

Abstract
Differentiated macrophages can self-renew in tissues and expand long-term in culture, but the gene regulatory mechanisms that accomplish self-renewal in the differentiated state have remained unknown. Here we show that in mice, the transcription factors MafB and c-Maf repress a macrophage-specific enhancer repertoire associated with a gene network controlling self-renewal. Single cell analysis revealed that, in vivo, proliferating resident macrophages can access this network by transient down-regulation of Maf transcription factors. The network also controls embryonic stem cell self-renewal but is associated with distinct embryonic stem cell-specific enhancers. This indicates that distinct lineage-specific enhancer platforms regulate a shared network of genes that control self-renewal potential in both stem and mature cells.
In many tissues of the body, differentiated cells are frequently replaced as part of homeostatic maintenance or in response to injury. Whereas in most cases this depends on tissue-specific stem cells, tissue macrophages can be maintained by local proliferation independently of hematopoietic stem cells (1–4), possibly by self-renewal mechanisms activated in mature macrophages (5). Unlike the few examples of differentiated normal cells that can transiently re-enter the cell cycle, such as hepatocytes, macrophages can also be expanded and maintained in long-term culture without transformation or loss of differentiation. This has been observed in macrophages with deletions of two core macrophage transcription factors (6), MafB and c-Maf (Maf-DKO macrophages) (7), or in cultures derived from fetal progenitors (8).
Understanding how regulatory programs are rewired to allow differentiated cells to self-renew is of considerable interest and self-renewing macrophages provide a unique opportunity to study this process. Genome-wide distribution of enhancer-associated histone modifications provides a reliable signature of cell identity (9–14) that has revealed macrophage specific enhancer repertoires (11,12) and tissue or activation state-dependent modifications (15–17). To identify the regulatory mechanism that enable macrophage self-renewal capacity to be integrated into the overall program of epigenetic macrophage identity, we therefore compared the enhancer repertoires of quiescent and self-renewing macrophages.
Absence of lineage independent self-renewal enhancers
To determine whether self-renewal in macrophages involves acquisition of dedicated, self-renewal specific enhancers, we therefore first compared the molecular enhancer signature defined by mono-methylated histone H3 at Lysine 4 (H3K4m1)(9, 13, 14) of self-renewing Maf-DKO and quiescent wild type (WT) bone marrow-derived macrophages (BMM) to several other cell types with limited proliferation or extended self-renewal capacity (Fig. S1). Surprisingly, our analysis revealed no common, lineage-independent repertoire of shared enhancer positions for the control of proliferation or self-renewal genes (Fig. S1A,B). We also compared genome-wide binding of the transcription factor PU.1, a key regulator of both macrophage and B-cell lineage identity that defines distinct enhancer positions in the genome of these two cell types (10–12). This revealed fewer differences in the position of H3K4m1+/PU.1+ enhancer peaks between Maf-DKO and WT BMM than between WT BMM and peritoneal macrophages (PM), and an equal distance of all macrophage populations to pro-B cells (Fig. S1C). This indicates that Maf-DKO macrophages can activate self-renewal but retain a macrophage-specific enhancer signature similar to WT BMM. Macrophage self-renewal thus does not appear to involve the acquisition of dedicated lineage-independent self-renewal enhancers or the loss of mature macrophage epigenetic identity.
Activation of a lineage specific subset of enhancers in self-renewing macrophages
Since self-renewing Maf-DKO macrophages showed no appreciable difference compared to quiescent macrophages with respect to genome-wide enhancer positions, we performed ChIP-seq analyses for activated enhancer marks, histone acetyl tranferase p300 and the histone modification mediated by this enzyme, acetylation of histone 3 lysine 27 (H3K27ac) (13, 17–19), to determine whether the activation status of these enhancers differed. We found that a large number of enhancers was activated specifically in Maf-DKO macrophages only (Maf-DKO-only) (Fig. 1A, red highlight), while only a small number of enhancers were activated specifically in WT BMM. Specifically, we calculated 7323 enhancer regions to be enriched for p300 binding and 7489 enriched for H3K27ac in Maf-DKO compared to WT BMM, whereas only 305 regions were enriched for p300 and 1923 for H3K27ac in WT BMM compared to Maf-DKO (Fig. 1B and S2). Further characterization of Maf-DKO-only regions revealed a typical H3K4m1+/H3K4m3low enhancer signature at these loci (Fig. 1A,C) and motif search analysis of the p300 and H3K27ac enriched Maf-DKO-only enhancer regions revealed the highest score for PU.1 binding motifs (logP-value= 6444) (Fig. S3A). Aggregate analysis of all PU.1-bound sites in Maf-DKO and WT macrophages confirmed that the large majority of DKO-only enhancers were bound by PU.1 (Fig. 1B,C,S3B), and that over 60% of these positions were also bound by PU.1 in WT macrophages (Fig. 1B,C,S3B). This PU.1 binding pattern is reminiscent of “poised” and “latent” enhancers previously described in non-stimulated and stimulated macrophages, respectively (17). Taken together, macrophages appear to possess a specific subset of largely poised macrophage-specific enhancers that is selectively activated in self-renewing Maf-DKO macrophages.
Fig. 1. A subset of lineage specific enhancers are enriched for activation marks in self-renewing Maf-DKO macrophages.
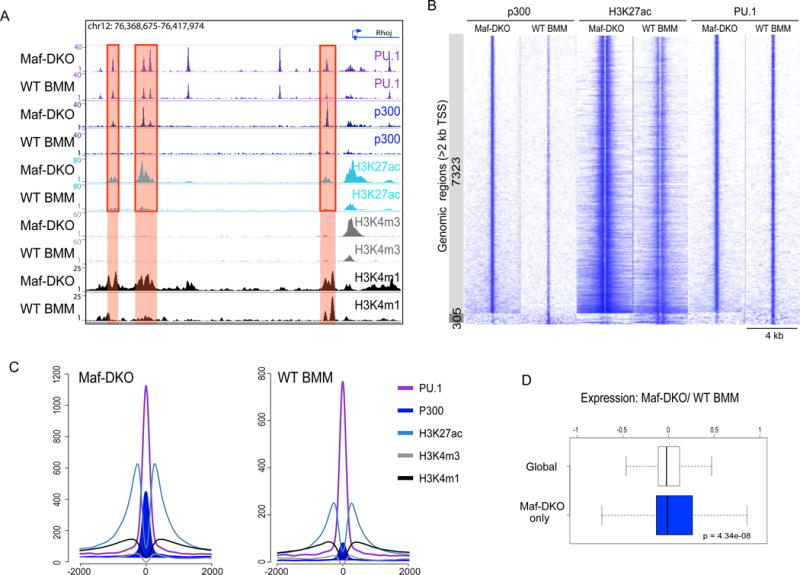
A) Representative predicted enhancer regions (red shading) with greater enrichment for enhancer activation marks p300 and H3K27ac in Maf-DKO versus WT BMM (red boxes). B) Direct alignment of p300, H3K27ac and PU.1 ChIP-Seq signals for enhancer regions differentially enriched for p300 marks in Maf-DKO (7323, light grey) and WT BMM (305, dark grey), centered and ranked on p300 signal. C) Aggregation plots showing average ChIP-seq signals for PU.1, H3K27ac, p300, H3K4m1 and H3K4m3 marks in Maf-DKO and WT BMM for p300 regions specifically enriched in Maf-DKO macrophages (depicted light grey in (B)). For each protein target, Chip-seq analysis was performed on at least two biological replicates, and results were reproducible in all cases. D) Microarray gene expression ratios of Maf-DKO versus WT BMM 2h after M-CSF stimulation, for total genes (white) or genes associated with Maf-DKO-only enhancers (blue). The box extends from the first to the third quartile with the whiskers denoting 1.5 times the interquartile range. Data were derived from three biological replicates.
Self-renewing macrophages activate a gene set also required for ES cell self-renewal
Under steady-state conditions, self-renewing Maf-DKO and WT BM macrophages have highly similar global gene expression profiles (7). However, it is possible that the global analysis hides relevant specific mechanisms and so we specifically selected the genes associated with Maf-DKO-only activated enhancers for further study, using the GREAT tool (20). We observed a significant and specific increase in the expression of these genes after stimulation with macrophage-colony stimulating factor (M-CSF), a cytokine required to maintain self-renewal(7) (Fig. 1D). This indicates that Maf-DKO macrophage activated enhancers include elements conferring functional responsiveness to M-CSF stimulation, potentially including those relevant to self-renewal. Gene Ontology (GO) analysis on Maf-DKO-only associated genes revealed immune and myeloid cell functions (Fig. S3C) but no groups associated with self-renewal activity. Because GO lacks self-renewal specific categories, we performed Gene Set Enrichment Analysis (GSEA) on the Maf-DKO-only associated genes, using both adult and embryonic stem cell-specific data modules enriched for self-renewal genes (21). This analysis revealed a significant enrichment of the embryonic but not the adult stem cell set in Maf-DKO over WT BM macrophage expression (Fig. 2A). Based on these results, we further focused on functionally validated genes identified in screens for self-renewal activity in ES cells (22–29). Although the identified list of 53 genes from these screens (Fig. 2B) is not likely to be exhaustive, it constitutes the largest functionally validated gene set available that should include core elements of self–renewal activity. The genes on this list fall into two categories, those affecting self-renewal only (25 genes) and those affecting both self-renewal and pluripotency as inferred by their influence on expression of Oct4 or nanog (28 genes) (Fig. 2B). Importantly, of the 25 genes in the self-renewal category, 15 (60%) were also associated with Maf DKO-only enhancers in macrophages, but of the 28 pluripotency genes only one (4%), Chd1, also had an activated enhancer signature in Maf-DKO macrophages (Figs. 2B, red type). Similar results were obtained when basing the selection of self-renewal genes on transcription factor cross-regulatory circuits and interactome data in ES cells (30): none of the factors in an ES cell transcriptional cross-regulatory circuit belonging to the nanog interactome (0/10) and 6/11 (55%) of genes in the network not interacting with nanog were activated in Maf-DKO macrophages (Fig. S4). Expression analysis showed increased expression in Maf-DKO compared to WT BM macrophages for all 16 self-renewal genes associated with Maf-DKO only activated enhancers, which was enhanced in response to M-CSF (Fig. 2C).
