

Abstract
Objective:
Identification of causative mutations in 3 consanguineous families (with 4 affected members) referred to our center with young-onset motor neuron disease and overlapping phenotypes resembling autosomal recessive juvenile amyotrophic lateral sclerosis (ARJALS) and autosomal recessive hereditary spastic paraplegia (ARHSP).
Methods:
Patients have a slowly progressive motor neuron disease with upper and lower motor neuron dysfunction. There is distal muscle weakness and atrophy associated with pyramidal signs. Whole-exome sequencing was performed on the patients and the unaffected parent samples to identify disease-causing mutations. Variants were prioritized according to their predicted pathogenicity and their relevance to the clinical phenotypes.
Results:
Five distinct homozygous mutations within the SPG11 gene were identified, 3 of which were novel and truncating: c.7155T>G/p.Tyr2385Ter, c.2250delT/p.Phe750Leufs*3, and c.1966_1967delAA/p.Lys656Valfs*11. The copresence of 2 distinct homozygous missense variations was observed in 2 families: c.6224A>G/p.Asn2075Ser and c.7132T>C/p.Phe2378Leu. The segregation of these variations in the family members was validated by Sanger sequencing.
Conclusions:
Four patients with juvenile-onset motor neuron disease with consanguineous parents were found to carry homozygous mutations in the SPG11 gene. Our findings confirm the overlapping phenotypes of SPG11-based ARJALS and ARHSP, indicating that these 2 entities may be the extreme phenotypes of the same disease continuum with many common features. This, in turn, confirms the difficult differential diagnosis of these 2 diseases in the clinic.
Autosomal recessive juvenile amyotrophic lateral sclerosis (ARJALS) is a rare form of amyotrophic lateral sclerosis (ALS) with adolescent onset. The recessive inheritance mode, early onset, and slow progression are major distinctions from classical ALS.1 ALS2, SETX, SPG11, and SIGMAR1 are the genes associated with ARJALS.2–5 Patients with ALS2 and SPG11 mutations often have a disease presentation similar to that of autosomal recessive hereditary spastic paraplegia (ARHSP). Hereditary spastic paraplegias (HSPs) are a heterogeneous group of disorders, clinically classified as pure or complicated. Pure and late-onset HSPs are commonly inherited dominantly, whereas ARHSPs are often associated with early and severe onset and complex phenotypes. Major symptoms of lower limb spasticity and weakness in pure HSPs may be accompanied by neurologic and nonneurologic manifestations such as cognitive impairment, cerebellar atrophy, polyneuropathy, thin corpus callosum (TCC), and amyotrophy. Today, HSPs are classified genetically depending on their specific loci, ranging from SPG1 to SPG72.6 In the Mediterranean basin, where consanguineous marriages are part of the culture, recessive disease is common. SPG11 mutations are the predominant cause of ARHSP-TCC.7,8 Several mutations have been described on SPG11 leading to loss of function of the spatacsin protein, which is essential to neuronal survival.9 Cognitive impairment and TCC are phenotype predictors of SPG11; polyneuropathy and cerebellar symptoms are other features. Recently, SPG11 mutations have been shown to give rise to juvenile ALS. SPG11-ARJALS is clinically characterized by a slowly progressive phenotype starting before the age of 20.10 HSP-TCC and SPG11-ARJALS have many similarities in clinical presentation, molecular genetics, and cellular pathology. This study evaluates the genotypes of Turkish ARJALS families by whole-exome sequencing (WES).
METHODS
Participants.
Standard protocol approvals, registrations, and patient consents.
This study analyzed 3 families. Clinical evaluations of all index cases and their family members were performed by expert neurologists. The diagnosis of clinically definite ALS was made according to the revised El Escorial criteria. Genetic counseling was offered, and informed consent was obtained from all individuals who participated in the study. The protocol was approved by the Ethics Committee on Research with Human Participants (INAREK) at Bogazici University, Istanbul, Turkey.
Families under investigation.
Family 1.
