

ABSTRACT
The association of host histones with parvoviral DNA is poorly understood. We analyzed the chromatinization and histone acetylation of canine parvovirus DNA during infection by confocal imaging and in situ proximity ligation assay combined with chromatin immunoprecipitation and high-throughput sequencing. We found that during late infection, parvovirus replication bodies were rich in histones bearing modifications characteristic of transcriptionally active chromatin, i.e., histone H3 lysine 27 acetylation (H3K27ac). H3K27ac, in particular, was located in close proximity to the viral DNA-binding protein NS1. Importantly, our results show for the first time that in the chromatinized parvoviral genome, the two viral promoters in particular were rich in H3K27ac. Histone acetyltransferase (HAT) inhibitors efficiently interfered with the expression of viral proteins and infection progress. Altogether, our data suggest that the acetylation of histones on parvoviral DNA is essential for viral gene expression and the completion of the viral life cycle.
IMPORTANCE Viral DNA introduced into cell nuclei is exposed to cellular responses to foreign DNA, including chromatinization and epigenetic silencing, both of which determine the outcome of infection. How the incoming parvovirus resists cellular epigenetic downregulation of its genes is not understood. Here, the critical role of epigenetic modifications in the regulation of parvovirus infection was demonstrated. We showed for the first time that a successful parvovirus infection is characterized by the deposition of nucleosomes with active histone acetylation on the viral promoter areas. The results provide new insights into the regulation of parvoviral gene expression, which is an important aspect of the development of parvovirus-based virotherapy.
INTRODUCTION
Nuclear chromatin is composed of DNA and histone proteins (1). The histone proteins assemble DNA into nucleosomes, the composition and spacing of which contribute to higher-order chromatin packing. The chromatin is organized into regions of less-condensed actively transcribed chromatin (euchromatin) and highly condensed transcriptionally repressed chromatin (heterochromatin). Epigenetic modifications of histone proteins have been shown to correlate with the spatial distribution of active and repressed chromatin (2, 3). The acetylation of lysine 9 or 27 of histone H3 (H3K9ac and H3K27ac, respectively) and trimethylation of lysine 4 (H3K4me3) correlate with transcriptional activity, while repressed chromatin is characterized by, e.g., trimethylation or dimethylation of the same H3 lysine residues (H3K9me3 and H3K27me3 as well as H3K9me2 and H3K27me2) (4–8).
Foreign DNA introduced into mammalian cells can be recognized as a threat by the host cell. The cellular responses to the foreign DNA, such as viral DNA, include the chromatinization of the entering DNA, leading to its transcriptional silencing (9–11). How viruses resist cellular chromatinization and silencing is known for only a few viruses (9, 12, 13). These include herpes simplex viruses (HSVs) (14–16), polyomaviruses (17, 18), adenoviruses (19), and cytomegaloviruses (20, 21), all of which encode proteins that promote transcriptionally activating histone modifications on chromatinized viral genomes during lytic infection.
In parvovirus infection, the nuclear entry of viral single-stranded DNA is followed by the formation of double-stranded replicative intermediates, the nuclear accumulation of viral proteins and DNA, and the formation of autonomous parvovirus-associated replication (APAR) bodies (22–25). Viral gene expression and DNA replication are dependent on the S phase of the cell cycle. In infected cells, the transcription of viral nonstructural protein 1 (NS1) at 4 h postinfection (p.i.) is followed by viral genome replication (26). Replication continues throughout the infection and leads to the production of viral capsids and their nuclear egress at 20 to 24 h p.i. The intranuclear chromatinization of the parvoviral genome, the modification of the assembled histones, and the effect of these events on viral gene expression are not well understood. To provide answers to these questions, we analyzed the infection of an autonomous protoparvovirus, canine parvovirus (CPV), by confocal imaging techniques and in situ proximity ligation assays (PLA) complemented with chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq). Our results demonstrated the enrichment of acetylated histones (H3K27ac) in close proximity to viral DNA-binding NS1 in APAR bodies and especially H3K27ac accumulated in the viral promoters. The inhibition of histone acetyltransferase (HAT) activity led to the interruption of the viral life cycle. These results reveal that histone acetylation on chromatinized parvoviral genomes is necessary for the expression of viral genes and successful progression of infection.
MATERIALS AND METHODS
Cells, viruses, and constructs.
Norden Laboratories feline kidney (NLFK; Quality Control of Pfizer Animal Health, Lincoln, NE) cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Paisley, United Kingdom) at 37°C in the presence of 5% CO2. CPV-2d isolates originated from an infectious plasmid clone (a gift from C. R. Parrish, Cornell University, Ithaca, NY [27]). The viruses had been isolated as described by Suikkanen et al. (28). For infection, the cells were inoculated with CPV (multiplicity of infection [MOI] of 1 to 2) and kept at 37°C until fixation. In order to synchronize infections, the cells were incubated on ice at 4°C for 20 min prior to virus addition. The cells then were inoculated on ice to allow for virus adsorption to occur for 30 min. The cells next were rinsed at 4°C with phosphate-buffered saline (PBS) containing 0.3% bovine serum albumin (BSA) followed by the addition of 37°C medium. The plasmid encoding fluorescent H3-enhanced green fluorescent protein (EGFP) was a generous gift from J. Langowski (German Cancer Research Center, Heidelberg, Germany). An NLFK cell line stably expressing H3-EGFP was established by transfection (TransIT-LT1 reagent; Thermo Fisher Scientific Inc., Waltham, MA) with an expression vector at 24 h after seeding. After 2 days, the DMEM was replaced by DMEM containing 1 mg/ml Geneticin (Sigma-Aldrich, St. Louis, MO). The cells then were seeded at different intervals until stable expression was observed by microscopy.
