

Abstract
Advances in digital pathology, specifically imaging instrumentation and data management, have allowed for the development of computational pathology tools with the potential for better, faster, and cheaper diagnosis, prognosis, and prediction of disease. Images of tissue sections frequently vary in color appearance across research laboratories and medical facilities due to differences in tissue fixation, staining protocols, and imaging instrumentation, leading to difficulty in the development of robust computational tools. To address this challenge, we propose a novel non-linear tissue-component discrimination (NLTD) method to automatically register the color space of histopathology images and visualize individual tissue components, independent of color differences between images. Our results show that the NLTD method could effectively discriminate different tissue components from different types of tissues prepared at different institutions. Further, we demonstrate that NLTD can improve the accuracy of nuclear detection and segmentation algorithms, compared to using conventional color deconvolution methods, and can quantitatively analyze immunohistochemistry images. Together, the NLTD method is objective, robust, and effective, and can be easily implemented in the emerging field of computational pathology.
Advances in imaging instrumentation and data management provide the foundation for computational approaches to analyze digitized images of tissue sections and derive objective, quantitative measurements at the tissue, cellular, subcellular, and molecular levels1. Computational pathology approaches offer a cost-effective platform to increase throughput, accuracy, and reliability of diagnoses of tissue samples2, 3. Further, the quantitative nature of computational pathology can be used in combination with other assays to improve pathologists’ knowledge of disease and help inform treatment strategies and further stratify patient prognosis. It has been shown that, by integrating information derived from computational pathology with a patient's clinical data, a better prognostic model can be derived for many diseases, including prostate cancer4-6, lung cancer7, breast cancer8-12, glioblastoma13, 14, basal cell carcinoma15, 16, and ovarian cancer17, 18.
One of central challenges of computational biology, which limits its large-scale applications, is that images of tissue sections frequently vary in color appearance across research laboratories and medical facilities due to differences in tissue fixation, staining protocols, and imaging instrumentation. The wide spectrum of image color appearance causes difficulty in robustly extracting the representative images of different tissue components, such as nuclei19. Previous studies have shown that technician variance or technique differences can lead to dramatic differences in staining20. For example, the conventional hematoxylin and eosin (H&E) staining techniques have been modified to reduce material use and processing time21 or to improve the contrast and detail in the digital image22. These technique differences provide some advantage to the pathologist, but also lead to variation in the staining of slides for use in computational pathology approaches that must be addressed.
Several stain normalization computational approaches- including color deconvolution23, histogram equalization24 and the use of the CMYK space25- have been developed to correct for the difference image appearance and facilitate the separation of tissue types19, 20. Of these approaches, color deconvolution is the most commonly used approach to extract nuclear and cellular images in both hematoxylin and eosin (H&E) and immunohistochemically (3,3' Diaminobenzidine, DAB) stained images2, 9, 23, 26-28. Color deconvolution utilizes the method of singular value decomposition (SVD), which seeks to linearly separate the color space to identify regions rich in each particular dye. However, a major disadvantage of color deconvolution is the requirement of prior knowledge for each dye's color spectrum in order to accurately visualize tissue components29. Due to color appearance difference between images, using the same stain vector across images will introduce variance in the representative image for each dye. Although there are automated methods to determine the stain vector for individual images, the additional processing step leads to significant increase in processing time across large image datasets30. Furthermore, color deconvolution only decouples the concentration of dye in the histo-pathological image, and further processing is needed to separate individual tissue components such as blood, nuclei, and extracellular matrix and cytoplasmic rich regions for quantification.
In this work, we propose a novel non-linear tissue-component discrimination (NLTD) method to automatically register the color space of histopathology images and obtain representative images for individual tissue components, such as nuclei or cytoplasm, irrespective of perceptual color differences between images. We demonstrate that the nuclei image obtained from NLTD display consistent appearance for histopathology images- including those with distinct color differences- taken from different tissues types and prepared at different institutions, including The Cancer Genome Atlas project (TCGA, http://cancergenome.nih.gov/). Importantly, the processing time of NLTD is highly comparable to the color deconvolution for small images, and much more efficient for large images, notably whole slide images. Further, we demonstrated that the nuclei images derived using NLTD produce highly accurate nucleus tracing and counting, and NLTD allows for quantitative analysis of antigen presence in immunohistochemical images. Together, we show that NLTD is an effective approach to obtain quantitative tissue component images that can be easily integrated in emerging computational pathology applications.
Methods
The non-linear tissue-component discrimination (NLTD) method consists of five main steps (Fig. 1a), detailed further here:
-
1)
Color joint-histogram creation
-
2)
Ridge detection
-
3)
Ridge set registration
-
4)
Transformation function creation
-
5)
Tissue component image creation
Figure 1. Brief overview of NLTD approach.