The index case is a 36-year-old man. His parents are first cousins; moreover, his mother was born to parents who were first cousins. The patient's father developed classical ALS with limb onset at the age of 50 and died within 5 years (prior to this study). The proband has a long-standing neurologic history dating to early childhood. At the age of 4, he was assessed for moderate psychomotor delay, including fine motor and language milestones. At the age of 23, he was reassessed because of frequent falling and imbalance with bilateral leg weakness followed by upper extremity weakness. Inborn hereditary metabolic diseases were excluded; mutation analyses for the major ataxia and HSP genes (FXN; ATXN 1, 2, 3, 6, 7, 17; AR; SPG4; SPG3A; and NIPA1) were negative. Neither direct sequencing nor fragment analysis showed any mutations in the major genes SOD1, C9orf72, TARDBP, FUS, ANG, and UBQLN2. WES was performed on the patient and his unaffected older brother and mother (figure 1).
Figure 1. Segregation of variants in family 1.
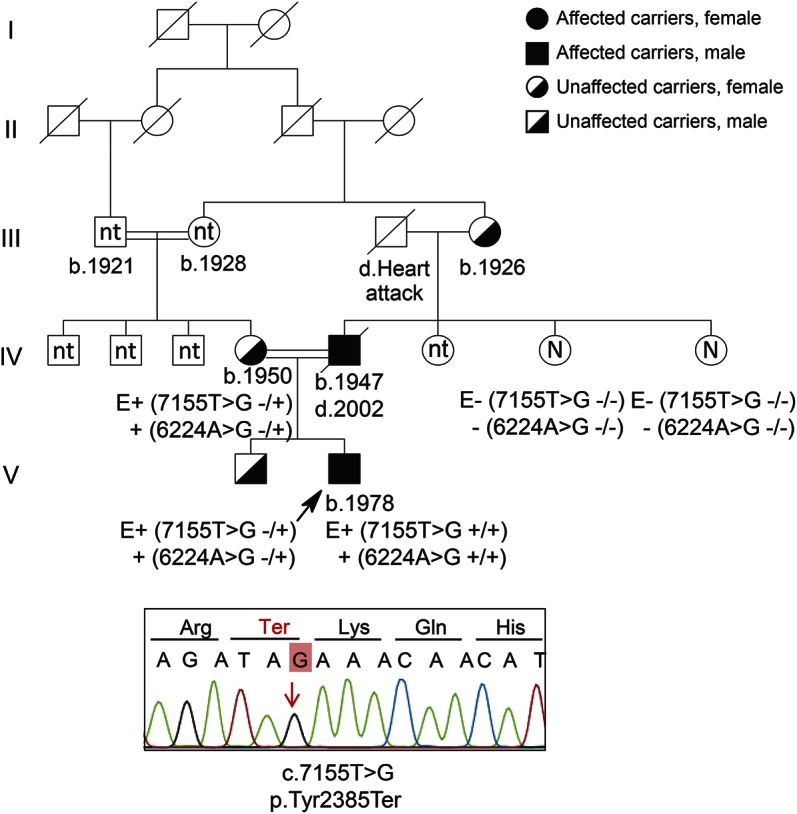
The 37-year-old patient was born from a consanguineous marriage. His father developed amyotrophic lateral sclerosis (ALS) at the age of 50 and died within 5 years. Unlike his son, his disease was possibly not the result of SPG11 pathology because its development and progression was the classical sporadic type of late-onset ALS. Sanger sequencing shows the truncation mutation for the index case. N = free of mutation; nt = not tested.
Family 2.
The probands are siblings from first-degree consanguineous parents. They had limb-onset disease at the ages of 14 (male) and 16 (female), with walking difficulties, twitching, spasticity, and fasciculation in legs. Both upper and lower motor neuron (UMN and LMN) involvement was present. Speech was dysarthric. There was no cognitive impairment and no optical complaints. The GAA expansion in the FXN gene was shown to be normal. Further mutation analyses for SOD1, TARDBP, FUS, ANG, NIPA1, REEP1, SPG3A, and SPG4 genes also did not reveal any pathogenic variation. The patients, now in their 30s, share a similar phenotype and have a slowly progressive motor neuron disease without any mental dysfunction. WES was performed on both affected siblings (figure 2).
