

Abstract
An approach to the validation of linker strategies for polyketide natural products with few or no obvious handles for linker attachment, and its application to dictyostatin are described. Analogs in which the C(6) and C(12) methyl groups were replaced by 4-azidobutyl groups were prepared and shown to retain the low nM potency of dictyostatin. Further, conjugation of the C(6)-analog with a cyclooctyne resulted in only minor attenuations in potency. Together, these results shed light on the binding of dictyostatin to β-tubulin, establish a validated linker strategy for dictyostatin, and set the stage for the synthesis and study of dictyostatin conjugates.
Graphical abstract
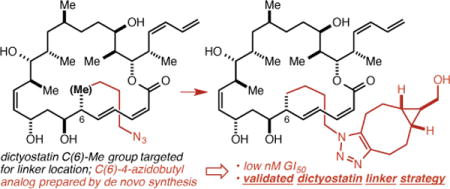
Strategies for the selective delivery of small molecule cancer chemotherapeutic agents to tumor cells (e.g. antibody-drug conjugates1) hold promise as a way to, in effect, increase their therapeutic index. A requirement for the drug in many of these approaches is a validated linker strategy,2 the most critical component of which is the identification of a site on the drug that may be modified without any deleterious impact on its activity. More broadly, the identification of such modifiable sites on bioactive natural products can facilitate chemical biology and mechanism of action studies and enable exploration of more novel linked constructs. Dictyostatin,3–5 for which we developed a synthesis that proceeds in 14 steps in the longest linear sequence,6 is a worthy candidate for linker strategy validation in that it is among the most potent of the microtubule-stabilizing agents (MSAs), retains significant potency against taxane-resistant cell lines, and has been shown to be a rare example of a brain-penetrant MSA.7
The principal challenge in the identification of modifiable sites on polyketide/polypropionate structures such as dictyostatin is that the hydroxyl groups may be the only readily modifiable groups (Figure 1a). Such an alcohol modification strategy would require the identification of an “innocent” hydroxyl group that is not critical for activity as well as a synthetic strategy to allow for selective modification of only that hydroxyl group. In this regard, we were aware of Paterson and Wright’s demonstration that the C(9)-OMe analog 1 mostly retains the low nM potency of the natural product,8 and mindful that the penultimate intermediate in our synthesis (2) is one in which the C(9)-OH group is, uniquely, unprotected (Figure 1a). Despite this, we rejected a C(9)-OH modification approach because we were concerned 1) that complex ether formation with our late stage intermediate might be difficult and 2) that acylation, the synthetically straightforward alternative, might be expected to subtly but significantly perturb the local electronic and steric structure and global conformation of the natural product9 as well as raise concerns about acyl group migration or cleavage in vivo (Figure 1b). Indeed, such concerns are not strictly hypothetical, as Paterson has demonstrated that analogs of a dictyostatin/discodermolide hybrid in which the C(7)- and C(9)-OH groups were acylated with taxoid sidechains were susceptible to both acyl migration and methanolysis, and were significantly less potent than the parent compound.10 Having rejected an alcohol acylation strategy, we became intrigued by the notion that the ideal approach would entail modifying one of the ubiquitous methyl groups to a linker-bearing linear alkyl group (Figure 1b). In most contexts, linear alkyl groups are electronically and sterically equivalent to methyl groups, and this approach would be expected to result in as minimal a perturbation of the structure and conformation of the natural product as possible, while also obviating any concerns about O-acyl migration or cleavage.
Figure 1.
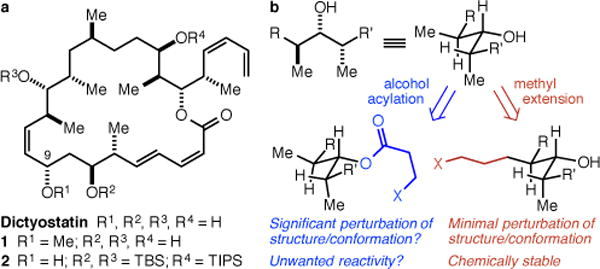
(a) Dictyostatin, Paterson’s C(9)-OMe analog 1, and the penultimate intermediate in our synthesis 2. (b) In crowded polypropionate arrays, alcohol acylation might result in significant structural perturbations, whereas methyl extension should not.
