

INTRODUCTION
Traumatic brain injury (TBI) is a major cause of mortality and morbidity, particularly at the two ends of the age spectrum, with large direct and indirect costs to society (see Ch. 1). The US Centers for Disease Control and Prevention (CDC) estimate that more than 1.7 million individuals in the US suffer a TBI annually (Faul et al., 2010), and the annual burden of TBI has been estimated at over US$60 billion based upon year 2000 dollars (Finkelstein et al., 2006). Yet even these numbers markedly underestimate the incidence and costs as the CDC data do not include reports of sports-related concussions (estimated incidence of 1.6–3.8 million per year (Langlois et al., 2006)) or military-related blast injuries (it is estimated that between 2000 and 2011 some 229 106 US service members suffered TBI in military conflict zones (Magnuson et al., 2012)). Globally, the incidence of TBI is also increasing, particularly in developing countries where road traffic accidents have increased as a result of greater motor vehicle use (Maas et al., 2008).
TBI is a highly complex disorder that is caused by both primary and secondary injury mechanisms (Loane and Faden, 2010) (see Ch. 5). Primary injury mechanisms result from the mechanical damage that occurs at the time of trauma to neurons, axons, glia and blood vessels as a result of shearing, tearing or stretching (see Ch. 7). Collectively, these effects induce secondary injury mechanisms that evolve over minutes to days and even months after the initial traumatic insult and result from delayed neurochemical, metabolic and cellular changes (Fig. 22.1) (see Ch. 42). These secondary injury events are thought to account for the development of many of the neurologic deficits observed after TBI, and their delayed nature suggests that there is a window for therapeutic intervention (pharmacologic or other) to prevent progressive tissue damage and improve functional recovery after injury. Implicated secondary injury mechanisms include disturbances of ionic homeostasis (Gentile and McIntosh, 1993), release of neurotransmitters (e.g., glutamate excitotoxicity) (Faden et al., 1989), mitochondrial dysfunction (Xiong et al., 1997), neuronal apoptosis (Yakovlev et al., 1997), lipid degradation (Hall et al., 2004), and initiation of inflammatory and immune responses (Morganti-Kossmann et al., 2007), among others. These neurochemical events induce toxic and proinflammatory molecules such as prostaglandins, oxidative metabolites, chemokines and proinflammatory cytokines, which lead to lipid peroxidation, blood–brain barrier (BBB) disruption, and the development of cerebral edema. The associated increase in intracranial pressure can contribute to local hypoxia and ischemia as well as secondary hemorrhage and herniation, leading to initiation and execution of multiple neuronal cell death mechanisms (Andriessen et al., 2010). Furthermore, secondary injury mechanisms may be highly interactive and often occur in parallel, thereby adding to the complexity of this disorder.
Fig. 22.1.
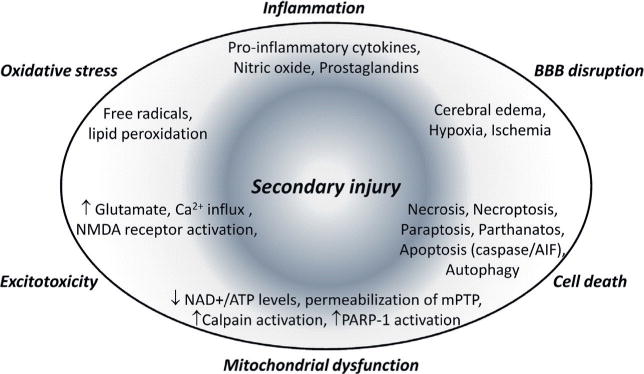
Secondary injury mechanisms after traumatic brain injury.
Considerable research has sought to elucidate secondary injury mechanisms in order to develop neuroprotective treatments. Although preclinical studies have suggested many promising pharmacologic agents, more than 30 phase III prospective clinical trials have failed to show significance for their primary end point (Narayan et al., 2002; Schouten, 2007; Maas et al., 2010). Most of these trials targeted single factors proposed to mediate secondary injury. But the complexity and diversity of secondary injury mechanisms have led to calls to target multiple delayed injury factors (Margulies and Hicks, 2009; Stoica et al., 2009; Vink and Nimmo, 2009), either by combining agents that have complementary effects or by using multipotential drugs that modulate multiple injury mechanisms. Whereas the multidrug approach has long been successfully employed for the treatment of cancer and infectious diseases, it is less likely to gain traction for neuroprotection because of the costs associated with establishing the efficacy of even a single agent. This recognition has led to the recent emphasis on multipotential treatments for TBI (Vink and Nimmo, 2009; Loane and Faden, 2010), several of which are now in clinical trials and others that are showing considerable promise in preclinical studies.
Neuroprotection approaches for both acute and chronic neurodegenerative disorders have historically been dominated by a neuronocentric view, in which modification of neuronal-based injury mechanisms is viewed as the primary focus of the neuroprotective strategy. However, increasing evidence in the literature underscores the importance of viewing injury more broadly to include endothelial cells, astrocytes, microglia, oligodendrocytes, and precursor cells. More recent neuroprotection approaches have recognized this complex structure and interplay, emphasizing therapeutic strategies that promote the recovery and optimal functioning of non-neuronal cells in addition to more directly inhibiting mechanisms of neuronal cell death (Stoica and Faden, 2010b). Thus, developing effective neuroprotective strategies for TBI requires an understanding of the complex cellular and molecular events that contribute to secondary injury. Mechanisms of neuronal cell death and post-traumatic neuroinflammation will be addressed in the following sections as well as a discussion on the many challenges translating promising preclinical neuroprotection therapeutic strategies to the clinic. Finally, we will critically review developing preclinical multipotential drug treatment strategies for TBI that show promise for successful clinical translation for head injury.
NEURONAL CELL DEATH: MORPHOLOGY VERSUS MECHANISM
Neuronal cell death is a major cause of neurologic dysfunction following TBI. For many years, it was believed that all or most cell death following brain trauma reflected a passive and unregulated form of neuronal death due to energy failure and related loss of ionic homeostasis, which was commonly called necrosis. However, over the past 15 years additional neuronal death phenotypes have been described based upon either morphologic or molecular features. Yet, despite the efforts of many research groups, defining a generally acceptable framework for the taxonomy of cell death mechanisms has been challenging. One reason is that key terms remain imprecisely defined (Majno and Joris, 1995; Levin, 1998; Kroemer et al., 2009; Stoica and Faden, 2010b; Galluzzi et al., 2012). For example, the term necrosis has been used by some pathologists not as a form of cell death, but rather to indicate the final stage shared by all types of cell death (Levin, 1998; Levin et al., 1999; Majno and Joris, 1999). In contrast, others have interpreted necrosis as a more specific phenotype of cell death characterized by morphologic features such as increased cell volume (oncosis), relatively unchanged nuclei, organelle swelling and rupture of the plasma membrane. This is distinguished from apoptosis, a type of cell death whose morphotype includes decreased cell volume (pyknosis), chromatin condensation with nuclear fragmentation (karyorrhexis) and release of vacuoles (apoptotic bodies) containing cytoplasm and comparatively intact organelles (Kroemer et al., 2009; Vandenabeele et al., 2010; Galluzzi et al., 2012). An additional challenge has been the struggle to eliminate the inaccurate yet widespread dichotomy between apoptosis as reflecting programmed, active/regulated cell death (which plays a role in physiologic development and homeostasis) and necrosis as reflecting “accidental” (non-programmed) and passive/unregulated cell death. This view was contradicted by early studies that demonstrated the presence of necrotic cell death morphology during physiologic development/homeostasis, and more recently by studies suggesting that cell death with necrotic features often involves active and regulated cellular activities (Majno and Joris, 1995; Bredesen, 2007; Bredesen, 2008; Stoica and Faden, 2010a). A limitation of classifying cell death based on morphologic features is that similar appearances can be found in the face of considerable mechanistic heterogeneity (Galluzzi et al., 2012). Because delineation of causal mechanisms of cell death is important for identifying therapeutic targets/agents it is understandable why the research community has advocated replacement of morphologic criteria with molecular-based mechanisms (Galluzzi et al., 2012).
A promising mechanism-based classification of cell death, which still retains morphologic considerations, was recently proposed by the Nomenclature Committee on Cell Death (Galluzzi et al., 2012). It groups cell death phenotypes that share key morphologic features but then proposes causal mechanisms to create more specific subgroups. For example, “apoptosis” is used to delineate several types of cell death that present key morphologic features of developmental apoptosis; these are subdivided into an “intrinsic” subgroup that has a mitochondrial initiation mechanism and an “extrinsic” subgroup that has a membrane receptor-based initiation mechanism.
Intrinsic apoptosis
Intrinsic apoptosis is triggered by mitochondrial outer membrane permeabilization (MOMP), which occurs either in response to pores mediated by BAX and BAK (proapoptotic members of the BCL-2 family) or to mitochondrial permeability transition (MPT) following opening of the permeability transition pore complex (Green and Kroemer, 2004; Galluzzi et al., 2012). MOMP results in the release from the mitochondrial intermembrane space of various cell death modulators such as cytochrome c, apoptosis-inducing factor (AIF), endonuclease G, Smac/DIABLO or Omi/HtrA2 (Daugas et al., 2000; Li et al., 2001; Suzuki et al., 2001; van Loo et al., 2002), as well as cessation of mitochondrial ATP synthesis, inhibition of the respiratory chain and increased reactive oxygen species (ROS) production (Galluzzi et al., 2012). Intrinsic apoptosis is further subdivided into caspase-dependent and caspase-independent forms (Galluzzi et al., 2012). Cytochrome c, along with Apaf-1 and dATP, form the “apoptosome” – a multiprotein complex that activates caspase-9 to initiate the caspase-3-dependent proteolytic cascade, which is considered a key executioner pathway of apoptotic cell death (Li et al., 1997; Galluzzi et al., 2012). AIF and endonuclease G act in a caspase-independent manner by translocating to the nucleus and mediating large-scale DNA fragmentation; these represent distinct executioner mechanisms of cell death (Daugas et al., 2000; Li et al., 2001).
Extrinsic apoptosis
Extrinsic apoptosis is a caspase-dependent type of cell death that is initiated by activation of specific plasma membrane receptors – including death receptors such as FAS/CD95, TNF-α receptor 1 and TRAIL receptors, or dependence receptors such as the netrin receptors. Death receptor stimulation results in caspase-8 activation, which, depending on cell type, leads to cell death either by directly cleaving and activating caspase-3 or by cleaving BID protein to generate the active fragment, tBID, that produces MOMP followed by apoptosome-mediated caspase-3 activation (Luo et al., 1998; Galluzzi et al., 2012).
Regulated necrosis
Importantly, the new nomenclature delineates regulated necrosis, which includes those types of cell death that share key morphologic features of necrosis yet have well-defined and regulated causal mechanisms. An example is necroptosis, a type of regulated necrosis controlled by RIP1 and RIP3 molecules (Vandenabeele et al., 2010; Galluzzi et al., 2012). It is initiated under certain conditions when signaling through death receptors such as TNF receptors is followed by activation of RIP1 or RIP3 kinase leading to the execution of the necrotic cell death (Vandenabeele et al., 2010). Paraptosis, a cell death process that occurs in response to growth receptor hyper-activation (e.g., insulin-like growth factor 1 receptor), also belongs to the regulated necrosis group. It shares key morphologic features of necrosis and its mechanisms are caspase-independent and involve activation of several members of the MAPK family (Sperandio et al., 2000; Bredesen, 2007). Parthanatos is a type of cell death first described by Dawson and colleagues following exposure of neurons to extensive DNA damage (Wang et al., 2009). Although parthanatos does not share all morphologic features of necrosis it is included in the group of regulated necrosis (Galluzzi et al., 2012). Parthanatos is a caspase-independent cell death mechanism that requires PARP-1 activation and its execution involves NAD+ and ATP depletion and AIF-dependent chromatinolysis (Wang et al., 2009). It is intriguing that AIF plays important roles as an executioner of cell death in both caspase-independent intrinsic apoptosis and parthanatos. These types of cell death not only have distinct morphologic features but may also reflect different mechanisms of AIF release from the mitochondria. Caspase-independent intrinsic apoptosis involves a pool of mitochondrial AIF that requires MOMP for release, whereas parthanatos uses a different pool of AIF molecules located on the cytosolic side of mitochondria which are released following interaction with PARP-1-produced poly(ADP-ribose) (Yu et al., 2009; Wang et al., 2011b).
Autophagy
A third major type of cell death with distinct morphologic features is linked to autophagy. Autophagy is a process involving lysosomal degradation of proteins and organelles that is controlled by genes from the Atg family. Under physiologic conditions it can have a protective role by generating amino acids and energy for the cell (Bredesen, 2008). The original description of “authophagic cell death” based exclusively on morphologic features such as large-scale cytoplasmic vacuolization appears to be inaccurate, as it lacked proof that activation of autophagy contributed to the execution of cell death (Bredesen, 2008; Galluzzi et al., 2012). More recent evidence suggests that in most cases the activation of autophagy represents a prosurvival effort on the part of the dying cell and that inhibition of autophagy increases cell death (Bredesen et al., 2006; Bredesen, 2007, 2008; Galluzzi et al., 2012). It has been suggested that the term “autophagic cell death” should only be used when in addition to the demonstration of increased autophagic flux there is evidence that inhibition of autophagy suppresses cell death (Galluzzi et al., 2012). Interestingly, in some cell death models inhibition of MOMP and/or caspase activation creates conditions where autophagy and the Atg genes may be involved in cell death (Bredesen, 2008), suggesting that autophagy might either be a cell death process working in parallel but secondary to apoptosis, or more likely acts as a compensatory mechanism initiated by inhibition of apoptosis (Bredesen, 2008).