Confocal microscopy.
For immunolabeling, cells seeded on round coverslips were infected with CPV and fixed at 8, 10, 12, 16, and 24 h p.i. with 4% paraformaldehyde (PFA; 15 min at room temperature). Viral proteins were detected with an NS1-specific monoclonal antibody (MAb) (generous gift from Caroline Astell) (29), an intact capsid MAb, and a polyclonal capsid-protein VP2 antibody (Ab; generous gifts from Colin R. Parrish) (27), followed by goat anti-mouse or anti-rabbit Alexa-555- or Alexa-633-conjugated secondary Abs (Molecular Probes, Life technologies, Grand Island, NY). Modified histones were labeled with rabbit Abs against H3K9me3 and H3K27ac (Abcam, Cambridge, MA) followed by goat anti-rabbit Alexa 633-conjugated secondary Abs. For deacetylation and hyperacetylation studies, cells were either pretreated for 30 min prior to virus inoculation or treated at 0 to 6 h p.i. with 0.1 mM anacardic acid prepared in 100% dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO). Cell viability was assessed using propidium iodide (Sigma) and annexin V Alexa 647 (Molecular Probes) staining by following the manufacturer's instructions (data not shown). All cells were fixed at 24 h p.i. with 4% PFA and permeabilized with 0.1% Triton X-100 in PBS supplemented with 1% BSA and 0.01% sodium azide. Imaging was done with an Olympus FV-1000 confocal microscope with the UPLSAPO 60× oil-immersion objective (numeric aperture, 1.35). EGFP was excited with a 488-nm argon laser, and fluorescence was collected with a 515/30-nm band-pass filter. Alexa 555 and the proximity ligation assay (PLA) reagent with a fluorophore, 594 nm, were excited with a 543-nm He-Ne laser, and fluorescence was collected with a 570-nm to 620-nm band-pass filter and a 560IF band-pass filter, respectively. Alexa 633 was excited with a 633-nm He-Ne laser, and the fluorescence was collected with a 647-nm long-pass filter. Image size was between 512 by 512 and 1,600 by 1,600 pixels with a pixel resolution of 66 to 69 nm. Deconvolution was performed with Huygens Essential software (SVI, Netherlands). The point-spread function was averaged, the iterative deconvolution was performed with a signal-to-noise ratio of 5, and the quality threshold was 0.01. Image analysis was done with ImageJ (30).
Correlation analysis was done with ImageJ using the JACoP plugin (31). Distance analysis of the fixed-cell samples was done first by making a Euclidian distance map from an image of interest and then plotting the original image against the distance map. The data from every cell were combined and arranged to 0.1-μm-wide bins based on the distance of the voxels from the nuclear envelope. The average intensity then was plotted for each bin. Distance maps were done in ImageJ with the exact Euclidian distance transform (3D) plugin, and the data analysis was done with an in-house Java code. Student's t test (two-tailed, unequal variance) was used to evaluate statistical significance in the change of the recovery time point values.
Analysis of protein interactions.
For in situ PLA (32), cells were grown on 8-well chamber slides (Nunc Lab-Tek II chamber slide system; Nalgene Nunc International, Penfield, NY) to 80 to 90% confluence and fixed with 4% PFA. PLA was done with a DuolinkII kit (Olink Bioscience, Uppsala, Sweden) and primary Abs against NS1 and H3K27ac. After labeling the cells with the primary Abs, the PLA probes (oligonucleotide-conjugated secondary anti-mouse and anti-rabbit IgGs diluted in 3% BSA in PBS) were incubated for 1 h at 37°C in a humidified chamber, followed by ligation and amplification according to the manufacturer's instructions. Samples were embedded in ProLong gold antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI). The specificity of the assay was confirmed using positive, negative, and technical control readings. Positive and negative controls consisted of infected and noninfected cells, respectively, labeled with anti-VP2 and intact capsid-specific Abs or anti-NS1 and anti-H3K27ac Abs. For a technical control, noninfected cells were labeled only with PLA probes. Quantitative analysis was done with ImageJ by determining the arithmetic mean of the total number of signals per cell. The PLA signal was normalized as PLA = NA − NT, where NA is the total number of nuclear PLA dots in infected and noninfected cells labeled with H3K27ac Ab and NS1 MAb and NT is the average number of nuclear PLA dots in the technical control. Student's t test (two-tailed, unequal variance) was used to evaluate statistical significance.
ChIP-seq and qPCR.