For guidance as to the selection of appropriately disposed methyl groups we turned to the extensive structure-activity relationship (SAR) data for dictyostatin reported by Curran11–14 and Paterson9,15,16 particularly as they related to the starkly contrasting models for the binding of dictyostatin in the taxane binding site advanced by Curran and Snyder17 and by Díaz and Jiménez-Barbero.18 More specifically, we took note of Curran’s demonstration that C(6)-epi dictyostatin is as potent as the natural product and Paterson’s demonstration that C(6)-normethyl dictyostatin is only slightly less potent (≤ 1 order of magnitude) than the natural product. According to Curran and Snyder, only their model (Figure 2a) is fully consistent with this SAR data, as it places the C(6)-methyl group in a solvent exposed position without contacts to the receptor. Conversely, the Díaz/Jiménez-Barbero model places the C(6)-methyl group deeper into the binding pocket and in van der Waals contact with Pro360, which Curran and Snyder contend is inconsistent with the SAR data because deletion or epimerization of the C(6)-methyl group would remove this contact with Pro360 and be expected to lead to a significant decrease in potency. The models lead to similar conclusions regarding the C(12)-methyl group, though here more caution is warranted in that the Curran/Snyder model locates it in proximity to the M-loop (the yellow loop at the bottom of Figure 2a), which undergoes significant conformational changes upon the binding of an MSA in the taxane binding pocket.19 Based on this analysis, we decided to target the C(6)- and C(12)- (4-azidobutyl) analogs 3 and 4 (Figure 2b), which we hoped would lead to a validated linker strategy for dictyostatin, and which would in the process provide support for the Curran and Snyder binding model.
Figure 2.
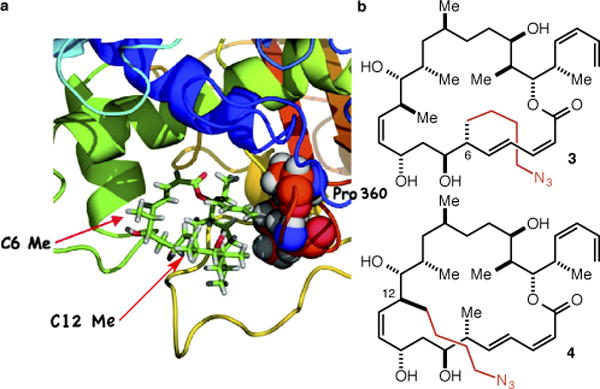
(a) The Curran/Snyder model for the binding of dictyostatin in the taxane binding site (reprinted with permission from J. Am. Chem. Soc. 2011, 133, 2427–2436, copyright 2011 American Chemical Society). (b) The C(6) and C(12) methyl extended analogs 3 and 4.
In our synthesis of dictyostatin,6 the C(6)-methyl group is installed in a Sc(OTf)3-catalyzed crotylation20 of aldehyde 5 to give ketone 6 (Scheme 1). To incorporate a 4-azidobutyl group instead, we employed a cross-metathesis reaction between allylsilane 7 and 6-chlorohex-1-ene using the 2nd generation Hoveyda-Grubbs catalyst21 (HG-II) to produce 8 (~3:1 E:Z), which was employed in situ in a Sc(OTf)3-catalyzed allylation of aldehyde 5.22,23 After ketal hydrolysis, 9 was isolated as a 3:1 mixture of diastereomers in 58% overall yield (90% ee for the major diastereomer). Subjection of 9 to a one-pot protection/bromination reaction gave 10, which was subjected to the Arbuzov/trans-esterification method we developed6 to give Still-Gennari-type β-ketophosphonate 11. Displacement of the chloride with NaN3 delivered 12, and Heck reaction with iodide 13 (TMSE = 2-trimethylsilyethyl) produced 14 in 58% overall yield. The diastereomers were separated at this stage, and we isolated 14 in 30% yield. Incorporation of 14 into the completed dictyostatin framework proved straightforward using our previously described synthesis.6 Thus, Still-Gennari-type coupling of 14 with previously described aldehyde 15 resulted in the isolation of pure Z-isomer 16 in 75% yield. Four additional steps (ester deprotection, macrolactonization,24 CBS reduction,25 and global deprotection) delivered the targeted C(6)-(4-azidobutyl) analog 3 (see the Supporting Information for details). Because we had previously prepared and stored a significant supply of aldehyde 15, this synthesis of 3 required just 10 total steps from 7, 5, and 6-chlorohex-1-ene.
Scheme 1.