(a) NLTD applied to an image of a hematoxylin and eosin (H&E) stained section (top) and immunohistochemically (IHC) stained image (bottom). Shown are a typical H&E image of a small artery, exhibiting multiple tissue components (nuclei (N), ECM-rich and cytoplasm (E), blood (B)) and a typical IHC image, stained for LINE-1 ORF1p expression31, exhibiting two tissue components (antigen (A) and nuclei (N)). The NLTD method is schematically shown in center. Briefly, the red-blue joint-histogram is first segmented to identify each region in the red-blue color space. The x-axis corresponds to each red color, the y-axis shows each blue-color, and the color-axis represents the frequency of each discrete color combination. Ridges for each tissue component are overlaid, on the RBJH. The ridge set is registered and transformed to yield the pseudo-colored transformation function for each component. The pseudo-colored grayscale images are shown for the nuclei, non-nuclei, and blood components (purple, pink, and red, respectively) in the far right box. (b-c) Grayscale correlation values for the red-blue joint histogram, blue-green joint histogram, and red-green joint-histogram, with a value of 1 corresponding to a completely correlated colorset. (b) Pancreatic cancer H&E dataset (n=45). (c) Ovarian immunohistochemistry dataset (n=81). (d) Separation of red-blue color space into individual tissue components: nuclei (purple box), extracellular matrix and cytoplasm rich (pink box).
Color joint-histogram creation
In a typical 8-bit tissue image, I, the color of an individual pixel, p, at location (xp, yp) is expressed by three intensities (rp,gp,bp), each of which ranges from 0 to 255, discretely, i.e.
| (1) |
For example, if all the intensities of a pixel are zero the resultant color is black; conversely, if all are 255, the resultant color is white. The color joint-histogram is a three dimensional histogram created by counting the occurrence of pixels at all different set of red, green, and blue intensities in an image. However, calculating every color combination in RGB color space and analyzing 3D RGB color joint histogram is a highly computationally intensive process. An 8-bit image can contain more than 16 million unique combinations. To reduce computational time, it would be advantageous to only consider two of the three color axes, reducing the number of unique combinations 256-fold
In a cohort of 45 H&E images, we found that blue and green color components are highly correlated within individual images (Fig. 1b). Furthermore, we also found that the red and green color components are highly correlated in a set of 81 immunohistochemically stained images (DAB)31 (Fig. 1c). These observations show that in both H&E and DAB images green color channel encodes highly correlative information to other color channel and implies that the red-blue color joint-histogram can be a representative simplification of the histo-pathological image RGB color space. The red-blue color joint histogram (RBJH) is a 2-dimensional matrix, created by counting the frequency (n) of pixels at different red (r) and blue (b) intensity values in the image (I), i.e.
| (2) |
The resultant RBJH can be visualized as a three dimensional surface, with the x and y axes corresponding to the red and blue color space values, respectively, and the z axis corresponding to the incidence rate for each R-B intensity combinations.
Ridge detection
The RBJH shows the most abundant color combinations in the red-blue color space for an image. In the RBJH, distinct populations of red-blue combinations are readily observed, corresponding to different tissue components (Fig. 1d). However, to detect and segment individual tissue components in the RBJH is challenging due to the elongated non-linear distribution of red-blue color combinations that complicates separation of the populations and , consequentially, common approaches, such as Gaussian mixture models or k-means clustering, do not work well. Gaussian mixture models fail because most images’ RBJH do not follow strictly Gaussian distributions, often having one major peak, along with a long sparse tail. Similarly, k-means clustering is not effective at detecting sparse areas in the RBJH. Additionally, both models require prior knowledge of the numbers of components present, which we have found can vary from 1 to 3 in most images. Successful extraction of individual tissue components’ locations in the red-blue color space needs to incorporate the asymmetric and elongated distribution of red-blue color combinations that is observed in the RBJH. Therefore, we propose to identify locations of major tissue components in the red-blue color space by tracing the location of ridges of distinct population in the RBJH (Fig. S2).
We first identified the major orientation of signal in RBJH using weighted principal component analysis (PCA) (Fig. S1a). PCA is applied to the each red and blue index of the RBJH, with the frequency (nr,b) associated with each color combination used as a weight. The principal component provides the major direction of the RBJH color space, which can be combined with the location of the global maximum of the RBJH to create a major axis (v0). Next, we identify the local maxima tangent along the major axis in RBJH (Fig. S1b). To ensure the all local maximum is detected, this routine is repeated along two other vectors at angles ±15 degrees of the major direction. A map of all local maxima identified is then created by counting the frequency of local maxima identified at each red-blue index (Fig. S1c). This map is further processed through morphological dilation and thinning operations to provide a binarized location of ridges for all distinct populations in RBJH (Fig. S1d).
Ridge set registration
To register the ridge set maps in RBJH to different tissue components, we developed a robust algorithm based on each ridge's proximity to specified reference color combination (Fig. S2a). Four reference color combinations are used: Red (r=255, b=0), Blue (r=0, b=255), Black (r=0, b=0), and White (r=255, b=255). The Euclidean distance transform32 is calculated for each reference color combination, and the minimum distance along each ridge is found. The component with the smallest distance from a reference color combination is determined to be the closest. In H&E staining, in general nuclei would be closer to black and blue, while extracellular matrix and cytoplasm rich areas would be closer to white. Similar logic can be applied to DAB chromagen staining, where antigen rich areas are closer to red than nuclei, which are closer to blue.