Cells were fixed with formaldehyde, and nuclei were isolated, lysed, and sonicated with a Covaris S220 ultrasonicator. The resulting nuclear extract was incubated overnight at 4°C with Dynal protein G beads preincubated with 5 μg of H3K27ac (ab4729) or H3 (ab1791) Abs. Beads were washed and bound complexes eluted, and cross-links were reversed by heating at 65°C. Immunoprecipitation (IP) and input DNA then were purified by a treatment with RNase A, proteinase K, and phenol-chloroform extraction. Before moving forward to ChIP-seq, precipitated DNA was analyzed by quantitative PCR (qPCR) using primers specific for gp1, gp2, and gp5 regions. Libraries were constructed from IP and input DNA by a NEBnext Ultra DNA library preparation kit for Illumina. DNA in the range of 150 to 350 bp was gel purified after PCR amplification. The library was quantified using an Agilent bioanalyzer and subjected to 50-bp single-end read sequencing with an Illumina HiSeq 2000 at EMBL Genecore, Heidelberg, Germany. Quality metrics for sequenced reads were gathered with FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adapter sequences were removed with cutadapt (33). Reads then were aligned with Bowtie2 (34) to cat (ICGSC Felis_catus 6.2) and CPV (NCBI reference sequence NC_001539.1) reference genomes. Sequencing reads of H3K27ac ChIP-seq from infected cells were normalized to CPV reads per genomic content (RPGC). File conversions to the BAM format were done with SAMtools (35), and visualizations were done with deepTools (36).
Accession number.
The ChIP-seq data reported in this paper have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE77785.
RESULTS
Temporal changes in localization of modified histones in infection.
To analyze the chromatinization of the CPV genome at various times postinfection (p.i.), we used H3-EGFP-expressing cells to identify the distribution of histone H3 and APAR bodies represented by NS1. The line profiles showed that at 12 h p.i. H3 was mostly located distinctly from NS1, whereas at 24 h p.i. increased colocalization of H3 with NS1 was observed (Fig. 1A and B). Similarly, Pearson correlation coefficient (PCC) analysis indicated the correlation of H3-EGFP with NS1 at 16 h p.i. (0.61 ± 0.29; n = 22; P value [Student's t test] at 16 and 24 h p.i. compared to results at 10 h p.i., <0.05) and 24 h p.i. (0.77 ± 0.21; n = 26; P < 0.01) but not at earlier times (Fig. 1G). To assess how H3-associated active and repressed chromatin is distributed in infected cells, we used H3K27ac and H3K9me3 as markers of active and repressed chromatin, respectively. Immunolabeling indicated that at 12 h p.i., distinct NS1 foci began to emerge, while H3K27ac and H3K9me3 showed a thorough nuclear distribution. The line profiles of H3K27ac and H3K9me3 with NS1 at 12 h p.i. indicated only a weak colocalization (Fig. 1C and E). However, at 24 h p.i. H3K27ac concentrated in the enlarging APAR bodies (Fig. 1D). Similarly, PCC of NS1 with H3K27ac was significantly higher at 16 h p.i. (0.83 ± 0.22; n = 18; P < 0.01) and at 24 h p.i. (0.87 ± 0.08; n = 23; P < 0.01) than at earlier times (Fig. 1G). No correlation of H3K9me3 with NS1 was observed by the line profile and PCC analyses (Fig. 1E to G). Colocalization of NS1 with H3 and H3K27ac correlated time dependently with the enlargement of APAR bodies. Quantitative 3D distribution analysis of H3K27ac and H3K9me3 as a function of distance from the nuclear rim indicated that the H3K27ac signal was located in the nuclear center in both infected (24 h p.i.) and noninfected cells (n = 20; 1,000 spots/cell counted) (Fig. 1H). In infected cells, the 0.7-μm-thick region at the nuclear periphery in particular was enriched in H3K9me3, and its intensity decreased toward the nuclear center. In noninfected cells, the H3K9me3 signal was distributed throughout the nucleus (n = 20; 1,000 spots/cell counted) (Fig. 1I).
FIG 1.

Intranuclear distribution of H3-EGFP, modified histones, and viral NS1 protein. (A and B) Confocal microscopy images of cells stably expressing H3-EGFP (cyan) at 12 (A) and 24 (B) h p.i. labeled with NS1 (yellow) antibody. (C to F) Infected cells at 12 h p.i. and 24 h p.i. labeled with antibodies for H3K27ac (cyan) (C and D), H3K9me3 (cyan) (E and F), and NS1 (yellow). In order to clarify the changes in colocalization, pseudocolor images are shown with intensity increasing from blue to yellow. Fluorescence line profile analysis of the intensity of H3-EGFP/H3K27ac/H3K9me3 (cyan) and NS1 (yellow) in a single optical section through the center of the nucleus is shown beside each image. Analysis was performed with ImageJ and the Plot RGB Profile plugin. Scale bars, 5 μm. (G) Quantitative colocalization analysis of H3K27ac, H3K9me3, and H3-EGFP with NS1. The mean PCC values and standard deviations (SD) are shown. Statistical significance of colocalization at 16 and/or 24 h p.i. compared to that at 10 h p.i. is shown (P values [Student's t test]: *, P < 0.05; **, P < 0.01). (H and I) Plots of intensity of H3K27ac (H) and H3K9me3 (I) in infected cells at 24 h p.i. and in noninfected cells as a function of distance from the nuclear envelope (NE). A.U., arbitrary units.
