 6:e74. doi: 10.1371/journal.pbio.0060074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hautefort I, Proença MJ, Hinton JCD. 2003. Single-copy green fluorescent protein gene fusions allow accurate measurement of Salmonella gene expression in vitro and during infection of mammalian cells. Appl Environ Microbiol 69:7480–7491. doi: 10.1128/AEM.69.12.7480-7491.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10. [Google Scholar]
- 29.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997. http://arxiv.org/abs/1303.3997.
- 30.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300. [Google Scholar]
- 32.Chaudhuri RR, Morgan E, Peters SE, Pleasance SJ, Hudson DL, Davies HM, Wang J, van Diemen PM, Buckley AM, Bowen AJ, Pullinger GD, Turner DJ, Langridge GC, Turner AK, Parkhill J, Charles IG, Maskell DJ, Stevens MP. 2013. Comprehensive assignment of roles for Salmonella typhimurium genes in intestinal colonization of food-producing animals. PLoS Genet 9(4):e1003456. doi: 10.1371/journal.pgen.1003456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paterson GK, Northern H, Cone DB, Willers C, Peters SE, Maskell DJ. 2009. Deletion of tolA in Salmonella Typhimurium generates an attenuated strain with vaccine potential. Microbiology 155:220–228. doi: 10.1099/mic.0.021576-0. [DOI] [PubMed] [Google Scholar]
- 34.Tacket CO, Sztein MB, Losonsky MB, Wasserman GA, Nataro SS, Edelman JP, Pickard R, Dougan D, Chatfield GSN, Levine MM. 1997. Safety of live oral Salmonella typhi vaccine strains with deletions in htrA and aroC aroD and immune response in humans. Infect Immun 65:452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dougan G, Chatfield CO, Pickard S, Bester D, O'Callaghan D, Maskell D. 1988. Construction and characterisation of vaccine strains of Salmonella harbouring mutations in two different aro genes. J Infect Dis 158:1329–1335. doi: 10.1093/infdis/158.6.1329. [DOI] [PubMed] [Google Scholar]
- 36.Hindle Z, Chatfield SN, Phillimore J, Bentley M, Johnson J, Cosgrove CA, Ghaem-Maghami M, Sexton A, Khan M, Brennan FR, Everest P, Wu T, Pickard D, Holden DW, Dougan G, Griffin GE, House D, Santangelo JD, Khan SA, Shea JE, Feldman RG, Lewis DJ. 2002. Characterization of Salmonella enterica derivatives harbouring defined aroC and Salmonella pathogenicity island 2 type III secretion system (ssaV) mutations by immunization of healthy volunteers. Infect Immun 70:3457–3467. doi: 10.1128/IAI.70.7.3457-3467.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.