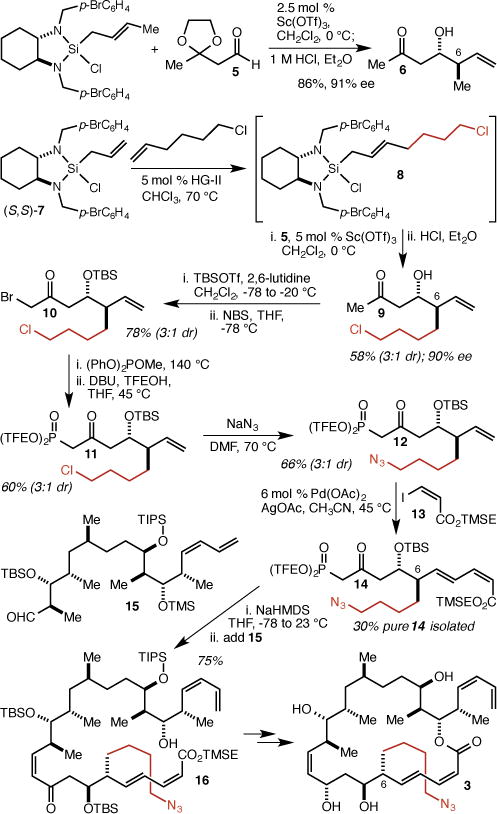
Synthesis of C(6) analog 3
To incorporate the 4-azidobutyl group at C(12), we adapted our recently reported two pot/three step protocol for the rapid and scalable synthesis of stereotriads.26 Thus, silylformylation of 6-chlorohex-1-yne gave aldehyde 17 which was directly crotylated with (S,S)-cis EZ-CrotylMix20 to give 18 in 90% yield and 95% ee (Scheme 2). Tamao oxidation/anti-diastereoselective tautomerization and protection of the aldehyde gave 19 in 66% yield over two steps. The remainder of the fragment synthesis followed our dictyostatin synthesis6 with the added azide displacement step, and produced, by way of intermediates 20, 21, and 22, iodide 23. Iodide 23 was then used to produce analog 4 (see the Supporting Information for details).
Scheme 2.
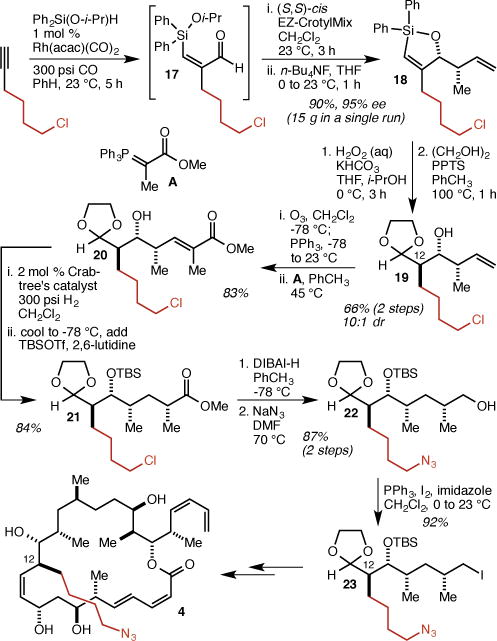
Synthesis of C(12) analog 4
Both because of the greater ease of its synthesis and because it was found to be more potent than the C(12) analog 4 (see below), the C(6) analog 3 was employed in model conjugation reactions. Treatment of 3 with 24, van Delft’s variant27 of Bertozzi’s cyclooctyne-based approach to metal-free click reactions,28,29 afforded triazole 25 in 83% yield as the expected 1:1 mixture of diastereomers (Scheme 3). A traceless Staudinger ligation30,31 was also carried out to give acetamide 26 in 35% yield.
Scheme 3.