Figure 2. Segregation of variants in family 2.
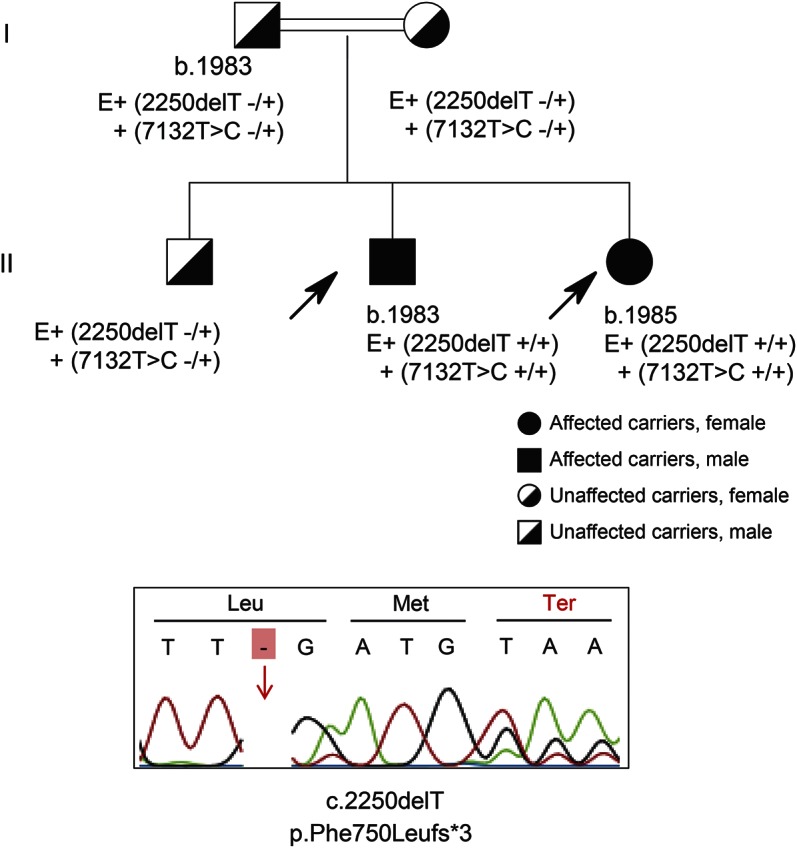
Both affected siblings have the same type of disease. The parents and the older brother carry the mutation in heterozygous form. Sanger sequencing shows the truncation mutation for 1 proband.
Family 3.
The index case, born from a second-degree consanguineous marriage, was reported to have slight walking difficulties at the age of 17, which worsened markedly in her 30s. She was only able to walk with assistance at the age of 40; speech difficulty and cognitive decline were apparent 10 years later. An older sister and a younger brother were reported to have died of a similar but more progressive pathology (age at onset around 20 and disease durations of 13 and 17 years, respectively) (figure 3). The recent neurologic examination of the 54-year-old woman showed marked dysarthria, weakness in the upper extremities, and total loss of muscle strength in proximal and distal lower limbs. Almost-complete contractions in ankles and elbows were present. Thenar and hypothenar muscles had progressive atrophy; deep tendon reflexes were hyperactive. Spasticity was marked. EMG showed neurogenic change in the tongue and lower and upper limbs, with fasciculation and fibrillation potentials and severe loss of motor units but normal motor and sensory conduction. She was not cooperative, and she had cognitive dysfunction (Mini-Mental State Examination score of 11); cranial MRI showed abundant cortical and cerebellar atrophy and thinning of the corpus callosum. WES was performed on the patient and her unaffected mother.
Figure 3. Segregation of variants in family 3.

Three of the siblings from a first-degree consanguineous marriage were affected with autosomal recessive juvenile amyotrophic lateral sclerosis. Only the proband (generation IV) was available to us; she carried the above-mentioned SPG11 mutation in homozygous form. Her mother and one of her sisters were carriers. A younger brother was free of mutation. N = free of mutation; nt = not tested.
