

Abstract
Nucleoporins (NUPs) are essential components of the nuclear pore complex (NPC).1 Only few diseases have been attributed to NPC dysfunction.2-4 Steroid resistant nephrotic syndrome (SRNS), a frequent cause of chronic kidney disease, is caused by dysfunction of glomerular podocytes.5 Here we identify in 8 families with SRNS mutations of NUP93, its interaction partner NUP205, or exportin5 (XPO5) as a hitherto unrecognized monogenic cause of SRNS. NUP93 mutations caused disrupted NPC assembly. NUP93 knockdown reduced the presence of NUP205 in the NPC and, reciprocally, a NUP205 mutation abrogated NUP93 interaction. We demonstrate that NUP93 and XPO5 interact with the signaling protein SMAD4, and that NUP93 mutations abrogated interaction with SMAD4. Significantly, NUP93 mutations interfered with BMP7-induced SMAD transcriptional reporter activity. We hereby demonstrate that mutations of NUPs cause a distinct renal disease, and reveal SMAD signaling as a novel disease mechanism of SRNS, opening a potential new avenue for treatment.
Results
Steroid-resistant nephrotic syndrome (SRNS) is a disease of the renal glomerular filter. It constitutes the second most frequent cause of end-stage kidney disease (ESKD) in the first 3 decades of life.6 Its renal histologic correlate is focal segmental glomerulosclerosis (FSGS), which invariably causes loss of renal function within a few years of onset requiring dialysis treatment or renal transplantation for survival. Over 30 monogenic genes lead to podocyte dysfunction if mutated, which revealed these glomerular epithelial cells as the critical site of SRNS.5,7 Disease gene identification also implicated multiple signaling pathways in the pathogenesis of SRNS.8-10 We recently demonstrated in a large cohort of 1,780 families with SRNS that in about 70% of cases a causative gene is unknown.11
To identify additional genes that cause SRNS if mutated we performed homozygosity mapping12 and whole exome sequencing13 in 160 families with SRNS. In three families (A1671, A1626, and A2241) (Fig. 1, Table 1, Supplementary Figs. 1 and 2) we detected 2 different homozygous missense mutations of the gene NUP93 (NM_014669.4) (p.Gly591Val and p.Tyr629Cys), which encodes the nuclear pore protein 931 (Table 1, Fig. 1a, d–e). By high-throughput exon sequencing11,14,15 in a worldwide cohort of 1,800 families with SRNS we detected 3 additional families (A2403, A3256, and A1394) with compound heterozygous truncating mutations or highly conserved missense mutations of NUP93 (Table 1, Fig. 1d–e, Supplementary Fig. 2). The variants p.Gly591Val and p.Tyr629Cys apparently represent European and Turkish founder alleles, respectively (Table 1). We show that the splice site mutation (c.1537+1G>A) detected in family A1394 (Table 1, Fig. 1d) leads to aberrant splicing with in-frame skipping of exon 13 (Supplementary Fig. 1B–E). NUP93 function is known to be essential for NPC assembly in S. cerevisiae,16,17 C. elegans, and D. rerio.18
Figure 1. Homozygosity mapping and whole exome sequencing (WES) identify recessive mutations of NUP93, NUP205, or XPO5 in 8 families with steroid resistant nephrotic syndrome.
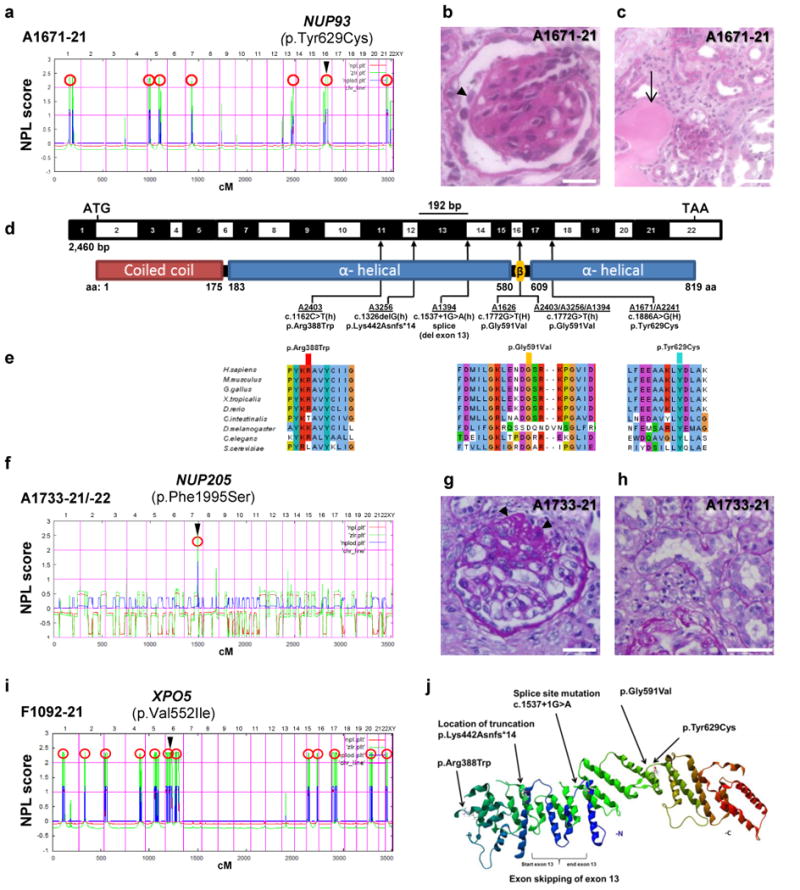
(a) Homozygosity mapping identifies seven recessive candidate loci (red circles) in family A1671 with steroid resistant nephrotic syndrome (SRNS), and WES identifies a homozygous mutation of NUP93 (p.Tyr629Cys). The NUP93 locus (arrowhead) is positioned within one of the maximum NPL peaks on chromosome 16q. (b–c) Renal histology of A1671-21 shows diffuse mesangial sclerosis (arrowhead) (b) and tubular dilation with protein casts (arrow) as well as tubular interstitial infiltration and fibrosis (c). (d) Exon (black-white) and protein domain (red-blue) structure of human NUP93 cDNA. Black bar denotes in-frame deletion of exon 13 (192 bp) resulting from a c.1537+1G>A mutation in family A1394-21. Three homozygous and three compound-heterozygous NUP93 mutations detected in 6 families with SRNS. Arrows indicate positions of mutations in relation to exons and protein domains. h, heterozygous; H, homozygous. (e) Conservation across evolution of altered amino acid residues for the three NUP93 missense mutations (p.Arg388Trp, p.Gly591Val, p.Tyr629Cys). (f) Genetic linkage mapping and WES in family A1733 with SRNS identifies a homozygous mutation in NUP205 (p.Phe1995Ser). (g–h) Renal histology sections of A1733 showing focal segmental glomerulosclerosis (FSGS). (i) Homozygosity mapping and WES in family F1092 with SRNS identifies a homozygous mutation in XPO5 (p.Val552Ile). (j) 3D structure of Nic96, the S. cerevisiae ortholog of human NUP93 (PDB 2QX5) lacking the coiled-coil domain. Scale bars are: (b,g) 25 μm, (c) 100 μm, and (h) 50 μm.