Fig. 2. Self-renewing Maf-DKO macrophages activate genes required for ES cell self-renewal.
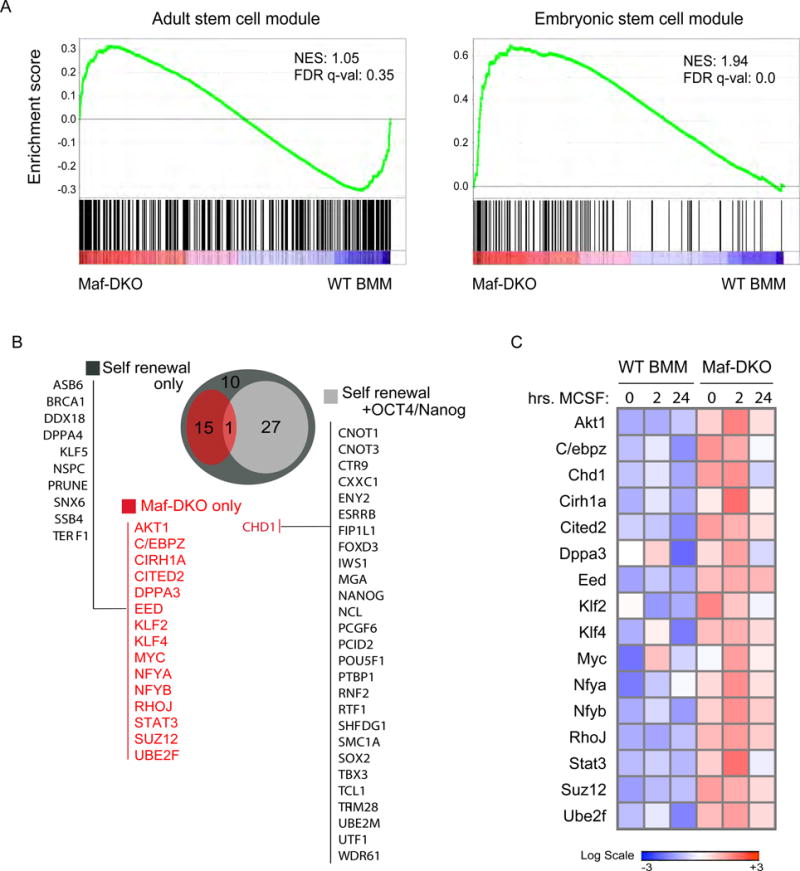
A) Gene set enrichment analysis (GSEA) using gene sets defined by Wong et al. (21) for adult tissue stem cells and core embryonic stem cell modules (Broad Institute MSigDB M1999 and M7079). comparing the expression of genes associated with Maf-DKO-only associated enhancers in Maf-DKO versus WT BMM. NES, Normalized enrichment score; FDR, false-discovery rate. B) Diagramm of overlapping group of genes associated with Maf-DKO-only activated enhancers (red) and functionally validated self-renewal or both self-renewal and pluripotency (self-renewal + Oct4/nanog) activity in ES cells demonstrated by gene inactivation screens (22–29). C) Gene expression by quantitative real-time PCR of self-renewal genes in Maf-DKO and WT BMM stimulated with M-CSF for the indicated times. Heatmap shows the average signal of technical replicates. Data are representative of four independent experiments.
Self-renewal genes are organized in a network and functionally important
To further investigate whether self-renewal genes in Maf-DKO macrophages were functionally integrated in a network of cross-regulated genes, we measured the effect of silencing individual self-renewal genes on the expression of other self-renewal genes. For 12 of the identified 16 self-renewal genes, validated shRNA vectors were available (31). In each knockdown, we measured the expression of 13 self-renewal genes associated with Maf-DKO-only macrophage enhancers and 4 additional self-renewal genes that might be regulated indirectly as part of a larger network. We also included probes against myeloid and housekeeping control genes (Fig. 3A). Knockdown of self-renewal genes only minimally influenced myeloid gene expression (Fig. 3A). By contrast, in all cases self-renewal-specific shRNAs substantially reduced not only the expression of their specific target, but also that of other self-renewal genes. These analyses revealed a network of self-renewal gene regulation with MYC and KLF2 as the two main nodes and KLF4 as a minor node (Fig. 3B).
Fig. 3. Self-renewal genes are organized in a network and functionally important for Maf-DKO macrophage proliferation.
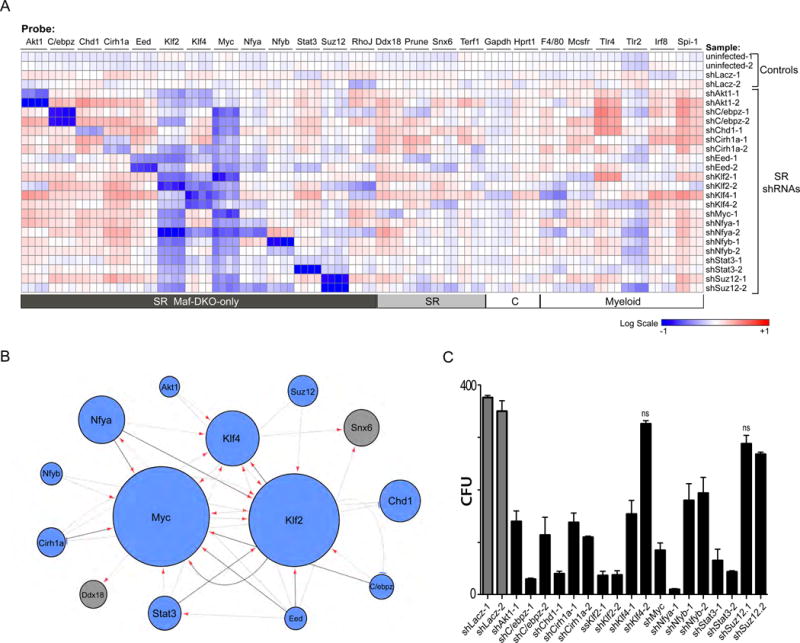
A) Gene expression analysis of Maf-DKO macrophages uninfected or infected with shLacZ control or shRNAs targeting self-renewal genes (rows) for self-renewal genes associated with Maf-DKO macrophage activated enhancers, not associated self-renewal genes (SR), house keeping (C) and macrophage specific (myeloid) genes (columns) using quadruplicate nano-fluidic real-time PCR on Fluidigm array. For each gene, the heatmap presents normalized values as percent change over average expression in non-infected and control lacZ shRNA infected cell samples. Data are representative of three independent experiments. B) A network model using an FDR-approach showing significant repression of an output target gene resulting from shRNA knockdown of a regulator gene, with darker lines denoting regulation in all replicates, arrows denoting repression and blue bars activation by shRNA. Circle size is a function of the number of times the target is affected by knockdown of other regulators. C) Number of colony forming units (CFU) obtained from equal numbers of Maf-DKO macrophages infected with shRNAs against control (shLacZ) or self-renewal gene targets after 12 days of culture in methocult medium containing M-CSF. The mean number of CFU for self-renewal gene shRNA infected populations is significantly different from the mean number of CFU for controls (One-way analysis of variance, P<0.05) unless indicated (ns= not significant). Error bars represent standard deviation (SD) of triplicate technical replicates and data are representative of 3 independent experiments.
To determine whether the identified network of self-renewal genes was functionally important for self-renewal activity in Maf-DKO macrophages, we analyzed the effect of silencing individual self-renewal genes on colony forming ability. We observed a significant reduction in Maf-DKO colony forming units (CFU) upon expression of at least one self-renewal gene-specific shRNA per gene compared to control (shLacz) for all vectors except Eed, which gave inconclusive results between different shRNA constructs and repeat experiments (Figs. 3C, S5). We did not observe any significant effect of self-renewal gene silencing on apoptosis, as indicated by Annexin V and intracellular propidium iodine labelling (Fig. S5B). The strongest effects on self-renewal were observed in the knockdowns of genes that occupy a central position in the network (KLF2/MYC). We observed weaker effects for several genes with peripheral positions (for example SUZ12), indicating that the knockdown of a single peripheral gene in the network might not be sufficient to compromise the activity of the entire self-renewal network.