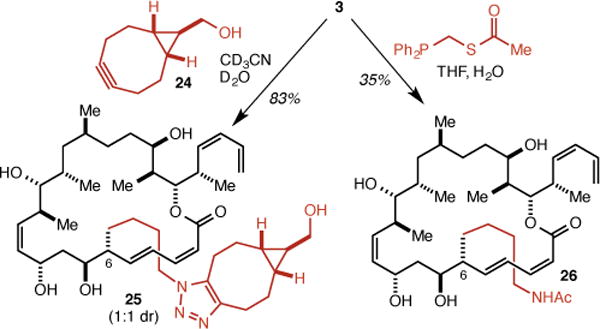
Model conjugations with 3
The C(6)- and C(12)-(4-azidobutyl) analogs 3 and 4 as well as triazole 25 and amide 26 were assayed for cell growth inhibition against four cell lines (PC3 (prostate), 1A9 (ovarian), DLD1 (colon), and A549 (lung)) alongside reference samples of synthetic dictyostatin and paclitaxel (Figure 3). Consistent with our predictions, the GI50 values for the C(6) analog 3 were found to be only slightly attenuated relative to dictyostatin in all four cell lines, while the C(12) analog 4 retains low nM potency as well, albeit with a somewhat more substantial drop in potency. Importantly, the activity of triazole 25 against the PC3 and 1A9 cell lines is only slightly diminished from that of azide 3, while more substantial (though not catastrophic) reductions in potency are observed in the DLD1 and A549 cell lines. Surprisingly, the potency of amide 26 is reduced by 1–2 orders of magnitude relative to azide 3 against all four cell lines. Thus, although the specific structure of the linking group (e.g. triazole, amide) can have an impact on potency in a somewhat cell line-dependent manner, the results for triazole 25 against the PC3 and 1A9 cell lines in particular constitute a convincing proof-of-concept that dictyostatin conjugates may be prepared from C(6)-(4-azidobutyl) analog 3 with only minor reductions in potency.
Figure 3.
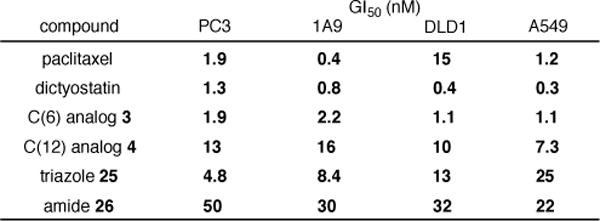
Cell growth inhibition GI50 values for 3, 4, 25, and 26 (values are the average over 2 or 3 experiments, see the Supporting Information for details).
Together, these results 1) establish a validated linker strategy for dictyostatin and 2) constitute compelling evidence in support of the Curran/Snyder binding model for dictyostatin. More broadly, our work demonstrates that even in cases where a synthetically convenient alcohol acylation strategy presents itself, the “methyl extension” strategy outlined here merits consideration, as it may be less likely to result in significant attenuations in potency and obviates any and all concerns about unwanted reactivity. Of course, it remains the case that this approach is more synthetic chemistry intensive, but in that regard we note that we have here demonstrated 1) that major improvements in step-economy and scalability such as in our dictyostatin synthesis can render this approach feasible in a far less time- and resource-intensive way, and 2) two ways in which our efficient and scalable polyketide synthesis methodologies may be adapted for the installation of 4-azidobutyl groups in place of methyl groups. We believe this strategy will prove applicable to other important polyketide/polypropionate natural products with few or no obvious handles for linker attachment, and will report our findings in this regard in due course.
Supplementary Material
Acknowledgments
This work was supported by NIGMS/NIH (GM058133), and was supported in part by the Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH.
Footnotes
Supporting Information. Experimental procedures, compound characterization data, and experimental details for the cell growth inhibition assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References
- 1.(a) Gerber H-P, Koehn FE, Abraham RT. Nat Prod Rep. 2013;30:625–639. doi: 10.1039/c3np20113a. [DOI] [PubMed] [Google Scholar]; (b) Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM. Drug Discovery Today. 2014;19:869–881. doi: 10.1016/j.drudis.2013.11.004. [DOI] [PubMed] [Google Scholar]; (c) Chari RVJ, Miller ML, Widdison WC. Angew Chem Int Ed. 2014;53:3796–3827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 2.Ducry L, Stump B. Bioconjugate Chem. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 3.Pettit GR, Cichacz ZA, Gao F, Boyd MR, Schmidt JM. J Chem Soc Chem Commun. 1994:1111–1112. [Google Scholar]