TABLE 1. Mutations in NUP93, NUP205 or XPO5 in 10 individuals from 8 families with steroid resistant nephrotic syndrome.
| Family -Indivi- dual |
Nucleotide change |
Amino acid change |
Exon (zygosity, Segregation) |
PPH2 score |
SIFT | Amino acid conservation to species |
Allelic loss of functiona (NPC localization/ depletion-addback/ SMAD interaction/ SMAD dependent transcription) |
EVS allele frequencies |
Gender | Ethnic origin |
Parental consan- guinity |
Age of onset (ESRD) |
Extrarenal manifestations |
Biopsy (at age) |
Therapy and response |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NUP93 | |||||||||||||||
| A2403 -21 |
c.1162C>T | p.Arg388Trp | 11 (het, p) | 0.940 | del | C. elegans | (+/-/+/-) | TT=0/TC=10/CC=6488 | F | Serbian | N | 6 yrs (6 yrs) |
HU | FSGS, tubulo-interstitial infiltrations, tubular dilation (6 yrs) | RTX (12 yrs) US: hyperechogenic kidneys |
| c.1772G>T | p.Gly591Val | 16 (het, m) | 1 | del | S. cerevisiae | (+/+/-/-) | TT=0/TG=2/GG=6496 | ||||||||
| A3256 -21 |
c.1326delG | p.Lys442Asn fs*14 | 12 (het, m) | NA | NA | NA | (-/-/-/-) | - | F | German | N | ∼3yrs (3 yrs) |
HU | FSGS, tubular atrophy, interstitial infiltrations (3yrs) | SRNS, CsA-PR, RTX (6 yrs)39 |
| c.1772G>T | p.Gly591Val | 16 (het, p) | 1 | del | S. cerevisiae | (+/+/-/-) | TT=0/TG=2/GG=6496 | ||||||||
| A1394 -21 |
c.1537+1G>A | del ex13 | 13 (het, m) | NA | NA | NA | (-/-/+/-) | AA=0/AG=1/GG=6496 | F | German | N | 3 yrs (4 yrs) |
HU, Marcus-Gunn-syndrome | FSGS, global sclerosis with immature glomeruli (3 yrs) | SRNS, RTX (4 yrs) |
| c.1772G>T | p.Gly591Val | 16 (het, p) | 1 | del | S. cerevisiae | (+/+/-/-) | TT=0/TG=2/GG=6496 | ||||||||
| A1626 -21 |
c.1772G>T | p.Gly591Val | 16 (hom, m, p) | 1 | del | S. cerevisiae | (+/+/-/-) | TT=0/TG=2/GG=6496 | M | Turkish | Y | 3 yrs (11 yrs) |
NP | FSGS (3 yrs) | SRNS, ACE-I |
| -22 | F | 6 yrs | NP | ACE-I | |||||||||||
| A1671 -21 |
c.1886A>G |
|
17 (hom, m) | 0.997 | del | S. cerevisiae | (+/+/-/-) | - | M | Turkish | Y | 1 yr (1 yr) |
HU | DMS/FSGS (15 mo) | SRNS, RTX (3 yrs) |
| A2241 -22 |
c.1886A>G |
|
17 (hom) | 0.997 | del | S. cerevisiae | (+/+/-/-) | - | M | Turkish | Y | 1 yr | HU | ND | SRNS |
| NUP205 | |||||||||||||||
| A1733 -21 |
c.5984T>C | p.Phe1995Ser | 43 (hom, m, p) | 0.888 | del | D. melanogaster | abrogation of NUP93 interaction | - | F | Turkish | N | 3 yrs (7 yrs) |
NP | FSGS (3.5 yrs) | RTX (7 yrs) SRNS |
| -22 | M | 2 yrs | BAV, AI, ARE | FSGS (3 yrs) | SPR | ||||||||||
| XPO5 | |||||||||||||||
| F1092 -21 |
c.1654G>A | p.Val552Ile | 15 (hom, m, p) | 0.45 | del | C. intestinalis | n/a | - | M | Turkish | Y | 2 yrs | Speech development delay | MCNS (2 yrs) No effacement | Primary SRNS with response to CsA |
ACE; angiotensin converting enzyme inhibitor; AI, Aortic insufficiency; ARE, Aortic root enlargement; BAV, bicuspid aortic valve; CsA, cyclosporin A; ESRD, end-stage renal disease; del, deleterious; F, female; FSGS, focal segmental glomerulosclerosis; het, heterozygous; HD, hemodialysis; hom, homozygous; HU, hematuria; m, maternal; M, male; MCNS, Minimal change Nephrotic syndrome; ND, not done; NP, not present; NPC, nuclear pore complex; p, paternal; PPH2 score humvar PolyPhen2 prediction score; PR, partial response; RTX, renal transplantation; SIFT, Sorting tolerant from intolerant; SPR, steroid partial response; SRNS, steroid resistant nephrotic syndrome; Y, yes; yrs, years; A Western European founder mutation is single underlined, and a Turkish founder mutation is double underlined.
