

Abstract
The oligoadenylate synthetase 1 (OAS1) enzyme acts as an innate sensor of viral infection and plays a major role in the defense against a wide diversity of viruses. Polymorphisms at OAS1 have been shown to correlate with differential susceptibility to several infections of great public health significance, including hepatitis C virus, SARS coronavirus, and West Nile virus. Population genetics analyses in hominoids have revealed interesting evolutionary patterns. In Central African chimpanzee, OAS1 has evolved under long-term balancing selection, resulting in the persistence of polymorphisms since the origin of hominoids, whereas human populations have acquired and retained OAS1 alleles from Neanderthal and Denisovan origin. We decided to further investigate the evolution of OAS1 in primates by characterizing intra-specific variation in four species commonly used as models in infectious disease research: the rhesus macaque, the cynomolgus macaque, the olive baboon, and the Guinea baboon. In baboons, OAS1 harbors a very low level of variation. In contrast, OAS1 in macaques exhibits a level of polymorphism far greater than the genomic average, which is consistent with the action of balancing selection. The region of the enzyme that directly interacts with viral RNA, the RNA-binding domain, contains a number of polymorphisms likely to affect the RNA-binding affinity of OAS1. This strongly suggests that pathogen-driven balancing selection acting on the RNA-binding domain of OAS1 is maintaining variation at this locus. Interestingly, we found that a number of polymorphisms involved in RNA-binding were shared between macaques and chimpanzees. This represents an unusual case of convergent polymorphism.
Keywords: OAS1, Polymorphism, Balancing selection, Macaque, Baboon
Introduction
Oligoadenylate synthetases (OASs) are members of the interferon pathway which plays an important role in innate antiviral defense (Goodbourn et al. 2000; Hovanessian 2007; Rebouillat and Hovanessian 1999). Upon infection with a viral pathogen, OAS enzymes are upregulated by interferon and act as viral sensors of infection in the cytosol by binding to, and being activated by, viral dsRNA. Upon activation, OAS enzymes synthesize 2′-5′-linked oligoadenylates using ATP and newly synthesized oligoadenylates as substrates. These oligoadenylates trigger the dimerization and activation of RNase L, which acts as an indiscriminate RNase, targeting both viral and cellular RNA for degradation, thus stopping protein synthesis and viral replication. In primates, the OAS gene family consists of three active members containing one (OAS1), two (OAS2), or three (OAS3) homologous domains (Justesen et al. 2000; Kumar et al. 2000). OAS1 has been shown to participate in the first line of defense against a wide range of viral infection including encephalomyocarditis virus, coxsackievirus B4, respiratory syncytial virus, West Nile virus, hepatitis C virus, and HIV-1 (Silverman 2007). Association studies have determined that variation at OAS1 correlates with disease severity of or susceptibility to several viral infections of great public health significance such as hepatitis C virus (Knapp et al. 2003; Zhao et al. 2013), SARS coronavirus (Hamano et al. 2005; He et al. 2006), West Nile virus (Bigham et al. 2011; Lim et al. 2009), and coxsackievirus A16 (Cai et al. 2014).
The crystal structure of the OAS1 protein has recently been solved (Donovan et al. 2013; Hartmann et al. 2003). OAS1 is a globular protein composed of two large lobes (N- and C-lobes) connected by a linker region. In between the two lobes lies the active site within a deep electronegative channel. On the directly opposite side of the enzyme lies a positively charged groove, the RNA-binding domain (RBD). Upon binding an RNA ligand, OAS1 undergoes a conformational change resulting in the activation of the enzyme (Donovan et al. 2013). Since OAS1 requires direct interaction with viral dsRNA for activation, it is likely that the evolution of this enzyme is affected by the evolution of viral pathogens or by the viral diversity to which an organism is exposed. Indeed, evolutionary analyses in rodents, primates, and bats strongly suggest that the evolution of the OAS1 protein is driven by host-pathogen interactions (Ferguson et al. 2008, 2012; Mozzi et al. 2015). In chimpanzees, for instance, OAS1 is extraordinarily variable and it was shown that OAS1 haplogroups predate the origin of hominoids (Ferguson et al. 2012). The retention of ancestral polymorphisms over such a long period of time is highly unusual and strongly suggests that OAS1 has evolved under balancing selection, a type of natural selection that maintains allelic variation in populations. In human, on the other hand, Mendez et al. (2012, 2013) failed to detect the signature of balancing selection, yet they made a very interesting observation: they showed that some contemporary human populations had acquired and retained OAS1 alleles of Neanderthal or Denisovan origin. However, it remains unknown if the persistence of these OAS1 alleles in modern human is adaptive, possibly conferring some advantage to the individuals carrying these alleles, or if this polymorphism is neutral.
We decided to further investigate the diversity of OAS1 in primates since evolutionary analyses can be of great help in answering essential questions concerning the antiviral function of immunoproteins (Cagliani and Sironi 2013; Fumagalli and Sironi 2014). Here, we analyze the diversity of OAS1 in baboons (g. Papio) and macaques (g. Macaca), two primate genera that have long served as models in infectious disease research (Misra et al. 2013; Palermo et al. 2013; Valdes et al. 2013; Wolf et al. 2006). We show that OAS1 harbors a very low diversity in baboons but a remarkably high variability in macaques, suggestive of the action of balancing selection. We demonstrate that balancing selection is principally acting on sites of the RNA-binding domain. Using in silico models of the OAS1 alloforms, we further show that these balanced polymorphisms are likely to affect the RNA-binding properties of the enzyme.
Materials and methods
Sampling
Genomic DNA from unrelated captive animals was acquired from the Southwest National Primate Research Center. The rhesus macaques (Macaca mulatta) sample includes six individuals of Indian origin and two of Chinese origin. The eight cynomolgus macaques (Macaca fascicularis) are of Indonesian origin. The seven olive baboons (Papio anubis) are of east African origin, and the six Guinea baboons (Papio papio), of west African origin (Boissinot et al. 2014). These baboon samples were previously analyzed for their mitochondrial and nuclear sequence variation, and we verified that their genetic diversity is representative of the diversity of their species of origin (Boissinot et al. 2014).
Sequencing and haplotype determination
The six exons constitutive of the entire protein-coding sequence of OAS1 were amplified independently by PCR using primers located in introns. The amplicons were Sanger-sequenced directly in both directions by the High-Throughput Genomics Unit at the University of Washington in Seattle. Geneious Pro 5.5.6 (created by Biomatters available at http://www.geneious.com) was used for sequence alignment, assembly, and heterozygote base-calls. We used PHASE v. 2.1 to determine haplotypes (Stephens and Scheet 2005; Stephens et al. 2001). Due to a high number of heterozygotes in some OAS1 reads, we ran PHASE using both individual exons and complete OAS1 cDNA sequences; these methods yielded identical results. For the analyses herein (except where otherwise mentioned), we have limited our analysis to the most common isoform (p40/42), consisting of exons 1 through 5. The sequences are deposited in GenBank under accession numbers KR260685-KR260713.
Evolutionary analyses
We calculated for each species the following summary statistics: h (the number of haplotypes), S (the number of segregating sites), π (the nucleotide diversity estimated as the average divergence between two alleles selected at random), and θ W, (the Watterson estimator of diversity, i.e., population mutation rate). We also calculated Tajima’s D to assess the impact of natural selection on variation (Tajima 1989). These calculations were performed using DnaSP v. 5 (Librado and Rozas 2009).