In summary, these data showed that in the infected cells both H3K27ac and H3-EGFP were enriched in APAR bodies. At the same time, H3K9me3 was found to accumulate in the nuclear periphery, an area known to harbor the layer of marginalized cellular chromatin. This suggests that the histones associated with the viral genome in the APAR area bear modifications that are characteristic of active gene expression.
Time-dependent intranuclear interplay of H3K27ac and NS1.
In order to address the nuclear interaction of H3K27ac with viral DNA-bound NS1, in situ PLA was performed. PLA is an immunodetection technique that generates a fluorescent signal only when two antigens of interest are within 40 nm of each other (32). To assess the nuclear interactions of H3K27ac and NS1, we analyzed infected cells by PLA at 8 to 24 h p.i. Noninfected control cells exhibited only a faint nuclear signal (PLA signal per cell, 1.1 ± 0.19; n = 111). At 8, 10, 12, and 16 h p.i., a time-dependent increase in the amount of punctate intranuclear PLA signals was detected (0.14 ± 0.16, n = 105; 0.81 ± 0.30, n = 92; 3.43 ± 0.91, n = 110; 19.46 ± 3.39, n = 90), with a maximal signal at 24 h p.i. (36.49 ± 3.97, n = 99) (Fig. 2A) in the nuclear interior with low levels of DAPI (Fig. 2B). The infection-induced compaction of the host chromatin and its dislocation into the nuclear periphery and around the nucleolus at 24 h p.i. were visualized with DAPI.
FIG 2.
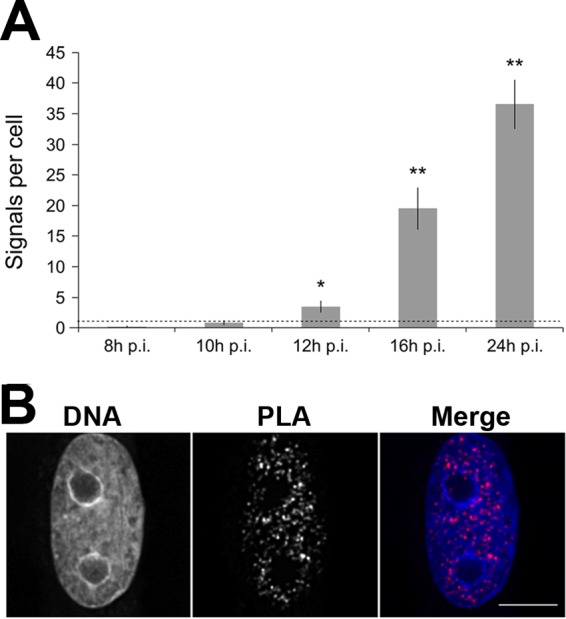
In situ PLA of H3K27ac interaction with NS1 in infected cells. (A) Number of PLA signals in infected cells at 8, 10, 12, 16, and 24 h p.i. The mean values of PLA signals ± standard errors from ∼100 cells per time point are shown. The negative control, PLA signal in noninfected cells, is shown by a dashed line. Statistical significance of PLA signals per cell at 12, 16, and 24 h p.i. compared to that of the negative control is shown (P values [Student's t test]: *, P < 0.05; **, P < 0.01). (B) Distribution of PLA signals in DAPI-stained nuclei at 24 h p.i. Scale bar, 10 μm.
In summary, these results demonstrated a time-dependent increase in the interaction or close proximity of H3K27ac with NS1 after 10 h p.i., followed by extensive interaction at 24 h p.i. This finding is consistent with the results of immunofluorescence and PCC analyses of infected cells, which showed increased colocalization and correlation of H3K27ac and NS1 during late stages of infection.
CPV promoters are rich in acetylated histones.
The prominent colocalization and interaction of NS1 with H3K27ac in the nucleus at 24 h p.i. prompted us to study whether the CPV genome per se is chromatinized with H3K27ac-enriched nucleosomes. To this end, ChIP-seq with H3K27ac Ab for the infected cells was performed. First, the success of ChIP was assessed with genome-wide occupancy of H3K27ac in infected cells showing a typical occupancy of this histone marker at ±2 kb around the transcription start sites (TSSs) of ∼35% of the genes (data not shown). We found that 9.23% of the total reads (2.23 × 107 reads) were aligned with the viral genome in a unique manner (0.00% in noninfected cells) (37). The alignment of these reads with the genome, after normalization to reads per CPV genomic content, revealed that H3K27ac is mostly enriched in the TSSs of P4 promoter-driven transcriptional units for NS1 and NS2 (gp1 loci) and P38-driven transcriptional units for VP1 and VP2 (gp2 loci) (Fig. 3A) (38).
FIG 3.