“-” indicates a defect of the allele in one of the 4 assays of NUP93 function: localization to the NPC (Fig. 3a,d), assembly of a minimal nuclear membrane (depletion-addback assay, Fig.3b-c), interaction with SMAD4 / importin7 (Fig. 4), SMAD4 nuclear translocation, or SMAD dependent transcription (Fig. 5).
Figure 2. Subcellular localization of NUP93 in podocytes and NUP93 knockdown resulting in reduced podocyte migration, proliferation, and impaired resistance to oxidative stress.
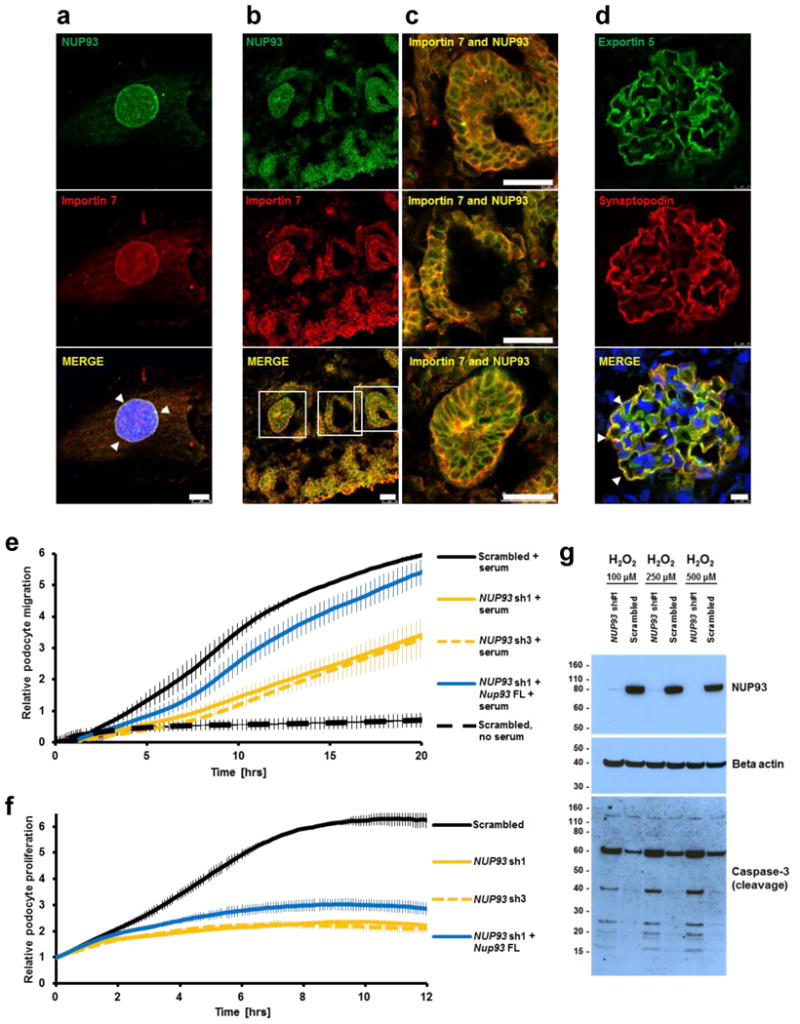
(a) In BMP7 treated human podocytes the nucleoporin NUP93 (green) and the nuclear transport factor importin7 (red) colocalize to a nuclear rim (arrow heads). (b–c) Neonatal rat kidney sections were stained with antibodies against NUP93 (green) and importin7 (red). Both proteins colocalize in podocyte precursor cells in different stages of glomerular development and other structures of the developing kidney. (c) Magnifications of the capillary loop and renal vesicle stage. (d) In adult rat glomerulus the nuclear exportin XPO5 partially colocalizes with the glomerular slit membrane marker synaptopodin (arrow heads). DAPI (blue) stains DNA. Scale bars are 10 μm in a, d and 25 μm in b, c. (e) Knockdown of NUP93 in human podocytes using two different shRNAs impairs podocyte migration (yellow curve vs. black curve). The decrease in podocyte migration was partially rescued by transfection of mouse Nup93 cDNA (blue curve). (f) Upon knockdown of NUP93 using two different shRNAs, the proliferation rate of human podocytes is severely impaired (yellow curve vs. black curve). Transfection of mouse Nup93 cDNA partially reversed the effect (blue curve). Experiments in e, f were performed in triplicates. Data points represent mean and standard deviation. (g) Addition of sub-lethal concentrations of H2O2 (100 μM, 250 μM, and 500 μM) to human podocyte culture does not induce apoptosis in control cells. In contrast, upon knockdown of NUP93 H2O2 induces apoptotic cell death as shown by increased cleavage of caspase-3.