In some cases, where the RBJH is more homogenous, it is possible that only one ridge is found (Fig. S2b). For H&E staining, in general, each image contains at least two distinct components- nuclei or extracellular matrix and cytoplasm rich components. The portion of ridge closer to white and red represents extracellular matrix and cytoplasm rich areas and the remaining portion of the ridge represents the nuclei-rich regions; this necessitates further segmentation of the ridge. To segment the ridge, the intensity profile (i.e. frequency of color combinations) of the RBJH along the ridge is first extracted. A peak, corresponding to the most frequently occurring color combination, commonly appears that represents the central location of extracellular matrix and cytoplasm rich regions on the ridge. Thus, we fit this intensity profile with a Gaussian distribution to measure the distribution of the extracellular matrix and cytoplasm rich areas along the ridge, and segment the single identified ridge using a distance of two standard deviations from the peak into two distinct ridges corresponding to nuclei or extracellular matrix and cytoplasm rich components. For DAB chromagen staining, the possibility of only one ridge being identified is most likely due to the lack of presence of antigen to probe in the tissue section and hence no further segmentation is needed.
Transformation function creation
We formulated tissue transformation functions (TF) to convert the red-blue color space to intensity of different tissue components (k=1,2,...,N). We assume the red-blue color space has different regions that exclusively correspond to different tissue components based on the proximity to each ridge in the ridge set. A watershed segmentation is applied to the ridge of the RBJH to identify regions of the red-blue color space that represent the unique tissue components (Fig. S3a). Additionally, the regions of the red-blue color space with the most absorption (i.e. lower r and/or b indices) correspond to the strongest signal within each tissue region. For each particular tissue component, the red and blue indices that are closest to the tissue's ridge indicate a higher likelihood of belonging to that tissue and also contribute to a stronger signal.
To account for these three factors, we developed a transformation function, TFk, that account for the tissue component's region in red-blue color space (fregion), its absorption (fabsorption), and the distance from each tissue component's ridge (fridge) (Fig. S3b), expressed by
| (3) |
k=1,2,...,N tissue component.
The tissue region function, fregion, uses the watershed segmentation result as a basis to exclude any part of the red-blue color space not belonging to the same tissue component. The region of the red-blue color space corresponding to the kth tissue component is defined as Wk. A Gaussian filter, g, can be applied to the edge of the region to allow for a smoother transition between components (Fig. S3b[i]), i.e.,
| (4) |
The absorption function, fabsorption , for kth tissue component is obtained by first calculating the Euclidean distance transform32 (Ddark) of all points in red-blue color space from the point with highest absorption (i.e. darkest) on the kth tissue component ridge (Rk), defined as the point on the ridge closest to black (r=0, b=0). To scale the distance with level of absorption of dye, the absorption function (Fig. S3b[ii]), is expressed by
| (5) |
The ridge function, fridge, is derived from the Euclidean distance transform32, D, which is the minimum distance of any point in the red-blue color space to a point on the ridge of the kth tissue component, Rk. To scale the likelihood with distance, the ridge function (Fig. S3b[iii]), is expressed by
| (6) |
Tissue component image creation
To obtain the kth tissue component image, Tk , the red and blue pixel intensities (rp, bp) at each location (xp,yp) in the original image, I, were used to create a grayscale image according to the transformation function, TFk, i.e.
| (7) |
Sample Acquisition
Histo-pathological images were acquired from pathologists at the Johns Hopkins University. The tissue samples were formalin-fixed and paraffin embedded. Tissue sections were fixed for 3 hours in formalin on tissue processor, followed by 1-2 hours of gross room fixation. Paraffin sections were cut at 5μm thickness. Sections were then stained with hematoxylin and eosin and digitized using a DP27 5MP color camera, Sections of pancreatic cancer, colon cancer, ovarian cancer, and glioblastoma were included. Immunohistochemical (3,3’-Diaminobenzidine (DAB)) stained tissue was acquired through an ovarian cancer tissue microarray, as described previously31. Additional tissue images were acquired from The Cancer Genome Atlas project (http://cancergenome.nih.gov) and published sources33, 34.
Nuclei Detection
To perform a comparison of segmentation results between color deconvolution and the NLTD method, a publicly available dataset33, including both tissue images and ground truth nuclei locations, was analyzed. For the NLTD method, the corresponding nuclei image was obtained and nuclei location is obtained using following procedure:
-
1)
Binarize each image using a dynamic threshold, calculated using Otsu's method35.
-
2)
Remove small objects based on a size threshold of 50 pixels.
-
3)
Watershed segmentation to separate clusters of nuclei.
The same segmentation approach was used for the color deconvolution image corresponding to the hematoxylin dye. For each segmented nucleus identified, the nearest ground-truth nucleus was found. If two segmented nuclei were attached to the same ground-truth nuclei, the nearest would be counted as a true positive (TP) and the other would be counted as a false positive (FP). Any segmented nucleus with the nearest ground-truth nuclei was more than one average cell diameter away was counted as a false positive (FP). Conversely, any ground-truth nucleus that did not have any segmented nuclei within one average cell diameter was counted as a false negative (FN).
