

Abstract
Transforming growth factor-beta1 (TGF-β1), a key member in the TGF-β superfamily, plays a critical role in the development of hepatic fibrosis. Its expression is consistently elevated in affected organs, which correlates with increased extracellular matrix deposition. SMAD proteins have been studied extensively as pivotal intracellular effectors of TGF-β1, acting as transcription factors. In the context of hepatic fibrosis, SMAD3 and SMAD4 are pro-fibrotic, whereas SMAD2 and SMAD7 are protective. Deletion of SMAD3 inhibits type I collagen expression and blocks epithelial-myofibroblast transition. In contrast, disruption of SMAD2 upregulates type I collagen expression. SMAD4 plays an essential role in fibrosis disease by enhancing SMAD3 responsive promoter activity, whereas SMAD7 negatively mediates SMAD3-induced fibrogenesis. Accumulating evidence suggests that divergent miRNAs participate in the liver fibrotic process, which partially regulates members of the TGF-β/SMAD signaling pathway. In this review, we focus on the TGF-β/SMAD and other relative signaling pathways, and discussed the role and molecular mechanisms of TGF-β/SMAD in the pathogenesis of hepatic fibrosis. Moreover, we address the possibility of novel therapeutic approaches to hepatic fibrosis by targeting to TGF-β/SMAD signaling.
Keywords: Epithelial-myofibroblast transition, hepatic fibrosis, miRNAs, TGF-β/SMAD
Introduction
Hepatic fibrosis (HF) is a chronic liver disease, and a leading cause of end-stage hepatocellular carcinoma (Dongiovanni et al. 2014), which is characterized by an increase in extracellular matrix (ECM) deposited around the sinusoidal cell layer in the space of Disse (Bataller and Brenner 2005; Gao and Bataller 2011). The increased fibrotic matrix is the result of an imbalance of ECM synthesis and degradation. Carbon tetrachloride (CCl4)-induced hepatic injury is a well-established model for the study of liver fibrogenesis. In the livers of CCl4-treated animals, severe morphological changes, such as fat degeneration, ballooning, necrosis and infiltration of inflammatory cells, are observed as compared with control animals. Masson’s trichrome staining shows that there is an increase in collagen I during rat liver fibrosis. Immunohistochemistry also shows that there is an increase in the abundance of p-SMAD2 and p-SMAD3 (Fig. 1) (Zhang et al. 2015). There are many mediators acting during the development of HF, such as growth factors (Sysa et al. 2009), mitogen-activated protein kinase (MAPK) (Qiang et al. 2006), leptin (Wang et al. 2009), and various integrins (Nadler et al. 2009). Of particular note is TGF-β1, a key mediator in the pathogenesis of HF (Bi et al. 2012). TGF-β1 activates SMAD-dependent and -independent pathways to exhibit its biological activities. It is well-known that TGF-β exerts its biological effects by activating downstream mediators SMAD2 and SMAD3, and is negatively regulated by an inhibitory SMAD7 (Lan and Chung 2011). SMADs also act as signal integrators and interact with other signaling pathways, such as the MAPK and NF-κB signaling pathways (Derynck and Zhang 2003; Sengupta et al. 2009). Recent research into microRNAs has demonstrated that TGF-β may regulate the expression of several microRNAs to influence fibrosis (Tian et al. 2008).
Figure 1.

The histology of hepatic fibrosis. Pathological observations of experimental rat liver sections stained with Masson’s trichrome staining in (A) vehicle control and (B) CCl4-treated liver tissue. The analysis shows that total collagen deposition is significantly increased in CCl4-induced hepatic fibrosis tissues. Compared with the vehicle control (C, E), an increase in positive staining for p-Smad2 (D) and p-Smad3 (F) immunohistochemistry is noted during rat liver fibrosis. Scale, 50 µm.
The present review focuses on the functional role and regulatory mechanisms of SMAD-dependent and -independent pathways that interact with the TGF-β pathway during the progression of HF. The therapeutic potential for targeting the related TGF-β/SMAD signaling pathways is also discussed.
The Pathological Process of HF
ECM Synthesis during the Development of Fibrosis
Chronic damage to the liver, including persistent infections, autoimmune reactions, allergic responses, chemical insults, and radiation, negatively impacts the liver’s wound healing response (Friedman 2004). Fibrosis is the end result of chronic inflammatory reactions and characterized by an excessive deposition of ECM of predominantly type I collagen. The excessive deposition of ECM disrupts the normal architecture of the liver and results in hepatic dysfunction. In response to liver injury, hepatic stellate cells (HSCs) become activated, which exhibit a myofibroblast-like phenotype. The activated HSC is primarily responsible for the excessive synthesis and deposition of ECM in the hepatic interstitium, leading to liver fibrogenesis (Shek and Benyon 2004).
Chronic inflammation and repair can trigger an excessive accumulation of ECM components, especially collagens, the major fibrous proteins in the ECM. The increased transcription of type I collagen genes, COL1A1 and COL1A2, contributes to this excessive tissue deposition of ECM proteins (Trojanowska et al. 1998; Uitto and Kouba 2000). Both collagen turnover and ECM remodeling are regulated by various MMPs and their inhibitors. The net accumulation of collagens in tissue fibrosis is a result of an imbalance between their synthesis and degradation (Verrecchia and Mauviel 2004).