Phenotypically, all 7 individuals of 6 families with recessive NUP93 mutations had SRNS that manifested early, i.e. between 1 and 6 years of age, and caused ESKD between ages 1 and 11 years (Table 1). Renal biopsy revealed FSGS or its developmental equivalent diffuse mesangial sclerosis (DMS) in the 5 individuals in whom a biopsy was performed (Table 1, Fig. 1b–c,g–h and Supplementary Fig. 3A). In addition, there was a renal tubular phenotype with proximal tubular dilation with protein casts and interstitial cell infiltrations (Fig. 1c, Supplementary Fig. 3B). Electron microscopy revealed partial podocyte foot process effacement (Supplementary Fig. 3C). It is known that glomerular developmental defects (diffuse mesangial sclerosis, DMS) and glomerular degenerative defects (focal segmental glomerular sclerosis, FSGS) can occur on a monogenic basis due to multiple allelism.19 However, in NUP93 mutations only one family had features of DMS, whereas 4 others had FSGS, making a glomerular developmental defect unlikely. One patient showed partial response to steroids, and two patients responded partially to CSA. A partial response to therapy with alternative agents is a rare but known feature of monogenic forms of nephrotic syndrome that is otherwise steroid resistant.20 However no genotype-phenotype correlation has been detected so far in these cases.
In addition, by genetic mapping (Fig. 1f) and whole exome sequencing in two siblings of family A1733 with early onset SRNS and FSGS we identified a homozygous missense mutation of the nucleoporin NUP205 (NM_015135.2) at a highly conserved amino acid residue (p.Phe1995Ser) (Fig. 1f, Table 1, Supplementary Fig. 2). Interestingly, NUP205 is a direct protein interaction partner of NUP93 within the inner ring of the NPC.17,21 Furthermore, by genetic mapping (Fig. 1i) and whole exome sequencing we identified a homozygous missense mutation of the nuclear export protein exportin5 (XPO5) (NM_020750.2) (p.Val552Ile) in individual F1092 with onset of SRNS at 2 years (Fig. 1i, Table 1, Supplementary Fig. 2). Remarkably, XPO5 is known to play a role in nuclear export in concert with nucleoporins.22-24
Nucleoporins (NUPs) are highly conserved eukaryotic proteins that form nuclear pore complexes (NPCs), huge macromolecular assemblies in the nuclear envelope which mediate the transport of proteins, RNAs and RNP particles between cytoplasm and the nuclear interior (Supplementary Fig. 4).25,17 NUPs function by interacting with transport receptors such as exportins and importins that shuttle their cargo through NPCs (Supplementary Fig. 4). While monogenic mutations in genes encoding NPC components have only been described in three other human diseases,2-4 we here implicate three NPC associated proteins in the pathogenesis of SRNS. We find, surprisingly, that very specific recessive mutations in genes that have a critical cellular function may cause a distinct renal disease phenotype.
To determine whether the two nucleoporins, NUP93 and NUP205, and the exportin XPO5 are expressed during renal development, we performed immunofluorescence (IF) studies in sections of fetal rat kidney. We observed that at the capillary loop stage of renal glomerular development NUP93, NUP205, and XPO5 are present in developing podocytes that are positive for the podocyte nuclear marker WT1 (Supplementary Fig. 5). Interestingly, in adult rat glomeruli, XPO5 colocalized in podocytes with synaptopodin, a marker of primary and secondary podocyte foot processes, in a pattern that has been described for many other products of genes that if mutated cause SRNS (Fig. 2d).8,26,27
To elucidate the role of the nucleoporin NUP93 in the pathogenesis of SRNS, we performed in-vitro functional assays in human immortalized podocytes. First, we studied podocyte migration, a well-established surrogate phenotype that is typically altered upon loss-of-function of genes that are involved in nephrotic syndrome.8-10 We show that knockdown of NUP93 using 2 different shRNAs resulted in an impaired migratory phenotype that was rescued by transfection of wildtype mouse Nup93 (Fig. 2e). Knockdown of the nucleoporin NUP153 in which we did not find mutations in individuals with SRNS did not affect podocyte migration (Supplementary Fig. 6). Furthermore, we found that upon knockdown of NUP93 the proliferation rate of human podocytes is strongly reduced (Fig. 2f). To assess oxidative stress resistance, we challenged human podocytes with sub-lethal concentrations of H2O2 (100 μM, 250 μM, and 500 μM). While H2O2 at these doses did not affect scrambled-control cells, it induced apoptotic cell death shown by increased cleavage of caspase-3 in NUP93 knockdown cells (Fig. 2g).
To assess whether the NUP93 mutations that we identified in individuals with SRNS interfere with nuclear pore complex (NPC) localization, we performed IF microscopy of the proteins of Myc tagged NUP93 constructs in human podocytes (Fig. 3a,c). Upon overexpression, wild type or some mutant constructs of NUP93 localized to the nuclear envelope, whereas constructs representing the truncating mutation p.Lys442Asnfs*14 and the splice site mutation p.del ex13, that we detected in individuals with SRNS (A3256 and A1394, respectively), failed to properly mark the nuclear envelope (Fig. 3a,c).
Figure 3. NUP93 mutations interfere with nuclear pore complex (NPC) assembly, and a NUP205 mutation affects NUP93-NUP205 interaction.