However, not all types of cell death can be integrated into this new framework because some share features of both apoptosis and necrosis. In these cases the causal mechanism becomes the main identifier. An example is “mitotic catastrophe,” a type of cell death initiated by aberrant mitosis (Galluzzi et al., 2012). It has therefore been proposed that the adjective “programmed” should be applied only to physiologic cell death occurring during development and/or tissue homeostasis and that the adjective “regulated” should indicate all types of cell death where molecular machinery plays a causal role. Here “regulated” appears to be synonymous with “active.” In this context, the historical term “passive” cell death, used to denote necrotic morphology following energy failure with loss of ionic homeostasis – usually in response to the most intense physicochemical insults – may retain some utility with regard to neuroprotection strategies because this form of cell death likely cannot be reversed (Galluzzi et al., 2012). In terms of this updated nomenclature, multiple types and mechanisms of neuronal cell death have been described following TBI.
CELL DEATH MECHANISMS IN TRAUMATIC BRAIN INJURY
Caspase-dependent neuronal cell death pathways
Multiple groups have shown that neuronal cell death involving caspase-dependent mechanisms occurs after experimental TBI (Rink et al., 1995; Yakovlev et al., 1997; Conti et al., 1998). Some studies have shown that FAS death receptors contributed to caspase activation following experimental and clinical TBI and that caspase-8 deletion protects against experimental TBI, thus supporting a role for extrinsic apoptosis after such injury (Qiu et al., 2002; Krajewska et al., 2011). Other studies have shown that caspase-9 rather than caspase-8 is predominantly activated in neurons following experimental TBI (Knoblach et al., 2002), supporting a role for caspase-dependent intrinsic apoptosis. Peptide-based caspase inhibitors have been used to examine the causal role of caspases in neuronal cell death in vitro (Eldadah et al., 1997; Gottron et al., 1997; Bilsland et al., 2002) and after experimental TBI (Yakovlev et al., 1997; Clark et al., 2000; Knoblach et al., 2002, 2004; Sullivan et al., 2002). In the rat lateral fluid percussion (LFP) TBI model treatment with zDEVD-fmk, a relatively selective caspase-3 inhibitor, inhibited neuronal apoptosis and improved early motor function recovery (Yakovlev et al., 1997), and treatment with zVAD-fmk, a pan-caspase inhibitor, improved both motor function recovery and cognitive function after TBI (Knoblach et al., 2002). Furthermore, administration of zDEVD-fmk also improved motor and cognitive function and reduced lesion volume in a mouse controlled cortical impact (CCI) model (Knoblach et al., 2004). In contrast, in a rat model of CCI plus moderate hypoxemia, administration of zDEVD-fmk did not improve functional recovery outcomes but it did reduce the contusion volume and neuronal loss in the hippocampus (Clark et al., 2000). A limitation of zDEVD-fmk and zVAD-fmk is that both peptides inhibit other proteases such as calpains in addition to caspases (Knoblach et al., 2004, 2005). The pan-caspase inhibitor boc-aspartyl(OMe)-fluoromethylketone (BAF) may provide greater specificity, as it is more selective than other peptide-based caspase inhibitors (Knoblach et al., 2005). However, in a rat CCI model BAF administration did not decrease the lesion volume (Sullivan et al., 2002); nor did it improve neurologic outcomes or reduce hippocampal neuronal loss after TBI (Clark et al., 2006). Thus, the neuroprotective effects of caspase inhibition following TBI have not been consistently validated across laboratories and TBI models. The observed inconsistencies may have multiple explanations including differences in experimental TBI models and species, the assortment of caspase inhibitors used and doses administered, and the different types of outcome measures used to assess the neuroprotective potential of the drugs. A recent study clarified the relative roles of caspase-dependent and caspase-independent apoptosis using a mouse CCI model (Piao et al., 2012). BAF treatment robustly attenuated caspase activation but the improvements in motor function recovery and reduction in the number of TUNEL+cells were modest. BAF-treated animals did not show a significant improvement in cognitive function and did not significantly reduce lesion volumes or attenuate hippocampal neuron cell loss. These findings, when combined with previous work, indicate that caspase-dependent mechanisms play a relatively modest role in secondary injury cascades after CCI in adult mice. It is also likely that the multiplicity of secondary injury processes minimize the therapeutic effectiveness of targeting caspase-dependent apoptosis in isolation (Piao et al., 2012).
Caspase-independent neuronal cell death pathways
Apoptosis-inducing factor
Apoptosis-inducing factor (AIF) is a flavoprotein that normally resides within the mitochondrial intermembrane space (Susin et al., 1999; Joza et al., 2001). Upon release into the cytosol, AIF translocates to the nucleus, interacts with chromatin and leads to peripheral chromatin condensation and large-scale DNA fragmentation (Zhang et al., 2002; Zhu et al., 2007b; Slemmer et al., 2008). Studies have shown that AIF-dependent neuronal cell death is present in multiple models of CNS injury (Zhang et al., 2002; Zhu et al., 2003; Slemmer et al., 2008; Osato et al., 2010; Stoica and Faden, 2010b). In contrast to other apoptosis-associated genes whose expression decreases with age (Yakovlev et al., 2001), AIF expression remains relatively stable during development (Zhu et al., 2003), suggesting that AIF-dependent cell death mechanisms may play a more significant role in the adult brain.
Cyclophilin A (CypA) is a cofactor required for AIF translocation to the nucleus (Cande et al., 2004). AIF physically interacts with CypA and forms an AIF/CypA complex, and studies have shown that this complex translocates to the nucleus in neurons following neonatal hypoxia-ischemia (Zhu et al., 2003, 2007a). Importantly, CypA knockout (CypA−/−) provides significant neuroprotection in this model. The mouse AIF hypomorphic mutant (Harlequin; Hq) confers protection to neurons against NMDA or glutamate neurotoxicity in vitro (Wang et al., 2004; Culmsee et al., 2005), and Hq mice are protected against brain injury induced by cerebral ischemia in vivo (Culmsee et al., 2005; Zhu et al., 2007b). Data generated using the Hq mouse model hinted that AIF and caspases may act through parallel subroutines, with caspase inhibition and decreased AIF expression having potentially additive neuroprotective effects (Zhu et al., 2007b). In a neonatal hypoxia-ischemia model, BAF treatment did not impact AIF translocation to the nucleus in injured neurons, suggesting the relative independence of caspase- and AIF-mediated pathways (Zhu et al., 2003). The role of AIF in TBI models has been less well studied. Zhang and colleagues used a rat CCI plus hypoxia model and demonstrated that AIF translocation to the nucleus in injured neurons of the cortex and hippocampus coincides with large-scale DNA fragmentation (Zhang et al., 2002), whereas Slemmer and colleagues reported that secondary contusion expansion after CCI was attenuated in Hq mice (Slemmer et al., 2008).
In contrast to caspase-mediated cell death which requires physiologic levels of ATP for caspase activation, AIF-mediated cell death can proceed under compromised bioenergetic conditions, as AIF-dependent chromatin condensation and large-scale DNA fragmentation are unaffected by ATP depletion (Chiarugi, 2005). In fact, in certain models of PARP-1-dependent cell death mitochondria release both AIF and cytochrome c, but while AIF translocates to the nucleus and executes its cell death mechanism, caspases are not activated because of the associated ATP depletion (Moubarak et al., 2007). Therefore, AIF may play a larger pathophysiologic role as part of caspase-independent intrinsic apoptosis and/or parthanatos following more severe brain injuries, which are associated with significant bioenergetic declines.
A recent study used cyclophilin A (CypA−/−) knockout mice to examine the causal role of AIF-dependent cell death pathway in TBI-induced neuronal cell death (Piao et al., 2012). The authors demonstrated that TBI-induced activation of the AIF pathway, including AIF translocation to the nucleus and the interaction of AIF/H2AX, was significantly reduced in CypA−/− mice. These animals also had a decreased number of TUNEL-positive (apoptotic) cells in the cortex and better long-term motor function recovery, suggesting that the AIF cell death pathway plays a significant role in secondary injury after TBI (Piao et al., 2012). The potential additive neuroprotective effects of targeting both caspase- and AIF-dependent pathways after TBI was also demonstrated in this study and is further addressed below.
Parthanatos
Parthanatos appears to contribute to the pathology of conditions ranging from stroke to neurodegeneration (Wang et al., 2009). Recent studies have shown that inhibitors of poly(ADP-ribose)-induced cell death (parthanatos) are neuroprotective against both NMDA-dependent excitotoxicity and stroke in vivo suggesting that parthanatos plays an important role in the secondary injury mechanisms in these models of CNS injury (Andrabi et al., 2011; Wang et al., 2011b). Importantly, systemic administration of PJ34, a selective PARP-1 inhibitor starting as late as 24 hours after controlled cortical impact resulted in improved motor function recovery in mice with TBI (Stoica et al., 2014). PJ34 treatment reduced the lesion volume, attenuated neuronal cell loss in the cortex and thalamus, and reduced microglial activation in the TBI cortex, suggesting that its neuroprotective effects after experimental TBI may be from multipotential actions on neuronal cell death and neuroinflammatory pathways (Stoica et al., 2014).
Necroptosis
Studies have demonstrated that inhibition of necroptosis using the RIP1-targeting chemical necrostatin-1 attenuates the histopathology and improves functional outcomes following experimental TBI in mice (You et al., 2008). These data suggest that necroptosis may play an important role in the pathogenesis of neuronal cell death and secondary injury associated functional deficits after TBI, but further studies are required to determine if its modulation represents a good candidate for neuroprotection.
Autophagic cell death
There is conflicting evidence about the role of autophagy after CNS injury. Certain studies find persistent autophagy activation after experimental TBI and speculate about its role without attempting to directly modulate autophagy (Lai et al., 2008; Liu et al., 2008; Sadasivan et al., 2008). Some have used pharmacologic modulation of autophagy and presented data that support a neuroprotective role of autophagy (autophagy inducers improve outcomes, autophagy inhibitors worsen outcomes) in models of focal cerebral ischemia (Yan et al., 2011) or experimental subarachnoid hemorrhage (Wang et al., 2012). In contrast, other studies have shown that inhibitors of autophagy improve outcomes after TBI (Luo et al., 2011) or severe global cerebral ischemia (Wang et al., 2011a) thereby suggesting that autophagy is a secondary injury mechanism in these models. The lack of agreement across these studies may reflect the distinct roles for autophagy in multiple models of CNS injury and/or the complex effects of autophagy-inhibiting drugs that are not sufficiently selective. Future studies are required to determine the role of autophagy after TBI and they should use multiple types of pharmacologic modulators and molecular tools to modulate the autophagy pathways.
Crosstalk between cell death programs
In most in vivo injury models multiple cell death pathways are activated. Furthermore, different cell death processes may be interactive and studies indicate that inhibition of one mechanism may reveal a different mechanism with the end result that cell death suffers only a phenotype change and/or delay (Galluzzi et al., 2012). The concept that alternative mechanisms become apparent primarily when the dominant mechanisms are attenuated has been previously discussed (Bredesen, 2008). One example of this hypothesis of mechanism multiplicity is illustrated by intrinsic apoptosis (Galluzzi et al., 2012). This type of cell death is centered around MOMP-dependent release from mitochondria of several cell death inducers including cytochrome c, a key activator of the caspase pathway as well as initiators of caspase-independent pathways such as AIF, EndoG, and others. These mechanisms are significantly redundant and in intrinsic apoptosis, unlike in extrinsic apoptosis, inhibition of caspase-dependent mechanisms often provide only temporary neuroprotection because caspase-independent mechanisms and/or ATP depletion that follows MOMP will continue to drive the cell death program (Volbracht et al., 2001; Galluzzi et al., 2012). The cellular environment also plays an important role in deciding which cell death program takes precedence (Stoica and Faden, 2010b). Caspase-dependent pathways require energy for their initiation and are dominant in conditions associated with a well preserved cellular bioenergetics status with caspase-independent pathways involving AIF taking a secondary role unless caspase inhibition occurs. In contrast, AIF may become a more major cell death pathway under bioenergetic deficient conditions. There is good evidence that AIF and caspases represent parallel cell death tracks and that strategies that target both have potentially additive therapeutic effects (Zhu et al., 2007b). This has been demonstrated in an experimental TBI model where inhibition of both caspase-dependent and AIF-dependent pathways resulted in improved neuroprotection compared to inhibition of either pathway in isolation (Piao et al., 2012). Autophagic and caspase-dependent cell death may also show a similar relationship (Bredesen, 2008). Thus, combined therapeutic strategies targeting more than one cell death pathway may in general provide better neuroprotection than inhibition of individual pathways.
A different type of crosstalk occurs in some models of necroptosis, where one cell death pathway is initiated only when another is inhibited (Galluzzi et al., 2012). Death receptor stimulation may lead to caspase activation and caspase-dependent cell death by blocking initiation of necroptosis through caspase-8-dependent cleavage/inactivation of RIP1 and RIP3 kinases (Declercq et al., 2011). In contrast, under conditions associated with inhibition of the caspase cascade, initiation of necroptosis is unhindered and necroptosis becomes the predominant cell death pathway (Declercq et al., 2011). This multiplicity of cell death pathways implies that the most successful therapeutic intervention should likely target multiple relevant mechanisms of cell death.