We used the Hudson-Kreitman-Aguade (HKA) test to determine the effect of selection at the OAS1 locus (Hudson et al. 1987). The HKA test evaluates selection by comparing the ratio of polymorphism to divergence at a locus of interest with the same ratio at one or more neutral loci. Under neutrality, this ratio should be the same across the genome but an excess of polymorphism relative to divergence at a specific locus could be interpreted as evidence for balancing selection while a deficit of polymorphism would indicate positive directional selection. Statistical significance is assessed using a X 2 goodness-of-fit test. As neutral regions of reference for the rhesus macaque, we used 30 randomly chosen loci that had previously been sequenced in 38 Indian and 9 Chinese unrelated individuals (Hernandez et al. 2007). For the cynomolgus macaque, we used 26 non-coding regions that were sequenced in 24 individuals from three Southeast Asian populations (Osada et al. 2010). For the two baboon species, we used the 12 neutral regions analyzed in Boissinot et al. (2014). As out-groups, we used human and gorilla homologous sequences. The multi-locus HKA test was performed using Jody Hey’s HKA software (available at https://bio.cst.temple.edu/∼hey/software/software.htm#HKA) or Sebas Ramos-Onsins HKAdirect software (http://bioinformatics.cragenomica.es/numgenomics/people/sebas/index.html).
We also used the McDonald-Kreitman test (MK), which compares the ratio of nonsynonymous to synonymous polymorphism within species to the ratio of nonsynonymous to synonymous fixed differences between species (McDonald and Kreitman 1991). Under neutrality, these ratios should be identical whereas an excess of nonsynonymous polymorphism indicates balancing selection and a deficit of nonsynonymous variation suggests directional positive selection. We used the online MK test program (http://bioinf3.uab.cat/mkt/MKT.asp), which includes a X 2 test for statistical validation (Egea et al. 2008). We also calculated the neutrality index (NI) simply defined as (number of polymorphic replacements/number of fixed replacements)/(number if polymorphic silent sites/number of fixed silent sites), which is equal to one under neutrality (Rand and Kann 1996).
The four-gamete test (Hudson and Kaplan 1985), implemented in DnaSP, was used to test for recombination and for identification of recombination break points. Linkage disequilibrium was evaluated using DnaSP by calculating the correlations for all pairs of single nucleotide polymorphisms (SNPs), statistical significance determined using a X 2 test with Bonferroni correction.
For each species, we constructed networks using the median-joining method (Bandelt et al. 1999) implemented in Network v. 4.6 (available at http://www.fluxus-engineering.com). We also created a maximum likelihood tree using the Tamura-Nei model of mutation, assessing its robustness with 1000 bootstrap iterations. The phylogenetic analysis was performed using MEGA 5.2.2 (Tamura et al. 2011).
PolyPhen-2 analysis
In order to pinpoint naturally occurring amino acid variants that may have significant effects on protein structure and function, we used PolyPhen-2 (Adzhubei et al. 2010). This software uses a naïve Bayesian classifier to identify SNPs that may alter protein structure and function. This iterative process combines properties mined from several sequence- and structure-based databases to determine the statistical likelihood of a replacement being benign to probably damaging (0.0–1.0 scale). Because PolyPhen-2 only operates with human protein sequences, we aligned our species’ OAS1 primary structure to human OAS1. Human OAS1 shares 94 % pairwise amino acid similarity with macaques and baboons, so editing a human OAS1 reference sequence (NCBI RefSeq: NP_001027581.1) to include each mutation was both simple and sufficient to garner useful output.
Creation and visualization of in silico protein models
To assess the structural and electrostatic changes associated with amino acid polymorphisms, we created in silico models of the OAS1 alleles found in baboons and macaques. The crystal structure of human OAS1 (hOAS1) with bound RNA and dATP has recently been published (Donovan et al. 2013). Considering the level of conservation of OAS1 between monkeys and humans at the amino acid level, it seems justified to use the solved structure of human OAS1 (PDB ID: 4IG8) to confidently model and annotate residues in the old world monkeys. Though taxonomically distant, we also found it valuable to use the porcine OAS1 (pOAS1) crystal structure (PDB ID: 1PX5), as it shares the overall structural fold (73 % pairwise, 86 % BLOSUM64 identity) and provides a template for modeling OAS1 without bound RNA. These templates allowed for creation of model sets representing before- and after-activation structural representations of the enzyme.
We used the modeling suite SWISS-MODEL (swissmodel.expasy.org) to generate three-dimensional models of primate OAS1 alleles. SWISS-MODEL uses a solved protein homolog as template, upon which the query sequence is framed. Model quality was assessed using two protein structure validation tools, Verify3D (Eisenberg et al. 1997) and ProSA-web (Sippl 1993; Wiederstein and Sippl 2007). Verify3D compares properties of primary structure to the structural environments modeled in the tertiary structure, producing quality scores in 21-residue windows across the amino acid sequence. ProSA-web assesses solvent-to-residue and intermolecular energies throughout the model to calculate an overall Z score (plotted amongst a database of solved proteins’ Z scores), as well as localized scores.
We used the molecular visualization platform PyMOL v. 1.7 (Schrödinger, LLC) to visualize and virtually analyze the OAS1 models created with and without RNA ligands. We created overlays of different OAS1 molecules to investigate how the solved and predicted model structures compare and to better understand, based on predicted position and orientation, the possible effects and importance of specific mutations. Unless otherwise noted, measures between nucleobases and amino acid side groups were made from the nearest non-hydrogen atom of the R-group to the nearest non-hydrogen atom of the base group of the ribonucleic acid (rather than to the RNA backbone). In order to map surface electrostatic characteristics onto models, we used the Adaptive Poisson-Boltzmann Solver (APBS) (Baker et al. 2001) plugin (written by Michael G. Lerner). APBS files were first created using PDB2PQR (Dolinsky et al. 2004) with the following settings: PARSE force field, internal naming scheme, optimized for H-bonding network, pH = 7.0.
Results
Polymorphism at OAS1
The four species of primates analyzed here present very different levels of polymorphism at the OAS1 gene (summarized in Table 1, Figs. 1 and 2). The highest variability was found in cynomolgus macaque with 48 SNPs (i.e., ∼44 SNPs/Kb) resulting in 16 different haplotypes (out of 16 sequenced haplotypes). A large number of SNPs was also found in the rhesus macaque (30 SNPs) resulting in nine different haplotypes. A significant fraction of SNPs are found at relatively high frequency (>0.25) in both species (13 and 12 SNPs in rhesus and cynomolgus, respectively), but within each species, a large number of private SNPs were detected: 22 in cynomolgus and 14 in rhesus. However, many of those SNPs that are private within species are in fact shared between species so that the actual number of singletons in macaques is 12 in cynomolgus and 3 in rhesus. The large number of private SNPs accounts for slightly negative, though non-significant, values for Tajima’s D (Table 1) in both species. We did not observe a single fixed difference between the two macaque species, which is not surprising considering their recent divergence (∼1 mya) and the occurrence of recent gene flow between them (Osada et al. 2008).
Table 1.