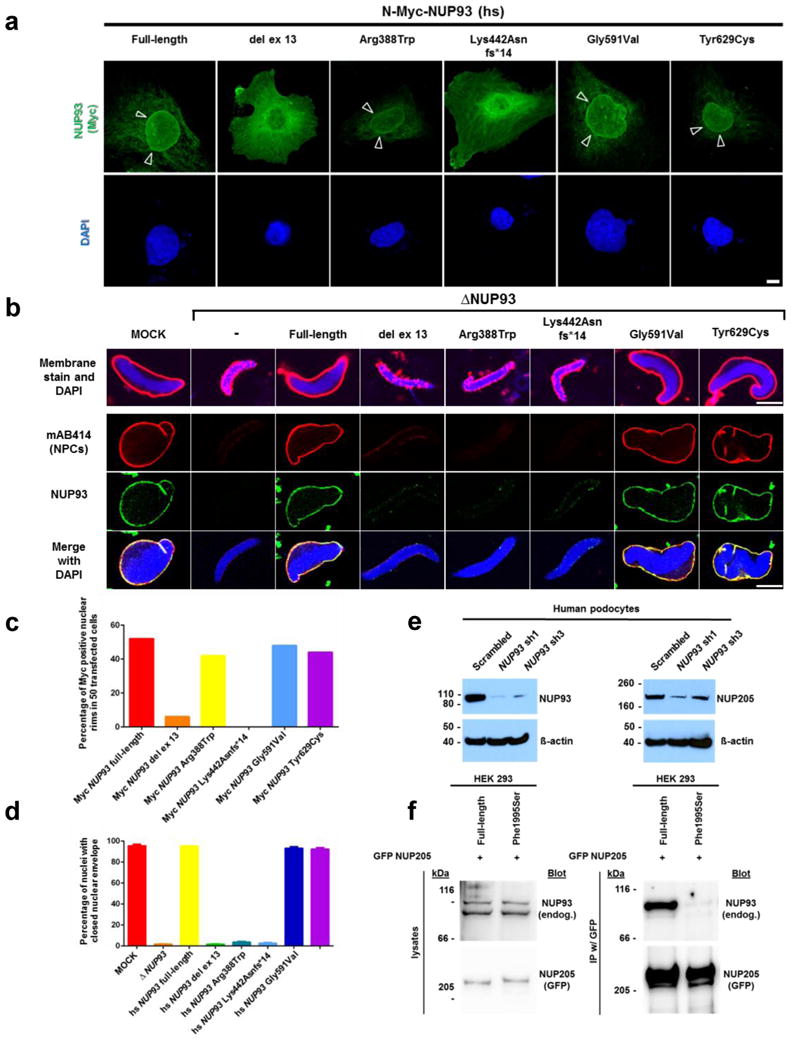
(a) Upon overexpression in human podocytes, Myc tagged NUP93 but not the splice site mutation del ex13 or the truncating mutation Lys442Asnfs*14, localize to the nuclear envelope (arrow heads). Scale bar 10 μm, see also c. (b) Mutations identified in individuals with SRNS fail to assemble an intact NPC in a depletion-addback assay in X. laevis egg extracts. Upon depletion of NUP93 nuclei fail to assemble properly. Addback of full-length or the mutant constructs Gly591Val or Tyr629Cys restore nuclear envelope and NPC assembly. The constructs of the in-frame deletion of exon 13 (del ex13), the missense mutation Arg388Trp, and the frameshift mutation Lys442Asnfs*14 lack this ability. Nuclear membranes are stained with DiIC18 (red, upper row), DNA with DAPI (blue), NPCs with mAB414 (second row, red), and α-Nup93 antibody (third row, green). Scale bar 10 μm, see also d. (c) Quantitation of nuclear localization data from (a) resulting from 50 transfected cells for each condition. (d) Quantitation of depletion-addback assay data from (b) presented as mean and standard deviation resulting from 100 nuclei each from three independent experiments. (e) Upon shRNA knockdown of NUP93 (left panel) immortalized human podocytes show a reduced expression level of NUP205 (right panel). (f) GFP tagged NUP205 precipitates endogenous NUP93 upon overexpression in HEK293 cells. A mutant allele of NUP205 identified in SRNS family A1733 (Phe1995Ser) lacks this interaction.
To further evaluate interference of mutations with nuclear envelope integrity, we performed a depletion-addback assay in Xenopus laevis egg extracts. In this assay formation of nuclear envelopes, indicated by a smooth membrane staining with DiIC18 as well as NPC formation (indicated by the antibody AB414) around sperm chromatin (Fig. 3b, upper and second rows), was faithfully reconstituted.28 Depletion of Nup93 abrogated nuclear envelope and pore formation,29,30 which was restored upon re-addition of wildtype protein or most mutants found in individuals with SRNS, but not by mutants del ex13, Arg388Trp, or Lys442Asnfs*14 (Fig. 3b,d). NUP93 tightly interacts with NUP20531 and, consistently, shRNA knockdown of NUP93 in podocytes caused depletion of NUP205 (Fig. 3e). Furthermore, we found that GFP tagged NUP205 interacts with endogenous NUP93 upon overexpression in HEK293 cells (Fig. 3f). The mutant NUP205 allele identified in family A1733 with SRNS, p.Phe1995Ser, abrogated this interaction (Fig. 3f). However, NUP93 mutations identified in families with SRNS did not abrogate the interaction with NUP205 (Supplementary Fig. 7).