Shared self-renewing genes have distinct enhancers in ES cells and macrophages
Our results indicated that self-renewal of Maf-DKO macrophages depends on an integrated network of cross-regulated self-renewal genes that are also employed for self-renewal in ES cells. To investigate how these very different cell types could access a similar gene network, we compared the gene regulatory mechanics underlying activation of self-renewal genes in Maf-DKO macrophages and ES cells. As expected from the previous data and the differentiated phenotype of macrophages, genes affecting both self-renewal and pluripotency in ES cells, such as Oct4/POU5F1, showed activated enhancers only in ES cells but not in macrophages (Fig. S6A). By contrast, active enhancer elements were found to be associated with the genes of the identified self-renewal network both in ES cells and Maf-DKO macrophages (Fig. 4). Interestingly, however, the two cell types use entirely distinct sets of enhancers to activate these genes. The examples for the central factors MYC, KLF2 and KLF4 (Fig. 4A) and further network elements (Fig. S6B) illustrate that there exist, for each gene, ES cell-specific H3K27ac+/p300+ enhancers (blue highlight) and macrophage-specific H3K27ac+/p300+ enhancers (red highlight). To address these observations quantitatively, we identified for each of the identified 16 self-renewal genes (Fig. 2B) the regulatory regions marked by H3K27ac in the three cell types (ES cells, Maf-DKO and WT BMM macrophages), using a multisample-calling strategy to capture all shared regions and strong cell type-specific regions (Fig. 4B). There were 152 such regions, ranging from one region per gene (4 genes) to 34 (Cited2), with an average of 9.5 and a median of 5. Genome browser inspection revealed almost completely non-overlapping sets of active enhancer regions in macrophages and ES cells (Fig. 4A, S6B). Consistent with these observations, we could identify two distinct enhancer clusters by k-means clustering (k = 2), where the most important characteristic that separated them is whether an enhancer is active in ES cells or in macrophages (Fig. 4B). The median normalized signal for enhancers in cluster 1 is 62-fold higher in ES cells than in Maf-DKO macrophages; conversely, in cluster 2 the median normalized signal is 16-fold higher in Maf-DKO macrophages than in ES cells. These patterns are mirrored at high statistical significance (P-values ranging from 1.4e-09 to 9.6e-16) in the ChIP-seq data from p300 and H3K4me1 (Mann-Whitney test), which had not been used in the clustering (Fig. 4B). Furthermore, enhancers associated with macrophage-specific self-renewal genes were 85% and 73% PU.1 positive in Maf-DKO and WT BM macrophages (Fig. 4B), indicating that macrophage self-renewal activates largely poised macrophage-specific enhancers. These robust results attest to the strong contrast among the clusters and their constituent regions, and support the model that the same self-renewal genes can be accessed by distinct lineage-specific enhancer elements in two different cell types with self-renewal capacity (Fig. 4C).
Fig. 4. Self-renewal genes are associated with distinct enhancers in ES cells and macrophages.
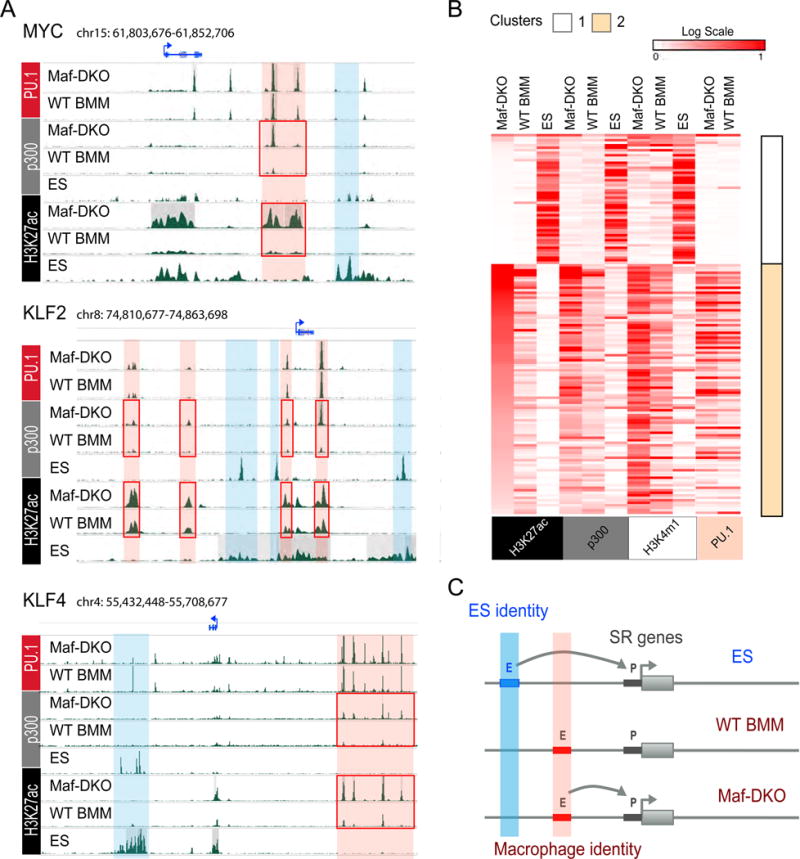
A) Genomic regions surrounding MYC, KLF2 and KLF4 genes showing distinct ES cell (blue) and macrophage (red) specific predicted enhancer regions with differential H3K27ac and p300 enrichment in Maf-DKO over WT BMM (red boxes). B) Heatmaps and k-means clustering (k=2) of Chip-seq signal of all H3K27ac+ regions associated with self-renewal genes in ES cells, Maf-DKO and WT BMM, including both differentially regulated Maf-DKO-only and non-differentially regulated regions. Corresponding regions are shown for p300, H3K4m1 and PU.1 (ES, Maf-DKO and WT BMM). C) Model based on panels A) and B) to describe tissue-specific macrophage and ES cell enhancer platforms associated with individual self-renewal genes.
MafB directly represses self-renewal enhancers in macrophages
To better understand the mechanism leading to the activation of self-renewal enhancers in macrophages, we re-expressed MafB in Maf-DKO macrophages. MafB expression resulted in the reduction of both colony size and number in CFU assays (Fig. 5A), strongly inhibited expression of self-renewal genes (Fig. 5B), and re-established low levels of p300 binding and H3K27Ac modification similar to WT BMM at both Maf-DKO-only (Fig. S7A) and self-renewal gene-associated enhancers (Fig. 5C). We further analyzed whether this rescue effect was due to a direct effect of MafB on self-renewal gene associated enhancers. Transcription factor binding sites enriched at Maf-DKO-only, and more specifically at self-renewal enhancers, included Maf (MARE) and related AP-1 family binding sites (Figs. S3A,S8). Furthermore, MafB can also bind directly to PU.1 (32) and might thus target PU.1-positive enhancers by protein-protein interactions in the absence of consensus MARE. Indeed, Chip-seq analysis for MafB in reconstituted Maf-DKO macrophages showed direct binding of MafB (Fig. 5D) to 65% of PU.1-positive DKO-only enhancers and 73% of self-renewal gene associated enhancers (Fig. 5E) as exemplified for the core factors of the network, Myc, KLF2 and KLF4 (Figs. 5F,S7B). Similar binding profiles were observed for ChIP-seq analysis of endogenous MafB in WT BMM (Fig. S9). Together this indicated that the activation of self-renewal is reversible and that the large majority of poised self-renewal associated macrophage enhancers are directly repressed by MafB binding.
Fig. 5. MafB inhibits macrophage self-renewal by direct repression of self-renewal gene enhancers.
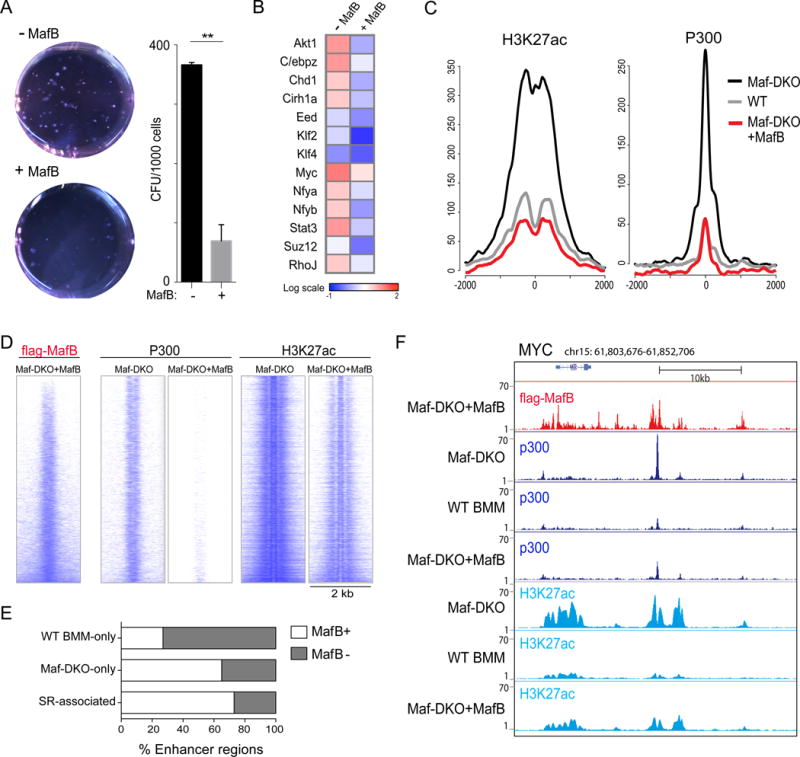
A) Colony assays for Maf-DKO macrophages expressing empty vector (-MafB) or a doxycycline-inducible flag-tagged MafB allele (+MafB) counted after 14 days of culture in methocult medium containing M-CSF and doxycycline (DOX), showing culture dishes (0.63x), and number of colony-forming units (CFU). Error bars represent the SD of two technical replicates and data are representative of three independent experiments. B) Expression of self-renewal genes in Maf-DKO macrophages (-MafB) and Maf-DKO macrophages expressing a doxycycline-inducible, flag-tagged MafB allele (+MafB) after 2 hours stimulation with M-CSF determined by nano-fluidic real-time PCR on Fluidigm array. Data are representative of three independent experiments. C) Aggregation plots showing average ChIP-seq signals for P300 and H3K27ac in Maf-DKO, WT BMM and Maf-DKO macrophages expressing a doxycycline-inducible, flag-tagged MafB allele in the presence of doxycycline (Maf-DKO+MafB) for the self-renewal associated enhancers regions (total=88 regions). D) Direct alignment of regions proximal to Maf-DKO-only enhancers for flag-MafB binding in Maf-DKO+MafB and corresponding regions in Maf-DKO and Maf-DKO+MafB macrophages for P300 and H3K27ac binding. E) Histogram showing the percent of WT BMM-only, Maf-DKO-only and self-renewal gene-associated enhancers bound by MafB as determined by ChIP-seq for flag-MafB in Maf-DKO+MafB cells. F) Genomic regions surrounding MYC gene with Chip-seq tracks as labelled.