EMT and EndoMT in the Development of Fibrosis
The key effector cells of fibrosis are the myofibroblasts, which serves as the primary collagen-producing cells when activated. HSCs are believed to be the major source of collagen-producing myofibroblasts in cirrhotic livers (Rygiel et al. 2008). Thus, the fibroblasts expressing alpha-smooth muscle actin (α-SMA) (myofibroblasts) may be derived from the transdifferentiation of quiescent HSCs. Fibroblasts are also generated from epithelial cells through epithelial-mesenchymal transition (EMT), which is the process of fully differentiated epithelial cells undergoing phenotypic transition into fully differentiated mesenchymal cells (fibroblasts or myofibroblasts) (Lee and Kay 2012; Thompson and Haviv 2011). During EMT, mature epithelial cells lose their original shape, cell-cell contact, and epithelial cell-specific protein expression, but gain the typical features of mesenchymal cells (Aldehni et al. 2009; Heinrich et al. 2012). The proteomic features of EMT include the loss of epithelial cell adhesion molecules, such as epithelial (E)-cadherin and zonula occludens-1 (ZO-1), which are replaced by the mesenchymal markers, α-SMA, matrix metalloproteinase (MMP)-2, MMP-9, collagens, and the intermediate filament protein vimentin (Bi et al. 2014; Zhang et al. 2012). This process leads to increased deposition of the ECM, which indicates that EMT is crucial in the pathology of liver fibrosis (Barnes et al. 2010). The generation of fibrogenic fibroblasts through EMT can occur through a variety of sources. In addition, endothelial-myofibroblast transition (EndoMT) has been implicated in fibrogenesis (Zeisberg et al. 2008; Zeisberg and Kalluri 2008). EMT of hepatocytes and cholangiocytes has been reported in patients and in mice with liver fibrosis (Syn et al. 2009; Zeisberg et al. 2007b). Evidence of EMT has also been reported in the kidney, lung, eye and serosal membranes, suggesting that EMT could be involved in the pathogenesis of fibrotic disorders in these organs (Willis et al. 2006). EMT in liver fibrosis causes a loss of epithelial cells, contributing to the parenchyma destruction seen in advanced fibrosis. Endothelial cells may also transit to mesenchymal cells, giving rise to (myo)fibroblasts in response to fibrogenic injury. EndoMT has been reported (Zeisberg et al. 2007a), but EndoMT is difficult to measure because there is not yet a specific marker for endothelial cells. EMT-derived myofibroblasts produce a variety of factors that are involved in the fibrotic process (Quan et al. 2006). Therefore, a therapeutic approach to disturbing their development may be an effective strategy for treating hepatic fibrosis.
TGF-β/SMAD Signaling Pathways
The TGF-β Superfamily
The TGF-β superfamily includes a large number of structurally and functionally related proteins, such as bone morphogenetic proteins (BMPs), activins, inhibins, growth differentiation factors (GDFs), and glial-derived neurotrophic factors (GDNFs) (Maribo et al. 1998; Massague and Wotton 2000). These members act as multifunctional regulators of a wide range of biological processes, such as morphogenesis, embryonic development, adult stem cell differentiation, immune regulation, wound healing, inflammation, and cancer (Boon et al. 2011; Kang et al. 2008). TGF-β family members (TGF-β1, -β2, and -β3) are induced and activated in a variety of fibrotic diseases (Govinden and Bhoola 2003). TGF-β1 was discovered in 1983 because of its ability to stimulate the growth of cultured rat fibroblasts in soft agar, and was regarded as the master cytokine in liver fibrogenesis (Drabsch and ten Dijke 2012). Release of TGF-β1 by necrotic hepatocytes during liver damage may be one of the first signals to activate adjacent quiescent HSCs, resulting in their transdifferentiation into myofibroblasts. TGF-β1 signaling inhibits HSC apoptosis, and induces HSCs to synthesize excessive amounts of matrix proteins, such as fibronectin, and collagen types I, III, and IV (Kanzler et al. 1999). In addition, TGF-β1 disturbs the production of matrix-degrading proteases and upregulates protease inhibitors, such as tissue inhibitor of metalloproteinase (TIMP) and plasminogen activator inhibitor. Thus, TGF-β1 promotes ECM production and inhibits its degradation (Dudas et al. 2001; Eddington et al. 2007). Furthermore, TGF-β1 modulates the expression of integrins in a manner that increases cellular adhesion to the matrix. In patients with HF, increased concentrations of TGF-β1 correlate with the severity of hepatic fibrosis, suggesting a link between TGF-β expression and progressive liver disease (Friedman 2008; Gressner and Weiskirchen 2006; Lee and Friedman 2011).
TGF-β1 promotes fibrogenesis via three mechanisms. Firstly, TGF-β1 inhibits ECM degradation by suppressing MMP and promoting the natural inhibitor TIMP. Secondly, TGF-β1 induces myofibroblast formation through tubular EMT. Thirdly, TGF-β1 induces matrix production through SMAD3-dependent or non-SMAD associated mechanisms (Border and Noble 1998; Lan 2003).
TGF-β Signaling through SMAD Proteins
SMAD proteins mediate intracellular TGF-β signaling. Members of the SMAD family can be classified into three groups according to their functions: 1) the receptor-regulated SMADs (R-SMADs), which include SMAD1, SMAD2, SMAD3, SMAD5, and SMAD8; 2) the common SMAD (Co-SMAD), of which there is only one member, SMAD4; 3) the inhibitory SMADs (I-SMADs), including SMAD6 and SMAD7. The R-SMADs bind to membrane-bound serine/threonine receptors, and are activated by their kinase activity. As a co-factor, the co-SMAD binds to the activated R-SMADs to form a complex that translocates into the nucleus. I-SMADs counteract the effects of R-SMADs, thus exerting an inhibitory effect on TGF-β superfamily signaling by various mechanisms (Blanco Calvo et al. 2009; Blank and Karlsson 2011).
Latent TGF-β1, which is called the latent precursor, binds to latent TGF-β-binding protein (LTBP). TGF-β1 is released from the latency-associated peptide (LAP) and LTBP when exposed to various factors, such as reactive oxygen species (ROS), plasmin or acid (Wang et al. 2005b). Upon activation, the mature TGF-β1 binds to type I and type II cell-surface receptor complexes. Each receptor complex is made of a small cysteine-rich extracellular region and an intracellular region consisting mainly of the kinase domains. In TGF-β/SMAD signaling, TGF-β1 initiates intracellular signaling by binding to the TGF-β receptor II (TβRII). Then, TGF-β1 activates the TGF-β receptor type I (TβRI) kinase, resulting in phosphorylation of SMAD2 and SMAD3. Subsequently, the activated SMAD2 and SMAD3 form oligomeric complexes with SMAD4. These oligomeric complexes translocate into the nucleus, where they regulate the transcription of target genes, including the induction of SMAD7 (Conidi et al. 2011; Meng et al. 2013; Xie et al. 2014).
Both TGF-β and BMP-7 share similar downstream SMAD signaling pathways but counter-regulate each other to maintain the balance of their biological activities. In mammals, TGF-β conveys intracellular signals through phosphorylation of SMAD2 and SMAD3. On the other hand, BMP-7 activates R-SMAD1/5/8. Phosphorylated R-SMADs will form heteromeric complexes with a common partner, SMAD4 (co-SMAD). The resultant SMAD4/SMAD1/5/8 and SMAD2/3/4 complexes regulate the transcription of two different sets of genes (Fig. 2) (Meng et al. 2013). Besides SMAD-dependent pathways, TGF-β1 also activates SMAD-independent pathways such as MAPK (West 2010), NF-kB, and phosphatidylinositol-3-kinase (PI3K) pathways (Derynck and Zhang 2003). This review focuses on the cross-talk between TGF-β/SMAD and other signaling pathways in the pathogenesis of HF.