Summary statistics for OAS1 coding sequences in macaque and baboons
| N a | h b | S c | π d (%) | θ e (%) | Tajima’s Df | NIg | HKAh | |
|---|---|---|---|---|---|---|---|---|
| Cynomolgus macaque | 16 | 16 | 45 | 1.10 | 1.30 | −0.541 | 1.72 (P = 0.16) | 0.001 |
| Rhesus macaque | 16 | 9 | 29 | 0.84 | 0.93 | −0.248 | 1.52 (P = 0.30) | 0.001 |
| Olive baboon | 14 | 5 | 7 | 0.16 | 0.20 | −1.001 | 1.08 (P = 1.00) | 0.847 |
| Guinea baboon | 12 | 1 | 0 | 0.00 | 0.00 | N/A | N/A | 0.518 |
aNumber of chromosomes
bNumber of different haplotypes
cNumber of segregating sites
dNucleotide diversity
eWatterson’s estimator of diversity
fTajima’s D (Tajima 1989)
gNeutrality index; P values calculated using a two-tailed Fisher exact test
hProbability that variation at OAS1 is not significantly different than variation at neutral loci
Fig. 1.
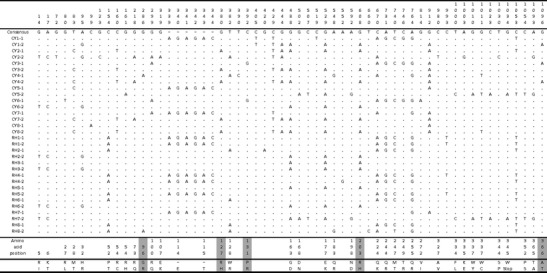
Variation at the OAS1 gene in cynomolgus and rhesus macaques. The numbers at the top correspond to the nucleotide position in an alignment of all macaque OAS1 alleles. The amino acid changes corresponding to each nonsynonymous mutation are indicated in the lower part of the figure. Amino acids changes that are recognized as damaging by PolyPhen-2 are shaded in grey
Fig. 2.
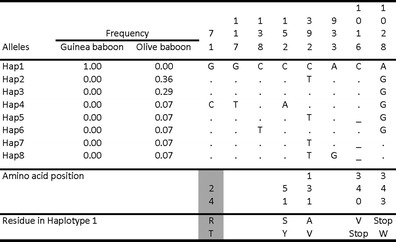
Variation at the OAS1 gene in Guinea and olive baboons. The numbers at the top correspond to the nucleotide position in an alignment of all baboon OAS1 alleles. The frequency of the haplotypes is indicated for each species. Replacements recognized as damaging by PolyPhen-2 are shaded in grey
Interestingly, the proportion of SNPs resulting in changes at the amino acid level is remarkably high in both macaque species: 36 (out of 48) and 22 (out of 30) SNPs are nonsynonymous in the cynomolgus and rhesus macaques, respectively. A premature stop codon in exon 5 was found in one haplotype in each species. In addition, an insertion of six nucleotides in the second exon, resulting in the addition of two amino acids, was found at a frequency of 0.2 and 0.4 in the cynomolgus and rhesus macaque, respectively. Consistent with the large number of SNPs in these two species, the nucleotide diversity is remarkably high, at 1.10 and 0.84 % in cynomolgus and rhesus, respectively. This is much higher than the diversity reported in protein-coding genes (∼0.175 to 0.27 %) and higher than the genomic average reported in these species (0.24–0.35 %) (Osada et al. 2010). The genetic diversity of macaques has been shown to vary by subspecies, geography, and sequencing coverage, but all published estimates are several-fold lower than those found at OAS1 (Ferguson et al. 2007; Osada et al. 2010).
By comparison, OAS1 in baboon harbors a very low variability (Fig. 2). We did not find a single SNP in the Guinea baboon and only seven SNPs in the olive baboon, resulting in nucleotide diversity of 0.00 and 0.16 %, respectively. Five of the seven SNPs correspond to private mutations. A single base pair deletion in exon 5 resulting in a premature stop codon was found in three haplotypes. Interestingly, all the Guinea baboon sequences have the same nonsense mutation in exon 5 identified in macaques. The low level of diversity in baboons is very similar to the genomic average in these species (0.023 and 0.14 % in Guinea and olive baboons, respectively; Boissinot et al. 2014).
Tests of selection
The high level of variation in macaque suggests that balancing selection is acting at this locus. We tested this hypothesis using the HKA test of selection which compares the ratio of polymorphism to divergence between OAS1 and a collection of non-coding, neutral regions that had previously been sequenced in the two macaques (Table 1). In both the rhesus and the cynomolgus macaque, we found that variation at OAS1 significantly exceeds what is expected given the divergence between macaque and human, confirming the balancing selection hypothesis (P = 0.001). It should be noted that the neutral regions of reference were sequenced in a geographically diverse sample of macaques; it is thus unlikely that neutral variation is underestimated when compared with OAS1 variation. As expected, the HKA test did not reveal any deviation from neutrality in baboons.
Another prediction of the balancing selection hypothesis is an excess of polymorphic replacements relative to silent mutations. The amount of polymorphism is correlated to the level of between-species divergence under neutrality, and this should be true for both replacement and silent sites. Thus, we compared the level of polymorphism at replacement and silent sites with the fraction of replacement and silent sites that are fixed between each of our focal species and an out-group. To this end, we calculated the parameter NI, which is predicted to be higher than 1 if balancing selection is acting at a locus (Table 1). Using human or gorilla as an out-group, we obtained values that were well above 1 for both macaque species (1.6 to 1.9 for cynomolgus and 1.4 to 1.7 for rhesus), although we were not able to statistically exclude neutrality using the MK test (P = 0.16 to 0.37 for cynomolgus and P = 0.30 to 0.56 for rhesus). By comparison, the NI values for the olive baboon ranged from 0.75 to 0.89 (P = 1.0).
Recombination and network analysis
The recombination analysis was performed for each species and for each genus. Rhesus macaques have fewer haplotypes, necessitating far less recombination, evidenced by the minimum number of recombination events predicted to be 2. In cynomolgus, nine events can minimally explain the current haplotypic structure. When we assess all macaque haplotypes together, the minimum number of recombination events is 12. We ran linkage disequilibrium analysis using DnaSP in order to test linkage between polymorphisms. Pooling all macaque sequences, linkage was found (P < 0.001) between 77 pairs of polymorphic sites. Nearly all linkage was found to be between physically close sites (located in the same exon). No linkage disequilibrium between distant sites that could be suggestive of epistatic interactions between polymorphic amino acids was found. The amount of recombination at the OAS1 locus is also apparent in the haplotype network (Fig. 3), which shows considerable reticulation. We did not find any evidence of recombination in olive baboon, which is not surprising considering the very small number of polymorphisms in this species.
Fig. 3.