Immunohistochemistry Scoring
A TMA of ovarian cancer tissue stained using an antibody for LINE-1 ORF1p31 was used to evaluate the utility of NLTD as an immunohistochemistry scoring aide. Each image in the TMA was separated into two images using the NLTD method, a nuclei-rich image and an antigen-rich image (Fig. S5). Pre-processing steps were performed in order to only analyze nuclei rich regions where antigen staining was present, and avoid background areas where no staining should occur. Briefly, the nuclei-rich image was segmented using Otsu's thresholding technique35. Small objects were removed from the image, followed by morphological opening and closing operations and another removal of small objects. After pre-processing, a transformation score was derived based on the ratio of antigen intensity to nuclei intensity (Eq. 8). Importantly, only antigen and nuclei intensity in the areas from the segmented, pre-processed image were counted.
| (8) |
Hardware and Software
All image processing was performed using MATLAB 2015 (Mathworks). To determine statistical significance, two-tailed t-tests were performed using Graphpad Prism 6. All computations were performed on Windows 7 Professional with an Intel Core i7-3820 processor and 16 GB RAM.
Statistics
To quantify the segmentation results, precision, recall, and F-Score statistics were used36. For this dataset it is not possible to assess accuracy or other statistics using true negative counts, since the classification system has no negative result included and only positive occurrences (i.e. nuclei) are identified. Each statistic is defined as follows:
| (9) |
| (10) |
| (11) |
Results
Overview of the non-linear tissue-component discrimination (NLTD) method
The NLTD method presented in this work consists of five major steps, as illustrated with an H&E-stained image and an immunohistochemically stained image in Fig. 1a. First, the red-blue color joint-histogram (RBJH) is created. This joint-histogram represents the frequency at which each red-and-blue pixel intensity combination occurs in a histo-pathological image, and serves as the basis for tissue-component discrimination. The RBJH is reduced to a set of curves representing the ridges, or local maxima, utilizing an iterative approach. This ridge set is further registered with corresponding individual tissue components (e.g. nuclei, extracellular matrix and cytoplasm rich, etc.). Further, the ridge set serves as a basis for the creation of a set of transformation functions used to create individual, grayscale images from the original image representative of each tissue component present in the image (see more details in Methods section). The resulting set of tissue component images can then be used for additional tissue processing and analysis, including nuclei detection algorithms and quantitative scoring of immunohistochemically stained samples. The MATLAB package is available upon request.
Robustness of NLTD
To demonstrate the robustness of the NLTD method, we applied NLTD to a set of histo-pathological images with wide range of apparent colors to show the uniformity in the images of nuclei extracted using the NLTD method (Fig. 2). The image set spans multiple tissue types, along with several different image sources: the Johns Hopkins School of Medicine; images from previous studies performed at University of California, Santa Barbara34 and the University of Berlin33; and publicly available images from the TCGA image database (http://cancergenome.nih.gov). The results show that even though the RBJH color space for different images have unique and different distributions, the NLTD method can successfully identify and register each tissue component and extract nuclei images consistently and robustly.
Figure 2. Application of NLTD across a wide variety of tissue types.

The NLTD method is applied on many different types of tissue. The original tissue image, RBJH, registered RBJH, and the nuclei component grayscale image (pseudo-colored purple) are shown (left to right). The registered RBJH shows a purple line for the nucleus component, a pink line for the ECM/cytoplasmic component, and a red line for the blood component. The sample tissue types are: (a) Colon cancer, (b) Kidney cancer, (c) Ovarian cancer, (d) Lung adenocarcinoma, (e) Gastric mucosa, (f) Astrocytoma, (g) Skin cutaneous melanoma, (h) Breast cancer.
Processing time
For most computational pathology applications, the time associated with processing each image and gleaning important information can quickly become a barrier with image size and/or lager cohort of images. Previous work has compared stain normalization processing time for smaller images (256×256, 512×512, and 1024×1024)29. Whole slide imaging, however, often results in much larger images (10000×10000 or greater), and it is important for image processing time to scale well with the size of each image. In our work, we compared color deconvolution, color deconvolution using Macenko's color normalization method37, and the NLTD approaches. Macenko's approach involves an additional preprocessing step to determine each individual image's optimal stain vector and uses the optimal stain vector for color deconvolution. Both color deconvolution approaches were faster than the NLTD method at small image size (up to 2500×2500), but, as the image size approached whole slide levels (15000×15000), the NLTD method was much faster than both color deconvolution based methods and took only a quarter of the time to process each image (Fig. 3). This result suggests that NLTD can more efficiently analyze larger images, which can be very useful for large datasets, such as TCGA. Since color deconvolution is more time efficient at smaller sizes, it is possible to partition one large image into many smaller images (i.e. one 10000×10000 image into one hundred 1000×1000 images). However, this additional processing step would still lead to an increase in processing time compared to the NLTD approach (3.8sec for NLTD on one 10000×10000 image, 11.85sec for color deconvolution on one hundred 1000×1000 images). Therefore, the NLTD method can be more efficiently applied to whole slide images and reduce the time needed to analyze large cohorts of images.
Figure 3. Processing time of NLTD and color deconvolution.