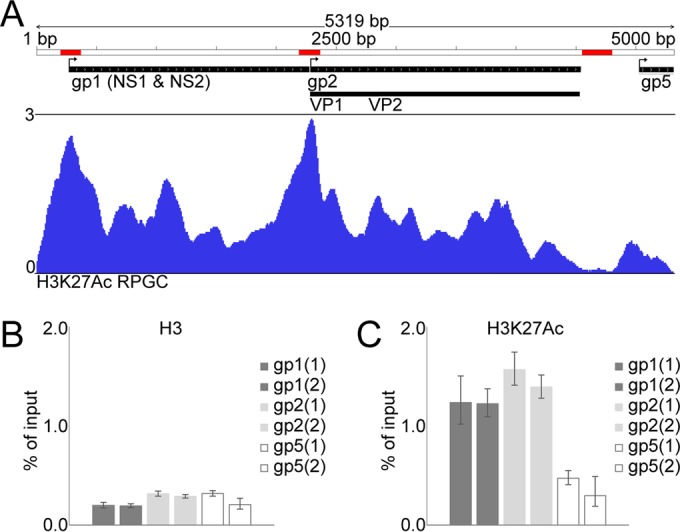
Enrichment of acetylated histones on gp1 and gp2 transcription start sites and effect on gene expression. (A) Histogram depicts occupancy of H3K27ac at CPV genome. Sequencing reads of H3K27ac ChIP-seq from infected cells at 24 h p.i. are normalized to RPGC. Bent arrows indicate TSSs of transcriptional units for NS1 and NS2 (gp1), VP1 and VP2 (gp2), and right-hand terminal gp5 loci, and red bars depict regions where qPCR primers anneal. ChIP-qPCRs for targeted measurements of H3 (B) and H3K27ac (C) levels at gp1 and gp2 TSSs and the VP2-gp5 interregion is shown as an enrichment of DNA relative to the level for input DNA. Two primer pairs for each of the three regions were used; columns represent the means ± SD from triplicate wells of two independent ChIP experiments.
The viral genome was mostly devoid of this histone marker toward the right end (gp5 loci). The location of gp1 loci was very similar to that of the P4 promoter of the parvovirus minute virus of mice (MVM; nucleotides 1 to 260), and the location of gp2 was similar to that of the CPV P38 promoter (1355 to 2260) (39). The targeted ChIP-qPCR of H3 with gp1-, gp2-, and gp5-specific primers showed that the CPV genome is thoroughly chromatinized with uniformly distributed H3 (Fig. 3B and data not shown). Finally, targeted ChIP-qPCR analysis confirmed the occupancy of H3K27ac in the genome (Fig. 3C).
In conclusion, our data verified that the parvoviral genome is chromatinized with histones during late stages of infection. Importantly, acetylated histones were observed to be enriched in viral promoters to allow for the transcriptional activation of the viral NS and VP genes.
Viral histone acetylation is important for infection progress.
In order to determine the importance of histone acetylation of parvoviral genomes during early stages of infection, the effect of the HAT inhibitor anacardic acid on viral protein synthesis was determined. Treatment with anacardic acid very early during infection (−0.5 to 1 h p.i.) resulted in a decrease in the percentage of cells with nuclear NS1 (∼5%) and VP2 (∼2%) at 24 h p.i. compared to the level for nontreated infected cells (68.0 and 50.3%, respectively; n ≥ 250). This suggests that the early inhibition of histone acetylation induced an almost complete block of infection. While the early treatment exerted a maximal effect, anacardic acid was able to inhibit the infection progress when introduced later, up to 3 h postinfection (Fig. 4A). Here, we cannot rule out the possibility that histone acetylation was not the only factor affecting viral protein production. Cell cycle progression also could be involved. Of note, the treatment did not affect cell viability (data not shown). Our results suggest that histone acetylation is required during very early infection for the production of viral proteins necessary for the efficient progress of infection.
FIG 4.

Importance of acetylation on progress of infection. (A) Percentage of cells showing viral NS1 and VP proteins in the nucleus in the cells exposed to the HAT inhibitor anacardic acid (0.1 mM). Cells were either pretreated for 30 min with the drugs prior to infection or treated at 0 to 6 h p.i., and the treatment continued until fixation at 24 h p.i. (B) Effect of anacardic acid treatment on localization of viral capsids. Images of cells representing four type categories based on the intracellular localization of capsids at 24 h p.i. and percentages of cells showing various types after treatment at 6 or 16 h p.i. The mean values ± SD are shown. (C) Distribution of H3K27ac compared to that of NS1 at 24 h p.i. in cells treated with anacardic acid at 16 or 20 h p.i. Scale bars, 10 μm.
We next studied if the anacardic acid-induced inhibition of histone acetylation affected late infection. For this, we divided the infected cells in four categories based on intracellular localization of capsids at 24 h p.i. The categories were (i) capsids localized into cytoplasmic endocytic vesicles due to inhibition of infection or secondary infection (24), (ii) capsids localized in discrete foci scattered throughout the nucleus, (iii) capsids in enlarged APAR bodies enriched with NS1, and (iv) capsids in both APAR bodies and the cytoplasm (egress initiated) (Fig. 4B). The localization of capsids next was determined at 24 h p.i. in cells exposed to anacardic acid at 6 h p.i. Studies indicated that levels of category II, III, and IV cells were reduced in treated cells (4.9%, 12.5%, and 21.2%; n ≥ 250) compared to the level of the nontreated infected control cells (7.9%, 26.8%, and 26.8%; n ≥ 250). Moreover, an increase in category I cells was detected in treated cells (61.1%; n ≥ 250) compared to the level in control cells (36.6%, 1.9%; n ≥ 250) (Fig. 4B). These results demonstrated that efficient progress of infection correlates with histone acetylation in APAR areas at late stages of infection.