Figure 2.

TGF-β1/SMAD signaling pathways in hepatic fibrosis. In hepatic fibrosis, TGF-β1 triggers SMAD2/3, whereas BMP7 activates SMAD1/5/8. Then the phosphorylated SMAD2/3 or SMAD1/5/8 form complexes with SMAD4 and shuttle into the nucleus to regulate gene transcription by binding to DNA sequences or cofactors. In addition, TGF-β1 also activates the inhibitory SMADs (SMAD7) to negatively regulate their signals. Abbreviations: BMPRI, bone morphogenetic protein receptor type I; p, phosphorylation.
TGF-β1/SMAD Signaling Pathways and Regulation in HF
Differential Roles of SMADs in HF
The balance of different SMAD isoforms is important for cell differentiation, stem cell maintenance, tissue homeostasis and tumorigenesis (Tao and Sampath 2010). An imbalance between positive and negative SMAD signaling pathways may play a vital role in the development of HF. SMADs have two conserved domains: N-terminal Mad homology 1 (MH1) and C-terminal Mad homology 2 (MH2) domains. Among R-SMADs and Co-SMADs, the MH1 domain is highly conserved. The MH2 domain is highly conserved among all SMADs (Moustakas et al. 2001; Singh et al. 2011).
Differential Roles of SMAD2 and SMAD3 in HF
The general structure of SMAD proteins is similar, with a few differences among the various SMAD categories. With structural similarity but functional diversity, SMAD2 and SMAD3 interact with each other to mediate TGF-β-triggered signaling transduction. Although SMAD2-deficient mice die early in development at embryonic day 9.5, SMAD3 KO mice are viable but usually die from defects in mucosal immunity at 1–6 months of age (Datto et al. 1999; Heyer et al. 1999). SMAD3 KO mice show diminished T-cell responsiveness to TGF-β. As SMAD3 binds to DNA directly, whereas SMAD2 does not, these two SMADs may have distinct effects on the regulation of target genes.
Whereas both SMAD2 and SMAD3 are strongly activated in liver fibrosis (Yao et al. 2012), only SMAD3 appears to be a key element in the signal transduction pathways responsible for fibrosis (Medeiros et al. 2004). A number of fibrogenic genes (collagens) and markers (α-SMA and E-cadherin) are SMAD3-dependent, and SMAD3 directly binds to DNA sequences that regulate these target genes (Latella et al. 2009; Masszi and Kapus 2011). In addition, TGF-β induces TIMP-1 by activating SMAD3, thus inhibiting ECM degradation. Overexpression of SMAD3 inhibits MMP-1 activity in fibroblasts. Knockdown of SMAD3 blocks EMT and attenuates renal fibrosis, inflammation, and apoptosis after unilateral ureteral obstruction (UUO) (Meng et al. 2010; Sato et al. 2003). All of these results suggest a pathogenic role for SMAD3 during TGF-β1–induced fibrosis.
In contrast, SMAD2 plays a protective role in hepatic fibrosis despite interacting with SMAD3 and sharing more than 90% structural similarity (Massague and Wotton 2000). Previous studies have demonstrated different functional roles of SMAD2 and SMAD3 in mediating the action of TGF-β1 (Phanish et al. 2006; Yang et al. 2003). These observations are further confirmed by our recent in vitro finding that knockdown of SMAD2 in human HSC LX-2 cells enhances SMAD3-dependent hepatic fibrosis by increasing phosphorylation, nuclear translocation, and collagen type I promoter binding of SMAD3 (Zhang et al. 2015). Therefore, SMAD2 plays a protective role in hepatic fibrosis by counter-regulating SMAD3 signaling.
Essential Role of SMAD4 in HF
SMAD4 interacts with SMAD2/3 and participates in the intracellular TGF-β signaling pathway. Because of the early embryonic lethality of SMAD4 knockout mice, functional research of SMAD4 in liver disease has been delayed (Hruska et al. 2000). However, a recent study in kidney-specific conditional SMAD4 knockout mice has found that disruption of the SMAD4 gene in kidney tubules decreases ECM synthesis in obstructed kidneys in vivo and in TGF-β1-treated kidney interstitial fibroblasts in vitro (Meng et al. 2012). Similarly, deletion of SMAD4 from mesangial cells results in the inhibition of TGF-β1-induced ECM deposition (Tsuchida et al. 2003). These results indicate that disruption of SMAD4 attenuates fibrosis by decreasing SMAD3-responsive promoter activity. SMAD4 plays an essential role in fibrosis disease by regulating the ability of SMAD3 to initiate transcription of its target genes.
Protective Role of SMAD7 in HF
It is now clear that TGF-β1 induces the expression of SMAD7, which, in turn, negatively regulates TGFβ1/SMAD signaling via two possible mechanisms. First, SMAD7 binds to TβRI, thereby preventing the recruitment and phosphorylation of R-SMADs. Second, SMAD7 recruits the E3 ubiquitin ligase SMAD ubiquitination regulatory factors (SMURFs) to SMAD2 and TβRI, which subsequently ubiquitinate and degrade these two proteins (Lin et al. 2000; Tan et al. 2008). It is important to note that SMAD7, an inhibitory SMAD, is induced by TGF-β1 to block the over activation of TGF-β signals via inhibition of TβRI and SMAD2/3, but that it does not simply enhance SMAD2 degradation and aid in hepatic fibrosis (Zhu et al. 1999; Kavsak et al. 2000).
SMAD7 is a potential master transcriptional repressor of HSC activation and hepatic fibrosis in vitro and in vivo. Deletion of SMAD7 promotes these processes, whereas overexpression of SMAD7 protects against them (Dooley et al. 2008; Hamzavi et al. 2008). Studies have shown that quiescent HSCs in culture display a negative feedback involving TGF-β1-dependent transient activation of SMAD7 expression, and this feedback mechanism is lost in myofibroblasts (Dooley et al. 2001; Stopa et al. 2000). Significantly decreased SMAD7 expression is seen in fibrotic livers during TGF-β1-induced HSC activation and in myofibroblast-like cells throughout chronic liver injury (Bian et al. 2014). Disruption of the SMAD7 gene enhances CCl4-dependent liver damage and fibrogenesis in mice (Hamzavi et al. 2008). Thus, SMAD7, like TGF-β, plays a role in EMT in hepatocytes (Zhu et al. 2011). Moreover, TGF-β1 treatment is able to induce migration of the cells, and this effect is significantly accelerated when SMAD7 is deleted.