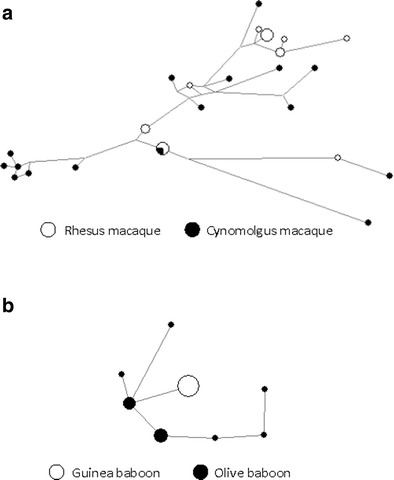
Haplotype networks of OAS1 alleles in macaques (a) and baboons (b) constructed using the median-joining method. Node size reflects haplotype frequency
In silico modeling of OAS1 alloforms
For the purposes of in silico modeling, we used only naturally occurring alleles, choosing those containing the largest proportion of high-frequency amino acid polymorphisms. Using these criteria, we selected alloforms Cy8-2 and Rh2-1 (Fig. 1) for the majority of in silico modeling and analysis. For the few polymorphisms of interest not represented, we modeled additional alloforms. SWISS-MODEL produced models of quality at, or very near, those of the solved human OAS1 protein (Verify3D overall average scores: 0.48 for hOAS1 and 0.46 to 0.48 for the predicted models; ProSA-web overall Z score: −9.73 for hOAS1 and −10.19 to −10.38 for the predicted models). A single loop that could not be crystallized for either porcine or human OAS1 (residues 120–131) was evaluated to be of low quality in all of our models. Despite this, they were sculpted nearly identically amongst alloforms (Fig. 4).
Fig. 4.
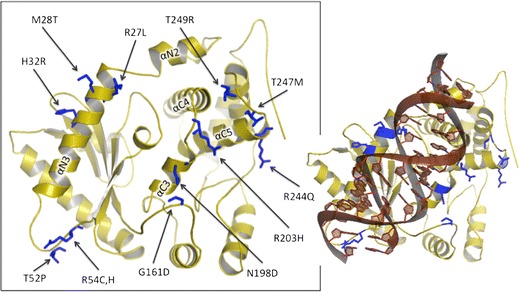
Ribbon representations of macaque OAS1 RNA-binding facet (Rh2-1 haplotype). Polymorphic residues are labeled and colored in blue. The alpha-helices participating in the RNA-binding domain are labeled. Right, dsRNA docked according to solved hOAS1 (PDB ID: 4IG8; Donovan 2013)
In analyzing the protein models, we have methodically judged the potential structural and functional effects of the polymorphic residues on an individual basis as well as collectively where relevant. We examined the impact of each amino acid replacement on (1) enzymatic function, (2) ligand-borne activation, and (3) RNA-binding influence. Salient characteristics considered were R-group biochemical properties (charge, hydrophobicity/polarity, size/structure, etc.), location, orientation (in relation to both neighboring residues and RNA), published research, and homology. In our analysis, we distinguished between polymorphisms in the RBD and those outside the RBD (Table 2) since pathogen-driven balancing selection predicts that regions of the protein that interact directly with viral RNA are most likely to contain the polymorphic sites.
Table 2.
Characteristics of the polymorphic amino acids in the RNA-binding region
| Amino acid position | Ancestral residue | Derived residue | Distance to closest RNA-binding site (in number of amino acids)a | Location on protein | Frequency of derived SNP in rhesus | Frequency of derived SNP in cynomolgus | ||
|---|---|---|---|---|---|---|---|---|
| 27 | R | Positive | L | Hydrophobic | 0 | αN3 | 0.00 | 0.06 |
| 28 | M | Hydrophobic | T | Polar | 1 | αN3 | 0.00 | 0.38 |
| 32 | H | Positive | R | Positive | 1, 2 | αN3 | 0.19 | 0.19 |
| 52 | P | Non-polar | T | Polar | 2 | Between αN3 and β1 | 0.56 | 0.00 |
| 54 | R | Positive | C | Polar | 0, 1 | Between αN3 and β1 | 0.00 | 0.31 |
| 54 | R | Positive | H | Positive | 0, 1 | Between αN3 and β1 | 0.00 | 0.06 |
| 163 | G | Polar | D | Negative | 3 | Between β5 and αC1 | 0.38 | 0.44 |
| 198 | N | Polar | D | Negative | 3 | αC3 | 0.06 | 0.06 |
| 203 | R | Positive | H | Positive | 1, 2, 2 | Between αC3 and αC4 | 0.31 | 0.44 |
| 247 | M | Hydrophobic | T | Polar | 2 | Between αC5 and αC6 | 0.50 | 0.19 |
| 249 | T | Polar | R | Positive | 0 | between αC5 and αC6 | 0.00 | 0.19 |
aA zero value indicates that the residue itself binds RNA. Multiple numbers indicate distances to several neighboring RNA-binding residues
Analysis of polymorphisms in the RNA-binding domain
The crystallized human OAS1 with docked RNA identified 20 amino acids that bond either electrostatically or via hydrogen bond to the RNA ligand (Donovan et al. 2013). Considering the high degree of overall conservation of sequence and structure and after meticulous comparison of hOAS1 overlaid with a variety of primate OAS1 models, we are confident that most, if not all, of these 20 sites serve homologous binding functions in primates as in human.
Polymorphisms R27L, M28T, and H32R are located within the same helix N3 comprising the major binding sites of the N-lobe of the RBD (Figs. 4 and 5a, b). Helix N3 notably takes part in the conformational change that OAS1 undergoes upon RNA-mediated activation (Donovan et al. 2013). These three polymorphic sites are situated on the surface of the protein and can affect RNA-binding affinity. Residue 27 participates in direct electrostatic binding to the docked RNA backbone, and the R27L mutation conveys significant electrostatic and hydrophobicity changes. PolyPhen-2, however, does not classify this change as damaging, possibly because leucine occupies this site in the hOAS2 and hOAS3 paralogs. For the H32R polymorphism, the surface electrostatic profile change appears quite dramatic as the arginine creates a far more electropositive surface than the histidine side group. While human OAS1’s solved structure, featuring histidine at this site, does not demonstrate direct ligand interaction here (Donovan et al. 2013), the polymorphic arginine has this potential.
Polymorphisms P52T, R54C, and R54H are found in a long and disordered linker between helix N3 and β strand 1 (Figs. 4 and 5a, c). They can presumably participate in RNA binding because of their proximity to docked RNA and because of the flexibility of the linker. P52 is highly conserved across taxa, but the threonine replacement would presumably allow for increased interaction with docked RNA by increasing the linker’s overall flexibility. Residue 54 participates in direct binding to RNA, and it is surprisingly variable within primates despite the otherwise high degree of conservation at this position amongst non-primates, as suggested by a BLASTP search. The surface electrostatics shows a significant effect of this polymorphism since R54 yields a broad electropositive surface to the protein while cysteine has a neutral effect. In addition, replacements at sites 52 and 54 may have an effect on residue S56, which binds directly to RNA nucleobase G17. The role of G17 is noteworthy because point mutation of G17 has been shown to decrease OAS1 activation 30-fold (Donovan et al. 2013). Presumably, even a small degree of interference with S56’s hydroxyl group would alter activation.
G163D (Fig. 5d) is located in a pliant and extensive linker between the N- and C-lobes (Fig. 5). The solved structure identifies only Q160 in this immediate region as contributing to RNA binding (via three hydrogen bonds). D163 partially occludes the deep positive groove created by helix C3, which features the conserved RNA-binding sites R197, K201, and Q202. The ancestral G163 amino acid increases overall linker flexibility and produces a concavity for the RNA backbone to nestle.
Fig. 5.