Finally, we studied whether the timing of viral histone acetylation was critical for the progress of late-stage infection. Here, the distribution of H3K27ac and NS1, positioned on the viral genome, was analyzed at 24 h p.i. in cells treated with anacardic acid. The inhibitor treatment at 16 h p.i. resulted in the exclusion of H3K27ac from the NS1-positive APAR bodies (Fig. 4C). This indicated that the viral genomes produced after drug treatment at 16 h p.i. were not acetylated. However, H3K27ac colocalized with NS1 after exposure to inhibitor at 20 h p.i. (Fig. 4C). These results suggested that histone acetylation on the majority of newly synthesized viral DNA occurs until 16 to 20 h p.i.
These inhibition studies provided evidence that the histone acetylation of intranuclear viral genomes early during infection plays an essential role in the production of viral proteins. Similarly, the acetylation of histones on newly synthesized viral genomes during active virus replication is necessary for the efficient accomplishment of infection. In summary, our findings demonstrate that viral histone acetylation is essential for both early and late steps in the parvoviral life cycle.
DISCUSSION
Although much is known about nuclear replication and gene expression of parvoviruses, little is known about the chromatinization of parvoviral genomes and histone modifications and the effect of these events on the progress of infection. For some DNA viruses, such as herpesviruses and adenoviruses, epigenetic mechanisms, including histone modifications, play an important role in the regulation of viral gene expression. During herpesvirus lytic infection, the viral genomes are associated with histones immediately after injection into the nucleus, and viral proteins are required to enhance histone acetylation to allow for efficient viral gene expression (40–44). Moreover, studies of infection by an adenovirus have shown that viral proteins mediate the transcriptional activation of viral promoter regions (45–47). In this work, we first observed that the progress of CPV infection was accompanied by the enrichment of H3 histones in the enlarged APAR body area. This is consistent with earlier studies showing nuclear chromatinization of adeno-associated parvoviruses, used as gene therapy vectors, and MVM genomes (48–51). We next addressed the existence of histone modifications in the parvoviral APAR bodies and revealed the accumulation of histones with modifications characteristic of transcriptionally active chromatin (H3K27ac). Moreover, our analysis demonstrated that H3K27ac was located in close proximity to the viral NS1 protein in the APAR bodies. This interaction likely is caused by the accumulation of NS1 on viral genomes because of its involvement in transcription and replication (52–54). NS1 of autonomous parvoviruses not only controls the viral activities but also regulates host gene expression through histone acetylation in cancer cells by recruiting innate proteins with HAT activity (55, 56). To date, evidence for the involvement of NS1 in the regulation of histone acetylation on parvoviral promoters has not been reported. Here, our PLA studies revealed that CPV NS1 was located in close proximity to H3K27ac in the enlarged APAR bodies. Our earlier studies indicated that CPV NS1 has two distinct binding sites in the viral genome (53, 54, 57). It is tempting to speculate that NS1 is involved in the histone acetylation of viral P4 and P38 promoters by recruiting host proteins with acetyltransferase activity. However, the specific role and interactions of NS1 in viral histone modifications remain to be determined. Our ChIP-seq analyses demonstrated for the first time that in the extensively chromatinized parvoviral genome, H3K27ac accumulated in viral P4 and P38 promoter areas. This suggests that parvoviral gene expression is regulated by histone acetylation in promoter areas. The importance of histone acetylation for the progress of infection was supported by our results showing that the inhibition of histone acetylation was accompanied by extensive repression of infection. Earlier studies showed that the formation of parvoviral gene transcription templates and intermediates for genome replication are temporally phased in infection (58–61). In line with this, our results suggest that parvoviruses regulate their gene expression via histone acetylation in a temporal fashion.
In summary, the CPV genome is chromatinized inside the nuclei of infected cells, and histone modifications associated with transcriptional activation are enriched in viral promoters. Our results highlight a critical role of epigenetic modification in the progression of the parvoviral life cycle.
ACKNOWLEDGMENTS
We are grateful to Klaus Hedman for comments on the manuscript. We thank The Sequencing Service GeneCore Sequencing Facility (EMBL, http://www.genecore.embl.de) for DNA sequencing.