Naturally low Maf levels in alveolar macrophages activate self-renewal enhancers
To further investigate whether our observations in Maf-DKO macrophages were directly relevant to the self-renewal capacity of genetically un-modified macrophages, we investigated alveolar macrophages (AM). AM are a population of adult resident macrophages with the well-characterized ability to autonomously self-renew (4) and which naturally express low levels of MafB and cMaf (6). Consistent with the constitutively low Maf levels compared to other macrophage populations (Fig. 6A,S10), we could expand AM in long-term liquid culture (Fig. 6B) and serially replate AM but not peritoneal macrophages (PM) in colony-forming assays without loss of replicative ability (Fig. 6C). Cultured alveolar macrophages also expressed generally increased levels of self-renewal network genes (Fig. 6D). Furthermore, ChIP-Seq analysis of epigenetic enhancer marks at the self-renewal gene associated enhancer regions showed comparable binding of PU.1 between AM, Maf-DKO and WT BM macrophages, but an enrichment of the activation mark p300 in AM similar to Maf-DKO macrophages (Fig. 6E,F). Statistical analysis confirmed a high correlation for PU.1 binding across all three populations but the highest correlation for p300 binding between AM and Maf-DKO macrophages (Fig. 6G). Thus, both experimental and natural reduction of MafB and c-Maf levels activates a set of poised macrophage-specific enhancers of the self-renewal gene network.
Fig. 6. The self-renewal gene network is activated in alveolar macrophages expressing naturally low levels of MafB and cMaf.
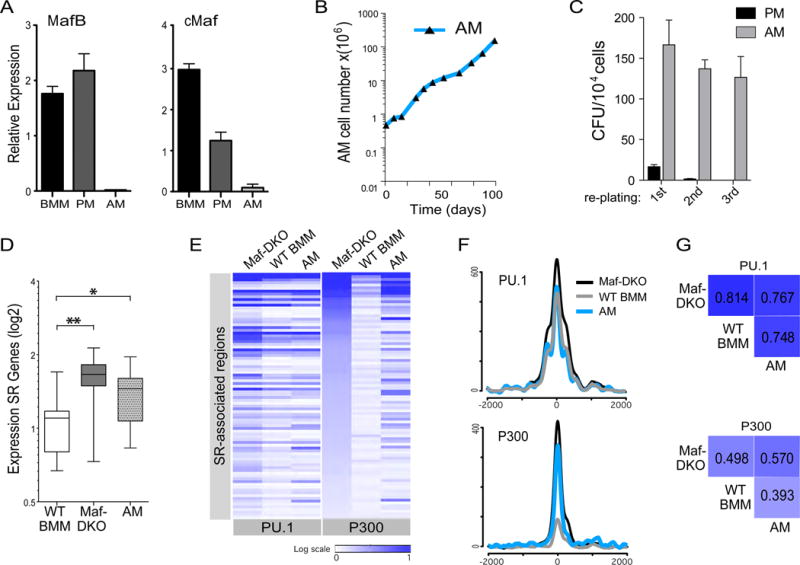
A) Expression of MafB and cMaf relative to HPRT1, measured by RT-QPCR, in short-term cultures of bone marrow macrophages (BMM), peritoneal macrophages (PM) and alveolar macrophages (AM). Data are representative of three independent experiments using biological replicates. B) Growth curve showing number of AM over time in liquid culture. Data are representative of two independent experiments. C) Number of colony forming units (CFU) counted at day 21 per 104 AM and PM plated in methocult medium after first plating, or after replating 104 cells washed out from first plating (2nd plating), or second plating (3rd plating). Data are representative of three independent experiments. D) Box plots showing average, inter-quartile and 5–95 percentile relative expression levels of all self-renewal genes in Maf-DKO, WT BMM and AM, measured by nano-fluidic real-time PCR on Fluidigm array. * p-value < 0.05; ** p-value < 0.01 based on an unpaired t-test. Data are based on the average signal from three biological replicates performed each with technical duplicates. E) Heatmap of ChIP-seq signal for all regions associated with self-renewal genes (total=88) in Maf-DKO, WT BMM and AM. Corresponding regions are shown for PU.1 and P300. F) Aggregation plots showing average ChIP-seq signals for PU.1 and P300 in Maf-DKO, WT BMM and AM for the self-renewal associated enhancers regions (total=88 regions). G) Pearson correlation matrix (PCA ranked log2 read number) for PU.1 and P300 binding to self-renewal gene associated enhancers (total =88 regions) based on ChIP-seq data for Maf-DKO, WT BMM, and AM. ChIP-seq analysis on AM for PU.1 and p300 was performed twice .
Low MafB levels activate self-renewal genes in resident macrophages in vivo
Many resident macrophage populations show a low level of local proliferation in vivo. Using immunofluorescence, we observed that the large majority of cycling, Ki67+ macrophages in the peritoneum, liver and spleen red pulp did not express MafB, whereas quiescent, Ki67 negative macrophages were nearly all MafB positive (Fig. 7A,B,S11, table S1), indicating that macrophage proliferation in vivo also involved reduced MafB levels. Resident macrophages can further expand massively by transient local proliferation in response to specific stimuli (5, 33), for example to M-CSF during infection of the peritoneum (34, 35), which can be mimicked by direct intraperitoneal injection of the cytokine (34, 35)(Fig. S12). To address whether under such conditions macrophages could access the self-renewal gene network by transient repression of MafB and/or cMaf, we measured both self-renewal gene and MafB/cMaf expression before and at various time points after M-CSF stimulation by single cell analysis of sorted resident peritoneal macrophages. Indeed, 1h after stimulation we observed a transient reduction of MafB (Fig. 7C) and to a much lesser extent of c-Maf but not of control myeloid genes (Fig. S13A). Reasoning that the behavior of resident peritoneal macrophages might be heterogeneous we could identify 3 distinct groups of cells by PCA analysis and k-means clustering (Fig. 7D). Whereas cells in cluster 3 were equally present before and after stimulation, cluster 2 type cells were strongly reduced and cluster 1 type cells strongly increased after M-CSF stimulation (Fig. 7E). Interestingly, cells in cluster 2 expressed high levels of MafB and c-Maf but were negative for nearly all self-renewal genes. By contrast, cells in cluster 1 showed low MafB levels and up-regulation of the large majority of self-renewal genes (Fig. 7F). Cluster 3 cells also showed high MafB levels (Fig. S13B). Direct comparison of the expression of MafB and Myc in individual cells revealed a large number of cells with high MafB and low Myc levels in cluster 2 and 3 (Fig. S13C). By contrast, a majority of cells in cluster 1 showed low/absent MafB and high levels of Myc (Fig. 7G). This analysis also demonstrated that a few cells with MafB low / Myc high profile already existed in the steady state but their number strongly increased after M-CSF stimulation (Fig. 7G).
Fig. 7. Single cell analysis of tissue macrophages in vivo reveals self-renewal gene network activation in MafB negative macrophages.
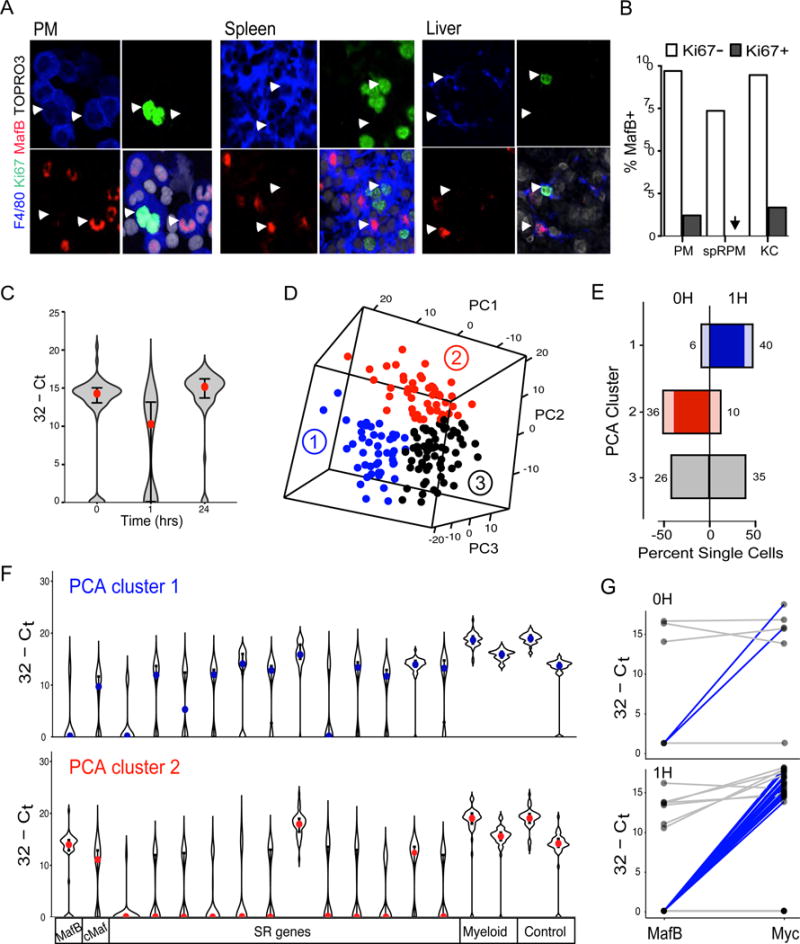
A) Immunofluorescent (IF) image of MafB and Ki67 positive peritoneal F4/80+ macrophages in cytospun peritoneal macrophages (PM), and tissue sections from spleen and Liver. White arrowheads point to examples of Ki67−/MafB+ and Ki67+/MafB− macrophages, F4/80+, in each tissue. Data are representative of three independent experiments and larger field images are shown in Fig. S11. B) Quantification of percent MafB+ cells in the Ki67- and Ki67+ fraction of F480+/SYTOX blue+ resident macrophages from peritoneum (PM) n=969, spleen red pulp macrophages (rpSPM) n=425 and liver Kupfer cells (KC) n=302, corresponding to IF images in Fig. 7A and S11. Absolute cell counts are shown in table S1. C) Violin plot showing expression of MafB across single PM isolated from a mouse at indicated time points after i.p. M-CSF injection, measured by nano-fluidic real-time PCR on Fluidigm array. Red dots mark the median value and error bars the interquartile range. D) Depiction in three-dimentional space of PCA analysis of single cell gene expression data for a k-means=3 of pooled data for 0 and 1h after MCSF injection. Distinct PCA clusters are distinguished by colors and numbers. E) Histogram showing the percent of single cells in each cluster shown in D) in the 0 and 1H time point datasets. Absolute numbers of cells in each group are indicated and the net change between 0 and 1H is shown in deeper color. F) Violin plots showing expression for Maf, self-renewal (SR), myeloid and control genes across cells in PCA cluster 1 (blue) and PCA cluster 2 (red) from D). Colored dots show the median value and error bars the interquartile range. G) Line diagrams showing individual cell comparison of MafB and Myc expression for PCA cluster 1 at 0 and 1H. Single cells with low MafB and high Myc expression are highlighted in blue. Consistent results to C-G) were obtained in a repeat experiment with cells sorted from a pool collected from 5 mice at each time point.