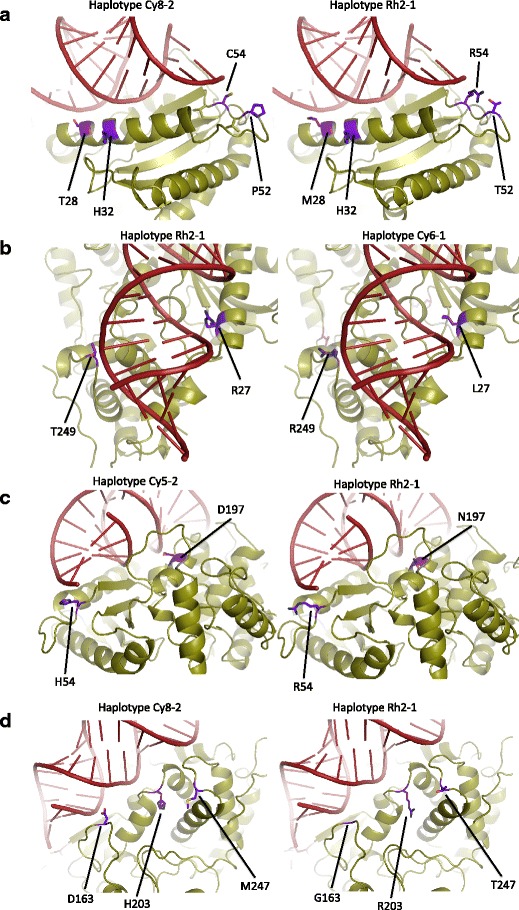
a–d Ribbon representations of macaque OAS1 with docked RNA. Four different haplotypes are shown from various perspectives. Polymorphic sites within the RNA-binding domain are colored purple
The remaining polymorphic residues of interest lie within the C-lobe. Variable residues N198D and R203H are located centrally along the lateral axis of the RNA-binding groove directly across from helix N3 (Fig. 5c, d). Although N198D is within the highly conserved helix C3, its positioning three and four residues away, place it cis to solved RNA binders K201 and Q202, superficial and facing the nucleic acid ligand. Negatively charged D198 can interfere with the electropositive character of the region conferred by R197 and K201. Immediately neighboring N198D is R197, a key player in conformational change, the side group of which exchanges position with that of K66 when RNA docks. This exchange is one of the major reconfigurations required for the active site to become functional (Donovan et al. 2013). However, we predict that N198D would not negatively impact this essential interchange because our structural and kinetic models show that its side group, before and after binding, is oriented external to the deep groove in which the conformational change physically occurs. In addition, several mammals (e.g., rodents, new world primates) have independently evolved this specific mutation (as detected in a BLASTP search) and PolyPhen-2 classifies this change as benign.
R203H lies in the middle of the linker region connecting helices C3 and C4 (Fig. 5d). The functional significance of this linker has been shown experimentally, as activity assays of either K201E or K206E mutations have shown decreased activity by two to three orders of magnitude (Hartmann et al. 2003). Neighboring are several residues identified as RNA binding, Q202, T205, and K204, with which 203 could interact. Residue 203 is situated at the minor groove of docked RNA, which can potentially allow for nucleobase recognition. PolyPhen-2 scores this replacement at 0.66, possibly damaging, most likely due to the ubiquitous conservation of arginine at this site. We predict that this mutation is likely to have a significant effect on RNA binding.
The most C’ polymorphic residues that play a part in the RBD are M247T and T249R, the latter of which forms hydrogen bonds with RNA (Fig. 5b, d). Both are located in a loop between helices C5 and C6, capable of interacting with RNA and each other. In addition, polymorphic amino acid 203 is situated very close to both 247 and 249, potentially allowing for interaction between these residues. Our models indicate that even accounting for the flexibility of the linker, only 249 appears to be situated such that it could take part in significant RNA binding at the minor groove. The electrostatic profile of the models shows that the T249R replacement considerably alters surface charge. Arginine greatly protrudes from the edge of the RBD with its highly positive side group. This region, at the edge of the RBD, may influence RNA ligand binding based on RNA length and higher order structure.
Analysis of macaque polymorphisms outside of the RNA-binding domain
The balancing selection hypothesis posits that selection acts on regions that interact directly with viral dsRNA, yet the enzymatic activity must not be hindered by polymorphic amino acids. To this end, we have examined the naturally occurring non-RBD mutations that might alter the essential mechanisms of activation and synthesis activity of OAS1. Outside of the RBD, there are 26 amino acid variants, and of these, only R127H, W128R, and P131R are predicted to noticeably affect the protein secondary structure in our models. The region from residues 121–126 in human and porcine OAS1 is uncharacterized due to poor crystallization and most likely corresponds to a disordered loop. PolyPhen-2 identifies R127H and P131R as possibly damaging with scores of 0.76 and 0.85, respectively, while W128R is completely benign (0.0 score). Verify3D calculated low-quality scores for this region (with nadir scores ranging from 0.00 to 0.23). Until this region is better characterized, it will be difficult to assess the functional significance, if any, of these three changes.
Polymorphism G96R is found in a bent helix on the opposite side of the protein from the RBD, but not in the catalytic domain. Secondary structure is not predicted to change with this replacement according to our models. However, the conservation of glycine at this position across mammals and the fact that PolyPhen-2 classifies this replacement as possibly damaging (0.79) suggest that this polymorphism could have some functional significance.
Focusing on the active site and the residues responsible for conformational change, we identified only one polymorphism, R73Q, located two residues from a metal-binding aspartic acid. Glutamine is relatively neutral, compared to arginine, but the terminal amide group may participate in some of the same interactions. We have also kinetically modeled this site to shift during the conformational change, so it could potentially interfere with activation or activity, yet PolyPhen-2 classifies this site as benign. The fact that human OAS2 has a glycine (an even more disparate residue) at the paralogous site leads us to believe that the mutation is not likely to impede enzymatic activity.
In addition to analyzing polymorphisms, it is of some value to note mutations that we do not observe. Of the many amino acid polymorphisms we identified in macaque OAS1, we do not have replacements predicted to disrupt any α-helices or β-sheets. We also found no polymorphic amino acids in our models with R-groups facing internally. Though macaque haplotypes do contain replacements with large hydrophobicity shifts, the position of each of these is located in superficial linkers or loops where they are least likely to interfere with known enzyme structure or function. This absence of harmful mutations is consistent with an evolutionary history of purifying selection at sites important for the activation and enzymatic activity of OAS1.
Discussion
We analyzed intra-specific variation at the OAS1 gene in four species of primates belonging to two primate genera that are widely used in infectious disease research. We found that the level of variation differs considerably among taxa. OAS1 exhibits a level of polymorphism far greater than the genomic average in macaques. Using the HKA test, we validated the hypothesis that this remarkably high level of variation is caused by balancing selection. We further showed that the region of the protein that interacts directly with viruses, the RNA-binding domain, contains a number of polymorphisms likely to affect the RNA-binding affinity of OAS1. These mutations however do not result in large surface electrostatic changes that would inhibit RNA binding and activation, and we did not observe mutations that would compromise essential enzymatic function and structure. Together, these lines of evidence strongly suggest that pathogen-driven balancing selection acting on the RBD is maintaining variation at this locus. In contrast, OAS1 in baboons exhibits a level of polymorphism comparable to, or lower than, the genomic average, suggesting that the selective pressure acting on OAS1 differs greatly between these two primate genera.