WES analysis.
Extraction of genomic DNA from peripheral blood was performed using the MagNA Pure DNA extraction kit. Paired-end sequencing was performed on Illumina HiSeq2000 and 2500 DNA sequencing platforms at the University of Massachusetts and The Scientific and Technological Research Council of Turkey. Raw reads were mapped to the reference genome (hg19) with the Burrows-Wheeler Aligner tool, and variant calling was performed with the Genome Analysis Toolkit (GATK) program. The ANNOVAR11 annotation tool was used for functional annotation of the variants, based on the RefSeq UCSC Gene, dbSNPv138, 1000 Genomes (April 2012 release), Exome Variant Server, and NHLBI GO Exome Sequencing Project (ESP) databases. KEGG and OMIM database information, which contains the association of genes with linked disorders, was added to the functionally annotated variant list. Variant filtration was first performed according to the mode of inheritance patterns in the families using VarSifter software.12 Noncoding and synonymous variants were excluded except for the splice junctions. Scores from prediction tools such as PolyPhen-2, SIFT, LRT, and MutationTaster were used to determine the possible effects of the mutations on the protein. Variants identified were confirmed by Sanger sequencing (primers available upon request).
RESULTS
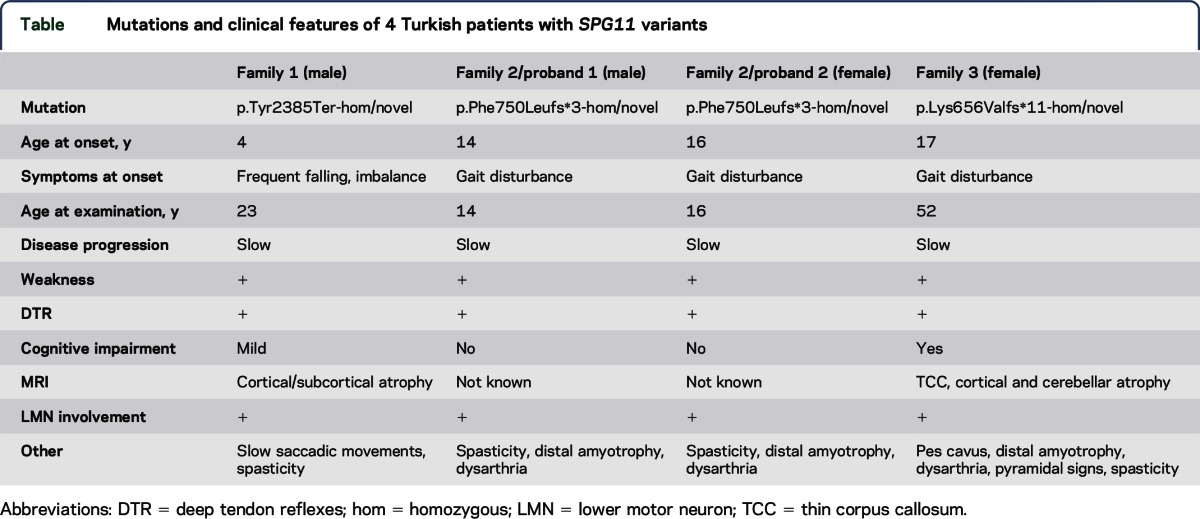
The table compiles the genotypes and the clinical features of the probands. Exome sequencing revealed 5 distinct homozygous SPG11 variants in the families investigated. Three were novel and 2 were previously reported.13–15 The novel mutations, 2 small deletions leading to frame-shift and 1 nonsense mutation, predict the truncation of the spatacsin protein with a loss-of-function type of mechanism in all 3 families. The truncation mutations were accompanied by homozygous missense variants on the same allele (in cis) in families 1 and 2. Both of these SPG11 missense variations were recently reported in 2 independent patients with HSP (c.6224A>G (p.Asn2075Ser), rs140824939, MAF = 0.0018, ExAC = 0.002586) and autosomal recessive spastic ataxia of Charlevoix-Saguenay (c.7132T>C (p.Phe2378Leu), rs150571352, MAF = 0.001, ExAC = 0.0001659) in heterozygous form; thus, their pathogenicity is not yet well established.16
Table.