Neuroinflammation after traumatic brain injury
Neuroinflammation is an important secondary injury mechanism that contributes to ongoing neurodegeneration and neurologic impairments associated with TBI. Post-traumatic neuroinflammation is characterized by glial cell activation, leukocyte recruitment, and upregulation of inflammatory mediators (Morganti-Kossmann et al., 2007). Although much research has focused on the detrimental effects of neuroinflammation on the injured brain, clear beneficial effects can be achieved if neuroinflammation is controlled in a regulated manner and for defined periods of time. Trauma to the brain results in rupture of the BBB, enabling recruitment of circulating neutrophils, macrophages, and lymphocytes to the injured site. The accumulation of blood-borne immune cells within the brain parenchyma has been reported in human TBI as well as animal models of brain trauma (Morganti-Kossmann et al., 2001). These cells release inflammatory mediators that mobilize glia and immune cells to the site of injury. In addition to the infiltration of immune cells, the activation of resident microglia plays a major role in the response to brain injury (Loane and Byrnes, 2010). Elegant in vivo two-photon microscopy imaging studies of fluorescently labeled microglia following a laser-induced injury demonstrated rapid proliferation and movement of ramified microglial cells to the site of injury in response to extracellular ATP released by the injured tissue (Davalos et al., 2005; Haynes et al., 2006). The microglial processes then fused to form an area of containment between healthy and injured tissues, suggesting that microglia may represent the first line of defense following traumatic injury (Davalos et al., 2005). However, when microglia become overactivated or reactive they can induce detrimental neurotoxic effects by releasing multiple cytotoxic substances, including proinflammatory cytokines (e.g., interleukin (IL)-1β, tumor necrosis factor a (TNF-α), and interferon a (IFN-γ)) and oxidative metabolites (e.g., nitric oxide, reactive oxygen and nitrogen species) (Block and Hong, 2005). Further, the release of proinflammatory cytokines and other soluble factors by activated microglia can significantly influence the subsequent activation of astrocytes and glial scar formation under pathologic conditions including CNS injury (Zhang et al., 2010).
Astrocyte activation (astrogliosis) is characterized by the increase of intermediate filaments, increased cell proliferation, and an accompanying cellular hypertrophy (Herrmann et al., 2008). Similar to microglia, reactive astrocytes can have detrimental and/or beneficial roles following CNS injury. Upon activation, astrocytes upregulate a number of neurotrophic factors (e.g., brain-derived neurotrophic factor (BDNF)) that support and protect against injury-induced cell death (Zhao et al., 2004). In addition, astrocytes play a crucial role in regulating extracellular glutamate levels, which can reduce glutamate excitotoxicity to neurons and other cells (Schousboe and Waagepetersen, 2005). Notably, impaired astrocyte performance exacerbates neuronal dysfunction following brain injury and transgenic ablation of reactive astrocytes increases neuronal cell death and promotes worse outcome after TBI (Myer et al., 2006); this may in part reflect the loss of ability to limit the influx of inflammatory cells. Following injury, hypertrophic astrocytes surround the lesion site and deposit an inhibitory extracellular matrix including chondroitin sulfate proteoglycans that contribute to the glial scar. This dense physical and chemical barrier inhibits axonal regeneration and prevents functional connections required for axonal growth and repair (Cafferty et al., 2007). On the one hand, astrocytes provide neurotrophic support and guidance for axonal growth following CNS injury, while on the other, prolonged astrogliosis and development of the glial scar may inhibit axon regeneration and impair functional recovery. Genetic studies using mice lacking the intermediate filament proteins GFAP and vimentin (GFAP−/−Vim−/−) showed that upregulation of astrocyte intermediate filament is a key step in activation of astrocytes and that reactive astrogliosis is important for the wound healing process (Pekny et al., 1999; Pekny and Nilsson, 2005). In brain injury models GFAP−/−Vim−/− mice show attenuated reactive astrogliosis with reduced hypertrophy of astrocyte processes and increased synaptic loss in the acute stage of the injury (Pekny et al., 1999; Wilhelmsson et al., 2004); however, the regeneration of neuronal synapses at a later stage is improved (Wilhelmsson et al., 2004) and this may be due to the loss of inhibitory chondroitin sulfate proteoglycans within the glial scar.
Microglia: mediators of the innate immune response to central nervous system injury
Microglia are the primary innate immune cells in the CNS. Under normal physiologic conditions these highly dynamic and motile cells are spread throughout the brain parenchyma and constantly survey their microenvironment for noxious agents and injurious processes (Nimmerjahn et al., 2005). They respond to extracellular signals and are responsible for clearing cellular debris and toxic substances by phagocytosis, thereby maintaining normal cellular homeostasis in the CNS (Hanisch and Kettenmann, 2007). Therefore, under nonpathologic conditions there is continuing low-level microglial activity in the CNS which is primarily involved in activity-dependent synaptic pruning (Tremblay et al., 2011; Schafer et al., 2012). However, in response to insult, infection or injury, microglia become dysregulated and highly activated which results in dramatic changes in cell morphology and behavior. Similar to peripheral innate immune cells microglia express pathogen recognition receptors (PRRs) such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs), and therefore respond to pathogen-associated molecular patterns (PAMPs) and endogenously produced danger-associated molecular patterns (DAMPs), which are secreted by damaged neurons and others cells of the CNS (Hanisch and Kettenmann, 2007). They also express receptors for a number of other factors that are released by damaged neurons, including ATP, glutamate, growth factors, and cytokines. Microglia respond to an array of molecules by secreting chemokines, pro- and anti-inflammatory cytokines, neurotrophins, and oxidative metabolites. Furthermore, microglia are antigen presenting cells, and upon activation they upregulate the expression of cell surface markers such as MHC class II and CD86 among others, as well as adhesion molecules and complement receptors (Lynch, 2009). A key to maintaining microglia in a quiescent state is the immunosuppressive potential of the brain microenvironment. In the healthy brain neurons express a number of immunosuppressive proteins such as CD200, CD47 and fractalkine, which interact with their receptors on microglia to maintain the microglia in a quiescent/nonactivated state (Harrison et al., 1998; Hoek et al., 2000; Lynch, 2009). In addition, soluble factors such as neurotrophins, anti-inflammatory cytokines and prostaglandins released locally in the brain by neurons, astrocytes and microglia downregulate the immune response thereby maintaining microglia in a quiescent form (Lynch, 2009).
Microglia, like peripheral macrophages, have multiple activation phenotypes (Gordon, 2003; Mantovani et al., 2004), and depending on the stimuli in their local microenvironment they can be polarized to have distinct molecular phenotypes and effector functions (Colton, 2009). For example, lipopolysaccharide (LPS) or the proinflammatory cytokine interferon-γ (IFN-γ) promote a “classic” (M1) phenotype, which produces high levels of proinflammatory cytokines and oxidative metabolites that are essential for host defense and phagocytic activity, but that also can cause damage to healthy cells and tissue (Lynch, 2009). Conversely, activating microglia in the presence of anti-inflammatory cytokines such as IL-4 or IL-10 promote “alternative” (M2a) or “acquired deactivated” (M2c) phenotypes respectively (Colton et al., 2006; Ponomarev et al., 2007), and both M2a and M2c phenotypes reduce M1 cytokines and other proinflammatory mediators (Gordon, 2003; Mantovani et al., 2004). It is thought that much like M2-polarized macrophages, M2 microglia can promote repair processes such as angiogenesis and extracellular matrix remodeling while also suppressing destructive immunity (Colton, 2009). In vitro studies have demonstrated that IL-4-polarized M2 macrophages promote extensive neurite elongation and outgrowth across inhibitory surfaces (Kigerl et al., 2009), indicating a role for M2 macrophages in repair. Microglia possess an M2 phenotype in the intact CNS (Ponomarev et al., 2007); however, following an acute insult or injury to the brain it is likely that M1 and M2 microglia exist in a state of dynamic equilibrium within the lesion microenvironment. Whether these cells differentiate into an M1 phenotype that exacerbates tissue injury or into an M2 phenotype that promotes CNS repair likely depends on the local signals in the lesion microenvironment. This is further complicated by the influence of infiltrating blood-borne macrophages of M1 or M2 phenotype and other infiltrating cells following CNS injury (Kigerl et al., 2009). However, understanding the molecular mechanisms that polarize and differentiate resident microglia or infiltrating macrophages towards an M2 phenotype, thereby promoting CNS repair while limiting inflammatory-mediated secondary injury cascades, may provide an opportunity for therapeutic intervention after CNS injury.
Involvement of pro- and anti-inflammatory cytokines in traumatic brain injury
The neuroinflammatory cascade activated in response to TBI is mediated by the release of pro- and anti-inflammatory cytokines and chemokines. Microglia are the primary source of these inflammatory mediators in the brain, and gene profiling studies in experimental models of TBI have shown that genes related to neuroinflammation and microglial activation are strongly upregulated in the acute phase after injury (Kobori et al., 2002; Natale et al., 2003; Raghavendra Rao et al., 2003). Consistent with the early activation profile of microglia following injury, there is rapid elevation of proinflammatory IL-1β within hours of TBI in both humans and rodents (Woodroofe et al., 1991; Fan et al., 1995; Winter et al., 2002). The damaging effects of IL-1β are mediated through interleukin 1 receptor type 1 (IL-1RI), which is expressed on microglia and neurons (Pinteaux et al., 2002; Lu et al., 2005b). This damage is not as a result of the cytokine itself, but rather its affect on activating other proinflammatory pathways such as TNF-α (Rothwell, 2003). Inhibiting IL-1β in experimental models of TBI has been shown to be neuroprotective and improve functional recovery (Toulmond and Rothwell, 1995; Basu et al., 2002; Tehranian et al., 2002; Lu et al., 2005a, b). The potent neurotoxic effects of IL-1β have been shown to be synergistically enhanced in the presence of TNF-α, suggesting that these important cytokines mediate post-traumatic neuroinflammation and brain damage (Chao et al., 1995). TNF-α levels rise within 1 hour of TBI, peak between 3 and 8 hours, and return to normal by 24 hours (Shohami et al., 1994; Fan et al., 1996; Stover et al., 2000). Consistently, TNF-α levels are elevated early after injury in the serum and cerebrospinal fluid (CSF) of severely injured TBI patients (Goodman et al., 1990; Ross et al., 1994). However, the role of TNF-α in the pathogenesis of TBI is somewhat controversial and complex in nature with different functional outcomes in the acute and delayed phases after TBI (Scherbel et al., 1999; Sullivan et al., 1999a). Initial preclinical neuroprotection studies targeting TNF-α showed considerable promise, with three different compounds (dexanabinol (HU-211), TNF-binding protein, pentoxifylline (PTX)) showing significant improvements in neurologic outcomes and reducing post-traumatic brain edema formation (Shohami et al., 1997). Mice lacking TNF-α had reduced deficits in the acute phase after TBI, but these were short lived and there were limited long-term neurologic improvements in this model (Scherbel et al., 1999). Another study demonstrated that mice lacking the TNF-α receptor had exacerbated tissue and BBB damage and impaired neurologic recovery after TBI (Sullivan et al., 1999a), suggesting that endogenous TNF-α may be neuroprotective. These genetic studies indicate that the function of TNF-α differs in the acute and delayed phase after TBI; immediately after injury TNF-α seems to act as a potent inflammatory mediator, but later it is a neurotrophic factor that is required for neuroprotection and repair. Unfortunately, a lack of understanding of the complex biphasic response of TNF-α to TBI may have been partly responsible for the failure of the TNF-α-targeted drug therapy, dexanabinol (HU-211), in phase III clinical trials for TBI (Maas et al., 2006).
Interestingly, anti-inflammatory cytokine levels are also changed after TBI. In humans, IL-10 and transforming growth factor-β1 (TGFβ1) levels are elevated acutely after injury (Csuka et al., 1999; Morganti-Kossmann et al., 1999), and experimental studies have shown that IL-10 has beneficial effects following trauma (Knoblach and Faden, 1998). These neuroprotective effects may be as a result of suppressed microglial activation, as IL-10 treatment has been shown to decrease production of proinflammatory cytokines (Kremlev and Palmer, 2005). Furthermore, treatment with the anti-inflammatory cytokine TGFb1 reduced lesion size and improved neurologic function after injury in rodent models (Tyor et al., 2002). Notably, the levels of IL-1 receptor antagonist (IL-1ra) are also elevated following TBI, and IL-1ra has significant anti-inflammatory properties. IL-1ra plays a major role in counteracting the biological effects of IL-1β, owing to its ability to bind to IL-1RI without initiating signal transduction (Allan et al., 2005). In experimental models of TBI, IL-1ra or IL-1β neutralization resulted in attenuated proinflammatory cytokine and chemokine production, reduced hippocampal damage and improved neurologic behavior after injury (Toulmond and Rothwell, 1995; Basu et al., 2002; Tehranian et al., 2002; Lu et al., 2005a, b).
Chronic microglial activation and neurodegeneration after traumatic brain injury
Chronic microglial activation is considered to be the most damaging response of microglia to injury (Block et al., 2007). DAMPs released by injured neurons after TBI interact with TLRs and other PRRs on activated microglia and trigger a vicious self-perpetuating cycle of damaging events that lead to prolonged and dysregulated microglial activation that drives pathogenic processes and neurodegeneration (Block et al., 2007; Loane and Byrnes, 2010). Human and animal studies indicate that microglia are chronically activated for weeks, months and even years after the initial brain trauma, and may contribute to chronic neurodegeneration and related neurologic deficits following injury (Smith et al., 1997a; Bramlett and Dietrich, 2002; Maxwell et al., 2006; Bendlin et al., 2008). Persistent long-term microglial activation has been demonstrated in animal models of TBI and is associated with increased expression of proinflammatory cytokines (e.g., IL-1β, TNF-α) (Holmin and Mathiesen, 1999). Notably, a recent clinical study utilizing the positron emission tomography ligand [11C] (R)PK11195 to assess chronic microglial activation in 10 patients studied more than 11 months after moderate to severe TBI reported significantly increased binding bilaterally at sites distant from areas of focal injury, such as thalamus, and correlated these changes with several measures of cognitive dysfunction (Ramlackhansingh et al., 2011). Furthermore, postmortem studies have also demonstrated increased microglial activation in the white matter of head injury survivors up to 16 years after TBI (Gentleman et al., 2004; Johnson et al., 2013). The experimental and clinical evidence now suggests that TBI should not be viewed as a static, acute neurodegenerative disorder. Instead, TBI initiates chronic biochemical processes leading to prolonged neuroinflammation and microglial activation, and as such there may be a longer therapeutic window for the treatment of head injury than traditionally accepted. In fact, a recent experimental study which used a pharmacologic treatment (mGluR5 agonist) to inhibit chronic microglial activation demonstrated that delayed treatment at 1 month postinjury significantly reduced both functional deficits and histologic outcomes through 4 months post-injury (Byrnes et al., 2012).