Balancing selection is believed to be rare in nature compared with positive directional selection (Fischer et al. 2014). In addition, selective sweeps of favorable alleles or drastic reductions in population size can erase the signature of balancing selection; thus, balanced polymorphisms are predicted to be transient, and long-term balancing selection exceedingly rare (Charlesworth 2006). Yet, recent studies in human have emphasized the importance of balancing selection as an efficient mechanism to maintain variation in populations (reviewed in Key et al. 2014), particularly at immunity genes. The most famous and best documented example of balancing selection concerns genes of the MHC system where alleles have been maintained in populations for millions of years, resulting in trans-specific polymorphisms (Klein et al. 1993; Lawlor et al. 1988; Mayer et al. 1988). In recent years, candidate gene analyses as well as genome-wide searches have identified additional loci that have evolved under balancing selection, including the ABO blood group (Leffler et al. 2013), the anti-retroviral TRIM5 gene (Cagliani et al. 2010), and the CCR5 gene (Bamshad et al. 2002). It has even been proposed that balancing selection is the main evolutionary force acting on innate immunity genes (Ferrer-Admetlla et al. 2008), a category of genes OAS1 belongs to.
Balancing selection had previously been demonstrated at OAS1 in central African chimpanzees (Ferguson et al. 2012). Comparable to what we find in macaques, chimpanzee OAS1 contains an extremely high level of polymorphism that far exceeds the genomic average as well as an excess of nonsynonymous polymorphisms. Furthermore, a number of polymorphic sites appeared to fall conspicuously in the RBD (Ferguson et al. 2012). In order to see if any of the replacement sites in macaque were shared with chimpanzee, we aligned the primary protein sequences as well as the structural models. The two main groups of alleles in chimpanzee differ by 15 polymorphic amino acids. Seven of these sites (28, 54, 96, 128, 163, 244, and 337) are also polymorphic in macaque. Surprisingly, six of these residues are polymorphic for exactly the same amino acids, including three RBD sites (28, 54, and 163), two of which (28 and 54) were identified as selected sites by Mozzi et al. (2015). Another five polymorphic amino acids in macaque immediately neighbor residues that are polymorphic in chimpanzee. The similarity between macaque and chimpanzee is demonstrated on Fig. 6, which shows RBD polymorphisms side by side. The similarity between these two primate taxa is striking and could suggest that these polymorphisms have been maintained by long-term balancing selection since macaque and chimpanzee split. This is however very unlikely since macaque and chimpanzee otherwise differ by 15 fixed amino acids and phylogenetic analyses demonstrate that chimpanzee and macaque alleles are reciprocally monophyletic (not shown). Instead, the similarity between chimpanzees and macaques represent an interesting case of convergent polymorphism.
Fig. 6.
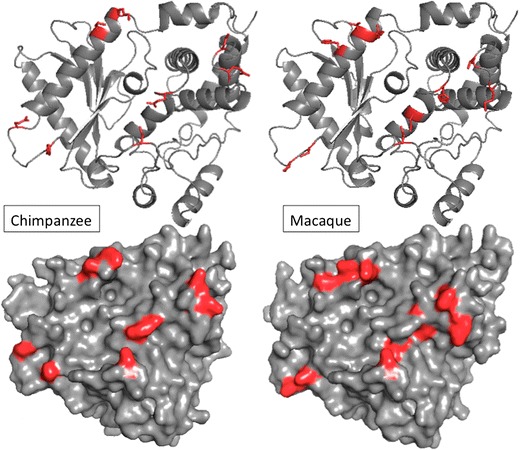
Ribbon and surface models of central African chimpanzee and macaque OAS1. Polymorphic residues found in each respective species within the RNA-binding domain are colored red
There are however two main differences between chimpanzee and macaque in the pattern of variation at OAS1. First, we did not detect in macaque an excess of intermediate frequency alleles that would have yielded positive values of Tajima’s D, whereas such an excess of intermediate frequency alleles was found in chimpanzee, resulting in positive and significant values of Tajima’s D. Second, we did not find evidence for linkage disequilibrium in macaque whereas very strong linkage, suggestive of epistatic interactions, was found in chimpanzee between mutations in exons 1, 3, and 4. Although differences in recombination rate could account for this pattern, it is more likely to reflect a difference in the age of the balanced polymorphism. Multi-locus selection takes a long time to generate linkage disequilibrium (Hedrick 2010), and Tajima’s D fails to detect balancing selection if selection is too recent (Garrigan and Hedrick 2003). Thus, it is most likely that balancing selection has not acted on OAS1 polymorphisms in macaque as long as it has in chimpanzee.
Since balancing selection has played such a significant role in the evolution of OAS1, it is somewhat surprising that we did not find any evidence for it in baboons. Two processes could have erased the signature of balancing selection in these species. First, a diversity-reducing demographic event, such as a population bottleneck, could have eliminated allelic diversity at OAS1. This is a possibility for the Guinea baboon, a species characterized by a remarkably low level of genetic variation (Boissinot et al. 2014). It is however very unlikely to be the case in olive baboons since this form has maintained a relatively large effective population size for most of its history (Boissinot et al. 2014). Second, a selective sweep resulting in the fixation of a highly favorable allele could have occurred in the ancestor of baboons. When compared to macaque, there are only three derived mutations that are fixed or nearly fixed in baboons at residues T24R, H25R, and Y51S. Residues 24 and 25 belong to the α-helix N3, which participates in the RBD, and residue 24 was shown to interact directly with the RNA ligand (Donovan et al. 2013). The fixation of a positively charged arginine in place of an uncharged threonine is undoubtedly a highly significant change that could have affected drastically the RNA-binding affinity of the baboon OAS1. Interestingly, residue 24 is polymorphic in chimpanzee (T/K) (Ferguson et al. 2012) and was identified as a site under positive selection in this species (Mozzi et al. 2015). The replacement at residue 25 is unlikely to have much effect since the sidegroup ostensibly orients away from the RBD and a mutation from arginine to histidine is a conservative change. The change from the hydrophobic tyrosine to the uncharged serine at position 51 could also have a functional impact due to its proximity to a group of six residues (residues 54 to 60) that bind directly to the RNA ligand. We speculate that an episode of positive directional selection acting on one of these three sites (or on a combination of them) could have erased the signature of balancing selection in baboons. This hypothesis will however require experimental validation.
The mechanisms responsible for the maintenance of balanced polymorphism at OAS1 remain unclear but could include heterozygote advantage (if heterozygotes are better protected against viral pathogens), frequency-dependent selection, or selection that varies in space and/or time. Whatever the exact mechanism, the persistence of amino acid polymorphisms, in particular at the RBD, strongly suggests that OAS1 alleles differ in their RNA-binding activity, most likely to achieve optimal recognition of specific viral RNAs. The RNA-binding specificity of OAS1 implied by our evolutionary analysis is not consistent with early molecular studies, which suggested that OAS1 is a non-specific sensor of viral infection, and with the involvement of the OAS1/RNase L pathway against a wide diversity of viral infection (Silverman 2007). More recent studies however have shown differential binding and activation of OAS1 based on RNA sequence, RNA secondary structure, or presence of 3′ single-stranded pyrimidine motifs (Deo et al. 2014; Hartmann et al. 1998; Kodym et al. 2009; Vachon et al. 2015). The dsRNA consensus sequences NNWW(N)nWGN (Kodym et al. 2009), AYAY(N)nCC, and UU(N)nACCC (Hartmann et al. 1998) have been identified experimentally, and it was shown that single or double SNPs in RNA ligands could alter OAS1 activation by degrees of magnitude. This is compelling evidence that minor changes to RNA sequences or to the RBD can profoundly affect OAS1’s antiviral response. It is thus plausible that the polymorphisms we identified can confer some level of specificity to different viral species. The present analysis, together with previous studies in chimpanzees and other mammals (Ferguson et al. 2012; Mozzi et al. 2015), provides a collection of candidate amino acids for future functional studies aimed at determining the molecular basis of the binding specificity of OAS1.