Mutations and clinical features of 4 Turkish patients with SPG11 variants
Family 1: c.7155T>G/p.Tyr2385Ter and c.6224A>G/p.Asn2075Ser.
The variants in exome analysis were filtered for being homozygous in the index case, heterozygous in the unaffected mother, and heterozygous/absent in the brother. Out of 1,196 exonic and splicing site variants (except the synonymous ones), 2 independent homozygous variations in the SPG11 gene, coexisting on the same allele, were identified in the index case: c.7155T>G/p.Tyr2385Ter and c.6224A>G/p.Asn2075Ser. The unaffected mother and older brother were shown to be heterozygous for both variations. The p.Tyr2385Ter variation in exon 38 of SPG11 is a novel mutation leading to a stop codon; this mutation is preceded by the homozygous p.Asn2075Ser missense variation in cis in exon 33 of the gene. Sanger sequencing showed the correct segregation of both mutations in family members. The paternal grandmother was found to be a carrier for both mutations, whereas the unaffected paternal aunts, sisters of the father of the index case, were completely free of the above 2 variations. Because the father, who died of classical ALS, was not available to us, we were not able to make the correlation between the genotypes of the index case and his father at this stage (figure 1).
Family 2: c.2250delT/p.Phe750Leufs*3 and c.7132T>C/p.Phe2378Leu.
In the affected siblings, the homozygous/compound heterozygous variants shared by both patients were selected. Exome sequencing analysis revealed 2 distinct SPG11 mutations in homozygous form in these patients. The p.Phe750Leufs*3 change in exon 12, resulting in a stop codon after 3 amino acids, is novel and accompanied by another variation (p.Phe2378Leu). Validation of both mutations was performed using Sanger sequencing in the whole family. The unaffected parents, obligate carriers, and the older brother were heterozygous for both mutations (figure 2).
Family 3: c.1966_1967delAA/p.Lys656Valfs*11.
In the index case and her unaffected mother, the variants were filtered for being homozygous in the patient and heterozygous in the mother, representing the autosomal recessive mode of inheritance. Among 985 exonic and splice site variations (excluding synonymous ones), the p.Lys656Valfs*11 mutation in exon 10 of the SPG11 gene was identified. The segregation was confirmed by Sanger sequencing in all available family members (figure 3).
DISCUSSION
We report 4 patients from 3 consanguineous marriages showing a slowly progressive juvenile motor neuron disease linked to SPG11 mutations. Probands screened for genes implicated in ataxias, HSP, ALS, and/or other neuromuscular diseases were found to be negative. Given the phenotypic heterogeneity that SPG11 mutations give rise to and the genetic heterogeneity of ARJALS and ARHSP (large genes and more than a dozen causative loci known so far), it is challenging to determine an optimal screening strategy for molecular diagnosis. These cases provide support for the value of WES as a diagnostic tool in the identification of neurodegenerative disorders, especially in consanguineous families with mendelian inheritance for which conventional methods are often insufficient. High-throughput methodologies are achieving widespread use as costs drop rapidly and are expected to become the gold standard in diagnostic strategies soon.
The analysis revealed 5 homozygous mutations in the SPG11 gene, 3 of which were novel and truncating and predicted to be damaging and disease causing. These mutations were absent in dbSNPv138, 1000 Genomes, EVS, ALS Exome Variant Server, and our in-house database including more than 100 exomes. In 2 families, surprisingly, the truncation mutations were accompanied by 2 distinct homozygous missense variations in cis, both previously reported in heterozygous form. We have not seen any indications of double mutations in the SPG11 gene resulting in ARJALS or ARHSP in the literature. On the other hand, in the presence of the disease-causing homozygous truncation mutations, the role of these missense mutations in homozygous form cannot be interpreted. Their importance in combination with the truncating mutations (despite possibly being disease-modifying on their own) is uncertain and remains to be elucidated. All prediction tools indicate both variations as benign, and the locations on the genome are not conserved among species. These variations could be benign polymorphisms in linkage disequilibrium with the corresponding truncation mutations; however, this needs to be further investigated in a larger patient cohort.