Aging also influences microglial activation (Conde and Streit, 2006b; Lucin and Wyss-Coray, 2009), and exacerbated microglial activation and astroglial responses to injury likely contribute to the enhanced susceptibility to and poor recovery from TBI in elderly patients (Galbraith, 1987; Pennings et al., 1993). In fact, experimental studies demonstrated that microglial activation was exaggerated and prolonged in aged mice when compared to adult mice (Sandhir et al., 2008), and these observations are consistent with reports of elevated microglial activation in the aged brain following injuries such as facial nerve axotomy (Conde and Streit, 2006a) and cerebral ischemia (Popa-Wagner et al., 2007). It has been proposed that microglia in the aged brain are “primed” to respond more rapidly, produce more pronounced inflammatory responses, and proliferate more vigorously than microglia in the young brain following TBI (Godbout et al., 2005; Conde and Streit, 2006b). Recent studies have revealed an altered relative balance between M1 and M2 microglial phenotypes and increased NADPH oxidase expression in microglia in the aged injured brain, and this exaggerated neuroinflammatory response was associated with increased cortical and hippocampal neurodegeneration (Kumar et al., 2013). Therefore, a hyperactivated and dysfunctional microglial response in the aged hippocampus may contribute to enhanced neuronal loss and worse cognitive outcomes in the elderly following brain trauma.
The immature brain is also particularly vulnerable to brain trauma during critical developmental phases that involve specific pathophysiologic responses such as prominent edema formation and resulting in significant motor and cognitive deficits (Cernak et al., 2010) (see Chs 15 and 41). Microglia may play a significant role in neuronal death in experimental models of pediatric brain injury (Tang et al., 2010). While elevated alcohol levels are associated with a higher incidence of brain trauma-inducing traffic accidents some studies have suggested that intoxicated patients have improved clinical outcomes at least in part due to reduced neuroinflammatory response to injury (Goodman et al., 2013). The protective effects of alcohol in brain trauma have been disputed by other experimental and clinical studies (Chen et al., 2012; He et al., 2013), which highlight the need for additional research on the molecular mechanisms and long-term effects of alcohol on outcomes after TBI.
Neuroprotection: challenges for clinical translation
Neuroprotective treatments that limit secondary injury mechanisms and/or improve behavioral outcome have been well established in multiple animal models of TBI. However, translation of promising experimental neuroprotective treatments to human injury have been very disappointing, with none of the pharmacologic treatments resulting in any consistent improvements in outcome in the clinic (Maas et al., 2010). Both conceptual issues and methodologic differences between preclinical research and clinical injury have undoubtedly contributed to these translational difficulties (see Ch. 29). The lack of success of neuroprotective drugs clinically has led investigators to identify potential factors contributing to such failures (see Ch. 49). These include (1) inadequate understanding of secondary injury mechanisms; (2) insufficient preclinical testing in multiple injury models, strains, species (including gyrencephalic), genders and ages; (3) lack of thorough investigation of pharmacokinetics and therapeutic brain concentrations; (4) failure adequately to examine therapeutic window and clinically relevant behavioral outcomes; (5) use of heterogeneous patient populations (see Ch. 3); (6) inadequate sample size; and (7) inadequate functional outcome measurements and biomarkers. But there are many other critical differences between clinical and preclinical studies that have potential relevance for translation. These include, among others: use of anesthesia in animal models with potential drug/anesthetic interactions; use of genetically identical populations and simplistic injury models with high consistency; post hoc deletion of animal subjects not meeting injury criteria versus the use of an intent-to-treat paradigm clinically. These differences have led some to question the value of animal models or to suggest highly regimented criteria for drugs considered for clinical trials, such as the STAIR criteria established for stroke neuroprotection studies (STAIR, 1999; Fisher et al., 2009) (see Ch. 8).
Numerous in vivo TBI models have been utilized to address potential mechanisms of secondary injury and to provide proof-of-principle support for specific treatment options. However, methodologic concerns have been raised with regard to clinical relevance, including choice of species, strain, or sex. For example, how well do models of brain trauma in rodents reflect injury in higher species? Notably, rodents have relatively small lissencephalic brains with less white matter than humans. Therefore, it has been suggested that potential therapies should be evaluated in multiple models including higher (gyrencephalic) species (Smith et al., 1997b). The porcine and human brains are gyrencephalic and have a similar distribution of white and gray matter. Furthermore, the stages of porcine brain development are comparable to those of the human brain, making the neonatal piglet an appropriate model for the infant brain (Sullivan et al., 2013). Although impacted by limitations such as high cost and complexity of behavioral tests, pig models continue to grow in importance as a tool in neuroscience, including the study of TBI (Sullivan et al., 2013).
Even within a given species, however, the same model can produce vastly different injury levels and outcomes across various strains (Fox et al., 1999). This potentially impacts studies using transgenic animals, in which outcome may reflect the type of backcrossing.
For many preclinical neuroprotection studies, questions arise about the specificity of the treatment target. Proclaimed specific modulators often have additional, and sometimes unexpected, effects on other pathways. Certain experimental strategies can be employed to address this concern, such as the use of structurally different modulators that share similar modulatory functions or parallel use of knockout models. However, such complementary experimental studies are not commonly performed. In addition, experimental models of TBI use anesthetized animals. However, anesthetics themselves are drugs that affect injury in different ways, and may serve to enhance or reduce cell death (Statler et al., 2006). Moreover, anesthetics may have considerable effects on the actions of the therapeutic drug, and these issues create potential problems for clinical translation. Animal models often use outcome measures with limited relevance to treatment effects in humans and few animal studies have examined therapeutic windows that correlate to likely treatment times in humans. Indeed, most animal studies generally employ either a pretreatment or very early post-treatment paradigm (i.e., <1 hour). In contrast, it is difficult to enter TBI patients into clinical trials before 6 hours, in part because of the informed consent issues. It is also rare for preclinical studies to examine pharmacokinetic or pharmacodynamic profiles of the drugs being examined, or brain levels of drugs in relation to therapeutic actions.
Preclinical study design and statistical analyses also differ considerably from those used clinically. Whereas clinical studies generally employ an intent-to-treat analysis, this is virtually never done preclinically. In addition, power analysis is not generally performed to determine the optimal animal sample size; rather, animal numbers are often determined either arbitrarily, or worse, the sample size may be increased incrementally during the study until statistical significance has been achieved. Randomization of treatment and blinding are also important factors that must be included in preclinical experimental design. Another problem is that clinical trials usually include a range of injury severities, whereas preclinical studies use well-defined, highly controlled animal models of preselected severity. They also preferentially use young healthy animals of a single sex to increase reproducibility, despite the fact that TBI is highly heterogeneous clinically. Further, TBI models typically examine the effects of local contusion or diffuse axonal injury in models that do not include significant secondary insults such as ischemia, hypoxia or associated systemic injuries, despite the fact that these insults are common in clinical TBI (Graham et al., 2005).
Thus, a number of conceptual and methodologic issues have undoubtedly contributed to the difficulty in translating promising neuroprotective treatments from animal to human. Nonetheless, many of these shortcomings can be addressed with improved preclinical screening of drugs. Recommendations regarding preclinical experimental design for the evaluation of neuroprotective agents for TBI have been made (Loane and Faden, 2010), and they build on the STAIR criteria for development of neuroprotective treatments for stroke/ischemia (STAIR, 1999; Fisher et al., 2009), and are in line with NIH guidelines for effective therapies for TBI (Saatman et al., 2008; Margulies and Hicks, 2009). Prehospital studies may focus on hyperacute treatment strategies that target early mechanisms such as glutamate release, but the methodologic and randomization issues for such studies in a disorder as heterogeneous as TBI would be major (see Ch. 23). Computational models will be an important tool aiding the design of novel and more effective pharmacologic interventions based on proven structures (Faden et al., 2003a, b, 2004). However, the heterogeneity of clinical TBI and the complex nature of the secondary injury process represent the most significant hurdles to future clinical trial successes. Comparative effectiveness studies in humans can be very important for agent development but depend upon evidence for an agent shown to be effective, which has not yet occurred for TBI (see Ch. 29). It is unlikely that targeting any single secondary injury factor will result in significant improvement in outcome after TBI. Therefore, simultaneous targeting of several injury factors using multipotential drugs may maximize the likelihood of developing a successful therapeutic intervention to improve outcome in TBI patients.
Multipotential drug treatment strategies for traumatic brain injury
Given the multifactorial nature of the secondary injury processes after trauma, it is unlikely that targeting any single factor will result in significant improvement in outcome after TBI in human injury (see Ch. 28). Conversely, simultaneous targeting of several injury factors using multipotential drugs (Table 22.1) may maximize the likelihood of developing a successful therapeutic intervention to improve outcome in TBI patients. Due to space limitations, only a limited number of promising pharmacologic agents currently under study can be detailed here. Several of the neuroprotective agents discussed have been shown to improve outcome in multiple models of CNS injury, target multiple secondary injury pathways, and are currently or likely to be studied in randomized clinical trials for head injury (Fig. 22.2). In addition, we also discuss some emerging multipotential strategies that show particular promise in preclinical TBI studies and warrant further investigation.
Table 22.1.
Multipotential drugs for traumatic brain injury
PARP, Poly (ADP-ribose) polymerase.
Fig. 22.2.
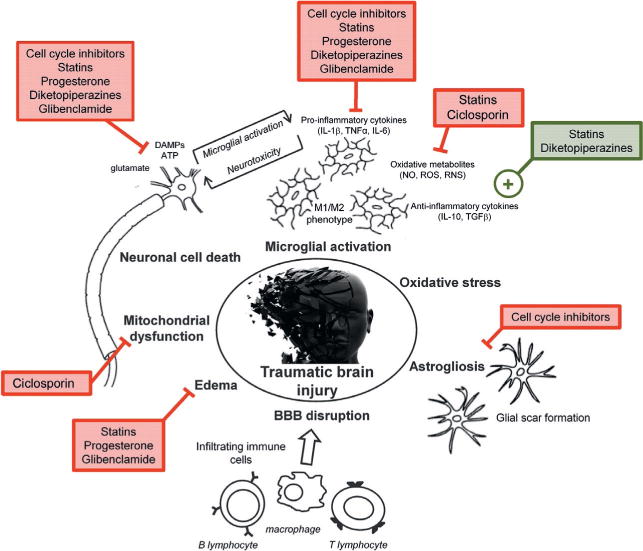
Multipotential drug treatment strategies for traumatic brain injury.
Statins
The 3-hydroxy-3-methyglutaryl coenzyme A (HMGCoA) reductase inhibitors (statins) are inhibitors of cholesterol biosynthesis, but have additional pleiotropic properties (Cucchiara and Kasner, 2001) that make them potentially attractive neuroprotective agents (Wible and Laskowitz, 2010). At the microvasculature level, statins increase endothelium-derived nitric oxide production (Eto et al., 2002), reduce vascular inflammation (Maeda et al., 2003), and limit hemorrhagic stroke (Delanty et al., 2001); after experimental TBI, they reduce post-traumatic hypoperfusion and rebound hyperemia (Wang et al., 2007). Statins protect cortical neurons from NMDA-induced excitotoxic death (Zacco et al., 2003), and improve neuronal survival in TBI models (Lu et al., 2004, 2007; Qu et al., 2005; Wang et al., 2007). They decrease apoptosis after trauma (Wu et al., 2008a), and favorably alter the ratio of antiapoptotic to proapoptotic factors (Lu et al., 2007). Statins may also promote the growth and differentiation of new neurons (Lu et al., 2007; Wu et al., 2008b); increased neurogenesis may reflect upregulation of neurotrophic factors such as brain-derived neurotrophic factor (BDNF) (Wu et al., 2008b) and vascular endothelial growth factor (VEGF) (Chen et al., 2005).
Statins exert potent anti-inflammatory effects, in part by decreasing the formation of isoprenoids. In TBI models, statins have been shown to limit production of proinflammatory mediators, glial cell activation and cerebral edema, while increasing BBB integrity (Chen et al., 2007, 2009; Wang et al., 2007). They decrease IL-1β (Chen et al., 2007, 2009), TNF-α (Chen et al., 2007, 2009; Wang et al., 2007), IL-6 (Wang et al., 2007; Chen et al., 2009) and intracellular adhesion molecule 1 (ICAM-1) (Chen et al., 2009) expression levels after TBI. Inhibition of the Toll-like receptor 4 and the nuclear factor κB (NF-κB) signaling pathways are potential mechanisms by which statins may modulate the inflammatory response (Chen et al., 2009).