Acknowledgments
The work was conducted in part with equipment from the Core Facilities for Imaging, Cellular and Molecular Biology at Queens College. This research was supported by the Professional Staff Congress-City University of New York grant 66642–00 44 to S.B. This investigation used resources that were supported by the Southwest National Primate Research Center grant P51 RR013986 from the National Center for Research Resources, National Institutes of Health, and that are currently supported by the Office of Research Infrastructure Programs through P51 OD011133.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Mummidi S, Gonzalez E, Ahuja SS, Dunn DM, Watkins WS, Wooding S, Stone AC, Jorde LB, Weiss RB, Ahuja SK. A strong signature of balancing selection in the 5′ cis-regulatory region of CCR5. Proc Natl Acad Sci U S A. 2002;99:10539–10544. doi: 10.1073/pnas.162046399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Bigham AW, Buckingham KJ, Husain S, Emond MJ, Bofferding KM, Gildersleeve H, Rutherford A, Astakhova NM, Perelygin AA, Busch MP, Murray KO, Sejvar JJ, Green S, Kriesel J, Brinton MA, Bamshad M. Host genetic risk factors for West Nile virus infection and disease progression. PLoS One. 2011;6:e24745. doi: 10.1371/journal.pone.0024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Alvarez L, Giraldo-Ramirez J, Tollis M. Neutral nuclear variation in Baboons (genus Papio) provides insights into their evolutionary and demographic histories. Am J Phys Anthropol. 2014;155:621–634. doi: 10.1002/ajpa.22618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagliani R, Sironi M. Pathogen-driven selection in the human genome. Int J Evol Biol. 2013;2013:204240. doi: 10.1155/2013/204240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagliani R, Fumagalli M, Biasin M, Piacentini L, Riva S, Pozzoli U, Bonaglia MC, Bresolin N, Clerici M, Sironi M. Long-term balancing selection maintains trans-specific polymorphisms in the human TRIM5 gene. Hum Genet. 2010;128:577–588. doi: 10.1007/s00439-010-0884-6. [DOI] [PubMed] [Google Scholar]
- Cai Y, Chen Q, Zhou W, Chu C, Ji W, Ding Y, Xu J, Ji Z, You H, Wang J. Association analysis of polymorphisms in OAS1 with susceptibility and severity of hand, foot and mouth disease. Int J Immunogene. 2014;41:384–392. doi: 10.1111/iji.12134. [DOI] [PubMed] [Google Scholar]
- Charlesworth D. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2006;2:e64. doi: 10.1371/journal.pgen.0020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo S, Patel TR, Dzananovic E, Booy EP, Zeid K, McEleney K, Harding SE, McKenna SA. Activation of 2′ 5′-oligoadenylate synthetase by stem loops at the 5′-end of the West Nile virus genome. PLoS One. 2014;9:e92545. doi: 10.1371/journal.pone.0092545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan J, Dufner M, Korennykh A. Structural basis for cytosolic double-stranded RNA surveillance by human oligoadenylate synthetase 1. Proc Natl Acad Sci U S A. 2013;110:1652–1657. doi: 10.1073/pnas.1218528110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea R, Casillas S, Barbadilla A. Standard and generalized McDonald-Kreitman test: a website to detect selection by comparing different classes of DNA sites. Nucleic Acids Res. 2008;36:W157–W162. doi: 10.1093/nar/gkn337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/S0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- Ferguson B, Street SL, Wright H, Pearson C, Jia Y, Thompson SL, Allibone P, Dubay CJ, Spindel E, Norgren RB., Jr Single nucleotide polymorphisms (SNPs) distinguish Indian-origin and Chinese-origin rhesus macaques (Macaca mulatta) BMC Genomics. 2007;8:43. doi: 10.1186/1471-2164-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson W, Dvora S, Gallo J, Orth A, Boissinot S. Long-term balancing selection at the West Nile virus resistance gene, Oas1b, maintains transspecific polymorphisms in the house mouse. Mol Biol Evol. 2008;25:1609–1618. doi: 10.1093/molbev/msn106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson W, Dvora S, Fikes RW, Stone AC, Boissinot S. Long-term balancing selection at the antiviral gene OAS1 in Central African chimpanzees. Mol Biol Evol. 2012;29:1093–1103. doi: 10.1093/molbev/msr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Admetlla A, Bosch E, Sikora M, Marques-Bonet T, Ramirez-Soriano A, Muntasell A, Navarro A, Lazarus R, Calafell F, Bertranpetit J, Casals F. Balancing selection is the main force shaping the evolution of innate immunity genes. J Immunol. 2008;181:1315–1322. doi: 10.4049/jimmunol.181.2.1315. [DOI] [PubMed] [Google Scholar]
- Fischer MC, Foll M, Heckel G, Excoffier L. Continental-scale footprint of balancing and positive selection in a small rodent (Microtus arvalis) PLoS One. 2014;9:e112332. doi: 10.1371/journal.pone.0112332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Sironi M. Human genome variability, natural selection and infectious diseases. Curr Opin Immunol. 2014;30:9–16. doi: 10.1016/j.coi.2014.05.001. [DOI] [PubMed] [Google Scholar]
- Garrigan D, Hedrick PW. Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution. 2003;57:1707–1722. doi: 10.1111/j.0014-3820.2003.tb00580.x. [DOI] [PubMed] [Google Scholar]
- Goodbourn S, Didcock L, Randall RE. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J Gen Virol. 2000;81:2341–2364. doi: 10.1099/0022-1317-81-10-2341. [DOI] [PubMed] [Google Scholar]
- Hamano E, Hijikata M, Itoyama S, Quy T, Phi NC, Long HT, Ha LD, Ban VV, Matsushita I, Yanai H, Kirikae F, Kirikae T, Kuratsuji T, Sasazuki T, Keicho N. Polymorphisms of interferon-inducible genes OAS-1 and MxA associated with SARS in the Vietnamese population. Biochem Biophys Res Commun. 2005;329:1234–1239. doi: 10.1016/j.bbrc.2005.02.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann R, Norby PL, Martensen PM, Jorgensen P, James MC, Jacobsen C, Moestrup SK, Clemens MJ, Justesen J. Activation of 2′-5′ oligoadenylate synthetase by single-stranded and double-stranded RNA aptamers. J Biol Chem. 1998;273:3236–3246. doi: 10.1074/jbc.273.6.3236. [DOI] [PubMed] [Google Scholar]
- Hartmann R, Justesen J, Sarkar SN, Sen GC, Yee VC. Crystal structure of the 2′-specific and double-stranded RNA-activated interferon-induced antiviral protein 2′-5′-oligoadenylate synthetase. Mol Cell. 2003;12:1173–1185. doi: 10.1016/S1097-2765(03)00433-7. [DOI] [PubMed] [Google Scholar]