It was recently shown that drosophila NUP93 and NUP205, apart from their known roles as scaffold nucleoporins in NPC assembly, also have cargo import functions and are essential for nuclear import of phosphorylated/activated SMADs (Supplementary Fig. 4).32 Interestingly, loss of function of other nucleoporins does not affect SMAD signaling.32 Following these previous findings, we performed further studies to define the role of NUP93 in SMAD signaling. Using immunofluorescence in human podocytes, we demonstrate that upon stimulation with BMP7 NUP93 colocalizes with the nuclear importer importin7 to a nuclear rim structure (Fig. 2a). Interestingly, both proteins also showed strong colocalization during different stages of renal development (Fig. 2b–c). Because of the described roles of NUP93 in SMAD signaling in drosophila, we explored whether NUP93 interacts with SMAD.32 We performed coimmunoprecipitation studies in HEK293 cells (Fig. 4). We demonstrate that SMAD4 interacts with endogenous NUP93 when overexpressed in HEK293 cells, which we confirmed for endogenous SMAD4 (Fig. 4a–b). Furthermore, we show that upon BMP7 stimulation NUP93 interacts with the phosphorylated/activated form of endogenous SMAD1/5 (Fig. 4d). Interestingly, the 3 NUP93 mutations p.Lys442Asnfs*14, p.Gly591Val, and p.Tyr629Cys that we found in patients with SRNS (Table 1, Fig. 1d–e) abrogated this interaction (Fig. 4c), likely reflecting their pathogenicity (Table 1). The abrogation of interaction was confirmed reciprocally (Supplementary Fig. 8).
Figure 4. NUP93, importin7, and exportin5 (XPO5) interact with SMAD proteins, and NUP93 mutations from individuals with SRNS abrogate the interaction with SMAD4 and importin7 upon coIP in HEK293 cells.
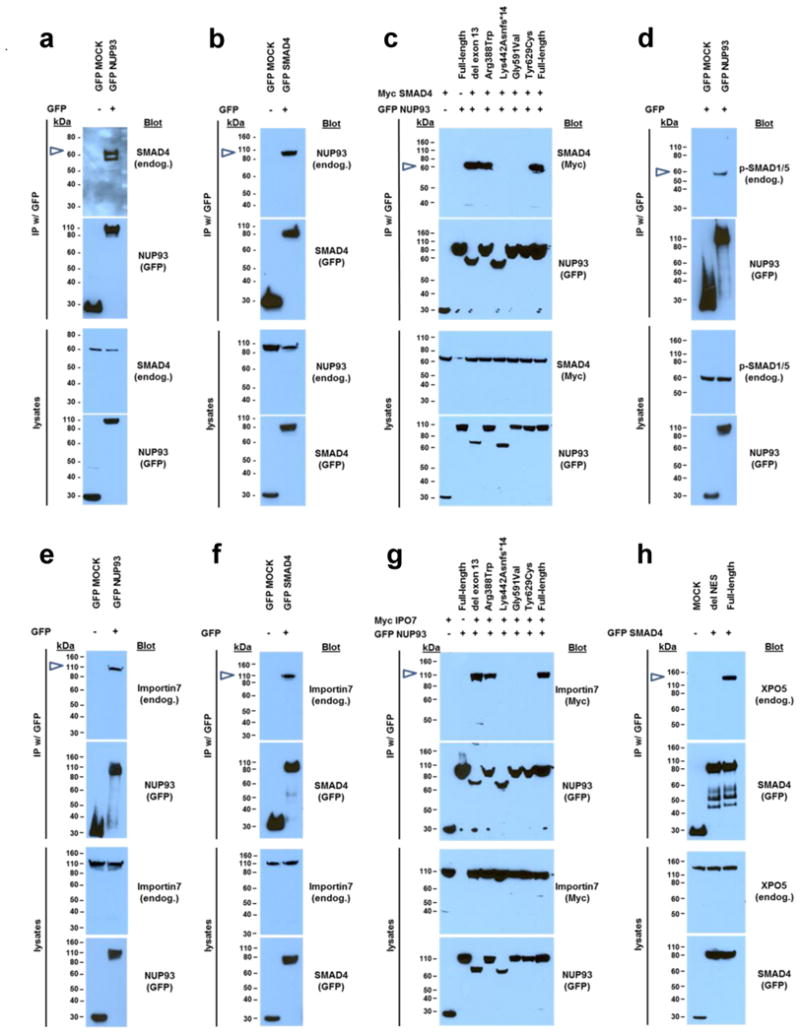
(a) C-terminally GFP tagged NUP93 precipitates endogenous SMAD4. (b) C-terminally GFP tagged SMAD4 precipitates endogenous NUP93. (c) Upon cooverexpression, GFP tagged NUP93 interacts with Myc tagged SMAD4. Mutations (Lys442Asnfs*14, Gly591Val, Tyr629Cys) identified in individuals with SRNS abrogate SMAD4 interaction. (d) Upon BMP7 stimulation C-terminally GFP-tagged NUP93 interacts with phosphorylated/activated SMAD1/5. (e) C-terminally GFP tagged NUP93 precipitates endogenous importin7. (f) C-terminally GFP tagged SMAD4 precipitates endogenous importin7. (g) Upon cooverexpression, GFP tagged NUP93 interacts with Myc tagged importin7. Mutations (Lys442Asnfs*14, Gly591Val, Tyr629Cys) identified in individuals with SRNS abrogate importin7 interaction. (h) GFP tagged SMAD4 precipitates endogenous XPO5. Deletion of the SMAD4 nuclear export signal (NES) (aa142-aa149) abrogates the interaction with XPO5. Coimmunoprecipitation experiments shown in a, b, d, e, and f were confirmed using N-terminally GFP-tagged fusion proteins.
Because of the suggested role of the karyopherin importin-7 (Msk) in NUP93 mediated nuclear import of SMAD,32,33 we explored whether the NUP93 mutations that we identified in individuals with SRNS affect BMP7 dependent SMAD signaling. We found that NUP93 and SMAD4 both interact with endogenous importin7 in humans (Fig. 4e–f). The same 3 mutations (p.Lys442Asnfs*14, p.Gly591Val, p.Tyr629Cys) that disrupted interaction with SMAD4 (Fig. 4c, Table 1, Fig. 1d–e) also abrogated interaction of NUP93 with importin7 (Fig. 4g). In addition, we found that NUP93 colocalizes importin7 in different stages of glomerular development and in podocyte precursor cells at the early capillary loop stage (Fig. 2b–c).