Preclinical studies demonstrate that statins target multiple secondary injury pathways and improve functional outcome after TBI (Lu et al., 2004, 2007; Qu et al., 2005; Chen et al., 2007, 2009; Wang et al., 2007, Wu et al., 2008a). Furthermore, the therapeutic window for this class of drugs is relatively large, with treatment 24 hours after TBI resulting in long-term functional improvements and reduced neuronal cell loss (Lu et al., 2004, 2007). Importantly, statins are well tolerated, easy to administer, have well-defined side-effects, are easily monitored, and have a long clinical track record in critically ill patients (Tseng et al., 2005). A small (n = 22) prospective, randomized, double-blind phase II clinical trial in TBI has been performed using rosuvastatin in patients with moderate TBI (GCS of 9–13). Treatment showed a modest improvement in TBI-associated amnesia and disorientation time scores within the first 2–3 weeks, although scores were similar in treatment and placebo groups at 3 months follow-up (Tapia-Perez et al., 2008). Further investigations are required for this class of drugs and other phase II clinical trials to evaluate rosuvastatin and atorvastatin in the treatment of head injury are planned. Of note, high dose statin treatment (atorvastatin) reduced the overall incidence of strokes and of cardiovascular events in a large scale double-blind, randomized, placebo-controlled clinical trial (ClinicalTrials.gov number, NCT00147602), despite a small increase in the incidence of hemorrhagic stroke (Amarenco et al., 2006).
Progesterone
Progesterone is a neurosteroid whose receptors are expressed in the CNS of both males and females (Camacho-Arroyo et al., 1994). Neuroprotective effects for progesterone have been reported in experimental spinal cord injury (SCI) (Gonzalez Deniselle et al., 2002), stroke (Jiang et al., 1996), and TBI (Roof and Hall, 2000). Roof and colleagues (1993) observed that female rats performed better than males in the Morris water maze after experimental TBI and that progesterone-treated male rats were less impaired in the task than vehicle-treated animals (Roof et al., 1994). These effects were associated with a reduction in neuronal cell death (He et al., 2004). Progesterone attenuates glutamate excitotoxicity (Smith, 1991), modulates apoptotic pathways (Djebaili et al., 2005; Yao et al., 2005), and decreases diffuse axonal injury (O’Connor et al., 2007); it also reduces membrane lipid peroxidation (Roof et al., 1997), possibly by upregulating superoxide dismutase (Moorthy et al., 2005). Treatment limits inflammation after TBI, attenuating NF-κB, p65 and TNF-α expression (Pettus et al., 2005; Pan et al., 2007). Progesterone also reduces edema after injury (Roof et al., 1996; O’Connor et al., 2007) through mechanisms that may include inhibition of Na+,K+-ATPase, modulation of vasopressin (Vink and Nimmo, 2009; Vink and van den Heuvel, 2010), or maintaining BBB function by upregulating P-glycoprotein (Cutler et al., 2007).
Although progesterone has been shown to have neuroprotective activity in models of experimental stroke, SCI, and TBI, recent reports question its effectiveness in trauma models. Importantly, a systematic review of progesterone treatment in CNS injury raised concerns about the methodologic quality of the TBI studies, and quantitative evaluation revealed possible experimental bias in these studies (Gibson et al., 2008). In addition, a highly regarded neurotrauma group failed to observe any protective effects of progesterone, at doses reported to be effective by others, in well characterized models of either TBI or SCI (Fee et al., 2007; Gilmer et al., 2008). Moreover, a clinically relevant therapeutic window has not been examined in most published neurotrauma studies, with the majority evaluating very early treatment times (i.e., 0–2 hours) after injury (Gibson et al., 2008).
Despite the limitations of the preclinical data, two randomized, double-blind, placebo-controlled phase II clinical trials for progesterone have been conducted (Wright et al., 2007; Xiao et al., 2008). Although these trials used different doses and treatment regimens, both indicated trends toward improved outcome in progesterone-treated patients. In the ProTECT II trial moderate to severe TBI patients (n = 100) were randomized to receive progesterone or lipid vehicle within 11 hours of injury (Wright et al., 2007), and the study was powered to detect predetermined safety measures such as hypotension, pneumonia, and hepatotoxicity. There were no differences in these safety measures between progesterone and placebo groups, although death within 30 days of injury was lower in the progesterone group. However, the number of patients studied was limited and randomization in the ProTECT II trial was 3:1, resulting in relatively few vehicle treated controls. In the other phase II trial only severe TBI patients (n = 159) were enrolled and randomized to receive progesterone or matching placebo within 8 hours of injury (Xiao et al., 2008). Patients who received progesterone had a higher favorable outcome, as measured by GOS at 3 months postinjury.
Based upon the positive trends in the phase II studies, two phase III multicenter clinical trials are now in process in moderate to severe TBI patients. The ProTECT III trial (NINDS/NIH) examines intravenous (IV) progesterone initiated within 4 hours of injury (with waiver of informed consent to allow such early intervention) and continued for 72 hours, which is then tapered off over 24 hours for a total of 96 hours infusion (n = 1140 moderate to severe TBI) (Stein and Wright, 2010). The SyNAPSE phase III trial (BHR Pharma) is a global, multicenter trial of BHR-100 (IV progesterone infusion) with treatment to be initiated within 8 hours of injury and duration of infusion is 120 hours (n = 1200 severe TBI patients) (Stein and Wright, 2010). Unfortunately, the ProTECT III trial was stopped early in 2014 after the independent Data and Safety Monitoring Board determined that the data indicated it was very unlikely that progesterone treatment would demonstrate better outcomes compared to a placebo control in this trial. The SyNAPSE phase III trial is nearing completion, and final results are expected in May 2014.
Ciclosporin
There are significant impairments of aerobic metabolism early after TBI (Vespa et al., 2005). Mitochondrial failure leads to energy and ionic imbalances, reduced brain ATP levels, changes in mitochondrial permeability transition, release of cytochrome c and induction of proapoptotic events (Mazzeo et al., 2009). The immunosuppressant drug cyclosporine A (CsA; INN ciclosporin) attenuates mitochondrial failure by binding to cyclophilin D and stabilizing the mitochondrial permeability transition pore (Szabo and Zoratti, 1991). Treatment with CsA reduced axonal damage in diffuse axonal injury models (Okonkwo and Povlishock, 1999; Okonkwo et al., 1999) and decreased lesion size following CCI TBI (Sullivan et al., 2000a, b). Improved outcome was associated with preserved mitochondrial function (Sullivan et al., 1999b) or structural integrity (Mbye et al., 2009; Okonkwo et al., 1999). CsA attenuates lipid peroxidation and free radical oxidative damage to mitochondrial proteins (Mbye et al., 2008, 2009). Given these multipotential effects and long therapeutic window (up to 24 hours (Sullivan et al., 2000a)), CsA appears to be an attractive candidate for clinical investigation.
A prospective randomized, placebo-controlled, double-blinded clinical trial of CsA was performed at two centers for severe human TBI. Patients treated with CsA showed significantly lower lactate/pyruvate ratios (Mazzeo et al., 2008), which may reflect improved metabolism (Vespa et al., 2005). Larger phase III clinical trials for CsA are in preparation. Potential advantages for CsA are that it is FDA approved for other uses and off-patent. However, CsA shows relatively poor brain penetration, has a biphasic drug-response curve, and prolonged use adversely impacts the immune system (Margulies and Hicks, 2009).
Diketopiperazines
Diketopiperazines are cyclized dipeptides that were developed through a rational drug design program based on the tripeptide thyrotropin-releasing hormone (TRH) (Faden et al., 2005a). TRH and TRH analogs inhibit multiple secondary injury factors and processes (Faden et al., 2005a). They were shown to be highly neuroprotective in experimental neurotrauma across many laboratories and a small clinical randomized study of TRH in human SCI was promising (Pitts et al., 1995). Four structurally different diketopiperazines demonstrated significant neuroprotective properties both in vitro and in animal TBI studies (Faden et al., 2005b). One of these (35b) showed effectiveness across TBI models and species. In neuronal cell cultures, 35b provided neuroprotection in multiple models of necrotic and apoptotic cell death (Faden et al., 2003b). Intravenous administration of 35b reduced both lesion volume and improved functional recovery after FPI in rats and CCI in mice (Faden et al., 2003a, b). The therapeutic window was at least 8 hours. Treatment also significantly reduced apoptotic cell death (Faden et al., 2003b). Effects appeared to be pleiotropic, with treatment reducing multiple potential secondary injury factors (cyclins, calpains, cathepsin), while upregulating various endogenous neuroprotective and neurotrophic factors (BDNF, HSP-70, HIF-1) (Faden et al., 2004, 2005b). In addition, data are available regarding pharmacokinetics, brain penetration after systemic administration and preclinical toxicology in the rat. Similar pleiotropic neuroprotective effects were reported with another diketopiperazine – cyclo-L-glycyl-L-2-allylproline (NNZ 2591) – in rats with hypoxic-ischemic brain injury (Guan et al., 2007). NNZ 2591 treatment improved functional recovery and long-term histologic outcomes, and reduced caspase-3 mediated apoptosis and microglial activation (Guan et al., 2007). Given their multipotential neuroprotective effects in experimental TBI models, their clinically relevant therapeutic window, and their safety profile, the diketopiperazines are attractive candidates for further clinical investigation.
SUR1-regulated NCCa-ATP channel inhibitors
Edema and progressive secondary hemorrhage are important secondary injury mechanisms that contribute to neurologic impairments in patients after TBI. Recent studies suggest that upregulation of sulfonylurea receptor 1 (SUR1)-regulated NCCa-ATP channels in microvascular endothelium play a key role in these secondary injury pathways (Simard et al., 2008). Pharmacologic blockade using the SUR1 inhibitor glibenclamide reduced edema, secondary hemorrhage, inflammation, apoptosis, and lesion size in experimental TBI and subarachnoid hemorrhage models (Simard et al., 2009a, b). In particular, glibenclamide treatment significantly reduced neuronal degeneration and apoptosis within the hippocampus after TBI and was highly effective in sparing rapid spatial learning in the Morris water maze test (Patel et al., 2010). Glibenclamide treatment is also neuroprotective in numerous models of preclinical stroke and neonatal hypoxia-ischemia (Simard et al., 2012). At present, a prospective multicenter, placebo-controlled, double-blind phase IIa trial of RP-1127 (glyburide for injection; Remedy Pharmaceuticals Inc., NY) is underway to test the effect of RP-1127 in patients with moderate-to-severe TBI (ClinicalTrials.gov Identifier: NCT01454154).
Cell cycle inhibitors
The cell cycle is upregulated in both mitotic (astrocytes and microglia) and postmitotic (neurons, oligodendrocytes) cells of the brain after CNS injury, and post-traumatic cell cycle activation is associated with caspase-mediated neuronal cell death and glial cell proliferation (Di Giovanni et al., 2005). Cell cycle inhibitors have been extensively studied for their role in cancer treatment, and inhibitors such as flavopiridol, roscovitine, and olomoucine have been shown to exert powerful neuroprotective effects in in vitro model systems for neuronal apoptosis (Padmanabhan et al., 1999; Verdaguer et al., 2004; Cernak et al., 2005), and they also produce potent inhibitory effects on the proliferation and activation of astrocytes and microglia (Cernak et al., 2005; Di Giovanni et al., 2005; Hilton et al., 2008). Given the multipotential properties of cell cycle inhibitors they are considered to be promising candidate drugs for the treatment of TBI.
In experimental studies treatment with flavopiridol, an inhibitor of all major cyclic-dependent kinases (CDKs) after LFP TBI in rats significantly reduced lesion volume, and improved long-term cognitive and sensorimotor recovery (Di Giovanni et al., 2005). In addition, flavopiridol treatment blocked caspase-mediated neuronal cell death and significantly reduced glial cell activation, and these changes were associated with the suppression of the cell cycle in neurons, astrocytes, and microglia within the injured cortex. Follow-up studies demonstrated that delayed treatment with flavopiridol as late as 24 hours postinjury resulted in significantly reduced TBI lesion volumes (Cernak et al., 2005). Roscovitine is a more selective cell cycle inhibitor, which acts specifically on CDKs 1, 2, and 5 (Meijer et al., 1997), and it too had significant neuroprotective actions in rat and mouse TBI models. In addition to improving functional recovery and reducing the TBI lesion, central and systemic administration of roscovitine markedly reduced microglial-mediated neuroinflammation and associated neurodegeneration up to 28 days postinjury (Hilton et al., 2008; Kabadi et al., 2012a).
In addition, a more potent second-generation roscovitine analog, CR8, provides significant neuroprotection in both mouse CCI and rat LFP experimental TBI models. Central administration of CR8, administered at 3 hours post-CCI, attenuated cell cycle activation pathways and reduced post-traumatic apoptotic cell death at 24 hours postinjury as well as significantly attenuated sensorimotor and cognitive deficits, decreased lesion volume, and improved neuronal survival in the cortex, CA3 and dentate gyrus region of the hippocampus and thalamus (Kabadi et al., 2012b). Significantly, systemic administration of CR8 markedly improved cognitive performance after TBI (Kabadi et al., 2012b). Administration of CR8 at 3 hours after LFP reduced markers of cell cycle activation, attenuated neuronal loss in the cortex, hippocampus and thalamus as well as limited cortical microglial and astrocyte activation (Kabadi et al., 2014). Furthermore, CR8 treatment attenuated sensorimotor and cognitive deficits, alleviated depressive-like symptoms, and decreased lesion volume (Kabadi et al., 2014). The role of cell cycle activation in post-traumatic secondary injury has also been confirmed using genetic deletion of cyclin d1, a key modulator of the cell cycle. Animals lacking cyclin d1 expression demonstrated significantly reduced microglial activation and improved histologic and functional outcomes after TBI (Kabadi et al., 2012c), thereby underpinning the key role that cell cycle activation plays in the neuroinflammatory response to TBI. Several cell cycle inhibitors have been studied in randomized clinical trials for cancer (Fischer and Gianella-Borradori, 2005). Although they are toxic when administered chronically, cell cycle inhibitors have required only single dose administration to exert powerful therapeutic effects in experimental TBI models. Therefore, given their clinically relevant therapeutic window and their multipotential neuroprotective effects, cell cycle inhibitors such as flavopiridol or roscovitine are attractive candidates for future clinical translation.