- He J, Feng D, de Vlas SJ, Wang H, Fontanet A, Zhang P, Plancoulaine S, Tang F, Zhan L, Yang H, Wang T, Richardus JH, Habbema JD, Cao W. Association of SARS susceptibility with single nucleic acid polymorphisms of OAS1 and MxA genes: a case–control study. BMC Infect Dis. 2006;6:106. doi: 10.1186/1471-2334-6-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. Genetics of populations. 4. Boston: Jones and Bartlett; 2010. [Google Scholar]
- Hernandez RD, Hubisz MJ, Wheeler DA, Smith DG, Ferguson B, Rogers J, Nazareth L, Indap A, Bourquin T, McPherson J, Muzny D, Gibbs R, Nielsen R, Bustamante CD. Demographic histories and patterns of linkage disequilibrium in Chinese and Indian rhesus macaques. Science. 2007;316:240–243. doi: 10.1126/science.1140462. [DOI] [PubMed] [Google Scholar]
- Hovanessian AG. On the discovery of interferon-inducible, double-stranded RNA activated enzymes: the 2′-5′oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev. 2007;18:351–361. doi: 10.1016/j.cytogfr.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justesen J, Hartmann R, Kjeldgaard NO. Gene structure and function of the 2′-5′-oligoadenylate synthetase family. Cell Mol Life Sci. 2000;57:1593–1612. doi: 10.1007/PL00000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key FM, Teixeira JC, de Filippo C, Andres AM. Advantageous diversity maintained by balancing selection in humans. Curr Opin Genet Dev. 2014;29:45–51. doi: 10.1016/j.gde.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Klein J, Satta Y, O’HUigin C, Takahata N. The molecular descent of the major histocompatibility complex. Annu Rev Immunol. 1993;11:269–295. doi: 10.1146/annurev.iy.11.040193.001413. [DOI] [PubMed] [Google Scholar]
- Knapp S, Yee LJ, Frodsham AJ, Hennig BJ, Hellier S, Zhang L, Wright M, Chiaramonte M, Graves M, Thomas HC, Hill AV, Thursz MR. Polymorphisms in interferon-induced genes and the outcome of hepatitis C virus infection: roles of MxA, OAS-1 and PKR. Genes Immun. 2003;4:411–419. doi: 10.1038/sj.gene.6363984. [DOI] [PubMed] [Google Scholar]
- Kodym R, Kodym E, Story MD. 2′-5′-Oligoadenylate synthetase is activated by a specific RNA sequence motif. Biochem Biophys Res Commun. 2009;388:317–322. doi: 10.1016/j.bbrc.2009.07.167. [DOI] [PubMed] [Google Scholar]
- Kumar S, Mitnik C, Valente G, Floyd-Smith G. Expansion and molecular evolution of the interferon-induced 2′-5′ oligoadenylate synthetase gene family. Mol Biol Evol. 2000;17:738–750. doi: 10.1093/oxfordjournals.molbev.a026352. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Ward FE, Ennis PD, Jackson AP, Parham P. HLA-A and B polymorphisms predate the divergence of humans and chimpanzees. Nature. 1988;335:268–271. doi: 10.1038/335268a0. [DOI] [PubMed] [Google Scholar]
- Leffler EM, Gao Z, Pfeifer S, Segurel L, Auton A, Venn O, Bowden R, Bontrop R, Wall JD, Sella G, Donnelly P, McVean G, Przeworski M. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science. 2013;339:1578–1582. doi: 10.1126/science.1234070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Lim JK, Lisco A, McDermott DH, Huynh L, Ward JM, Johnson B, Johnson H, Pape J, Foster GA, Krysztof D, Follmann D, Stramer SL, Margolis LB, Murphy PM. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog. 2009;5:e1000321. doi: 10.1371/journal.ppat.1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer WE, Jonker M, Klein D, Ivanyi P, van Seventer G, Klein J. Nucleotide sequences of chimpanzee MHC class I alleles: evidence for trans-species mode of evolution. EMBO J. 1988;7:2765–2774. doi: 10.1002/j.1460-2075.1988.tb03131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Mendez FL, Watkins JC, Hammer MF. Global genetic variation at OAS1 provides evidence of archaic admixture in Melanesian populations. Mol Biol Evol. 2012;29:1513–1520. doi: 10.1093/molbev/msr301. [DOI] [PubMed] [Google Scholar]
- Mendez FL, Watkins JC, Hammer MF. Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol Biol Evol. 2013;30:798–801. doi: 10.1093/molbev/mst004. [DOI] [PubMed] [Google Scholar]
- Misra A, Thippeshappa R, Kimata JT. Macaques as model hosts for studies of HIV-1 infection. Front Microbiol. 2013;4:176. doi: 10.3389/fmicb.2013.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzi A, Pontremoli C, Forni D, Clerici M, Pozzoli U, Bresolin N, Cagliani R, Sironi M (2015) OASes and STING: adaptive evolution in concert. Genome Biol Evol [DOI] [PMC free article] [PubMed]
- Osada N, Hashimoto K, Kameoka Y, Hirata M, Tanuma R, Uno Y, Inoue I, Hida M, Suzuki Y, Sugano S, Terao K, Kusuda J, Takahashi I. Large-scale analysis of Macaca fascicularis transcripts and inference of genetic divergence between M. fascicularis and M. mulatta. BMC Genomics. 2008;9:90. doi: 10.1186/1471-2164-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada N, Uno Y, Mineta K, Kameoka Y, Takahashi I, Terao K. Ancient genome-wide admixture extends beyond the current hybrid zone between Macaca fascicularis and M. mulatta. Mol Ecol. 2010;19:2884–2895. doi: 10.1111/j.1365-294X.2010.04687.x. [DOI] [PubMed] [Google Scholar]
- Palermo RE, Tisoncik-Go J, Korth MJ, Katze MG. Old world monkeys and new age science: the evolution of nonhuman primate systems virology. ILAR J. 2013;54:166–180. doi: 10.1093/ilar/ilt039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Rebouillat D, Hovanessian AG. The human 2′,5′-oligoadenylate synthetase family: interferon-induced proteins with unique enzymatic properties. J Interferon Cytokine Res. 1999;19:295–308. doi: 10.1089/107999099313992. [DOI] [PubMed] [Google Scholar]
- Silverman RH. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon VK, Calderon BM, Conn GL. A novel RNA molecular signature for activation of 2′-5′ oligoadenylate synthetase-1. Nucleic Acids Res. 2015;43:544–552. doi: 10.1093/nar/gku1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes I, Gil L, Castro J, Odoyo D, Hitler R, Munene E, Romero Y, Ochola L, Cosme K, Kariuki T, Guillen G, Hermida L. Olive baboons: a non-human primate model for testing dengue virus type 2 replication. Int J Infect Dis. 2013;17:e1176–e1181. doi: 10.1016/j.ijid.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf RF, Papin JF, Hines-Boykin R, Chavez-Suarez M, White GL, Sakalian M, Dittmer DP. Baboon model for West Nile virus infection and vaccine evaluation. Virology. 2006;355:44–51. doi: 10.1016/j.virol.2006.06.033. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Kang H, Ji Y, Chen X. Evaluate the relationship between polymorphisms of OAS1 gene and susceptibility to chronic hepatitis C with high resolution melting analysis. Clin Exp Med. 2013;13:171–176. doi: 10.1007/s10238-012-0193-6. [DOI] [PMC free article] [PubMed] [Google Scholar]