- 4.Isbrucker RA, Cummins J, Pomponi SA, Longley RE, Wright AE. Biochem Pharmacol. 2003;66:75–82. doi: 10.1016/s0006-2952(03)00192-8. [DOI] [PubMed] [Google Scholar]
- 5.Paterson I, Britton R, Delgado O, Wright AE. Chem Commun. 2004:632–633. doi: 10.1039/b316390c. [DOI] [PubMed] [Google Scholar]
- 6.Ho S, Bucher C, Leighton JL. Angew Chem Int Ed. 2013;52:6757–6761. doi: 10.1002/anie.201302565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunden KR, Gardner NM, James MJ, Yao Y, Trojanowski JQ, Lee VM-Y, Paterson I, Ballatore C, Smith AB., III ACS Med Chem Lett. 2013;4:886–889. doi: 10.1021/ml400233e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paterson I, Gardner NM, Poullennec KG, Wright AE. Bioorg Med Chem Lett. 2007;17:2443–2447. doi: 10.1016/j.bmcl.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 9.Hung DT, Nerenberg JB, Schreiber SL. J Am Chem Soc. 1996;118:11054–11080. [Google Scholar]
- 10.Paterson I, Naylor GJ, Gardner NM, Guzmán E, Wright AE. Chem Asian J. 2011;6:459–473. doi: 10.1002/asia.201000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukui Y, Brückner AM, Shin Y, Balachandran R, Day BW, Curran DP. Org Lett. 2006;8:301–304. doi: 10.1021/ol0526827. [DOI] [PubMed] [Google Scholar]
- 12.Shin Y, Fournier J-H, Brückner AM, Madiraju C, Balachandran R, Raccor BS, Edler MC, Hamel E, Sikorski RP, Vogt A, Day BW, Curran DP. Tetrahedron. 2007;63:8537–8562. doi: 10.1016/j.tet.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung W-H, Harrison C, Shin Y, Fournier J-H, Balachandran R, Raccor BS, Sikorski RP, Vogt A, Curran DP, Day BW. J Med Chem. 2007;50:2951–2966. doi: 10.1021/jm061385k. [DOI] [PubMed] [Google Scholar]
- 14.Zhu W, Jiménez M, Jung W-H, Camarco DP, Balachandran R, Vogt A, Day BW, Curran DP. J Am Chem Soc. 2010;132:9175–9187. doi: 10.1021/ja103537u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paterson I, Gardner NM, Naylor GJ. Pure Appl Chem. 2009;81:169–180. [Google Scholar]
- 16.Paterson I, Gardner NM, Guzmán E, Wright AE. Bioorg Med Chem. 2009;17:2282–2289. doi: 10.1016/j.bmc.2008.10.084. [DOI] [PubMed] [Google Scholar]
- 17.Jogalekar A, Damodaran K, Kriel FH, Jung W-H, Alcaraz AA, Zhong S, Curran DP, Snyder JP. J Am Chem Soc. 2011;133:2427–2436. doi: 10.1021/ja1023817. [DOI] [PubMed] [Google Scholar]
- 18.Canales A, Matesanz R, Gardner NM, Andreu JM, Paterson I, Díaz JF, Jiménez-Barbero J. Chem Eur J. 2008;14:7557–7569. doi: 10.1002/chem.200800039. [DOI] [PubMed] [Google Scholar]
- 19.Prota AE, Bargsten K, Zurwerra D, Field JJ, Fernando Díaz J, Altmann K-H, Steinmetz MO. Science. 2013;339:587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 20.Kim H, Ho S, Leighton JL. J Am Chem Soc. 2011;133:6517–6520. doi: 10.1021/ja200712f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
- 22.Goldberg SD, Grubbs RH. Angew Chem Int Ed. 2002;41:807–810. doi: 10.1002/1521-3773(20020301)41:5<807::aid-anie807>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 23.Huber JD, Perl NR, Leighton JL. Angew Chem Int Ed. 2008;47:3037–3039. doi: 10.1002/anie.200705621. [DOI] [PubMed] [Google Scholar]
- 24.Shiina I, Kubota M, Oshiumi H, Hashizume M. J Org Chem. 2004;69:1822–1830. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]
- 25.Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 26.Foley CN, Leighton JL. Org Lett. 2014;16:1180–1183. doi: 10.1021/ol500051e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dommerholt J, Schmidt S, Temming R, Hendriks LJA, Rutjes FPJT, van Hest JCM, Lefeber DJ, Friedl P, van Delft FL. Angew Chem Int Ed. 2010;49:9422–9425. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agard NJ, Prescher JA, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 29.Lutz J-F. Angew Chem Int Ed. 2008;47:2182–2184. doi: 10.1002/anie.200705365. [DOI] [PubMed] [Google Scholar]
- 30.Nilsson BL, Kiessling LL, Raines RT. Org Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 31.Saxon E, Armstrong JI, Bertozzi CR. Org Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.