A wide variety of phenotypic expression has been reported with SPG11 mutations, including diversity in presentation and clinical appearance, commonly seen in complex forms of ARHSP. This is reflected by the genetic heterogeneity: 72 different gene loci involving different inheritance patterns are associated with HSP. Genetic heterogeneity results in diverse clinical phenotypes, altering several cellular functions involved in neuronal metabolism. To make the picture more complex, 1 HSP locus, SPG11, was recently reported to be associated with a young-onset form of ALS, ARJALS, further broadening its clinical expression in 2 independent studies.17,18 This is another study demonstrating that loss of spatacsin results in an overlapping phenotype, with features of both ARJALS and complicated SPG11-based ARHSP. These overlapping phenotypes not only blur the line between both diseases but also emphasize the complexity of correct clinical differential diagnosis of diseases linked to spatacsin.
The chief characteristic that defines ALS is progressive weakness from neurodegeneration of the UMN and LMN. Signs of both UMN and LMN dysfunction in one or more body regions are obvious in the clinical examination. Axonal neuropathy associated with anterior horn signs is indicative of LMN degeneration and can clinically mask ALS when muscle wasting is prominent.8,19 Although in our 3 families the phenotypes have more pronounced overlapping features than those reported in earlier studies, e.g., mental retardation and TCC, there are several clinical manifestations that point toward ARJALS rather than ARHSP. Although both UMN and LMN clinical signs were present, the LMN signs predominated. The presence of bulbar signs and the absence of sensory involvement and ocular abnormalities, prominent features of ARHSP, definitely excluded the diagnosis of ARHSP in our patients. In the special case of family 1, because we do not have a sample from the father, we can only speculate that the phenotype of our proband is the consequence of the first-cousin marriage of his parents rather than a vertical transmission from his father, whose ALS cannot be explained by SPG11. This is also supported by the very different phenotype of the index case and the fact that his father was reported to have died of classical rapidly progressive adult-onset ALS.
Most of the currently known proteins involved in HSP have a biological function in axonal transport, membrane trafficking, or biogenesis of mitochondria. Spatacsin is ubiquitously expressed in the nervous system, prominently in the cerebellum, cerebral cortex, hippocampus, and pineal gland. The protein seems to be essential to neuronal survival. A recent study demonstrated the crucial role of spatacsin in axonal maintenance and cargo trafficking, especially in anterograde transport.20 Given the common finding of axonal involvement in both disorders, SPG11 genetic variants may contribute to a common pathway in ARHSP and ARJALS. No clear boundaries are observed among the several clinical phenotypes of both diseases, as they probably represent continuing points on the same spectrum. In addition, the link between HSP, ALS, Alzheimer disease, and Parkinson disease was further confirmed in a recent study using the random walk distance algorithm in the HSP interactome.21
Our knowledge of the clinical, genetic, and neuropathologic aspects of motor neuron diseases has dramatically increased in recent years. There is compelling evidence that the level of complexity is far greater than assumed. Several pleiotropic gene mutations impair different biochemical pathways, resulting in distinct clinical and pathologic phenotypes. We hope that the mystery of why the same gene mutations lead to different clinical outcomes will soon be unraveled.