Vimentin is a well-recognized marker for EMT, as is E-cadherin for epithelial cells (Kalluri and Weinberg 2009). TGF-β1-induced reduction of E-cadherin and upregulation of vimentin are profoundly enhanced by SMAD7 deletion. SMAD7 deficiency is also able to enhance TGF-β1-induced EMT in hepatocytes. Overexpression of SMAD7 in mouse liver could attenuate TGF-β1 signaling and decrease CCl4-provoked liver fibrosis (Dooley et al. 2008). These data indicate that SMAD7 is protective against TGF-β1 signaling.
Cross-talk between SMADs and Other Signaling Pathways in HF
Although the R-SMADs can be phosphorylated by kinase domains on membrane-bound receptors, they can also be phosphorylated by kinases involved in other pathways. Both MAPK and NF-κB pathways are the major cross-talk pathways related to SMADs signaling pathway. The MAPK pathway has been shown to be involved in HSC proliferation, and may amplify the number of fibrogenic cells and promote fibrogenic response. The MAPK pathway contains the c-Jun N-terminal kinase (JNK), the extracellular signal-regulated kinases (ERKs), and p38 kinase. JNK has been shown to positively regulate cell proliferation in several cell types, including HSC (Schreiber et al. 1999). Inhibiting JNK activity in quiescent HSCs or in culture-activated HSCs using a dominant-negative form of JNK prevented HSC proliferation (Schnabl et al. 2001; Schreiber et al. 1999). ERK is activated by platelet-derived growth factor (PDGF) stimulation of HSCs, both in vitro and in vivo (Gentilini et al. 2000; Ross and Hill 2008). Pharmacological ERK inhibitors positively regulate cell growth, demonstrating the importance of this signaling pathway in HSC proliferation (Pages et al. 1993). In contrast, activation of p38 kinase inhibits HSC multiplication in either quiescent or activated HSCs (Schnabl et al. 2001).
The crosstalk between the TGF-β1/SMAD7 and NF-κB signaling pathway is also important in the development of liver inflammation. Loss of SMAD7 enhances NF-κB-driven inflammation (Fukasawa et al. 2004; Ng et al. 2005). In contrast, overexpression of SMAD7 effectively attenuates inflammatory cell infiltration and suppresses the production of inflammatory cytokines during the pathogenesis of various diseases (Ka et al. 2007; Ka et al. 2012). The underlying mechanism of SMAD7-mediated inflammation attenuation may be due to increased expression of IκBα, an inhibitor of NF-κB (Azuma et al. 1999; Wang et al. 2005a).
Regulation of Relationship between microRNA and TGF-β1/SMAD in HF
MicroRNAs (miRNAs) are small, noncoding RNAs, 18–24 nucleotides in length, that regulate gene expression by binding to mRNAs to interfere with the process of translation (Szabo and Bala 2013). As an evolutionarily conserved species, miRNAs are involved in many biological processes including regulation of apoptosis, development, signal transduction, cell proliferation, and immune defense (Spizzo et al. 2009).
miRNAs target and regulate essentially all biological processes and cell types, including those in the liver, and influence gene expression in virtually all cellular processes. Numerous reports have demonstrated that alterations in intracellular miRNAs correlate with various liver diseases, such as miR-155, miR-132, miR-21, miR-26a, and miR-217. miR-155 was shown to be elevated in Kupffer cells after alcohol feeding, and TNF was identified as a target of miR-155 to promote liver inflammation (Bala et al. 2011). Induction of miR-132 was also reported in the liver and Kupffer cells after chronic alcohol administration in mice (Bala and Szabo 2012). In addition, various miRNAs control hepatocyte proliferation. miR-21 regulates various genes involved in the cell cycle and DNA synthesis (Ng et al. 2012). In contrast to miR-21, miR-26a is shown to be downregulated after partial hepatectomy and promotes hepatocyte proliferation (Zhou et al. 2012). miR-217 has an anti-proliferative role, and methylation of its promoter region results in its decreased expression during liver regeneration (Pan et al. 2012).
Growing evidence indicates that miRNAs participate in liver fibrotic process and activation of HSCs, and miRNAs play a role through targeting SMAD proteins in the liver. miR-199a has a critical role in EMT and promotes liver fibrosis by enhancing the expression of fibrotic genes such as those that encode α1 procollagen, collagenase 3, and metalloproteinase inhibitor 1 (Murakami et al. 2011). At the same time, overexpression of miR-200 inhibits SMAD3 activity and attenuates TGF-β1-induced fibrosis. This finding suggests that miR-200a is not only regulated by TGF-β1/SMAD3, but also interacts with SMAD3 to exhibit its functional activities (Wang et al. 2011). Recently, the miR-454 family has been reported to be up-regulated in human colorectal cancer tissues and cell lines by targeting SMAD4. The level of the miR-454 was down-regulated in fibrotic livers. On the contrary, the levels of α-SMA and SMAD4 expression were all upregulated. Since there is a tendency for SMAD4 expression to oppose that of miR-454 in hepatic fibrosis, it has been suggested that miR-454 may be involved in the progression of liver fibrosis through the TGF-β1/SMAD4 pathway (Zhu et al. 2014). Overexpression of miR-146a suppresses TGF-β1-induced HSC proliferation and increases both HSC apoptosis and the expression of α-SMA, at least, in part, via decreasing the expression of SMAD4 (He et al. 2012). TGF-β1 induces miR-33a expression, and miR-33a stimulates HSC fibrogenic activation by targeting the inhibitory SMAD7 (Huang et al. 2015). In addition, SMAD7 is also targeted by miR-21, further suggesting that SMAD7 is targeted during miRNA-associated fibrosis progression (Marquez et al. 2010). These findings indicate the complex relationship between TGF-β1/SMADs and miRNAs under pathophysiological conditions (Fig. 3).
Figure 3.