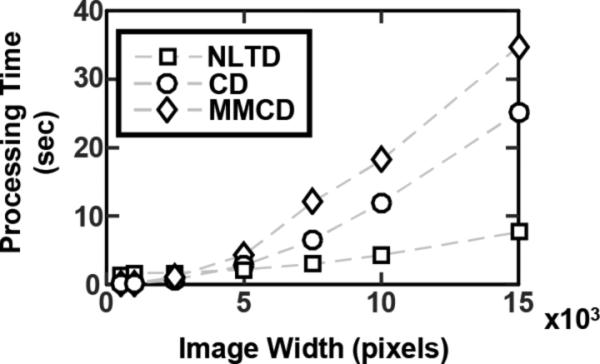
Comparison time for processing of images of various sizes using three different color normalization techniques: NLTD (squares), color deconvolution (CD, circles), and color deconvolution using Macencko's method of automated stain vector determination (MMCD, diamonds). Each image used was a three-dimensional RGB image, with side lengths defined by the x-axis. Processing time is shown on the y-axis in seconds as the median of 10 runs for each method at each image size.
Improving Nuclei detection with NLTD
Nuclei detection in histo-pathological images has been critical and often used in computational pathology approaches to develop prognostic and diagnostic models7-9, 11, 12, 17, 33. Currently, color deconvolution (CD) is commonly used to extract a representative nuclei image (corresponding to the hematoxylin dye levels) to apply nuclei detection algorithms9, 28-30, 33, 38. Here, we show that using the nuclei image derived from the NLTD method improves the detection of nuclei over the color deconvolution approach. We first evaluated the contrast of individual nuclei images created from both the NLTD and the color deconvolution method (Fig. 4 a-c). By examining the intensity profile along one axis across nuclei, we found that the nuclei image obtained from NLTD has a substantial decrease in intensity at the periphery of the nucleus compared to nuclei images from color deconvolution. This result suggests that the implementation of segmentation algorithms to the NLTD nuclei image would be less sensitive to the intensity threshold value and hence could lead to improvements in the accuracy and robustness of nuclei segmentation algorithms.
Figure 4. Evaluation of NLTD method.

(a-c) Nuclei intensity comparison between NLTD and color deconvolution approaches. Representative nuclei from several tissue types are shown, along with the NLTD and color deconvolution nuclei transformations. The intensity of each color space is integrates along the dotted lines shown, with the NLTD intensity shown in purple and the color deconvolution intensity in brown. Intensity values are normalized linearly between 0 and 1, with 0 corresponding to the minimum value in the input image, and 1 corresponding to the maximum. (d) Typical breast cancer image33. (e) Example of segmentation results from Otsu thresholding of the nuclei NLTD color space. Detected nuclei are overlaid on top of the image from panel (d). True positives are represented by a green dot, false positives by a red dot, and false negatives by a yellow dot. (f) Precision, (g) sensitivity, and (h) F-score values for segmentation results from 35 images. (i) Receiver operating characteristic curve for change in segmentation parameterization (threshold value) for nuclei detection. Recall (sensitivity) is shown on the x-axis, with precision shown on the y-axis. Results from NLTD method are shown in black, with color deconvolution (CD) shown in gray.
To quantitatively examine the performance of the NLTD and color deconvolution methods in nuclei detection and segmentation, we applied previous proposed detection algorithm (see more details in Methods section) after applying both color normalization methods (NLTD and CD) to a published set of 35 images33. This dataset included nuclei locations that had been previously registered by a pathologist and were used as ground truth (Fig. 4d-e).
To assess each method, the precision, sensitivity, and F-score were measured. High precision and sensitivity are both valuable in a nuclei detection system. A system that lacks precision will lead to unnecessary calculation and validation by an observer with too many nuclei identifies. Conversely, a system that is not sensitive will miss many nuclei and potentially distort the values of nuclei counts or exclude rare nuclei events, such as mitotic or atypical nuclei. The F-score provides the harmonic mean between sensitivity and precision and serves as an overall measure of how accurate the system is.
Among the 35 images tested, we found that, overall, the images normalized using NLTD have significantly higher sensitivity in detecting nuclei than the corresponding color deconvolution images (NLTD=0.868; CD=0.753), but slightly lower precision (NLTD=0.938; CD=0.976) (Fig. 4f-h). The overall accuracy, as represented by the F-score, for NLTD images is 0.860 and is significantly higher than the color deconvolution images (F-score = 0.805). The slightly lower precision in our NLTD system correlates to an over-detection, with more nuclei identified by the NLTD detection system than the ground truth. The higher sensitivity, however, means that the NLTD detection system leaves fewer ground truth nuclei undetected. Together, these results suggest that the NLTD method is able to provide more accurate nuclei segmentation results, compared with conventional color deconvolution methods.