BMP7 plays a crucial role in renal development.34 Furthermore, growing experimental evidence suggests that BMP7 is an important mediator of renal response to injury, and has protective effects in multiple animal models of acute and chronic renal injury.35 BMP7 balances the profibrotic effects of TGF-β, and it has been shown that loss of SMAD4 promotes renal fibrosis and inflammation.36 In addition, BMP7 inhibits apoptotis and promotes podocyte survival in experimental models of diabetic nephropathy.37 Since, podocytes and collecting duct cells were shown to be the primary target cells of BMP7 in the kidney,38 we explored whether the NUP93 mutations that we identified in individuals with SRNS affect BMP7 dependent SMAD signaling. We demonstrate that knockdown of NUP93 in human podocytes using two different shRNAs disrupts BMP7 dependent activation of SMAD signaling as demonstrated by reduced nuclear accumulation of SMAD4 after BMP7 stimulation (Fig. 5a). An equivalent result was seen when using HEK 293 (Supplementary Fig. 9). Consistently, expression of mouse wildtype Nup93 but none of the 5 mutants detected in individuals with SRNS rescued the BMP7 dependent nuclear translocation of endogenous SMAD4 in knockdown cells (Fig. 5b, Supplementary Fig. 9B).
Figure 5. Knockdown of NUP93 interferes with BMP7-induced activation of SMAD signaling.
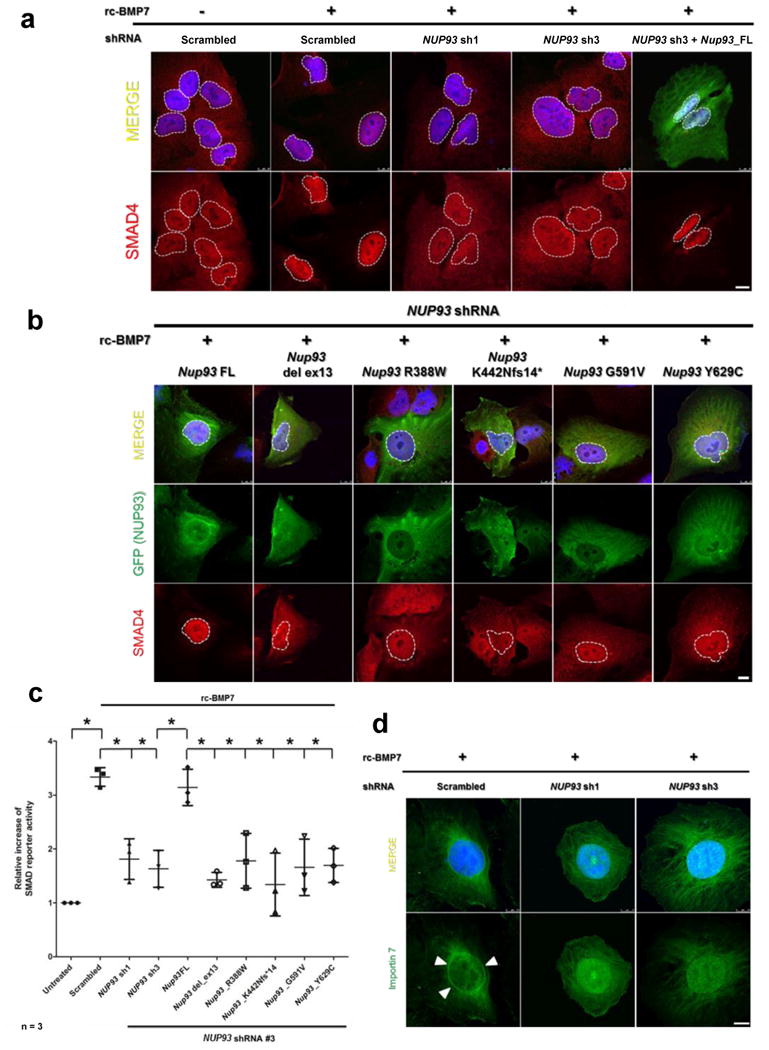
(a) Stimulation of human podocytes with recombinant BMP7 (rc-BMP7) (100 ng/μl) induces nuclear accumulation of SMAD4 in control cells (columns 1 vs. 2). Upon knockdown of NUP93 using two different shRNAs (sh1 and sh3) BMP7 fails to induce efficient nuclear translocation of SMAD4 (columns 3, 4). Transfection of full-length wildtype mouse Nup93 cDNA restores this effect (column 5). White dotted lines mark the borders of the nuclear area. Endogenous SMAD4 is stained in red. (b) Transfection of full-length Nup93 restores BMP7-induced nuclear accumulation of SMAD4 in NUP93 knockdown podocytes (column 1). In contrast, cells transfected with clones reflecting all 5 SRNS mutantions do not show efficient nuclear translocation of SMAD4 (columns 2-6). (c) In a luciferase reporter assay, treatment of HEK293 cells with BMP7 increases SMAD reporter activity. Knockdown of NUP93 with two different shRNAs (sh1 and sh3) reduces SMAD reporter activity in response to BMP7 stimulation. Transfection of mouse full-length Nup93 restores a cellular response that is compatible to control cells. In contrast, clones reflecting all 5 SRNS mutations fail to rescue the defects in BMP7-SMAD4 signaling. Data result from three independent experiments each, and are presented as mean and standard deviation. * Indicates statistical significance (p<0.05). (d) Upon stimulation of human podocytes with BMP7, endogenous importin7 (green) accumulates in a nuclear rim pattern (arrow heads). Knockdown of NUP93 by two shRNAs (sh1 and sh3) inhibits this rim formation. Scale bars are 10 μm. Nuclei are stained with DAPI (blue).