CONCLUSION
TBI is a highly complex disorder, which is characterized by multiple interacting secondary injury cascades. The focus on highly selective “laser-guided” neuroprotective strategies has given way to the concept of multipotential drugs that modulate multiple secondary injury pathways. The potential limitations of using single models and species for preclinical screening of neuroprotective agents has been increasingly underscored, as have the methodologic differences between clinical and preclinical trials. At the same time, there has been increasing attention directed toward methodologic issues in clinical trial design and analysis. In the future, it will be important to better facilitate bidirectional translational research between preclinical and clinical investigators – which should serve to improve both approaches to animal modeling and the design of clinical trials. Future advances in clinical data sharing (e.g., using common data elements) should improve TBI classification in ways that may lead to delineation of specific patient subgroups that may benefit from better targeted neuroprotective strategies.
References
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Amarenco P, Bogousslavsky J, Callahan A, 3rd, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549–559. doi: 10.1056/NEJMoa061894. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kang HC, Haince JF, et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med. 2011;17:692–699. doi: 10.1038/nm.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriessen TM, Jacobs B, Vos PE. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med. 2010;14:2381–2392. doi: 10.1111/j.1582-4934.2010.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Krady JK, O’Malley M, et al. The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J Neurosci. 2002;22:6071–6082. doi: 10.1523/JNEUROSCI.22-14-06071.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendlin BB, Ries ML, Lazar M, et al. Longitudinal changes in patients with traumatic brain injury assessed with diffusion-tensor and volumetric imaging. Neuroimage. 2008;42:503–514. doi: 10.1016/j.neuroimage.2008.04.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland J, Roy S, Xanthoudakis S, et al. Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci. 2002;22:2637–2649. doi: 10.1523/JNEUROSCI.22-07-02637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong J. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 2002;103:607–614. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- Bredesen DE. Key note lecture: toward a mechanistic taxonomy for cell death programs. Stroke. 2007;38:652–660. doi: 10.1161/01.STR.0000257802.82826.a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE. Programmed cell death mechanisms in neurological disease. Curr Mol Med. 2008;8:173–186. doi: 10.2174/156652408784221315. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes KR, Loane DJ, Stoica BA, et al. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation. 2012;9:43. doi: 10.1186/1742-2094-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Yang SH, Duffy PJ, et al. Functional axonal regeneration through astrocytic scar genetically modified to digest chondroitin sulfate proteoglycans. J Neurosci. 2007;27:2176–2185. doi: 10.1523/JNEUROSCI.5176-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Arroyo I, Perez-Palacios G, Pasapera AM, et al. Intracellular progesterone receptors are differentially regulated by sex steroid hormones in the hypothalamus and the cerebral cortex of the rabbit. J Steroid Biochem Mol Biol. 1994;50:299–303. doi: 10.1016/0960-0760(94)90135-x. [DOI] [PubMed] [Google Scholar]
- Cande C, Vahsen N, Kouranti I, et al. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- Cernak I, Stoica B, Byrnes KR, et al. Role of the cell cycle in the pathobiology of central nervous system trauma. Cell Cycle. 2005;4:1286–1293. doi: 10.4161/cc.4.9.1996. [DOI] [PubMed] [Google Scholar]
- Cernak I, Chang T, Ahmed FA, et al. Pathophysiological response to experimental diffuse brain trauma differs as a function of developmental age. Dev Neurosci. 2010;32:442–453. doi: 10.1159/000320085. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Ehrlich L, et al. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang C, Jiang H, et al. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab. 2005;25:281–290. doi: 10.1038/sj.jcbfm.9600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SF, Hung TH, Chen CC, et al. Lovastatin improves histological and functional outcomes and reduces inflammation after experimental traumatic brain injury. Life Sci. 2007;81:288–298. doi: 10.1016/j.lfs.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Chen G, Zhang S, Shi J, et al. Simvastatin reduces secondary brain injury caused by cortical contusion in rats: possible involvement of TLR4/NF-kappaB pathway. Exp Neurol. 2009;216:398–406. doi: 10.1016/j.expneurol.2008.12.019. [DOI] [PubMed] [Google Scholar]
- Chen CM, Yi HY, Yoon YH, et al. Alcohol use at time of injury and survival following traumatic brain injury: results from the National Trauma Data Bank. J Stud Alcohol Drugs. 2012;73:531–541. doi: 10.15288/jsad.2012.73.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A. “Simple but not simpler”: toward a unified picture of energy requirements in cell death. FASEB J. 2005;19:1783–1788. doi: 10.1096/fj.05-4200rev. [DOI] [PubMed] [Google Scholar]
- Clark R, Kochanek P, Watkins S, et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74:740–753. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- Clark RS, Nathaniel PD, Zhang X, et al. boc-Aspartyl(OMe)-fluoromethylketone attenuates mitochondrial release of cytochrome c and delays brain tissue loss after traumatic brain injury in rats. J Cereb Blood Flow Metab. 2006;27:316–326. doi: 10.1038/sj.jcbfm.9600338. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, et al. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde JR, Streit WJ. Effect of aging on the microglial response to peripheral nerve injury. Neurobiol Aging. 2006a;27:1451–1461. doi: 10.1016/j.neurobiolaging.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Conde JR, Streit WJ. Microglia in the aging brain. J Neuropathol Exp Neurol. 2006b;65:199–203. doi: 10.1097/01.jnen.0000202887.22082.63. [DOI] [PubMed] [Google Scholar]
- Conti AC, Raghupathi R, Trojanowski JQ, et al. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csuka E, Morganti-Kossmann MC, Lenzlinger PM, et al. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6 TNF-alpha TGF-beta1 and blood–brain barrier function. J Neuroimmunol. 1999;101:211–221. doi: 10.1016/s0165-5728(99)00148-4. [DOI] [PubMed] [Google Scholar]
- Cucchiara B, Kasner SE. Use of statins in CNS disorders. J Neurol Sci. 2001;187:81–89. doi: 10.1016/s0022-510x(01)00529-9. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, et al. Apoptosis-inducing factor triggered by poly(ADP-Ribose) polymerase and bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler SM, Cekic M, Miller DM, et al. Progesterone improves acute recovery after traumatic brain injury in the aged rat. J Neurotrauma. 2007;24:1475–1486. doi: 10.1089/neu.2007.0294. [DOI] [PubMed] [Google Scholar]
- Daugas E, Nochy D, Ravagnan L, et al. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–123. doi: 10.1016/s0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Declercq W, Takahashi N, Vandenabeele P. Dual face apoptotic machinery: from initiator of apoptosis to guardian of necroptosis. Immunity. 2011;35:493–495. doi: 10.1016/j.immuni.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Delanty N, Vaughan CJ, Sheehy N. Statins and neuroprotection. Expert Opin Investig Drugs. 2001;10:1847–1853. doi: 10.1517/13543784.10.10.1847. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Movsesyan V, Ahmed F, et al. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A. 2005;102:8333–8338. doi: 10.1073/pnas.0500989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebaili M, Guo Q, Pettus EH, et al. The neurosteroids progesterone and allopregnanolone reduce cell death gliosis and functional deficits after traumatic brain injury in rats. J Neurotrauma. 2005;22:106–118. doi: 10.1089/neu.2005.22.106. [DOI] [PubMed] [Google Scholar]
- Eldadah BA, Yakovlev AG, Faden AI. The role of CED-3-related cysteine proteases in apoptosis of cerebellar granule cells. J Neurosci. 1997;17:6105–6113. doi: 10.1523/JNEUROSCI.17-16-06105.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Kozai T, Cosentino F, et al. Statin prevents tissue factor expression in human endothelial cells: role of Rho/Rho-kinase and Akt pathways. Circulation. 2002;105:1756–1759. doi: 10.1161/01.cir.0000015465.73933.3b. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, et al. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Faden AI, Fox GB, Di X, et al. Neuroprotective and nootropic actions of a novel cyclized dipeptide after controlled cortical impact injury in mice. J Cereb Blood Flow Metab. 2003a;23:355–363. doi: 10.1097/01.WCB.0000046144.31247.33. [DOI] [PubMed] [Google Scholar]
- Faden AI, Knoblach SM, Cernak I, et al. Novel diketopiperazine enhances motor and cognitive recovery after traumatic brain injury in rats and shows neuroprotection in vitro and in vivo. J Cereb Blood Flow Metab. 2003b;23:342–354. doi: 10.1097/01.WCB.0000046143.31247.FD. [DOI] [PubMed] [Google Scholar]
- Faden AI, Knoblach SM, Movsesyan VA, et al. Novel small peptides with neuroprotective and nootropic properties. J Alzheimers Dis. 2004;6:S93–S97. doi: 10.3233/jad-2004-6s603. [DOI] [PubMed] [Google Scholar]
- Faden AI, Knoblach SM, Movsesyan VA, et al. Novel neuroprotective tripeptides and dipeptides. Ann N Y Acad Sci. 2005a;1053:472–481. doi: 10.1111/j.1749-6632.2005.tb00057.x. [DOI] [PubMed] [Google Scholar]
- Faden AI, Movsesyan VA, Knoblach SM, et al. Neuroprotective effects of novel small peptides in vitro and after brain injury. Neuropharmacology. 2005b;49:410–424. doi: 10.1016/j.neuropharm.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Fan L, Young PR, Barone FC, et al. Experimental brain injury induces expression of interleukin-1 beta mRNA in the rat brain. Brain Res Mol Brain Res. 1995;30:125–130. doi: 10.1016/0169-328x(94)00287-o. [DOI] [PubMed] [Google Scholar]
- Fan L, Young PR, Barone FC, et al. Experimental brain injury induces differential expression of tumor necrosis factor-alpha mRNA in the CNS. Brain Res Mol Brain Res. 1996;36:287–291. doi: 10.1016/0169-328x(95)00274-v. [DOI] [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, et al. Traumatic Brain Injury in the United States: Emergency Department Visits Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta, GA: 2010. [Google Scholar]
- Fee DB, Swartz KR, Joy KM, et al. Effects of progesterone on experimental spinal cord injury. Brain Res. 2007;1137:146–152. doi: 10.1016/j.brainres.2006.12.024. [DOI] [PubMed] [Google Scholar]
- Finkelstein EA, Corso PS, Miller TR. Incidence and Economic Burden of Injuries in the United States. Oxford University Press; New York: 2006. [Google Scholar]
- Fischer PM, Gianella-Borradori A. Recent progress in the discovery and development of cyclin-dependent kinase inhibitors. Expert Opin Investig Drugs. 2005;14:457–477. doi: 10.1517/13543784.14.4.457. [DOI] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox GB, Levasseur RA, Faden AI. Behavioral responses of C57BL/6 FVB/N and 129/SvEMS mouse strains to traumatic brain injury: implications for gene targeting approaches to neurotrauma. J Neurotrauma. 1999;16:377–389. doi: 10.1089/neu.1999.16.377. [DOI] [PubMed] [Google Scholar]
- Galbraith S. Head injuries in the elderly. Br Med J (Clin Res Ed) 1987;294:325. doi: 10.1136/bmj.294.6568.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile NT, Mcintosh TK. Antagonists of excitatory amino acids and endogenous opioid peptides in the treatment of experimental central nervous system injury. Ann Emerg Med. 1993;22:1028–1034. doi: 10.1016/s0196-0644(05)82746-5. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Leclercq PD, Moyes L, et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Gray LJ, Bath PM, et al. Progesterone for the treatment of experimental brain injury; a systematic review. Brain. 2008;131:318–328. doi: 10.1093/brain/awm183. [DOI] [PubMed] [Google Scholar]
- Gilmer LK, Roberts KN, Scheff SW. Efficacy of progesterone following a moderate unilateral cortical contusion injury. J Neurotrauma. 2008;25:593–602. doi: 10.1089/neu.2007.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, et al. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Gonzalez Deniselle MC, Lopez Costa JJ, Gonzalez SL, et al. Basis of progesterone protection in spinal cord neurodegeneration. J Steroid Biochem Mol Biol. 2002;83:199–209. doi: 10.1016/s0960-0760(02)00262-5. [DOI] [PubMed] [Google Scholar]
- Goodman JC, Robertson CS, Grossman RG, et al. Elevation of tumor necrosis factor in head injury. J Neuroimmunol. 1990;30:213–217. doi: 10.1016/0165-5728(90)90105-v. [DOI] [PubMed] [Google Scholar]
- Goodman MD, Makley AT, Campion EM, et al. Preinjury alcohol exposure attenuates the neuroinflammatory response to traumatic brain injury. J Surg Res. 2013;184:1053–1058. doi: 10.1016/j.jss.2013.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gottron FJ, Ying HS, Choi DW. Caspase inhibition selectively reduces the apoptotic component of oxygen-glucose deprivation-induced cortical neuronal cell death. Mol Cell Neurosci. 1997;9:159–169. doi: 10.1006/mcne.1997.0618. [DOI] [PubMed] [Google Scholar]
- Graham DI, Adams JH, Murray LS, et al. Neuropathology of the vegetative state after head injury. Neuropsychol Rehabil. 2005;15:198–213. doi: 10.1080/09602010443000452. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Guan J, Mathai S, Harris P, et al. Peripheral administration of a novel diketopiperazine NNZ 2591 prevents brain injury and improves somatosensory-motor function following hypoxia-ischemia in adult rats. Neuropharmacology. 2007;53:749–762. doi: 10.1016/j.neuropharm.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, et al. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- Hamby AM, Suh SW, Kauppinen TM, et al. Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke. 2007;38:632–636. doi: 10.1161/01.STR.0000250742.61241.79. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- He J, Hoffman SW, Stein DG. Allopregnanolone a progesterone metabolite enhances behavioral recovery and decreases neuronal loss after traumatic brain injury. Restor Neurol Neurosci. 2004;22:19–31. [PubMed] [Google Scholar]