Overall, our results demonstrate that MafB/cMaf down-regulation and concomitant activation of a self-renewal gene network is a hallmark of proliferating resident macrophages in culture and in vivo. Natural or experimentally induced constitutively low levels of MafB and c-Maf also enable long-term continuous self-renewal of adult macrophages in culture, as observed for alveolar or Maf-DKO macrophages, respectively. Although inactivation of type I interferon signalling has also been associated with increased macrophage proliferation(36), we could not identify a direct link of Maf activity to this pathway. Furthermore repression of interferon β by MafB has been reported (37), which is the opposite of what would be expected, if Mafs acted through interferons in limiting macrophage proliferation.
We show that a network of genes that governs macrophage self-renewal overlaps substantially with that controlling self-renewal in ES cells. Remarkably, the regulatory mechanism by which activation of these genes is accomplished involves almost entirely separate sets of enhancers in the two cell types. The identified enhancer architecture associated with self-renewal genes in macrophages is already present in quiescent cells and can become activated when direct repression by Maf transcription factors is relieved. In summary, we have shown that self-renewal activity can be activated from an intrinsic cell type-specific enhancer repertoire in differentiated cells. Our findings provide a general molecular rationale for the compatibility of self-renewal and differentiated cell functions, and may also be more generally relevant for the activation of self-renewal activity in other differentiated cell types with therapeutic potential.
Materials and Methods
Cell Culture
Maf-DKO macrophages were grown sub-confluently in macrophage growth medium (Dulbecco’s modified medium (DMEM) with 20% L-cell sup, 1% sodium pyruvate and 1% L-Glutamate) as described (7). WT BMM macrophages were differentiated from total mouse bone marrow for 15 days in macrophage growth medium, selecting for adherent cells every 4 days. For time course analysis of self-renewal gene expression in Maf-DKO and WT BMM macrophages, cells were seeded 18h prior to time 0h in growth medium without L-cell sup and at time 0h, 100 ng/ml recombinant MSCF (Peprotech) was added to culture medium and cells were harvested for RNA isolation and analysis at 0, 1, 2, and 24 hours.
For culture of AM, alveolar lavages were pooled from ten 1mL 37°C BAL washes (PBS without Mg++/Ca++, 2mM EDTA, 2% FBS (GE Healthcare) per mouse and stored on ice. RBC lysis was then performed at room temperature (RT) for 3 minutes (RBC Lysis Buffer, Invitrogen). Cells were plated at a density of 1.1 million cells per 10cm bacterial petri dish in complete medium (RPMI, 10 % FCS,1 % Pen/Strep, 1 % Pyruvate, 1 % Glutamate) supplemented with 1 % GM-CSF supernatant from J558L cells transfected with murine GM-CSF cDNA.
MafB expression in Maf-DKO Macrophages
The pRetroX-Tet-On Advanced (Clontech, Cat.: 632104) and the pRetroX-Tight-Pur (632104) retroviral backbones were used for generating the flag-tagged MafB inducible Maf-DKO macrophages. For this, Neor was replaced by Hygror and Puror by GFP in pRetroX-Tet-On Advanced and pRetroX-Tight-Pur Retroviruses, respectively. Maf-DKO macrophages were first infected with the pRetroX-Tet-On-hygror retrovirus and selected for 2 weeks in medium containing 200μg/mL of Hygromycin B (Lifetechnologies, Cat: 10687-010). Selected Maf-DKO macrophages were next infected with the empty pRetroX-Tight-GFP or the pRetroX-Tight-GFP containing the flag-tagged MafB gene cloned downstream of the modified Tet-responsive promoter. GFP positive Maf-DKO macrophages were then FACS sorted and cultured in absence of doxycycline.
Peritoneal Macrophages Stimulation and Isolation
Either 200ul PBS (control) or 20ug of recombinant MCSF (rhMCSF, Novartis) was injected into the peritoneums (i.p.) of wild type, C57/B6J 8–10 week old mice at indicated times prior analysis. For cell cycle analysis, mice were also injected i.p. with 2 mg of BrdU (BD Pharmingen) dissolved in PBS, as per manufacturers instructions 4h prior to analysis. To harvest peritoneal cells mice were sacrificed and subsequently injected with 10mL ice-cold PBS containing 2mM EDTA (Sigma). Intra-peritoneal washouts were collected in 50mL Falcon tubes. Red cell lysis was performed on total washouts (RBC Lysis Buffer, Invitrogen) prior to staining for cell surface markers and cell cycle analysis or FACS sorting. Remaining mononuclear cells were then stained using the following antibody cocktail in the presence of FcBlock (BD Biosciences): CD11b-PE-Cy7 (BD Biosciences), MHC-II-FITC, B220-APC-Cy7, F4/80-PE (all from eBioscience) and Fixable Aqua Dead-V500 (Life Technologies) and either sorted for single cell analysis or further processed for cell cycle analysis.
Cell Cycle Analysis
Bromodeoxyuridine (BrdU) analysis was performed using a BrdU Flo Kit (BD Pharmingen) according to manufacturer’s instructions and with the following modifications: DNaseI digestion was performed for 90 minutes and intracellular staining for 60 minutes and anti-BrdU-AlexaFluor 647 Ab (Invitrogen, clone MoBU-1) was substituted for anti-BrdU provided in the kit. For Ki67 staining cells were harvested and treated as for BrdU staining but substituting an anti-Ki67-V450 antibody (eBiosciences, clone SolA15) for final incubation step. Cells were analysed using an LSRII and FlowJo software (Tree Star).
shRNA infections
Lentiviral vector particles were produced at the Centre International de Recherche en Infectiologie (CIRI) U1111/UMR5308 Inserm-CNRS-UCBL - ENS de Lyon (Lyon, France) by tri-transfection of plasmids harbouring the packaging construct, the transfer vector (31) and the envelope-expressing construct into producer cells using calcium chloride methodology. Virus was concentrated after transfection, viral supernatants were harvested and used directly for infections or stored at −80°C.
Maf-DKO macrophages were seeded subconfluently in 12 well dishes 24 hours prior to infection. The next day, 8μg/ml of polybrene was incubated for 1.5h at 37°C, 5% CO2 with viral supernatants and 2ml/well/infection was used to replace media on Maf-DKO macrophages. Spin infections were carried out at 25°C, 2h, 2500rpm. Viral supernatants were removed immediately after spin infection and replaced with macrophage growth medium. Cells were then further incubated at 37°C, 5% CO2 for 72 hours prior to harvesting and divided into fractions for apoptosis, colony assay, RNA and DNA isolation.
Apoptosis was measured using the AnnexinV Apoptosis Detection Kit (eBioscience) according to the manufacturers instructions. To control for infection genomic DNA was isolated using DNeasy Blood and Tissue Kit (Qiagen) from infected Maf-DKO cells at 72h post-infection. QPCR was performed to detect the quantity PURO cassette contained in the lentiviral vector relative to a control genomic amplicon (actin TSS) and to ensure similar infection efficiencies across all populations (S6). Reactions were run in a 7500 Real-Time PCR System (Applied Biosystems, USA) including dissociation curves to validate unique amplicons. RNA was extracted from infected cells and processed for gene expression profiling.
Colony Assays
For shRNA infected cells, 5000 cells were counted from each population of infected cells and mixed with 1ml of methocult medium (M3231, Stem Cell Technologies) with the addition of 100ng/ml recombinant MCSF (Peprotech), plated in duplicate and grown at 37°C, 5% CO2. For experiments using MafB-inducible Maf-DKO cells, 1000 cells were plated and 1μg/ml doxycycline (Sigma) was added in addition to MCSF where indicated. The number of colony forming units (CFU) was counted at Day 12 after plating.
Maf-DKO macrophages expressing empty vector (-MafB) or a doxycycline-inducible, flag-tagged MafB allele (+MafB) were plated at 1000 cell per ml of Methocult medium, as above, with the addition of 1 μg/ml doxycycline (Sigma) to Methocult.