REFERENCES
- 1.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Venkatesh S, Workman JL. 2015. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 3.Kornberg RD. 1974. Chromatin structure: a repeating unit of histones and DNA. Science 184:868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stützer A, Fischle W, Bonaldi T, Pasini D. 2014. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell 53:49–62. doi: 10.1016/j.molcel.2013.10.030. [DOI] [PubMed] [Google Scholar]
- 5.Smolle M, Workman JL. 2013. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta 1829:84–97. doi: 10.1016/j.bbagrm.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res 21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKittrick E, Gafken PR, Ahmad K, Henikoff S. 2004. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci U S A 101:1525–1530. doi: 10.1073/pnas.0308092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santos-Rosa H, Schneider R, Bannister AJ, Sheriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 9.Knipe DM. 2015. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 479:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cereghini S, Yaniv M. 1984. Assembly of transfected DNA into chromatin: structural changes in the origin-promoter-enhancer region upon replication. EMBO J 3:1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B. 2014. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog 10(11):e1004503. doi: 10.1371/journal.ppat.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orzalli MH, Knipe DM. 2014. Cellular sensing of viral DNA and viral evasion mechanisms. Annu Rev Microbiol 68:477–492. doi: 10.1146/annurev-micro-091313-103409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riu E, Chen Z-Y, Xu H, He C-Y, Kay M. 2007. Histone modifications are associated with the persistence or silencing of vector-mediated transgene expression in vivo. Mol Ther 15:1348–1355. doi: 10.1038/sj.mt.6300177. [DOI] [PubMed] [Google Scholar]
- 14.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 15.Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, Margolis D, Nieto A, Nevels M, Parks RJ, Kristie TM. 2013. Snapshots: chromatin control of viral infection. Virology 435:141–156. doi: 10.1016/j.virol.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Opdenbosch N, Favoreel H, Van de Walle GR. 2012. Histone modifications in herpesvirus infections. Biol Cell 104:139–164. doi: 10.1111/boc.201100067. [DOI] [PubMed] [Google Scholar]
- 17.Milavetz B, Kallestad L, Gefroh A, Adams N, Woods E, Balakirshnan L. 2012. Virion-mediated transfer of SV40 epigenetic information. Epigenetics 7:528–534. doi: 10.4161/epi.20057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang CY, Shen CH, Wang M, Chen PL, Chan MW, Hsu PH, Chang D. 2015. Global profiling of histone modifications in the polyomavirus BK virion minichromosome. Virology 483:1–12. doi: 10.1016/j.virol.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Giberson AN, Davidson AR, Parks RJ. 2012. Chromatin structure of adenovirus DNA throughout infection. Nucleic Acids Res 40:2369–2376. doi: 10.1093/nar/gkr1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nevels M, Paulus C, Shenk T. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc Natl Acad Sci U S A 101:17234–17239. doi: 10.1073/pnas.0407933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berns K, Parrish CR. 2007. Parvoviridae, p 2437–2477. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 23.Reed AP, Jones EV, Miller TJ. 1988. Nucleotide sequence and genome organization of canine parvovirus. J Virol 62:266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cotmore SF, Tattersall P. 1998. High-mobility group 1/2 proteins are essential for initiating rolling-circle-type DNA replication at a parvovirus hairpin origin. J Virol 72:8477–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cziepluch C, Lampel S, Grewenig A, Grund C, Lichter P, Rommelaere J. 2000. H-1 parvovirus-associated replication bodies: a distinct virus-induced nuclear structure. J Virol 74:4807–4815. doi: 10.1128/JVI.74.10.4807-4815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ihalainen TO, Willman SF, Niskanen EA, Paloheimo O, Smolander H, Laurila JP, Kaikkonen MU, Vihinen-Ranta M. 2012. Distribution and dynamics of transcription-associated proteins during parvovirus infection. J Virol 86:13779–13784. doi: 10.1128/JVI.01625-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parrish CR. 1991. Mapping specific functions in the capsid structure of canine parvovirus and feline panleukopenia virus using infectious plasmid clones. Virology 183:195–205. doi: 10.1016/0042-6822(91)90132-U. [DOI] [PubMed] [Google Scholar]
- 28.Suikkanen S, Sääjärvi K, Hirsimäki J, Välilehto O, Reunanen H, Vihinen-Ranta M, Vuento M. 2002. Role of recycling endosomes and lysosomes in dynein-dependent entry of canine parvovirus. J Virol 76:4401–4411. doi: 10.1128/JVI.76.9.4401-4411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeung DE, Brown GW, Tam P, Russnak RH, Wilson G, Clark-Lewis I, Astell CR. 1991. Monoclonal antibodies to the major nonstructural nuclear protein of minute virus of mice. Virology 181:35–45. doi: 10.1016/0042-6822(91)90467-P. [DOI] [PubMed] [Google Scholar]
- 30.Abràmoff MD, Magalhães PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int 11:36–41. [Google Scholar]
- 31.Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalisation analysis in light microscopy. J Microsc 224:13–232. [DOI] [PubMed] [Google Scholar]