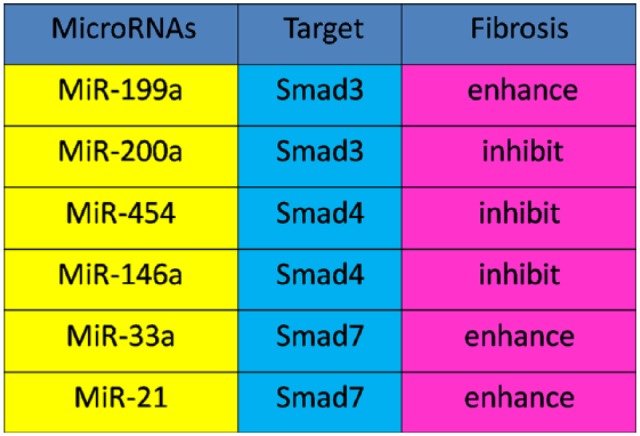
The relationship between microRNA and TGF-β1/SMAD in hepatic fibrosis. In hepatic fibrosis, miR-146a, miR-454 and miR200a negatively correlate with SMAD4 and SMAD3, respectively. In addition, miR-199a, and miR-33a and miR-21 are positively associated with SMAD3 and SMAD7, respectively.
The Relationship between Long Non-coding RNA and Fibrosis
Non-coding RNAs (ncRNAs) include small ncRNAs and various classes of long ncRNAs (lncRNAs). LncRNAs, defined by non-protein coding transcripts longer than 200 nucleotides, are a new class of ncRNAs found to be pervasively transcripted in the genome (Szabo and Bala 2013). The Arid2-IR is one of the most highly up-regulated lncRNAs present following UUO in the kidneys of wild-type mice, with the progressive renal fibrosis and inflammation able to be largely suppressed in the diseased kidney of SMAD3 knockout mice. Arid2-IR deletion from medullary thymic epithelial cells (mTECs) or from the UUO kidney did not influence TGF-β/SMAD3 signaling or renal fibrosis. Thus, Arid2-IR may play a functional role in renal fibrosis (Zhou et al. 2015). Hence, we surmise that lncRNAs may be useful as a therapeutic target for fibrosis disease.
Therapeutic Strategies
Considering the central role of TGF-β1 signaling in the pathogenesis of liver inflammation and fibrosis, targeting TGF-β1 signaling may represent a novel therapeutic strategy for liver diseases. To combat TGF-β-induced fibrosis, neutralizing TGF-β antibodies, with antisense TGF-β oligodeoxynucleotides, soluble human TβRII and specific inhibitors against TβR kinases can be used to block upstream TGF-β signaling and attenuate fibrosis (Lan and Chung 2012). However, these potential therapies may also increase liver inflammation by inhibiting the general anti-inflammatory property of TGF-β1.
With recent advances in the understanding of the negative effects of inhibiting TGF-β signaling, we propose to target downstream mediators of the TGF-β signaling pathway, especially SMAD3. Since SMAD3 plays such a critical role in mediating the pathobiology of fibrotic disease, inhibition of SMAD3 signaling could be a potential target for fibrotic disease intervention. Additionally, SMAD7 is an effective inhibitor of TGF-β signaling and is a key regulator of TGF-β-induced fibrogenesis. Overexpression of SMAD7 not only inhibits the activation of SMAD2/3, but also blocks NF-κB signaling in fibrosis and during inflammation, suggesting that SMAD7 may be a potential therapeutic agent. TGF-β/SMAD signaling is a major contributor to the development of HF. In the context of HF, SMAD3 is pathogenic, whereas SMAD2 and SMAD7 are protective. Understanding the specific roles of ligands–receptors and antagonists, as well as the cross-talk between intracellular signaling pathways at transcriptional and posttranscriptional levels, including SMA-dependent miRNAs in the pathogenesis of liver fibrosis, would enable us to develop specific and effective therapeutics for HF.
Footnotes
Author Contributions: The authors contributed as follows: Design, Lei Zhang; manuscript preparation, Fengyun Xu and Changwei Liu; manuscript editing, Fengyun Xu and Changwei Liu; and manuscript review, Lei Zhang
Competing Interests: The authors declared no potential competing interests with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Chinese National Natural Science Foundation Project (81100302) and Intercollegiate Key Projects of Nature Science of Anhui Province (KJ2011A174).
References
- Aldehni F, Spitzner M, Martins JR, Barro-Soria R, Schreiber R, Kunzelmann K. (2009). Bestrophin 1 promotes epithelial-to-mesenchymal transition of renal collecting duct cells. J Am Soc Nephrol 20:1556-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma M, Motegi K, Aota K, Yamashita T, Yoshida H, Sato M. (1999). TGF-beta1 inhibits NF-kappaB activity through induction of IkappaB-alpha expression in human salivary gland cells: a possible mechanism of growth suppression by TGF-beta1. Exp Cell Res 250:213-222. [DOI] [PubMed] [Google Scholar]
- Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, Szabo G. (2011). Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem 286:1436-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bala S, Szabo G. (2012). MicroRNA Signature in Alcoholic Liver Disease. Int J Hepatol 2012:498232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes EA, Kenerson HL, Jiang X, Yeung RS. (2010). Tuberin regulates E-cadherin localization: implications in epithelial-mesenchymal transition. Am J Pathol 177:1765-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. (2005). Liver fibrosis. J Clin Invest 115:209-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi WR, Xu GT, Lv LX, Yang CQ. (2014). The ratio of transforming growth factor-beta1/bone morphogenetic protein-7 in the progression of the epithelial-mesenchymal transition contributes to rat liver fibrosis. Genet Mol Res 13:1005-1014. [DOI] [PubMed] [Google Scholar]
- Bi WR, Yang CQ, Shi Q. (2012). Transforming growth factor-beta1 induced epithelial-mesenchymal transition in hepatic fibrosis. Hepatogastroenterology 59:1960-1963. [DOI] [PubMed] [Google Scholar]
- Bian EB, Huang C, Wang H, Chen XX, Zhang L, Lv XW, Li J. (2014). Repression of Smad7 mediated by DNMT1 determines hepatic stellate cell activation and liver fibrosis in rats. Toxicol Lett 224:175-185. [DOI] [PubMed] [Google Scholar]
- Blanco Calvo M, Bolos Fernandez V, Medina Villaamil V, Aparicio Gallego G, Diaz Prado S, Grande Pulido E. (2009). Biology of BMP signalling and cancer. Clin Transl Oncol 11:126-137. [DOI] [PubMed] [Google Scholar]
- Blank U, Karlsson S. (2011). The role of Smad signaling in hematopoiesis and translational hematology. Leukemia 25:1379-1388. [DOI] [PubMed] [Google Scholar]
- Boon MR, van der Horst G, van der Pluijm G, Tamsma JT, Smit JW, Rensen PC. (2011). Bone morphogenetic protein 7: a broad-spectrum growth factor with multiple target therapeutic potency. Cytokine Growth Factor Rev 22:221-229. [DOI] [PubMed] [Google Scholar]
- Border WA, Noble NA. (1998). Evidence that TGF-beta should be a therapeutic target in diabetic nephropathy. Kidney Int 54:1390-1391. [DOI] [PubMed] [Google Scholar]