AM were plated at 10,000 cells per 1mL of Methocult M3231 (StemCell Technologies) containing 100 ng/ml rGM-CSF (Peprotech) and colonies were counted after 3 weeks.
All experiments were performed with n=2 replicates and results were reproduced at least 3 times independently.
In situ immunofluorescent cell staining
PM were harvested with refrigerated PBS and 10,000 cells were loaded onto Shandon cytocentrifuge chambers and centrifuged at 1000 rpm for 3 min. Slides were air-dried, fixed with refrigerated 4% PFA for 10 minutes at RT, and washed once with PBS. Spleen and liver were harvested after perfusion with PBS and freshly embedded in TissueTeck® (OCT™ Compound) prior to freezing. Serial frozen sections (8μm) were fixed with refrigerated 4% PFA for 10 min minutes and washed 3 times for 5 minutes with PBS at RT.
Slides were then permeabilized with Triton-X 0.05% (Sigma, cat. 9002-93-1) for ten minutes and washed 3 times for 5 minutes with PBS. Unspecific antigens were blocked for 1 hour with PBS containing 1% FCS, 2% BSA and 1% goat serum prior to staining. After blocking, slides were incubated for 1h at RT with rabbit anti mouse MafB (Bethyl IHC-101) diluted at 1:50 and rat anti mouse KI67 (eBioscience 14-5698-82) diluted at 1:50 in blocking solution. Slides were subsequently washed 3 times for 10 minutes with PBS at RT and incubated with the secondary antibody donkey anti-rat A488 (Invitrogen A31572) diluted 1:500 and donkey anti-rabbit A555 (Invitrogen A21208) diluted 1:500 in PBS. Slides were again washed 3 times for 10 minutes with PBS at RT and, finally, incubated for 1h with F4/80 A647 (Serotec MCA497A647) diluted at 1:50. A last wash series of 3 times for 10 minutes with PBS at RT was performed before mounting with Prolong Gold (Thermo Fischer, cat. P36930) with SYTOX Blue diluted at 1:1000 (Thermo Fischer, cat. S11348) for nuclear staining. Images were analyzed on a Zeiss LSM510 confocal microscope.
ChIP-seq
Cells cultured in plates were fixed by the addition of 1/10 volume of freshly made cross-linking solution (11% formaldehyde, 100mM NaCl, 1mM EDTA, 0.5 mM EGTA, 50mM Hepes pH 7.8) directly to cell medium and incubation at room temperature (RT) for 10 minutes. Formaldehyde was then quenched for 5 minutes at RT by the addition of 2.5M Glycine solution to a final concentration of 125mM. Fixed cells were washed twice with cold PBS, scraped off the plate, counted, and transferred in 50ml Falcon tubes. Cells were then pelleted by centrifugation at 700g for 5 minutes at 4°C, snap-frozen in liquid nitrogen and stored at −80°C for storage or shipment on dry-ice.
Each batch of 100 millions cells was lysed by adding 10 ml of ChIP Lysis Buffer (Santa Cruz Biotechnology, sc-45000) or RIPA buffer (1×PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing Protease Inhibitor Cocktail (PIC). One complete PIC tablet (Roche, REF 11873580001) for 50 ml buffer or 1 mini PIC tablet (Roche, REF 11836153001) for 10ml buffer was used. After rotating the cell tube for 10–15 minutes at 4°C, nuclei were pelleted by centrifugation at 700g for 5 minutes at 4°C. Nuclei pellet was resuspended in 5ml of Santa Cruz Biotechnology ChIP Lysis Buffer High Salt (sc-45001) or RIPA buffer (1×PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing PIC. Aliquots of 1ml containing 20 million nuclei were transferred into 1.5 ml low-bind microfuge tubes. The nuclei suspension was either sonicated immediately or snap-frozen in liquid nitrogen and stored at −80°C.
The nuclei/chromatin suspension in each ml was sonicated (Sonics Vibra Cell, Model CV188, with Stepped Tip 1/8”-630-0422) on ice in 4°C cold room. Sonication was performed with 9 cycles of 30 seconds (s) ON at 60% intensity and 30 s OFF for chromosome modifications, H3k4me1 and H3K27ac, or with 8 cycles for transcription factors, P300 and PU.1. The sheared chromatin was centrifuged at 14krpm for 10–15 minutes, then transferred as in the supernatant into a new 1.5 tube and kept on ice for ChIP.
For ChIP, 50 ul Invitrogen Dynabeads (7×108 beads/ml) were used for 20 millions cells, either anti-rabbit IgG (Cat. no. 112.03D) or anti-mouse IgG (Cat. no. 112.01D) depending on the source of the first antibody. The beads were first washed with PBS/BSA/PIC buffer (1× PBS, 5mg/ml BSA (Sigma, A3059-10G, Fraction V), Roche PIC (1 mini tablet for 10 ml or 1 complete tablet for 50 ml, added before use)) then washed beads were thoroughly resuspended in 1ml of PBS/BSA/PIC buffer in a 1.5 ml microfuge tube. Tube was placed on a magnetic rack for 1 minute, then the supernatant was removed. Wash was repeated twice by resuspending beads in 1 ml PBS/BSA/PIC buffer, rotating for 5 minutes and supernatant was removed as above. The beads were then coated with antibody by resuspending in 250 ul PBS/BSA/PIC buffer in each tube, adding 5 ug antibody, rotating overnight at 4 °C. The following antibodies were used: H3K4me1 (Abcam, ab8895), H3K4m3 (Abcam ab8580), H3K27ac (Abcam, ab4729), P300 (Santa Cruz Biotechnology, sc-585), PU.1 (Santa Cruz Biotechnology, sc-352), MafB (Bethyl IHC-101) and Flag M2 antibody (Sigma). After overnight incubation with antibody, the beads were pelleted by placing the tube on a magnetic rack for 1 minute followed by removing the supernatant. The beads were then washed 3 times with PBS/BSA/PIC buffer.
ChIP was carried out by adding the sheared chromatin suspension to the antibody-coated beads, rotating overnight at 4 °C, and followed by a series of washes. The beads were first washed once with 1ml of low salt wash Buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100), then twice with high salt wash buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100), three times with LiCl wash buffer (10 mM Tris-HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 1% GEPAL CA630, 1% Na-Deoxyholate), and twice with TE buffer. ChIP DNA was eluted by adding 200 ul of ChIP Elution Buffer (Santa Cruz Biotechnology, sc-45003, or 1%SDS/0.1 M NaHCO3.) to each bead tube, vortexing to resuspend beads, and incubation at 65°C for 1 hour with vortexing every 15 minutes. After centrifugation for 3 minutes at RT and placing on a magnetic rack for 1 minute, ChIP DNA in the supernatant was transferred to a new tube.
Reversing of crosslinks was done by incubation at 65°C overnight, and DNA was extracted with an equal volume of Phenol:Chloroform:IAA (24:24:1). After centrifugation at 14krpm for 3 minutes, ChIP DNA in the supernatant was transferred to a 2.0 ml low-bind tube. More DNA was extracted by adding 100 ul water to the Phenol:Chloroform phase and repeating the exaction. DNA extract was combined and mixed with 5 volumes of PBI (from Qiagen MinElute PCR Purification Kit, Cat# 28004). The pH of the mixture was adjusted with 3M NaAc pH5.2 to lower than 7.5 before apply to the column. The mixture turned back to yellow color, which is important for DNA to bind to Qiagen column. DNA was purified according to manufacturer’s instructions, and eluted in 30 ul buffer EB. DNA concentration was measured with Qubit. 30ng (P300, PU.1, MafB and Flag-MafB) to 100ng (H3K4me1 and H3K27ac) DNA were used to make each library.
ChIP-seq libraries were made using adaptors from Illumina (FC102-1001) and other enzymes and reagents from NEB, following the Illumina ChIP-seq protocol with some minor modifications. ChIP DNA was end repaired by T4 DNA polymerase (M0203S/L), Klenow DNA polymerase (M0210S/L), and T4 PNK (M0201S/L), and purified with Qiagen MinElute PCR Purification Kit. A-base was then added to the 3′ end by Klenow Fragment, 3′ to 5′ exo- (NEB, Cat# M0212S/L) and dATP. The Illumina adaptors (1ul of 1:10 dilution) were subsequently added to both ends by DNA Ligase (M2200S/L). The adaptor-ligated DNA was sized selected for 200–400bp fragments by 2% low-melting agarose gel (Lonza, Cat# 50080), followed by purification by Qiagen MinElute Gel Extraction Kit (Cat# 28604). Library DNA was amplified in 100 ul reaction by Phusion PCR Master Mix (NEB, Cat# F-531S), primers SolexaPCR_F (5′- AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC T -3′) and SolexaPCR_R (5′- CAA GCA GAA GAC GGC ATA CGA GCT CTT CCG ATC T -3′). Libraries of H3K4me1 and H3K27ac were amplified by pre-denaturing at 98°C for 30 sec followed by 10 cycles of (98°C / 10s, 65°C / 30s 72°C / 30s), and extra 5 min at 72°C at the end. For the P300 and PU.1 libraries, 12 cycles of PCR were used. After library DNA was purified, library concentration was measured by Qubit, and size distribution was determined by Bioanalyzer. Each ChIP-seq library was sequenced on 1 lane of Illumina GAIIx or Illumina HiSeq 2000 with 1×36 bases read length. Biological replicates from independently derived Maf-DKO macrophage lines from 2 different mice were sequenced, one of which was determined to be tetraploid. Comparison of ChIP-seq and gene expression data revealed no substantive differences between the two lines. Sequencing statistics for all samples are included (S5).