- He M, Liu WG, Wen L, et al. Influence of acute ethanol intoxication on neuronal apoptosis and Bcl-2 protein expression after severe traumatic brain injury in rats. Chin J Traumatol. 2013;16:136–139. [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton GD, Stoica BA, Byrnes KR, et al. Roscovitine reduces neuronal loss glial activation and neurologic deficits after brain trauma. J Cereb Blood Flow Metab. 2008;28:1845–1859. doi: 10.1038/jcbfm.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- Holmin S, Mathiesen T. Long-term intracerebral inflammatory response after experimental focal brain injury in rat. Neuroreport. 1999;10:1889–1891. doi: 10.1097/00001756-199906230-00017. [DOI] [PubMed] [Google Scholar]
- Jiang N, Chopp M, Stein D, et al. Progesterone is neuroprotective after transient middle cerebral artery occlusion in male rats. Brain Res. 1996;735:101–107. doi: 10.1016/0006-8993(96)00605-1. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Byrnes KR, et al. Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. J Cereb Blood Flow Metab. 2012a;32:137–149. doi: 10.1038/jcbfm.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Hanscom M, et al. CR8 a selective and potent CDK inhibitor provides neuroprotection in experimental traumatic brain injury. Neurotherapeutics. 2012b;9:405–421. doi: 10.1007/s13311-011-0095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Loane DJ, et al. Cyclin D1 gene ablation confers neuroprotection in traumatic brain injury. J Neurotrauma. 2012c;29:813–827. doi: 10.1089/neu.2011.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Loane DJ, et al. CR8 a novel inhibitor of CDK limits microglial activation astrocytosis neuronal loss and neurologic dysfunction after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2014;34:502–513. doi: 10.1038/jcbfm.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. Poly(ADP-ribose) polymerase-1 promotes microglial activation proliferation and matrix metalloproteinase-9-mediated neuron death. J Immunol. 2005;174:2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- Kauppinen TM, Suh SW, Berman AE, et al. Inhibition of poly(ADP-ribose) polymerase suppresses inflammation and promotes recovery after ischemic injury. J Cereb Blood Flow Metab. 2009;29:820–829. doi: 10.1038/jcbfm.2009.9. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblach SM, Faden AI. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimentaltraumatic brain injury. Exp Neurol. 1998;153:143–151. doi: 10.1006/exnr.1998.6877. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Nikolaeva M, Huang X, et al. Multiple caspases are activated after traumatic brain injury: evidence for involvement in functional outcome. J Neurotrauma. 2002;19:1155–1170. doi: 10.1089/08977150260337967. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Alroy DA, Nikolaeva M, et al. Caspase inhibitor z-DEVD-fmk attenuates calpain and necrotic cell death in vitro and after traumatic brain injury. J Cereb Blood Flow Metab. 2004;24:1119–1132. doi: 10.1097/01.WCB.0000138664.17682.32. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Huang X, Vangelderen J, et al. Selective caspase activation may contribute to neurological dysfunction after experimental spinal cord trauma. J Neurosci Res. 2005;80:369–380. doi: 10.1002/jnr.20465. [DOI] [PubMed] [Google Scholar]
- Kobori N, Clifton GL, Dash P. Altered expression of novel genes in the cerebral cortex following experimental brain injury. Brain Res Mol Brain Res. 2002;104:148–158. doi: 10.1016/s0169-328x(02)00331-5. [DOI] [PubMed] [Google Scholar]
- Krajewska M, You Z, Rong J, et al. Neuronal deletion of caspase 8 protects against brain injury in mouse models of controlled cortical impact and kainic acid-induced excitotoxicity. PLoS One. 2011;6:e24341. doi: 10.1371/journal.pone.0024341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremlev SG, Palmer C. Interleukin-10 inhibits endotoxin-induced pro-inflammatory cytokines in microglial cell cultures. J Neuroimmunol. 2005;162:71–80. doi: 10.1016/j.jneuroim.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Stoica BA, Sabirzhanov B, et al. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–1411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Hickey RW, Chen Y, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540–550. doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- Levin S. Apoptosis necrosis or oncosis: what is your diagnosis? A report from the Cell Death Nomenclature Committee of the Society of Toxicologic Pathologists. Toxicol Sci. 1998;41:155–156. doi: 10.1006/toxs.1997.2432. [DOI] [PubMed] [Google Scholar]
- Levin S, Bucci TJ, Cohen SM, et al. The nomenclature of cell death: recommendations of an ad hoc Committee of the Society of Toxicologic Pathologists. Toxicol Pathol. 1999;27:484–490. doi: 10.1177/019262339902700419. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Liu CL, Chen S, Dietrich D, et al. Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28:674–683. doi: 10.1038/sj.jcbfm.9600587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Goussev A, Chen J, et al. Atorvastatin reduces neurological deficit and increases synaptogenesis angiogenesis and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma. 2004;21:21–32. doi: 10.1089/089771504772695913. [DOI] [PubMed] [Google Scholar]
- Lu KT, Wang YW, Wo YY, et al. Extracellular signal-regulated kinase-mediated IL-1-induced cortical neuron damage during traumatic brain injury. Neurosci Lett. 2005a;386:40–45. doi: 10.1016/j.neulet.2005.05.057. [DOI] [PubMed] [Google Scholar]
- Lu KT, Wang YW, Yang JT, et al. Effect of interleukin-1 on traumatic brain injury-induced damage to hippocampal neurons. J Neurotrauma. 2005b;22:885–895. doi: 10.1089/neu.2005.22.885. [DOI] [PubMed] [Google Scholar]
- Lu D, Qu C, Goussev A, et al. Statins increase neurogenesis in the dentate gyrus reduce delayed neuronal death in the hippocampal CA3 region and improve spatial learning in rat after traumatic brain injury. J Neurotrauma. 2007;24:1132–1146. doi: 10.1089/neu.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, et al. Bid a Bcl2 interacting protein mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Luo CL, Li BX, Li QQ, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. NSC. 2011;184:54–63. doi: 10.1016/j.neuroscience.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- Maas AI, Murray G, Henney H, 3rd, et al. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised placebo-controlled clinical trial. Lancet Neurol. 2006;5:38–45. doi: 10.1016/S1474-4422(05)70253-2. [DOI] [PubMed] [Google Scholar]
- Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7:115–126. doi: 10.1016/j.nurt.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Kawane T, Horiuchi N. Statins augment vascular endothelial growth factor expression in osteoblastic cells via inhibition of protein prenylation. Endocrinology. 2003;144:681–692. doi: 10.1210/en.2002-220682. [DOI] [PubMed] [Google Scholar]
- Magnuson J, Leonessa F, Ling GS. Neuropathology of explosive blast traumatic brain injury. Curr Neurol Neurosci Rep. 2012;12:570–579. doi: 10.1007/s11910-012-0303-6. [DOI] [PubMed] [Google Scholar]
- Majno G, Joris I. Apoptosis oncosis and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- Majno G, Joris I. Commentary: on the misuse of the term “necrosis”: a step in the right direction. Toxicol Pathol. 1999;27:494. doi: 10.1177/019262339902700422. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Margulies S, Hicks R. Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26:925–939. doi: 10.1089/neu.2008.0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell WL, Mackinnon MA, Smith DH, et al. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol. 2006;65:478–488. doi: 10.1097/01.jnen.0000229241.28619.75. [DOI] [PubMed] [Google Scholar]
- Mazzeo AT, Alves OL, Gilman CB, et al. Brain metabolic and hemodynamic effects of cyclosporin A after human severe traumatic brain injury: a microdialysis study. Acta Neurochir (Wien) 2008;150:1019–1031. doi: 10.1007/s00701-008-0021-7. discussion 1031. [DOI] [PubMed] [Google Scholar]
- Mazzeo AT, Beat A, Singh A, et al. The role of mitochondrial transition pore and its modulation in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol. 2009;218:363–370. doi: 10.1016/j.expneurol.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Sullivan PG, et al. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811 a non-immunosuppressive cyclosporin A analog. Exp Neurol. 2008;209:243–253. doi: 10.1016/j.expneurol.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Carrico KM, et al. Comparative neuroprotective effects of cyclosporin A and NIM811 a nonimmunosuppressive cyclosporin A analog following traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:87–97. doi: 10.1038/jcbfm.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, et al. Biochemical and cellular effects of roscovitine a potent and selective inhibitor of the cyclin-dependent kinases cdc2 cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Moorthy K, Sharma D, Basir SF, et al. Administration of estradiol and progesterone modulate the activities of antioxidant enzyme and aminotransferases in naturally menopausal rats. Exp Gerontol. 2005;40:295–302. doi: 10.1016/j.exger.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Hans VH, Lenzlinger PM, et al. TGF-beta is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. J Neurotrauma. 1999;16:617–628. doi: 10.1089/neu.1999.16.617. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Otto VI, et al. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165–177. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, Bye N, et al. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Moubarak RS, Yuste VJ, Artus C, et al. Sequential activation of PARP-1 calpains and Bax is essential in AIF-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, et al. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Narayan RK, Michel ME, Ansell B, et al. Clinical trials in head injury. J Neurotrauma. 2002;19:503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale JE, Ahmed F, Cernak I, et al. Gene expression profile changes are commonly modulated across models and species after traumatic brain injury. J Neurotrauma. 2003;20:907–927. doi: 10.1089/089771503770195777. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- O’Connor CA, Cernak I, Johnson F, et al. Effects of progesterone on neurologic and morphologic outcome following diffuse traumatic brain injury in rats. Exp Neurol. 2007;205:145–153. doi: 10.1016/j.expneurol.2007.01.034. [DOI] [PubMed] [Google Scholar]
- Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- Okonkwo DO, Buki A, Siman R, et al. Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport. 1999;10:353–358. doi: 10.1097/00001756-199902050-00026. [DOI] [PubMed] [Google Scholar]
- Osato K, Sato Y, Ochiishi T, et al. Apoptosis-inducing factor deficiency decreases the proliferation rate and protects the subventricular zone against ionizing radiation. Cell Death Dis. 2010;1:e84. doi: 10.1038/cddis.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan J, Park DS, Greene LA, et al. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci. 1999;19:8747–8756. doi: 10.1523/JNEUROSCI.19-20-08747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan DS, Liu WG, Yang XF, et al. Inhibitory effect of progesterone on inflammatory factors after experimental traumatic brain injury. Biomed Environ Sci. 2007;20:432–438. [PubMed] [Google Scholar]
- Patel AD, Gerzanich V, Geng Z, et al. Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury. J Neuropathol Exp Neurol. 2010;69:1177–1190. doi: 10.1097/NEN.0b013e3181fbf6d6. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, et al. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol. 1999;145:503–514. doi: 10.1083/jcb.145.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings JL, Bachulis BL, Simons CT, et al. Survival after severe brain injury in the aged. Arch Surg. 1993;128:787–793. doi: 10.1001/archsurg.1993.01420190083011. discussion 793–794. [DOI] [PubMed] [Google Scholar]
- Pettus EH, Wright DW, Stein DG, et al. Progesterone treatment inhibits the inflammatory agents that accompany traumatic brain injury. Brain Res. 2005;1049:112–119. doi: 10.1016/j.brainres.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Piao CS, Loane DJ, Stoica BA, et al. Combined inhibition of cell death induced by apoptosis inducing factor and caspases provides additive neuroprotection in experimental traumatic brain injury. Neurobiol Dis. 2012;46:745–758. doi: 10.1016/j.nbd.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinteaux E, Parker LC, Rothwell NJ, et al. Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J Neurochem. 2002;83:754–763. doi: 10.1046/j.1471-4159.2002.01184.x. [DOI] [PubMed] [Google Scholar]
- Pitts LH, Ross A, Chase GA, et al. Treatment with thyrotropin-releasing hormone (TRH) in patients with traumatic spinal cord injuries. J Neurotrauma. 1995;12:235–243. doi: 10.1089/neu.1995.12.235. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, et al. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa-Wagner A, Carmichael ST, Kokaia Z, et al. The response of the aged brain to stroke: too much too soon? Curr Neurovasc Res. 2007;4:216–227. doi: 10.2174/156720207781387213. [DOI] [PubMed] [Google Scholar]
- Qiu J, Whalen MJ, Lowenstein P, et al. Upregulation of the Fas receptor death-inducing signaling complex after traumatic brain injury in mice and humans. J Neurosci. 2002;22:3504–3511. doi: 10.1523/JNEUROSCI.22-09-03504.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C, Lu D, Goussev A, et al. Effect of atorvastatin on spatial memory neuronal survival and vascular density in female rats after traumatic brain injury. J Neurosurg. 2005;103:695–701. doi: 10.3171/jns.2005.103.4.0695. [DOI] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Dhodda VK, Song G, et al. Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis. J Neurosci Res. 2003;71:208–219. doi: 10.1002/jnr.10486. [DOI] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Rink A, Fung KM, Trojanowski JQ, et al. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- Roof RL, Hall ED. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17:367–388. doi: 10.1089/neu.2000.17.367. [DOI] [PubMed] [Google Scholar]
- Roof RL, Zhang Q, Glasier MM, et al. Gender-specific impairment on Morris water maze task after entorhinal cortex lesion. Behav Brain Res. 1993;57:47–51. doi: 10.1016/0166-4328(93)90060-4. [DOI] [PubMed] [Google Scholar]
- Roof RL, Duvdevani R, Braswell L, et al. Progesterone facilitates cognitive recovery and reduces secondary neuronal loss caused by cortical contusion injury in male rats. Exp Neurol. 1994;129:64–69. doi: 10.1006/exnr.1994.1147. [DOI] [PubMed] [Google Scholar]
- Roof RL, Duvdevani R, Heyburn JW, et al. Progesterone rapidly decreases brain edema: treatment delayed up to 24 hours is still effective. Exp Neurol. 1996;138:246–251. doi: 10.1006/exnr.1996.0063. [DOI] [PubMed] [Google Scholar]