- Conidi A, Cazzola S, Beets K, Coddens K, Collart C, Cornelis F, Cox L, Joke D, Dobreva MP, Dries R, Esguerra C, Francis A, Ibrahimi A, Kroes R, Lesage F, Maas E, Moya I, Pereira PN, Stappers E, Stryjewska A, van den Berghe V, Vermeire L, Verstappen G, Seuntjens E, Umans L, Zwijsen A, Huylebroeck D. (2011). Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGFbeta/BMP signaling in vivo. Cytokine Growth Factor Rev 22:287-300. [DOI] [PubMed] [Google Scholar]
- Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. (1999). Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol 19:2495-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425:577-584. [DOI] [PubMed] [Google Scholar]
- Dongiovanni P, Romeo S, Valenti L. (2014). Hepatocellular carcinoma in nonalcoholic fatty liver: role of environmental and genetic factors. World J Gastroenterol 20:12945-12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, Ueberham E, Gebhardt R, Kanzler S, Geier A, Breitkopf K, Weng H, Mertens PR. (2008). Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology 135:642-659. [DOI] [PubMed] [Google Scholar]
- Dooley S, Streckert M, Delvoux B, Gressner AM. (2001). Expression of Smads during in vitro transdifferentiation of hepatic stellate cells to myofibroblasts. Biochem Biophys Res Commun 283:554-562. [DOI] [PubMed] [Google Scholar]
- Drabsch Y, ten Dijke P. (2012). TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev 31:553-568. [DOI] [PubMed] [Google Scholar]
- Dudas J, Kovalszky I, Gallai M, Nagy JO, Schaff Z, Knittel T, Mehde M, Neubauer K, Szalay F, Ramadori G. (2001). Expression of decorin, transforming growth factor-beta 1, tissue inhibitor metalloproteinase 1 and 2, and type IV collagenases in chronic hepatitis. Am J Clin Pathol 115:725-735. [DOI] [PubMed] [Google Scholar]
- Eddington KM, Dolcos F, Cabeza R, KR RK, Strauman TJ. (2007). Neural correlates of promotion and prevention goal activation: an fMRI study using an idiographic approach. J Cogn Neurosci 19:1152-1162. [DOI] [PubMed] [Google Scholar]
- Friedman SL. (2008). Mechanisms of hepatic fibrogenesis. Gastroenterology 134:1655-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL. (2004). Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol 1:98-105. [DOI] [PubMed] [Google Scholar]
- Fukasawa H, Yamamoto T, Togawa A, Ohashi N, Fujigaki Y, Oda T, Uchida C, Kitagawa K, Hattori T, Suzuki S, Kitagawa M, Hishida A. (2004). Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc Natl Acad Sci U S A 101:8687-8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Bataller R. (2011). Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141:1572-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentilini A, Marra F, Gentilini P, Pinzani M. (2000). Phosphatidylinositol-3 kinase and extracellular signal-regulated kinase mediate the chemotactic and mitogenic effects of insulin-like growth factor-I in human hepatic stellate cells. J Hepatol 32:227-234. [DOI] [PubMed] [Google Scholar]
- Govinden R, Bhoola KD. (2003). Genealogy, expression, and cellular function of transforming growth factor-beta. Pharmacol Ther 98:257-265. [DOI] [PubMed] [Google Scholar]
- Gressner AM, Weiskirchen R. (2006). Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med 10:76-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamzavi J, Ehnert S, Godoy P, Ciuclan L, Weng H, Mertens PR, Heuchel R, Dooley S. (2008). Disruption of the Smad7 gene enhances CCI4-dependent liver damage and fibrogenesis in mice. J Cell Mol Med 12:2130-2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Huang C, Sun X, Long X-r, Lv X-w, Li J. (2012). MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell Signal 24:1923-1930. [DOI] [PubMed] [Google Scholar]
- Heinrich EL, Walser TC, Krysan K, Liclican EL, Grant JL, Rodriguez NL, Dubinett SM. (2012). The inflammatory tumor microenvironment, epithelial mesenchymal transition and lung carcinogenesis. Cancer Microenviron 5:5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer J, Escalante-Alcalde D, Lia M, Boettinger E, Edelmann W, Stewart CL, Kucherlapati R. (1999). Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc Natl Acad Sci U S A 96:12595-12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruska KA, Guo G, Wozniak M, Martin D, Miller S, Liapis H, Loveday K, Klahr S, Sampath TK, Morrissey J. (2000). Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am J Physiol Renal Physiol 279:F130-143. [DOI] [PubMed] [Google Scholar]
- Huang CF, Sun CC, Zhao F, Zhang YD, Li DJ. (2015). miR-33a levels in hepatic and serum after chronic HBV-induced fibrosis. J Gastroenterol. 50:480-490. [DOI] [PubMed] [Google Scholar]
- Ka SM, Huang XR, Lan HY, Tsai PY, Yang SM, Shui HA, Chen A. (2007). Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J Am Soc Nephrol 18:1777-1788. [DOI] [PubMed] [Google Scholar]
- Ka SM, Yeh YC, Huang XR, Chao TK, Hung YJ, Yu CP, Lin TJ, Wu CC, Lan HY, Chen A. (2012). Kidney-targeting Smad7 gene transfer inhibits renal TGF-beta/MAD homologue (SMAD) and nuclear factor kappaB (NF-kappaB) signalling pathways, and improves diabetic nephropathy in mice. Diabetologia 55:509-519. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. (2009). The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Saunier EF, Akhurst RJ, Derynck R. (2008). The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat Cell Biol 10:654-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P, Rose-John S, zum Buschenfelde KH, Blessing M. (1999). TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol 276:G1059-1068. [DOI] [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. (2000). Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6: 1366-1375. [DOI] [PubMed] [Google Scholar]
- Lan HY. (2003). Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr Opin Nephrol Hypertens 12:25-29. [DOI] [PubMed] [Google Scholar]
- Lan HY, Chung AC. (2012). TGF-beta/Smad signaling in kidney disease. Semin Nephrol 32:236-243. [DOI] [PubMed] [Google Scholar]
- Lan HY, Chung AC. (2011). Transforming growth factor-beta and Smads. Contrib Nephrol 170:75-82. [DOI] [PubMed] [Google Scholar]
- Latella G, Vetuschi A, Sferra R, Catitti V, D’Angelo A, Zanninelli G, Flanders KC, Gaudio E. (2009). Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int 29:997-1009. [DOI] [PubMed] [Google Scholar]