NLTD for quantitative immunohistochemistry (IHC) analysis
In addition to providing a platform for image appearance normalization and nuclei detection, the NLTD method can be used as a companion diagnostic for analysis of immunohistochemical labeling quantitatively and objectively. The intensity level of DAB chromagen labeling is used to access the level of antigen presence in tissue sample by pathologists. We applied our method to an ovarian cancer tissue microarray cohort that had been immunolabeled for L1ORF1p, a cytoplasm-localizing protein associated with cancer31 (Fig. 5 and Fig. S4). Each tissue sample in this cohort was scored by a trained pathologist using a discrete scoring system (0, 1, 2, or 3). A score of 0 indicates no significant protein expression whereas a score of 3 was given for high expression. We applied our NLTD method to individual tissue images of the TMA to create component images for antigen rich regions and nuclei rich regions. These images were then used to calculate an overall score corresponding to the level of antigen, normalized by nuclei intensity (see more details in Methods section). Our results showed a strong correlation (Spearman's ρ = 0.8122) between our automated scoring platform and the scoring by the pathologist. Minor overlap exists between tissues with a score of 1 and 2 but both high expression (3) and very low expression (0) scores were well stratified. This result shows the utility of our NLTD method as a nonparametric tool to assess immunolabeling.
Figure 5. NLTD method as a quantitative descriptor for IHC.
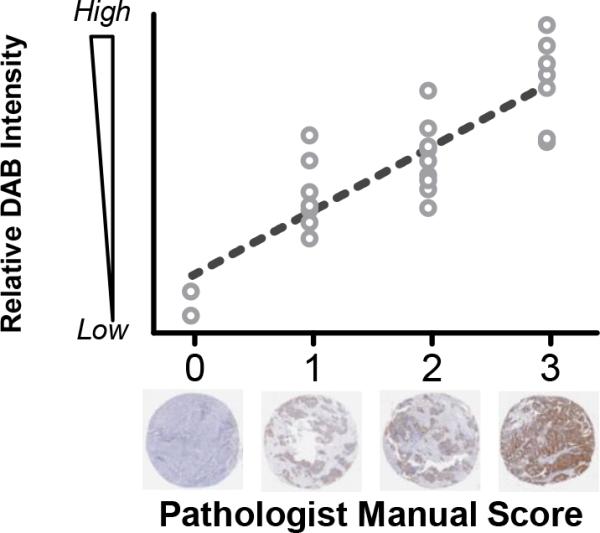
Ovarian tissue samples were stained with an antibody for LINE-1 ORF1p and manually scored by a pathologist31 on a discrete scale of 0 (no expression) to 3 (high expression). A quantitative score is calculated using the NLTD grayscale images. The scores correlate well, with a Pearson r = 0.85.
Discussion
Color deconvolution23, and other associated methods9, 26-29, 37, are routinely used for dye separation in histo-pathological images, but are limited by difference in dye appearance between images, potentially time consuming automated image processing, and a need for further post-processing to identify specific tissue components, such as nuclei. The NLTD approach presented here is able to bypass these limitations, specifically the requirement of prior knowledge of color information for different batches of histo-pathological images. The NLTD approach makes no inherent assumptions about the histo-pathological image's color space, and yields consistent, batch-invariant tissue component separation in histo-pathological images. We demonstrate that the NLTD method can successfully identify nuclei for a wide variety of histo-pathological images despite large variations in the perceptual color space (Fig. 2). Importantly, no prior knowledge or user input is required, as our algorithm will automatically register locations of for each tissue component, and the method can be used across multiple batches of images without additional user input. Therefore, NLTD method can be seamlessly integrated in computational pathology pipelines that aim to analyze large cohorts of images, such as the TCGA project (http://cancergenome.nih.gov/) or Human Protein Atlas Project39. The TCGA project also provides the opportunity to link morphological features of the histo-pathological images with genomic information, with potential for better understanding of what effect the changes in gene expression can have on the morphology of the tissue.
The tissue component images created through the NLTD method can be readily analyzed to yield additional information, such as nuclei information and immunohistochemical grading. We found that the NLTD method performs nuclei segmentation better than the color deconvolution approach. The segmentation approach presented here based on a simple implementation of Otsu's thresholding, but more refined approaches, as mentioned in reviews of computational pathology19, 20, should lead to greater accuracy using tissue component images from the NLTD method. We have demonstrated that the nuclei component images generated using the NLTD method have greater separation of signal from background compared to color deconvolution, suggesting simpler processes for nuclei edge detection can be used and lead to significantly reduced segmentation times. Nuclei detection requires very fast computation since an individual tissue image can have millions of nuclei, leading to large increases in total processing time with each additional nuclei detection step.
The field of computational pathology is rapidly growing, and there are many opportunities for computational approaches to provide additional prognostic and diagnostic information that cannot be provided by pathologists alone40-42. The NLTD method presented here provides a framework that can be easily implemented for many different applications, including nuclei detection and immunohistochemistry grading. In addition to these applications, NLTD could be used as a visualization tool to normalize tissue appearance across batches, provide texture information for abundance of certain tissue components in a sample, or identify rare occurrences in whole slide images, such as mitotic nuclei. Further, the NLTD method requires no prior knowledge of an image's color space and requires no parameterization from the user, which can allow for pathologists or medical technicians to apply this approach without requiring more sophisticated knowledge that may be needed for optimization methods or complex, linear algebraic approaches. Together, the proposed NLTD method presents an opportunity to establish a pipeline for classification and analysis of histo-pathological images that, in combination with pathologists’ expertise, can lead to better diagnosis and treatment planning for patients in the future.
Supplementary Material
Acknowledgements
The authors acknowledge funding from the National Cancer Institute (U54CA143868) and American Heart Association (12POST12050638).