- Roof RL, Hoffman SW, Stein DG. Progesterone protects against lipid peroxidation following traumatic brain injury in rats. Mol Chem Neuropathol. 1997;31:1–11. doi: 10.1007/BF02815156. [DOI] [PubMed] [Google Scholar]
- Ross SA, Halliday MI, Campbell GC, et al. The presence of tumour necrosis factor in CSF and plasma after severe head injury. Br J Neurosurg. 1994;8:419–425. doi: 10.3109/02688699408995109. [DOI] [PubMed] [Google Scholar]
- Rothwell N. Interleukin-1 and neuronal injury: mechanisms modification and therapeutic potential. Brain Behav Immun. 2003;17:152–157. doi: 10.1016/s0889-1591(02)00098-3. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Duhaime AC, Bullock R, et al. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivan S, Dunn WA, Jr, Hayes RL, et al. Changes in autophagy proteins in a rat model of controlled cortical impact induced brain injury. Biochem Biophys Res Commun. 2008;373:478–481. doi: 10.1016/j.bbrc.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Sandhir R, Onyszchuk G, Berman NE. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp Neurol. 2008;213:372–380. doi: 10.1016/j.expneurol.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherbel U, Raghupathi R, Nakamura MS, et al. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schousboe A, Waagepetersen HS. Role of astrocytes in glutamate homeostasis: implications for excitotoxicity. Neurotox Res. 2005;8:221–225. doi: 10.1007/BF03033975. [DOI] [PubMed] [Google Scholar]
- Schouten JW. Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr Opin Crit Care. 2007;13:134–142. doi: 10.1097/MCC.0b013e3280895d5c. [DOI] [PubMed] [Google Scholar]
- Shohami E, Novikov M, Bass R, et al. Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–619. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- Shohami E, Gallily R, Mechoulam R, et al. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72:169–177. doi: 10.1016/s0165-5728(96)00181-6. [DOI] [PubMed] [Google Scholar]
- Simard JM, Woo SK, Bhatta S, et al. Drugs acting on SUR1 to treat CNS ischemia and trauma. Curr Opin Pharmacol. 2008;8:42–49. doi: 10.1016/j.coph.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JM, Geng Z, Woo SK, et al. Glibenclamide reduces inflammation vasogenic edema and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2009a;29:317–330. doi: 10.1038/jcbfm.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JM, Kilbourne M, Tsymbalyuk O, et al. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J Neurotrauma. 2009b;26:2257–2267. doi: 10.1089/neu.2009.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JM, Woo SK, Schwartzbauer GT, et al. Sulfonylurea receptor 1 in central nervous system injury: a focused review. J Cereb Blood Flow Metab. 2012;32:1699–1717. doi: 10.1038/jcbfm.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slemmer JE, Zhu C, Landshamer S, et al. Causal role of apoptosis-inducing factor for neuronal cell death following traumatic brain injury. Am J Pathol. 2008;173:1795–1805. doi: 10.2353/ajpath.2008.080168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SS. Progesterone administration attenuates excitatory amino acid responses of cerebellar Purkinje cells. Neuroscience. 1991;42:309–320. doi: 10.1016/0306-4522(91)90377-z. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Pierce JE, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997a;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Xu BN, et al. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J Neuropathol Exp Neurol. 1997b;56:822–834. [PubMed] [Google Scholar]
- Sperandio S, De Belle I, Bredesen DE. An alternative nonapoptotic form of programmed cell death. Proc Natl Acad Sci U S A. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAIR (Stroke Therapy Academic Industry Roundtable) Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- Statler KD, Alexander H, Vagni V, et al. Comparison of seven anesthetic agents on outcome after experimental traumatic brain injury in adult male rats. J Neurotrauma. 2006;23:97–108. doi: 10.1089/neu.2006.23.97. [DOI] [PubMed] [Google Scholar]
- Stein DG, Wright DW. Progesterone in the clinical treatment of acute traumatic brain injury. Expert Opin Investig Drugs. 2010;19:847–857. doi: 10.1517/13543784.2010.489549. [DOI] [PubMed] [Google Scholar]
- Stoica BA, Faden AI. Programmed neuronal cell death mechanisms in CNS injury. In: Fujikawa DG, editor. Acute Neuronal Injury: The Role of Excitotoxic Programmed Cell Death Mechanisms. Springer; 2010a. pp. 169–199. [Google Scholar]
- Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010b;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica B, Byrnes K, Faden AI. Multifunctional drug treatment in neurotrauma. Neurotherapeutics. 2009;6:14–27. doi: 10.1016/j.nurt.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Loane DJ, Zhao Z, et al. PARP-1 inhibition attenuates neuronal loss microglia activation and neurological deficits after traumatic brain injury. J Neurotrauma. 2014;31:758–772. doi: 10.1089/neu.2013.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover JF, Schoning B, Beyer TF, et al. Temporal profile of cerebrospinal fluid glutamate interleukin-6 and tumor necrosis factor-alpha in relation to brain edema and contusion following controlled cortical impact injury in rats. Neurosci Lett. 2000;288:25–28. doi: 10.1016/s0304-3940(00)01187-3. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, et al. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999a;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999b;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Hicks RR, et al. Dose-response curve and optimal dosing regimen of cyclosporin A after traumatic brain injury in rats. Neuroscience. 2000a;101:289–295. doi: 10.1016/s0306-4522(00)00380-8. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson M, Scheff SW. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp Neurol. 2000b;161:631–637. doi: 10.1006/exnr.1999.7282. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Keller JN, Bussen WL, et al. Cytochrome c release and caspase activation after traumatic brain injury. Brain Res. 2002;949:88–96. doi: 10.1016/s0006-8993(02)02968-2. [DOI] [PubMed] [Google Scholar]
- Sullivan S, Friess SH, Ralston J, et al. Behavioral deficits and axonal injury persistence after rotational head injury are direction dependent. J Neurotrauma. 2013;30:538–545. doi: 10.1089/neu.2012.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Chuang LF, Doi RH, et al. Kappa-opioid receptors on lymphocytes of a human lymphocytic cell line: morphine-induced up-regulation as evidenced by competitive RT-PCR and indirect immunofluorescence. Int Immunopharmacol. 2001;1:1733–1742. doi: 10.1016/s1567-5769(01)00083-2. [DOI] [PubMed] [Google Scholar]
- Szabo I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991;266:3376–3379. [PubMed] [Google Scholar]
- Tang M, Alexander H, Clark RS, et al. Minocycline reduces neuronal death and attenuates microglial response after pediatric asphyxial cardiac arrest. J Cereb Blood Flow Metab. 2010;30:119–129. doi: 10.1038/jcbfm.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Perez JH, Sanchez-Aguilar M, Torres-Corzo JG, et al. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758) J Neurotrauma. 2008;25:1011–1017. doi: 10.1089/neu.2008.0554. [DOI] [PubMed] [Google Scholar]
- Tehranian R, Andell-Jonsson S, Beni SM, et al. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J Neurotrauma. 2002;19:939–951. doi: 10.1089/089771502320317096. [DOI] [PubMed] [Google Scholar]
- Toulmond S, Rothwell NJ. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995;671:261–266. doi: 10.1016/0006-8993(94)01343-g. [DOI] [PubMed] [Google Scholar]
- Tremblay ME, Stevens B, Sierra A, et al. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng MY, Czosnyka M, Richards H, et al. Effects of acute treatment with pravastatin on cerebral vasospasm autoregulation and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: a phase II randomized placebo-controlled trial. Stroke. 2005;36:1627–1632. doi: 10.1161/01.STR.0000176743.67564.5d. [DOI] [PubMed] [Google Scholar]
- Tyor WR, Avgeropoulos N, Ohlandt G, et al. Treatment of spinal cord impact injury in the rat with transforming growth factor-beta. J Neurol Sci. 2002;200:33–41. doi: 10.1016/s0022-510x(02)00113-2. [DOI] [PubMed] [Google Scholar]
- Van Loo G, Saelens X, Van Gurp M, et al. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 2002;9:1031–1042. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Verdaguer E, Jimenez A, Canudas AM, et al. Inhibition of cell cycle pathway by flavopiridol promotes survival of cerebellar granule cells after an excitotoxic treatment. J Pharmacol Exp Ther. 2004;308:609–616. doi: 10.1124/jpet.103.057497. [DOI] [PubMed] [Google Scholar]
- Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25:763–774. doi: 10.1038/sj.jcbfm.9600073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink R, Nimmo AJ. Multifunctional drugs for head injury. Neurotherapeutics. 2009;6:28–42. doi: 10.1016/j.nurt.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink R, Van Den Heuvel C. Substance P antagonists as a therapeutic approach to improving outcome following traumatic brain injury. Neurotherapeutics. 2010;7:74–80. doi: 10.1016/j.nurt.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volbracht C, Leist M, Kolb SA, et al. Apoptosis in caspase-inhibited neurons. Mol Med. 2001;7:36–48. [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu SW, Koh DW, et al. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lynch JR, Song P, et al. Simvastatin and atorvastatin improve behavioral outcome reduce hippocampal degeneration and improve cerebral blood flow after experimental traumatic brain injury. Exp Neurol. 2007;206:59–69. doi: 10.1016/j.expneurol.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol. 2009;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Xia Q, Chu KT, et al. Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: a widely used inhibitor of autophagy. J Neuropathol Exp Neurol. 2011a;70:314–322. doi: 10.1097/NEN.0b013e31821352bd. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Haince JF, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos) Science Signal. 2011b;4:ra20. doi: 10.1126/scisignal.2000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Shi XY, Yin J, et al. Role of autophagy in early brain injury after experimental subarachnoid hemorrhage. J Mol Neurosci. 2012;46:192–202. doi: 10.1007/s12031-011-9575-6. [DOI] [PubMed] [Google Scholar]
- Wible EF, Laskowitz DT. Statins in traumatic brain injury. Neurotherapeutics. 2010;7:62–73. doi: 10.1016/j.nurt.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, et al. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 2004;24:5016–5021. doi: 10.1523/JNEUROSCI.0820-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter CD, Iannotti F, Pringle AK, et al. A microdialysis method for the recovery of IL-1beta IL-6 and nerve growth factor from human brain in vivo. J Neurosci Methods. 2002;119:45–50. doi: 10.1016/s0165-0270(02)00153-x. [DOI] [PubMed] [Google Scholar]
- Woodroofe MN, Sarna GS, Wadhwa M, et al. Detection of interleukin-1 and interleukin-6 in adult rat brain following mechanical injury by in vivo microdialysis: evidence of a role for microglia in cytokine production. J Neuroimmunol. 1991;33:227–236. doi: 10.1016/0165-5728(91)90110-s. [DOI] [PubMed] [Google Scholar]
- Wright DW, Kellermann AL, Hertzberg VS, et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:391–402. 402 e1–2. doi: 10.1016/j.annemergmed.2006.07.932. [DOI] [PubMed] [Google Scholar]
- Wu H, Lu D, Jiang H, et al. Increase in phosphorylation of Akt and its downstream signaling targets and suppression of apoptosis by simvastatin after traumatic brain injury. J Neurosurg. 2008a;109:691–698. doi: 10.3171/JNS/2008/109/10/0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lu D, Jiang H, et al. Simvastatin-mediated upregulation of VEGF and BDNF activation of the PI3K/Akt pathway and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008b;25:130–139. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- Xiao G, Wei J, Yan W, et al. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care. 2008;12:R61. doi: 10.1186/cc6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Gu Q, Peterson PL, et al. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J Neurotrauma. 1997;14:23–34. doi: 10.1089/neu.1997.14.23. [DOI] [PubMed] [Google Scholar]
- Yakovlev AG, Knoblach SM, Fan L, et al. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev AG, Ota K, Wang G, et al. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci. 2001;21:7439–7446. doi: 10.1523/JNEUROSCI.21-19-07439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Zhang H, Bai X, et al. Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res. 2011;1402:109–121. doi: 10.1016/j.brainres.2011.05.049. [DOI] [PubMed] [Google Scholar]
- You Z, Savitz SI, Yang J, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao XL, Liu J, Lee E, et al. Progesterone differentially regulates pro- and anti-apoptotic gene expression in cerebral cortex following traumatic brain injury in rats. J Neurotrauma. 2005;22:656–668. doi: 10.1089/neu.2005.22.656. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang Y, Frydenlund DS, et al. Outer mitochondrial membrane localization of apoptosis-inducing factor: mechanistic implications for release. ASN Neuro. 2009;1:275–281. doi: 10.1042/AN20090046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacco A, Togo J, Spence K, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicity. J Neurosci. 2003;23:11104–11111. doi: 10.1523/JNEUROSCI.23-35-11104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen J, Graham SH, et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181–191. doi: 10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]
- Zhang D, Hu X, Qian L, et al. Astrogliosis in CNS pathologies: is there a role for microglia? Mol Neurobiol. 2010;41:232–241. doi: 10.1007/s12035-010-8098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Alam S, Oppenheim RW, et al. Overexpression of glial cell line-derived neurotrophic factor in the CNS rescues motoneurons from programmed cell death and promotes their long-term survival following axotomy. Exp Neurol. 2004;190:356–372. doi: 10.1016/j.expneurol.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Zhu C, Qiu L, Wang X, et al. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem. 2003;86:306–317. doi: 10.1046/j.1471-4159.2003.01832.x. [DOI] [PubMed] [Google Scholar]
- Zhu C, Wang X, Deinum J, et al. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J Exp Med. 2007a;204:1741–1748. doi: 10.1084/jem.20070193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Wang X, Huang Z, et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007b;14:775–784. doi: 10.1038/sj.cdd.4402053. [DOI] [PubMed] [Google Scholar]