- Lee JG, Kay EP. (2012). NF-kappaB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Invest Ophthalmol Vis Sci 53:1530-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee UE, Friedman SL. (2011). Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol 25:195-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Liang M, Feng XH. (2000). Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem 275:36818-36822. [DOI] [PubMed] [Google Scholar]
- Maribo H, Olsen EV, Barton-Gade P, J, Møller A. (1998). Comparison of dehiding versus scalding and singeing: effect on temperature, pH and meat quality in pigs. Meat Sci 50:175-189. [DOI] [PubMed] [Google Scholar]
- Marquez RT, Bandyopadhyay S, Wendlandt EB, Keck K, Hoffer BA, Icardi MS, Christensen RN, Schmidt WN, McCaffrey AP. (2010). Correlation between microRNA expression levels and clinical parameters associated with chronic hepatitis C viral infection in humans. Lab Invest 90:1727-1736. [DOI] [PubMed] [Google Scholar]
- Massague J, Wotton D. (2000). Transcriptional control by the TGF-beta/Smad signaling system. EMBO J 19:1745-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masszi A, Kapus A. (2011). Smaddening complexity: the role of Smad3 in epithelial-myofibroblast transition. Cells Tissues Organs 193:41-52. [DOI] [PubMed] [Google Scholar]
- Medeiros AI, Sa-Nunes A, Soares EG, Peres CM, Silva CL, Faccioli LH. (2004). Blockade of endogenous leukotrienes exacerbates pulmonary histoplasmosis. Infect Immun 72:1637-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XM, Chung AC, Lan HY. (2013). Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond) 124:243-254. [DOI] [PubMed] [Google Scholar]
- Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P, Ju W, Bottinger EP, Lan HY. (2010). Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol 21:1477-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XM, Huang XR, Xiao J, Chung AC, Qin W, Chen HY, Lan HY. (2012). Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int 81:266-279. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH. (2001). Smad regulation in TGF-beta signal transduction. J Cell Sci 114:4359-4369. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Toyoda H, Tanaka M, Kuroda M, Harada Y, Matsuda F, Tajima A, Kosaka N, Ochiya T, Shimotohno K. (2011). The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS One 6:e16081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler EP, Patterson D, Violette S, Weinreb P, Lewis M, Magid MS, Greco MA. (2009). Integrin alphavbeta6 and mediators of extracellular matrix deposition are up-regulated in experimental biliary atresia. J Surg Res 154:21-29. [DOI] [PubMed] [Google Scholar]
- Ng R, Song G, Roll GR, Frandsen NM, Willenbring H. (2012). A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J Clin Invest 122:1097-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng YY, Hou CC, Wang W, Huang XR, Lan HY. (2005). Blockade of NFkappaB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int Suppl: S83-91. [DOI] [PubMed] [Google Scholar]
- Pages G, Lenormand P, L’Allemain G, Chambard JC, Meloche S, Pouyssegur J. (1993). Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci U S A 90:8319-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C, Chen H, Wang L, Yang S, Fu H, Zheng Y, Miao M, Jiao B. (2012). Down-regulation of MiR-127 facilitates hepatocyte proliferation during rat liver regeneration. PLoS One 7:e39151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, Dockrell ME. (2006). The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J 393:601-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang H, Lin Y, Zhang X, Zeng X, Shi J, Chen YX, Yang MF, Han ZG, Xie WF. (2006). Differential expression genes analyzed by cDNA array in the regulation of rat hepatic fibrogenesis. Liver Int 26:1126-1137. [DOI] [PubMed] [Google Scholar]
- Quan TE, Cowper SE, Bucala R. (2006). The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep 8:145-150. [DOI] [PubMed] [Google Scholar]
- Ross S, Hill CS. (2008). How the Smads regulate transcription. Int J Biochem Cell Biol 40:383-408. [DOI] [PubMed] [Google Scholar]
- Rygiel KA, Robertson H, Marshall HL, Pekalski M, Zhao L, Booth TA, Jones DE, Burt AD, Kirby JA. (2008). Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest 88:112-123. [DOI] [PubMed] [Google Scholar]
- Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. (2003). Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 112:1486-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabl B, Bradham CA, Bennett BL, Manning AM, Stefanovic B, Brenner DA. (2001). TAK1/JNK and p38 have opposite effects on rat hepatic stellate cells. Hepatology 34:953-963. [DOI] [PubMed] [Google Scholar]
- Schreiber M, Kolbus A, Piu F, Szabowski A, Möhle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF. (1999). Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev 13:607-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta U, Ukil S, Dimitrova N, Agrawal S. (2009). Expression-based network biology identifies alteration in key regulatory pathways of type 2 diabetes and associated risk/complications. PLoS One 4:e8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shek FW, Benyon RC. (2004). How can transforming growth factor beta be targeted usefully to combat liver fibrosis? Eur J Gastroenterol Hepatol 16:123-126. [DOI] [PubMed] [Google Scholar]
- Singh P, Wig JD, Srinivasan R. (2011). The Smad family and its role in pancreatic cancer. Indian J Cancer 48:351-360. [DOI] [PubMed] [Google Scholar]
- Spizzo R, Rushworth D, Guerrero M, Calin GA. (2009). RNA inhibition, microRNAs, and new therapeutic agents for cancer treatment. Clin Lymphoma Myeloma 9 Suppl 3:S313-318. [DOI] [PubMed] [Google Scholar]
- Stopa M, Anhuf D, Terstegen L, Gatsios P, Gressner AM, Dooley S. (2000). Participation of Smad2, Smad3, and Smad4 in transforming growth factor beta (TGF-beta)-induced activation of Smad7. THE TGF-beta response element of the promoter requires functional Smad binding element and E-box sequences for transcriptional regulation. J Biol Chem 275:29308-29317. [DOI] [PubMed] [Google Scholar]