ChIP-seq Analysis
While DNANexus pipeline was used for peak detection algorithms (38) and alignments of raw data (uploaded fastq files), the aligned data (BAM files) were subsequently downloaded and processed with a custom R pipeline for bioinformatics analyses. A threshold based on the number of sequenced tags (Nseq) has been set up as Nseq/7,000,000. All regions with a number of repeated tags (with identical sequences/coordinates) above this threshold were filtered out to remove possible sequencing and/or alignment artefacts. In order to accurately represent and further process the ChIP-seq and input control signals, the Watson and Crick strand tags need to be merged after elongation/size extension to the gel purified fragment size. The optimal elongation size of each ChIP-seq experiment was estimated in-silico by employing a step-wise 10bp chromosomal sequence tag shifting and score multiplication. Tag coordinates were subsequently elongated according to this estimated DNA fragment size, corresponding to the tag shift maximizing the score. Then, a nucleotide score representing the genome-wide overlap of elongated tags was computed across both strands. Wiggle files for genome-wide scores were generated following a binning step, calculating the average enrichment score every 50bp.
When input experiments were available, enrichment scores from WIG files were scaled and used as follow. Due to the size of the genome and relative low frequencies of binding events, we assumed that most (>90%) of the obtained scores from the ChIP-seq experiments represent a background (BG) level. We therefore used the genome-wide average score in each experiment to estimate the BG level. Using these average scores, it is possible to rescale the scores accordingly, acting as normalization and reducing the inter-experiment differences due to effects of different sequencing depths and/or fragment sizes.
We employed an input subtraction step for each experiment using the normalized file for the input control. This not only allows for correction of overrepresentations of certain genomic regions due to possible (un-) favorable events during sonication and/or DNA sequencing, but also serves to reduce the signal/noise ratio especially for experiments with low enrichment values.
H3K4me1 enriched regions were extracted from all samples (Maf-DKO, BMM, PEM, pro-B, ES, Liver, MEF, annotation files available)(12, 13). All regions close to annotated TSS (<2kb) were omitted and the union of resulting regions was used as a reference to extract enrichment scores of H3K4me1 derived from all samples. R scripts were developed and used for retrieving bin scores around the center of these annotations (+/− 2kb). These scores and their original genomic coordinates were used to re-center values around the H3K4me1 enriched regions using linear interpolation. In total, 1000 points were interpolated for each selected region, which resulted in a 1000 column matrix. These matrices were loaded, viewed, and color-scaled according to sample read depth using Java TreeView (39). Finally, these matrices were assembled by sample.
A large selection of regions enriched for H3K27ac and H3K4me1 regions was performed on WT and DKO samples. All regions in the close vicinity to any annotated TSS (<2kb) were ignored and the union of all regions has been defined as putative active enhancers (annotation file available). The binned enrichments of PU.1 samples in these regions were merged and compared among tissues using a non-parametric spearman correlation method.
Heatmaps were created with the same strategy as for SupFig. 1A (but focused on regions isolated with previously described isolated peaks). For each reference experiment (H3K27ac or P300), the regions were sorted by increasing average enrichment score in the regions, and the other heatmaps were respecting this initial sorting for corresponding experiments.
For the heatmap in Fig. 4B, SRA files for ES cell chip-seq data (13) were converted into FASTQ format before being uploaded and processed on DNANexus platform. Two replicate H3K27ac ES cell chip-seq BAM files were merged. BAM files were then processed by DNAnexus as described above. All reads from BAM files with MAPQ (MAPping Quality) value less than 10 were filtered out to keep only confidently mapped reads. The numbers of reads in each relevant region were counted according to their start position. Each read with a start position outside the region was not taken into account whatever the strand considered.
The read counts matrix was normalized by considering each mark independently from the other ones. For each antibody, the number of reads for each region was normalized relative to the total number of confidently mapped reads of the least-sequenced sample. Since these regions are not homogeneous in length, counts were normalized on the size of the region in Kilo base pair (Kbp). Next, considering each antibody independently, counts exceeding the 95th percentile were set to this value. This was done to avoid a lack of constraints on the figure. Values were then set to the range 0–1, still considering each antibody independently. Values were sorted according to the Maf-DKO region’s H3K27ac level. Finally a 2-means clustering (k=2) was made on H3K27ac marks for the three cell types (WT, Maf-DKO, ES).
All analysis were performed using Samtools, R (v 2.14) and gene-e (v 3.0.202)
Motif Search at Enhancer Loci
Motifs analysis was performed using HOMER2 (http://homer.salk.edu/homer/) on 10 232 DKO-only peaks (the union of H3K27ac and P300 selection based on enrichment in Maf-DKO versus WT BMM) and 88 self-renewal gene associated peaks from within the DKO-only selection. HOMER2, inter alia, screens for enrichment of known motifs. HOMER perl script findMotifsGenome.pl was used with the mm9 mouse genome as a background (random genomic sequences sampled according to GC content of input sequences).
GSEA Analysis
Maf-DKO vs WT log2 ratios were computed for each ProbeSet from normalized data by RMA method (Robust Multi-Array Average). H3K27ac DKO-only regions and P300 DKO-only peaks were associated with nearby genes via GREAT (http://bejerano.stanford.edu/great/public/html/index.php).
Union of DKO-only enhancers associated genes were then considered. 7499 Maf-DKO vs WT ratios of these non-unique DKO-only enhancer associated genes (a gene can have multiple associated ProbeSets) were selected for GSEA (Gene Set Enrichment analysis, www.broad.mit.edu/gsea). As Genesets, we used two « stem-cell modules ». An « adult tissue stem » module regrouping genes co-ordinately up-regulated in a compendium of mouse adult tissue stem cells and a second module comprised of genes co-ordinately up-regulated in a compendium of mouse embryonic stem cells shared with the ESC-like module (21). These two sets were extracted from the public Molecular Signatures Database (MSigDB, http://www.broadinstitute.org/gsea/msigdb/index.jsp).
Single Cell and Bulk Cell Population Gene Expression Profiling and Analysis
Single cells were sorted using the autoclone module on an AriaIII sorter (Becton Dickinson) directly into 96-well plates in the CellsDirect Reaction Mix (Invitrogen). Individual cell lysis, cDNA synthesis and amplification were performed according to Fluidigm Advanced Development Protocol by single-cell microfluidic real-time PCR using Dynamic Array IFCs (Biomark Fluidigm). Pre-amplified products (20 cycles) were diluted fivefold before analysis with Universal PCR Master Mix and inventoried TaqMan gene expression assays (ABI) in 96.96 Dynamic Arrays on a BioMark System (Fluidigm). For single cell analysis Ct values were calculated from the system’s software (BioMark Real-time PCR Analysis; Fluidigm) and filtered according to a set of quality control rules outlined below.
For single cell gene expression level analysis the pre-processing of the raw data was the following: For each gene, including controls, genes with a difference of duplicate CtValues > = 2.0 were considered inconsistent and removed. If the control gene (GAPDH) was not expressed or filtered out the whole sample was removed. If CtCall = FAILED but the sample showed expression of at least one other gene, it was considered as not expressed and Ctvalue was set to 31.9. The relative expression values were calculated according the following formula:
R scripts with ggplot2 package were used to construct violin plots and lineplots on relative expression values. Shown results were representative of 2 independent experiments.
RGL package was used for principal component analysis to cluster relative expression values on a 3D principal component space. PCA coordinates were clustered using the k-means method (Hartigan, J. A. and Wong, M. A. (1979). A K-means clustering algorithm. Applied Statistics 28, 100–108] using R script).
For bulk cell populations, total RNA was isolated from cells using the Quiagen RNAeasy kit and treated with RNAse-free DNAseI (Qiagen) prior to elution from columns. For gene expression profiling, RNA from Maf-DKO macrophages was extracted using RNeasy mini Kit (Qiagen) and QIAshredder columns (Qiagen) according to manufacturer’s instructions. On column DNA digestion was performed. 50 ng of total RNA was used for reverse transcription to cDNA using SuperScriptTM II RT (Invitrogen), Oligo(dT) primers (invitrogen) and RNaseOUT (Invitrogen). Specific gene target amplification was performed according to Fluidigm protocols. Selected TaqMan Gene Expression assays (20×) were pooled and diluted with water 100 fold, so that each assay is at a final concentration of 0.2× in the pooled assay mix. For pre-amplification of each cDNA sample, 1.25 μl of the respective cDNA, 1.25 μl of pooled assay mix and 2.5 μl of TaqMan PreAmp Master Mix (Applied Biosystems 4391128) were combined to a final volume of 5 μl in one well of a 96 well qPCR plate. cDNA samples were amplified using the following program: 1) 95°C, 10 minutes; 2) 95°C, 15 seconds; 3) 60°C, 4 minutes; repeat steps 2) and 3) 14 times. Amplified cDNA samples were diluted 1:5 using 20 μl of water. For Fluidigm 96.96 Dynamic Array IFC analysis, 5 μl of each cDNA sample and 5 μl of each TaqMan probe (20×) were loaded on the chip.
Microfluidic real-time PCR using Dynamic Array IFCs (Biomark Fluidigm) was performed with Universal PCR Master Mix and inventoried TaqMan gene expression assays (ABI) in 96.96 Dynamic Arrays on a BioMark System (Fluidigm). Ct values were calculated from the system’s software (BioMark Real-time PCR Analysis; Fluidigm) and filtered according to a set of quality control rules outlined below.