- 32.Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius K-J, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 33.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 34.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. 2014. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christensen J, Storgaard T, Viuff B, Aasted B, Alexandersen S. 1993. Comparison of promoter activity in Aleutian mink disease parvovirus, minute virus of mice, and canine parvovirus: possible role of weak promoters in the pathogenesis of Aleutian mink disease parvovirus infection. J Virol 67:1877–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cotmore SF, Tattersall P. 1987. The autonomously replicating parvoviruses of vertebrates. Adv Virus Res 33:91–174. doi: 10.1016/S0065-3527(08)60317-6. [DOI] [PubMed] [Google Scholar]
- 40.Hancock MH, Cliffe AR, Knipe DM, Smiley JR. 2010. Herpes simplex virus VP16, but not ICP0, is required to reduce histone occupancy and enhance histone acetylation on viral genomes in U2OS osteosarcoma cells. J Virol 84:1366–1375. doi: 10.1128/JVI.01727-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalamvoki M, Roizman B. 2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc Natl Acad Sci U S A 107:17721–17726. doi: 10.1073/pnas.1012991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang J, Kent JR, Placek B, Whelan KA, Hollow CM, Zeng P-Y, Fraser NW, Berger SL. 2006. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J Virol 80:5740–5746. doi: 10.1128/JVI.00169-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120–128. doi: 10.1016/j.gene.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 44.Nitzsche A, Paulus C, Nevels M. 2008. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J Virol 82:11167–11180. doi: 10.1128/JVI.01218-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Morral N, Engel DA. 2007. Transcription releases protein VII from adenovirus chromatin. Virology 369:411–422. doi: 10.1016/j.virol.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 46.Komatsu T, Haruki H, Nagata K. 2011. Cellular and viral chromatin proteins are positive factors in the regulation of adenovirus gene expression. Nucleic Acids Res 39:889–901. doi: 10.1093/nar/gkq783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komatsu T, Nagata K. 2012. Replication-uncoupled histone deposition during adenovirus DNA replication. J Virol 86:6701–6711. doi: 10.1128/JVI.00380-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ben-Asher E, Bratosin S, Aloni Y. 1982. Intracellular DNA of the parvovirus minute virus of mice is organized in a minichromosome structure. J Virol 41:1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marcus-Sekura CJ, Carter BJ. 1983. Chromatin-like structure of adeno-associated virus DNA in infected cells. J Virol 48:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doerig C, McMaster G, Sogo J, Bruggmann H, Beard P. 1986. Nucleoprotein complexes of minute virus of mice have a distinct structure different from that of chromatin. J Virol 58:817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Penaud-Budloo M, Le Guiner C, Nowrouzi A, Toromanoff A, Chérel Y, Chenuaud P, Scmidt M, von Kalle C, Rolling F, Moullier P, Snyder RO. 2008. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J Virol 82:7875–7885. doi: 10.1128/JVI.00649-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cotmore SF, Gottlieb RL, Tattersall P. 2007. Replication initiator protein NS1 of the parvovirus minute virus of mice binds to modular divergent sites distributed throughout duplex viral DNA. J Virol 81:13015–11327. doi: 10.1128/JVI.01703-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niskanen EA, Ihalainen TO, Kalliolinna O, Häkkinen MM, Vihinen-Ranta M. 2010. Effect of ATP binding and hydrolysis on dynamics of canine parvovirus NS1. J Virol 84:5391–5403. doi: 10.1128/JVI.02221-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niskanen EA, Kalliolinna O, Ihalainen TO, Häkkinen M, Vihinen-Ranta M. 2013. Mutations in DNA binding and transactivation domains affect the dynamics of parvovirus NS1 protein. J Virol 87:11762–11774. doi: 10.1128/JVI.01678-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iseki H, Shimizukawa R, Sugiyama F, Kunita S, Iwama A, Onodera M, Nakauchi H, Yagami K. 2005. Parvovirus nonstructural proteins induce an epigenetic modification through histone acetylation in host genes and revert tumor malignancy to benignancy. J Virol 79:8886–8893. doi: 10.1128/JVI.79.14.8886-8893.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohshima T, Yoshida E, Nakajima T, Yagami K, Fukamizu A. 2001. Effects of interaction between parvovirus minute virus of mice NS1 and coactivator CBP on NS1- and p53-transactivation. Int J Mol Med 7:49–54. [DOI] [PubMed] [Google Scholar]
- 57.Ihalainen TO, Niskanen EA, Jylhävä J, Turpeinen T, Rinne J, Timonen J, Vihinen-Ranta M. 2007. Dynamics and interactions of parvoviral NS1 protein in the nucleus. Cell Microbiol 9:1946–1959. doi: 10.1111/j.1462-5822.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 58.Clemens KE, Pintel DJ. 1988. The two transcription units of the autonomous parvovirus minute virus of mice are transcribed in a temporal order. J Virol 62:1448–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schoborg RV, Pintel DJ. 1991. Accumulation of MVM gene products is differentially regulated by transcription initiation, RNA processing and protein stability. Virology 181:22–34. doi: 10.1016/0042-6822(91)90466-O. [DOI] [PubMed] [Google Scholar]
- 60.Tullis G, Schoborg RV, Pintel DJ. 1994. Characterization of the temporal accumulation of minute virus of mice replicative intermediates. J Gen Virol 75:1633–1646. doi: 10.1099/0022-1317-75-7-1633. [DOI] [PubMed] [Google Scholar]
- 61.Lieberman PM. 2006. Chromatin regulation of virus infection. Trends Microbiol 14:132–140. doi: 10.1016/j.tim.2006.01.001. [DOI] [PubMed] [Google Scholar]