Abbreviations
- H&E
hematoxylin and eosin
- IHC
Immunohistochemistry
- DAB
3,3' Diaminobenzidine
- NLTD
Non-Linear Tissue-component Discrimination
- TCGA
The Cancer Genome Atlas
- RBJH
Red-Blue Joint-Histogram
- PCA
Principal Component Analysis
Footnotes
Disclosure/Duality of Interest
The authors declare no competing financial interests.
Supplementary information is available at Laboratory Investigation's website.
References
- 1.Hipp J, Flotte T, Monaco J, et al. Computer aided diagnostic tools aim to empower rather than replace pathologists: Lessons learned from computational chess. Journal of pathology informatics. 2011;2:25. doi: 10.4103/2153-3539.82050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Sousa EMF, Wang X, Jansen M, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nature medicine. 2013;19(5):614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 3.Beck AH, Sangoi AR, Leung S, et al. Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Science translational medicine. 2011;3(108):108ra113–108ra113. doi: 10.1126/scitranslmed.3002564. [DOI] [PubMed] [Google Scholar]
- 4.Kwak JT, Hewitt SM, Sinha S, et al. Multimodal microscopy for automated histologic analysis of prostate cancer. BMC cancer. 2011;11:62. doi: 10.1186/1471-2407-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doyle S, Feldman M, Tomaszewski J, et al. A boosted bayesian multiresolution classifier for prostate cancer detection from digitized needle biopsies. Biomedical Engineering, IEEE Transactions on. 2012;59(5):1205–1218. doi: 10.1109/TBME.2010.2053540. [DOI] [PubMed] [Google Scholar]
- 6.Monaco JP, Tomaszewski JE, Feldman MD, et al. High-throughput detection of prostate cancer in histological sections using probabilistic pairwise Markov models. Medical image analysis. 2010;14(4):617–629. doi: 10.1016/j.media.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamilton PW, Wang Y, Boyd C, et al. Automated tumor analysis for molecular profiling in lung cancer. 2015 doi: 10.18632/oncotarget.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan Y, Failmezger H, Rueda OM, et al. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Science translational medicine. 2012;4(157) doi: 10.1126/scitranslmed.3004330. [DOI] [PubMed] [Google Scholar]
- 9.Veta M, Kornegoor R, Huisman A, et al. Prognostic value of automatically extracted nuclear morphometric features in whole slide images of male breast cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2012;25(12):1559–1565. doi: 10.1038/modpathol.2012.126. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Pécot T, Zynger DL, et al. Identifying survival associated morphological features of triple negative breast cancer using multiple datasets. Journal of the American Medical Informatics Association. 2013;20(4):680–687. doi: 10.1136/amiajnl-2012-001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L-W, Qu A-P, Yuan J-P, et al. Computer-based image studies on tumor nests mathematical features of breast cancer and their clinical prognostic value. 2013 doi: 10.1371/journal.pone.0082314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong F, Irshad Humayun, Oh Eun-Yeong, et al. Computational Pathology to Discriminate Benign from Malignant Intraductal Proliferations of the Breast. PLoS ONE. 2014;9(12) doi: 10.1371/journal.pone.0114885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper LA, Kong J, Gutman DA, et al. Novel genotype-phenotype associations in human cancers enabled by advanced molecular platforms and computational analysis of whole slide images. Laboratory investigation; a journal of technical methods and pathology. 2015;95(4):366–376. doi: 10.1038/labinvest.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper LA, Kong J, Gutman DA, et al. Integrated morphologic analysis for the identification and characterization of disease subtypes. Journal of the American Medical Informatics Association. 2012;19(2):317–323. doi: 10.1136/amiajnl-2011-000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Díaz G, Romero E. Micro-structural tissue analysis for automatic histopathological image annotation. Microscopy research and technique. 2012;75(3):343–358. doi: 10.1002/jemt.21063. [DOI] [PubMed] [Google Scholar]
- 16.Haggerty JM, Wang XN, Dickinson A, et al. Segmentation of epidermal tissue with histopathological damage in images of haematoxylin and eosin stained human skin. BMC medical imaging. 2014;14(1):7. doi: 10.1186/1471-2342-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kothari S, Phan JH, Osunkoya AO, et al. Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine. ACM; 2012. Biological interpretation of morphological patterns in histopathological whole-slide images. pp. 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janowczyk A, Chandran S, Singh R, et al. High-throughput biomarker segmentation on ovarian cancer tissue microarrays via hierarchical normalized cuts. Biomedical Engineering, IEEE Transactions on. 2012;59(5):1240–1252. doi: 10.1109/TBME.2011.2179546. [DOI] [PubMed] [Google Scholar]
- 19.Irshad H, Veillard A, Roux L, et al. Methods for Nuclei Detection, Segmentation, and Classification in Digital Histopathology: A Review— Current Status and Future Potential. Biomedical Engineering, IEEE Reviews in. 2014;7:97–114. doi: 10.1109/RBME.2013.2295804. [DOI] [PubMed] [Google Scholar]