Gene filter: (1) for each gene, including controls, data with CtCall = FAILED and CtQuality < threshold were removed. (2) For each gene, including controls, only CtValues < 2.0 * the lowest CtValue of no RT or no RNA controls were considered, to filter out very low expression genes and inefficient probes. (3) For each gene, including controls, values with a difference ≥2.0 between Ctmax and Ctmin of replicates were considered inconsistent and removed. (4) If a gene was removed according to filters (1–3) from uninfected or control shRNA samples it was removed from all samples. Sample filter: (1) if the control gene (GAPDH) was not expressed or was removed according to gene filters (1–3), the whole sample was removed. (2) if a control gene (GAPDH) had a Ctvalue ≥ 2.0 different from average Ctvalue of control genes in all samples, the whole sample was removed to eliminate non specific shRNA and normalization artefacts. For the heatmap each Ct value was normalized against GAPDH using the following formula: relative expression Sample = 2^(mean(Ctgapdh)−CtSample)×100. For each gene, heatmaps in Fig 2C present normalized values as percent change over average expression in noninfected and control lacZ shRNA infected cell samples.
We furthermore calculated z-scores for each gene of shRNA infected cell samples based on uninfected and lacZ control shRNA infected samples, as described(31). Briefly, we defined a statistic z for each observation oij of transcript i in each shRNA experiment j:
where mi and si are, respectively, the mean and variance of the expression of transcript i in the control experiments (uninfected and lacZ shRNAs).
We used two FDR-based approaches to obtain confidence estimates of the observed z-scores. First we defined a per gene confidence score by using the variation of that gene’s expression in the control shRNA experiments. Permuted scores zijk as a null distribution and the FDR for a given z-score zij for gene i in experiment j are given as:
where n is the number of shRNA experiments and the confidence for z is conf(z) = 1-FDR(z). P={c| zic >z; c=1,…n)}
In the second approach we defined a per sh-RNA confidence score for each measurement of a self-renewal gene by calculating a FDR based on the variation of expression in GAPDH and myeloid genes. Formally, we let zij,…, znj be the scores for the jth experiment (shRNA), and assumed the first l transcripts were control transcripts whose expression did not change in response to any component. We defined where now and are, respectively, the mean and variance of the z-scores of the control transcripts 1, …, l in the jth shRNA experiment. We performed l permutations as described above, by swapping each observed z-scores with a control transcript score and computing z, then computing an FDR as above. For the construction of the network model we considered only values with a FDR < 0.05 and where at least half of the replicates fulfilled these criteria. Circle sizes reflect the number of times the expression of a gene is affected by the perturbation of another gene, scoring each gene and shRNA replicate separately.
For time-course analysis of self-renewal gene expression in Maf-DKO and WT BMM macrophages, values were obtained by subtracting the considered gene mean across all the samples from an individual raw score and then dividing the difference by the considered gene standard deviation across all the samples.
Statistical analysis was performed using R (v2.14), heatmaps were created using the software gene-e (Broad Institute) and network diagrams with Cytoscape (v3.0.2).
Construction of Self-Renewal Gene Network
To investigate the potential for cross-regulation between self-renewal genes, data from quadruplicate nano-fluidic real-time PCR on Fluidigm array, presented in Fig. 2C, was re-analysed and Z-score computations were used to decide if a given shRNA regulated it’s specific target mRNA and whether this regulation affected the expression of other genes (control or self-renewal genes). Computations were performed according to a « per gene » and a « per shRNA » computation method as described above and with the following additional considerations: 1) With the exception of Myc and Chd1, where only one shRNA was found to effectively knockdown expression of said target, all self-renewal genes were effectively targeted by two shRNAs. Therefore, to balance the number of replicates per samples so as not to undercount the number of regulations relative to other shRNAs, all the regulations where shMyc and shChd1 were involved were counted twice for subsequent computations. 2) The regulation of a gene upon expression of a given shRNA was not taken into account unless greater than two replicates for gene expression were significantly regulated whatever the computation method considered. This concerned only 13/86 regulations that were subsequently filtered out for the « per gene » computation and none for the « per shRNA computation ». 3) All auto-regulations (shGene-x regulating Gene-x) were considered trivial and were removed. 4) Baseline expression of all genes was determined based on average expression levels in control samples.
Together, these data were used to construct a gene regulation network (depicted in Fig. 2D). The size of the bubble for each gene in the network was drawn relative to the number of times the gene was regulated by an shRNA for which it was not the direct target. A line was drawn to connect genes when the shRNA regulated the expression of a non-target RNA at least 2/4 times tested for at least one of two shRNAs in both computations. Given that there are 2 shRNA per gene, 4 gene expression replicates and 2 computation methods, 16 relations could be evaluated for a given shRNA. When all 16 regulations were significant and were the same, lines are drawn in black, otherwise they are drawn in grey. The red end of the arrows indicates a repression of the gene by the shRNA whereas the blue end indicates activation. Genes that were targeted only by shRNA having significance in only one computation (single nodes) were not represented.
Supplementary Material
Acknowledgments
We thank Noah Spies for advice on statistical analysis and Stephanie Vargas-Aguilar for reagents. The data presented in this manuscript are tabulated in the main paper and in the supplementary materials. We gratefully acknowledge grants from the Agence Nationale de la Recherche (ANR-11-BSV3-026-01) and Institut National du Cancer (13-10/405/AB-LC-HS). ES received fellowships from the EU Marie Curie program and La Fondation de France and ES and PD were supported by La Ligue Nationale Contre le Cancer (Equipe labellisée). KM was supported by a Human Frontier Science Program long-term fellowship and Stiftung Charité, LG by the Fondation ARC pour la recherche sur le cancer, JCA by the Fondation pour la Recherche Médicale (AJE20130728183) and AS by the National Institutes of Health / NIA (P01AG036695). MS is an Einstein-BIH fellow and Fondation pour la Recherche Médicale (DEQ20110421320) and INSERM-Helmholtz group leader.
References
- 1.Jenkins SJ, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011 Jun 10;332:1284. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012 Apr 6;336:86. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 3.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013 Jan 24;38:79. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hashimoto D, et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity. 2013 Apr 18;38:792. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013 Nov 22;342:1242974. doi: 10.1126/science.1242974. [DOI] [PubMed] [Google Scholar]
- 6.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012 Sep 30;13:1118. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aziz A, Soucie E, Sarrazin S, Sieweke MH. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science. 2009 Nov 6;326:867. doi: 10.1126/science.1176056. [DOI] [PubMed] [Google Scholar]
- 8.Fejer G, et al. Nontransformed, GM-CSF-dependent macrophage lines are a unique model to study tissue macrophage functions. Proc Natl Acad Sci U S A. 2013 Jun 11;110:E2191. doi: 10.1073/pnas.1302877110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009 May 7;459:108. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Natoli G. Maintaining cell identity through global control of genomic organization. Immunity. 2010 Jul 23;33:12. doi: 10.1016/j.immuni.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Ghisletti S, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010 Mar 26;32:317. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010 May 28;38:576. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010 Nov 24; doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Natoli G, Andrau JC. Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet. 2012;46:1. doi: 10.1146/annurev-genet-110711-155459. [DOI] [PubMed] [Google Scholar]
- 15.Gosselin D, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014 Dec 4;159:1327. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavin R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014 doi: 10.1016/j.cell.2014.11.018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ostuni R, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013 Jan 17;152:157. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009 Feb 12;457:854. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011 Feb 10;470:279. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010 May;28:495. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong DJ, et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008 Apr 10;2:333. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanova N, et al. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006 Aug 3;442:533. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 23.Zhang JZ, et al. Screening for genes essential for mouse embryonic stem cell self-renewal using a subtractive RNA interference library. Stem Cells. 2006 Dec;24:2661. doi: 10.1634/stemcells.2006-0017. [DOI] [PubMed] [Google Scholar]
- 24.Pritsker M, Ford NR, Jenq HT, Lemischka IR. Genomewide gain-of-function genetic screen identifies functionally active genes in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2006 May 2;103:6946. doi: 10.1073/pnas.0509861103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall J, et al. Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell. 2009 Dec 4;5:597. doi: 10.1016/j.stem.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Jiang J, et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. 2008 Mar;10:353. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 27.Gaspar-Maia A, et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009 Aug 13;460:863. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu G, et al. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009 Apr 1;23:837. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding L, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell. 2009 May 8;4:403. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Macarthur BD, Ma’ayan A, Lemischka IR. Systems biology of stem cell fate and cellular reprogramming. Nat Rev Mol Cell Biol. 2009 Oct;10:672. doi: 10.1038/nrm2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amit I, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009 Oct 9;326:257. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bakri Y, et al. Balance of MafB and PU.1 specifies alternative macrophage or dendritic cell fate. Blood. 2005 Apr 1;105:2707. doi: 10.1182/blood-2004-04-1448. [DOI] [PubMed] [Google Scholar]
- 33.Gentek R, Molawi K, Sieweke MH. Tissue macrophage identity and self-renewal. Immunol Rev. 2014 Nov;262:56. doi: 10.1111/imr.12224. [DOI] [PubMed] [Google Scholar]
- 34.Davies LC, et al. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat Commun. 2013;4:1886. doi: 10.1038/ncomms2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jenkins SJ, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. 2013 Oct 21;210:2477. doi: 10.1084/jem.20121999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamilton JA, Whitty GA, Kola I, Hertzog PJ. Endogenous IFN-alpha beta suppresses colony-stimulating factor (CSF)-1-stimulated macrophage DNA synthesis and mediates inhibitory effects of lipopolysaccharide and TNF-alpha. J Immunol. 1996 Apr 1;156:2553. [PubMed] [Google Scholar]
- 37.Motohashi H, Igarashi K. MafB as a type I interferon rheostat. Nat Immunol. 2010 Aug;11:695. doi: 10.1038/ni0810-695. [DOI] [PubMed] [Google Scholar]
- 38.Valouev A, et al. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods. 2008 Sep;5:829. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004 Nov 22;20:3246. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.