ACKNOWLEDGMENT
The authors gratefully acknowledge the support of the Suna and Inan Kirac Foundation and Bogazici University Research Funds. The authors thank Asli Gundogdu Eken, Irmak Sahbaz, Gokcin Kara, Gizem Tanriver, Ekin Su Akdemir, and Pinar Kavak for their excellent technical assistance; Dr. Serkan Alkan and Nesli Ece Sen for the critical reading of the manuscript; and the patients, their families, and the Turkish ALS Association for their invaluable cooperation.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- ARHSP
autosomal recessive hereditary spastic paraplegia
- ARJALS
autosomal recessive juvenile amyotrophic lateral sclerosis
- HSP
hereditary spastic paraplegia
- LMN
lower motor neuron
- TCC
thin corpus callosum
- UMN
upper motor neuron
- WES
whole-exome sequencing
AUTHOR CONTRIBUTIONS
Ceren Iskender screened for common genes in motor neuron diseases and revised the manuscript. Ece Kartal performed the analyses of exome sequencing data and compiled the manuscript. Fulya Akcimen assisted in the revision of the manuscript. Cemile Kocoglu performed the analyses of exome sequencing data. Dr. Aslihan Ozoguz screened for common genes in motor neuron diseases. Dr. Dilcan Kotan evaluated the patients. Dr. Mefkure Eraksoy evaluated the patients. Dr. Yesim Parman evaluated the patients. Dr. A. Nazli Basak designed the study and worked on the manuscript.
STUDY FUNDING
This study was funded by the Suna and Inan Kirac Foundation and Bogazici University Research Funds.
DISCLOSURE
Ceren Iskender, Ece Kartal, Fulya Akcimen, Cemile Kocoglu, and Dr. Aslihan Ozoguz have received research support and fellowship from the Suna and Inan Kirac Foundation and report no disclosures. Dr. Dilcan Kotan, Dr. Mefkure Eraksoy, Dr. Yesim Parman, and Dr. A. Nazli Basak report no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Ben Hamida M, Hentati F, Ben Hamida C. Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis). Conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain 1990;113:347–363. [DOI] [PubMed] [Google Scholar]
- 2.Al-Saif A, Al-Mohanna F, Bohlega S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol 2011;70:913–919. [DOI] [PubMed] [Google Scholar]
- 3.Chance PF, Rabin BA, Ryan SG, et al. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Hum Genet 1998;62:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hentati A, Bejaoui K, Pericak-Vance MA, et al. Linkage of recessive familial amyotrophic lateral sclerosis to chromosome 2q33-q35. Nat Genet 1994;7:425–428. [DOI] [PubMed] [Google Scholar]
- 5.Hentati A, Ouahchi K, Pericak-Vance MA, et al. Linkage of a commoner form of recessive amyotrophic lateral sclerosis to chromosome 15q15-q22 markers. Neurogenetics 1998;2:55–60. [DOI] [PubMed] [Google Scholar]
- 6.Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 2014;261:518–539. [DOI] [PubMed] [Google Scholar]
- 7.Stevanin G, Santorelli FM, Azzedine H, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 2007;39:366–372. [DOI] [PubMed] [Google Scholar]
- 8.Stevanin G, Azzedine H, Denora P, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2008;131:772–784. [DOI] [PubMed] [Google Scholar]
- 9.Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci 2012;35:25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen S, Sayana P, Zhang X, Le W. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener 2013;8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics 2012;28:599–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pensato V, Castellotti B, Gellera C, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 2014;137:1907–1920. [DOI] [PubMed] [Google Scholar]
- 14.Liew WKM, Ben-Omran T, Darras BT, et al. Clinical application of whole-exome sequencing: a novel autosomal recessive spastic ataxia of Charlevoix-Saguenay sequence variation in a child with ataxia. JAMA Neurol 2013;70:788. [DOI] [PubMed] [Google Scholar]
- 15.Özoğuz A, Uyan Ö, Birdal G, et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging 2015;36:1764 e9–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Exome Aggregation Consortium (ExAC). Cambridge, MA: Available at: http://exac.broadinstitute.org. Accessed August 25, 2015. [Google Scholar]
- 17.Orlacchio A, Babalini C, Borreca A, et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010;133:591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daoud H, Zhou S, Noreau A, et al. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging 2012;33:5–9. [DOI] [PubMed] [Google Scholar]
- 19.Ravits J, Appel S, Baloh RH, et al. Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph Lateral Scler Frontotemporal Degener 2013;14(suppl 1):5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pérez-Brangulí F, Mishra HK, Prots I, et al. Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum Mol Genet 2014;23:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014;343:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]