- 20.McCann MT, Ozolek JA, Castro CA, et al. Signal Processing Magazine. 1. Vol. 32. IEEE; 2015. Automated Histology Analysis: Opportunities for signal processing. pp. 78–87. [Google Scholar]
- 21.Falkeholm L, Grant CA, Magnusson A, et al. Xylene-free method for histological preparation: a multicentre evaluation. Laboratory investigation. 2001;81(9):1213–1221. doi: 10.1038/labinvest.3780335. [DOI] [PubMed] [Google Scholar]
- 22.Martina JD, Simmons C, Jukic DM. High-definition hematoxylin and eosin staining in a transition to digital pathology. Journal of pathology informatics. 2011:2. doi: 10.4103/2153-3539.86284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruifrok A, Johnston D. Quantification of Histochemical Staining by Color Deconvolution. Analytical and Quantitative Cytology and Histology. 2001;23(4):291–299. [PubMed] [Google Scholar]
- 24.Kothari S, Phan JH, Moffitt R, et al. Biomedical Imaging: From Nano to Macro. IEEE International Symposium on. IEEE; 2011. 2011. Automatic batch-invariant color segmentation of histological cancer images. pp. 657–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pham N-A, Morrison A, Schwock J, et al. Quantitative image analysis of immunohistochemical stains using a CMYK color model. Diagnostic pathology. 2007;2(1):8. doi: 10.1186/1746-1596-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khan AM, El-Daly H, Rajpoot NM. Pattern Recognition (ICPR), 2012 21st International Conference on. IEEE; 2012. A gamma-gaussian mixture model for detection of mitotic cells in breast cancer histopathology images. pp. 149–152. [Google Scholar]
- 27.Veta M, Huisman A, Viergever MA, et al. Biomedical Imaging: From Nano to Macro. IEEE International Symposium on. IEEE; 2011. 2011. Marker-controlled watershed segmentation of nuclei in H&E stained breast cancer biopsy images. pp. 618–621. [Google Scholar]
- 28.Veta M, van Diest PJ, Kornegoor R, et al. Automatic nuclei segmentation in H&E stained breast cancer histopathology images. PLoS ONE. 2013;8(7):e70221. doi: 10.1371/journal.pone.0070221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan AM, Rajpoot N, Treanor D, et al. A Non-Linear Mapping Approach to Stain Normalisation in Digital Histopathology Images using Image-Specific Colour Deconvolution. IEEE Transactions on Biomedical Engineering. 2014;61(6) doi: 10.1109/TBME.2014.2303294. [DOI] [PubMed] [Google Scholar]
- 30.Hoffman RA, Kothari S, Wang MD. Comparison of Normalization Algorithms for Cross-Batch Color Segmentation of Histopathological Images. 2014:194–197. doi: 10.1109/EMBC.2014.6943562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodic N, Sharma R, Sharma R, et al. Long Interspersed Element-1 Protein Expression Is a Hallmark of Many Human Cancers. Am J Pathol. 2014;184(5):1280–1286. doi: 10.1016/j.ajpath.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fabbri R, Costa LDF, Torelli JC, et al. 2D Euclidean distance transform algorithms: A comparative survey. ACM Computing Surveys (CSUR) 2008;40(1):2. [Google Scholar]
- 33.Wienert S, Heim D, Saeger K, et al. Detection and segmentation of cell nuclei in virtual microscopy images: a minimum-model approach. Scientific reports. 2012;2:503. doi: 10.1038/srep00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drelie Gelasca E, Obara B, Fedorov D, et al. A biosegmentation benchmark for evaluation of bioimage analysis methods. BMC bioinformatics. 2009;10:368. doi: 10.1186/1471-2105-10-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otsu N. A threshold selection method from gray-level histograms. Automatica. 1975;11(285-296):23–27. [Google Scholar]
- 36.Powers DMW. Evaluation: From Precision, Recall and F-Meausure to ROC, Informedness, Markedness & Correlation. Journal of Machine Learning Technologies. 2011;2(1):37–63. [Google Scholar]
- 37.Macenko M, Niethammer M, Marron JS, et al. A Method for Normalizing Histology Slides for Quantitative Analysis. 2009 Ieee International Symposium on Biomedical Imaging: From Nano to Macro. 2009;1 and 2:1107–1110. [Google Scholar]
- 38.Chen J-M, Qu A-P, Wang L-W, et al. New breast cancer prognostic factors identified by computer-aided image analysis of HE stained histopathology images. Scientific reports. 2015:5. doi: 10.1038/srep10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uhlen M, Oksvold P, Fagerberg L, et al. Towards a knowledge-based human protein atlas. Nature biotechnology. 2010;28(12):1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 40.Bouzin C, Lamba Saini M, Khaing K-K, et al. Digital pathology : elementary, rapid and reliable automated image analysis. Histopathology. 2015 doi: 10.1111/his.12867. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 41.Louis DN, Gerber GK, Baron JM, et al. Computational pathology: an emerging definition. Archives of pathology & laboratory medicine. 2014;138(9):1133. doi: 10.5858/arpa.2014-0034-ED. [DOI] [PubMed] [Google Scholar]
- 42.Roth KA, Almeida JS. Coming into Focus: Computational Pathology as the New Big Data Microscope. The American journal of pathology. 2015;185(3):600–601. doi: 10.1016/j.ajpath.2015.01.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.