- Syn WK, Jung Y, Omenetti A, Abdelmalek M, Guy CD, Yang L, Wang J, Witek RP, Fearing CM, Pereira TA, Teaberry V, Choi SS, Conde-Vancells J, Karaca GF, Diehl AM. (2009). Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 137:1478-1488 e1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sysa P, Potter JJ, Liu X, Mezey E. (2009). Transforming growth factor-beta1 up-regulation of human alpha(1)(I) collagen is mediated by Sp1 and Smad2 transacting factors. DNA Cell Biol 28:425-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Bala S. (2013). MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol 10:542-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan R, He W, Lin X, Kiss LP, Liu Y. (2008). Smad ubiquitination regulatory factor-2 in the fibrotic kidney: regulation, target specificity, and functional implication. Am J Physiol Renal Physiol 294:F1076-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S, Sampath K. (2010). Alternative splicing of SMADs in differentiation and tissue homeostasis. Dev Growth Differ 52:335-342. [DOI] [PubMed] [Google Scholar]
- Thompson EW, Haviv I. (2011). The social aspects of EMT-MET plasticity. Nat Med 17:1048-1049. [DOI] [PubMed] [Google Scholar]
- Tian Z, Greene AS, Pietrusz JL, Matus IR, Liang M. (2008). MicroRNA-target pairs in the rat kidney identified by microRNA microarray, proteomic, and bioinformatic analysis. Genome Res 18:404-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowska M, LeRoy EC, Eckes B, Krieg T. (1998). Pathogenesis of fibrosis: type 1 collagen and the skin. J Mol Med (Berl) 76:266-274. [DOI] [PubMed] [Google Scholar]
- Tsuchida K, Zhu Y, Siva S, Dunn SR, Sharma K. (2003). Role of Smad4 on TGF-beta-induced extracellular matrix stimulation in mesangial cells. Kidney Int 63:2000-2009. [DOI] [PubMed] [Google Scholar]
- Uitto J, Kouba D. (2000). Cytokine modulation of extracellular matrix gene expression: relevance to fibrotic skin diseases. J Dermatol Sci 24 Suppl 1:S60-69. [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Mauviel A. (2004). TGF-beta and TNF-alpha: antagonistic cytokines controlling type I collagen gene expression. Cell Signal 16:873-880. [DOI] [PubMed] [Google Scholar]
- Wang B, Koh P, Winbanks C, Coughlan MT, McClelland A, Watson A, Jandeleit-Dahm K, Burns WC, Thomas MC, Cooper ME, Kantharidis P. (2011). miR-200a Prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes 60:280-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Leclercq I, Brymora JM, Xu N, Ramezani-Moghadam M, London RM, Brigstock D, George J. (2009). Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 137:713-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Huang XR, Li AG, Liu F, Li JH, Truong LD, Wang XJ, Lan HY. (2005a). Signaling mechanism of TGF-beta1 in prevention of renal inflammation: role of Smad7. J Am Soc Nephrol 16:1371-1383. [DOI] [PubMed] [Google Scholar]
- Wang W, Koka V, Lan HY. (2005b) .Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton) 10:48-56. [DOI] [PubMed] [Google Scholar]
- West J. (2010). Cross talk between Smad, MAPK, and actin in the etiology of pulmonary arterial hypertension. Adv Exp Med Biol 661:265-278. [DOI] [PubMed] [Google Scholar]
- Willis BC, duBois RM, Borok Z. (2006). Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc 3:377-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Zhang Z, van Dam H, Zhang L, Zhou F. (2014). Regulation of TGF-beta Superfamily Signaling by SMAD Mono-Ubiquitination. Cells 3:981-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YC, Piek E, Zavadil J, Liang D, Xie D, Heyer J, Pavlidis P, Kucherlapati R, Roberts AB, Bottinger EP. (2003). Hierarchical model of gene regulation by transforming growth factor beta. Proc Natl Acad Sci U S A 100:10269-10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao QY, Xu BL, Wang JY, Liu HC, Zhang SC, Tu CT. (2012). Inhibition by curcumin of multiple sites of the transforming growth factor-beta1 signalling pathway ameliorates the progression of liver fibrosis induced by carbon tetrachloride in rats. BMC Complement Altern Med 12:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. (2008). Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19:2282-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. (2007a). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13:952-961. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Kalluri R. (2008). Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci 13:6991-6998. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. (2007b). Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 282:23337-23347. [DOI] [PubMed] [Google Scholar]
- Zhang L, Liu C, Meng XM, Huang C, Xu F, Li J. (2015). Smad2 protects against TGF-beta1/Smad3-mediated collagen synthesis in human hepatic stellate cells during hepatic fibrosis. Mol Cell Biochem 400:17-28. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wei J, Wang H, Xue X, An Y, Tang D, Yuan Z, Wang F, Wu J, Zhang J, Miao Y. (2012). Epithelial mesenchymal transition correlates with CD24+CD44+ and CD133+ cells in pancreatic cancer. Oncol Rep 27:1599-1605. [DOI] [PubMed] [Google Scholar]
- Zhou J, Ju W, Wang D, Wu L, Zhu X, Guo Z, He X. (2012). Down-regulation of microRNA-26a promotes mouse hepatocyte proliferation during liver regeneration. PLoS One 7:e33577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Huang XR, Yu J, Yu X, Lan HY. (2015). Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal Inflammation. Mol Ther 23:1034-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, He X, Duan Y, Chen J, Wang J, Sun X, Qian H, Feng J, Sun W, Xu F, Zhang L. (2014). Expression of microRNA-454 in TGF-beta1-stimulated hepatic stellate cells and in mouse livers infected with Schistosoma japonicum. Parasit Vectors 7:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HJ, Sizeland AM. (1999). Smad7 Differentially Regulates Transforming Growth Factor b-mediated Signaling Pathways. J Biol Chem 274:32258-32264. [DOI] [PubMed] [Google Scholar]
- Zhu L, Wang L, Wang X, Luo X, Yang L, Zhang R, Yin H, Xie D, Pan Y, Chen Y. (2011). Hepatic deletion of Smad7 in mouse leads to spontaneous liver dysfunction and aggravates alcoholic liver injury. PLoS One 6:e17415. [DOI] [PMC free article] [PubMed] [Google Scholar]