Upon shRNA knockdown of NUP93 in HEK293 cells BMP7 treatment failed to induce SMAD dependent reporter activity when compared to scrambled control (Fig. 5c). Reciprocally, whereas transfection of wild type full-length mouse Nup93 (under NUP93 knockdown) restored BMP7 dependent SMAD reporter activity, none of the constructs reflecting the 5 mutations detected in individuals with SRNS rescued SMAD4 dependent transcription (Fig. 5c). Furthermore, upon treatment of human podocytes with BMP7, importin-7 localizes to a nuclear rim structure in control cells (Fig. 2a, Fig. 5d). Knockdown of NUP93 by two different shRNAs inhibited this rim formation (Fig. 5d). Knockdown of the nucleoporin NUP188 in which we did not find mutations in individuals with SRNS did not reduce SMAD reporter activity in our assay (Supplementary Fig. 10).
We demonstrated loss of function for all NUP93 mutations detected in individuals with SRNS. NUP93 mutations either disrupt NPC integrity (Fig. 3a–c) (mutations p.del ex13, p.Arg388Trp, and p.Lys442Asnfs*14) or abrogate the interaction of NUP93 with SMAD4 (Fig. 4c) or importin7 (Fig. 4g) (mutations p.Lys442Asnfs*14, p.Gly591Val, p.Tyr629Cys) (see middle column of Table 1). In all individuals with mutations in NUP93 there was a defect in SMAD signaling, suggesting SMAD signaling as the relevant pathogenic pathway shared by these mutations. Furthermore, we show that XPO5, which we found defective in another individual with SRNS (Fig. 1i, Table 1) also interacts with SMAD4 (Fig. 4h).
In summary, we here demonstrate that specific mutations of NUP93, NUP205, or XPO5 cause a distinct entity of SRNS and we introduce the terms “NPHS11”, “NPHS12”, and “NPHS13”, respectively. We thereby link NPC associated proteins to a new pathogenic pathway for SRNS. Furthermore, we implicate BMP7 dependent SMAD signaling as a novel disease mechanism of SRNS, thereby potentially opening new approaches towards its therapy.
Online Methods
Research subjects
We obtained blood samples and pedigrees following informed consent from individuals with SRNS. Approval for human subjects research was obtained from Institutional Review Boards of the University of Michigan, and Boston Children's Hospital. The diagnosis of steroid-resistant nephrotic syndrome was based on published clinical criteria.
Linkage analysis
For genome-wide homozygosity mapping the GeneChip® Human Mapping 250k StyI Array from Affymetrix was used. Non-parametric LOD scores were calculated using a modified version of the program GENEHUNTER 2.140,41 through stepwise use of a sliding window with sets of 110 SNPs and the program ALLEGRO42 in order to identify regions of homozygosity as described12,43 using a disease allele frequency of 0.0001 and Caucasian marker allele frequencies. For graphical presentation (Fig. 1a, f, i, Suppl. Fig. 1) non-parametric lod scores (NPL) were calculated and plotted across the human genome. The x-axis shows Affymetrix 250K StyI array SNP positions on human chromosomes concatenated from p-ter (left) to q-ter (right). Genetic distance is given in cM.
Whole exome sequencing
Whole exome sequencing (WES) and variant burden analysis was performed as described previously44. In brief, genomic DNA was isolated from blood lymphocytes and subjected to exome capture using Agilent SureSelect™ human exome capture arrays (Life technologies™) followed by next generation sequencing on the HiSeq Illumina™ sequencing platform as previously described.
Mutation calling
Sequence reads were mapped against the human reference genome (NCBI build 37/hg19) using CLC Genomics Workbench (version 6.5.1) software (CLC bio). Variants with minor allele frequencies <1% in the dbSNP (Version 137) database were selected and annotated for impact on the encoded protein and for conservation of the reference base and amino acid among orthologs across phylogeny. Mutation calling was performed by geneticists/cell biologists, who had knowledge of the clinical phenotypes and pedigree structure, as well as experience with homozygosity mapping and exome evaluation.
High-throughput mutation analysis by array-based multiplex PCR and NGS
We used PCR-based 48.48 Access Array microfluidic technology (Fluidigm™) with consecutive next generation sequencing. We applied a 12-fold primer multiplexing approach allowing PCR-based amplification for 48 DNA samples simultaneously in 576 amplicons.14,15 A total of 1,800 individuals with nephrotic syndrome were analyzed, among them subset of 500 individuals with proteinuria and hematuria. After amplification of all targeted coding and splice site regions, sample-derived products were indexed with 384 different 10 bp-barcodes in a subsequent PCR. Finally, 2 × 250bp paired-end sequencing was performed on an Illumina™ MiSeq instrument. Bioinformatic analysis was conducted using CLC-Genomics-Workbench™ software. Potential mutations were confirmed by Sanger sequencing and evaluated for segregation. Primer sequences for targeted exon sequencing are provided in Suppl. Table 1.
cDNA and splice mutation
RNA of A1394-21 was purified from whole blood (purelink, Invitrogen) and cDNA was synthesized using a high fidelity RT-PCR system (Agilent Technologies). RT-PCR was performed on cDNA from A1394-21 and from healthy controls using exonic primers flanking exon 13. PCR products were Sanger sequenced and analyzed on an agarose gel to test for exon skipping (Suppl. Fig. 1).
cDNA cloning
Human NUP93 full-length cDNA was subcloned by PCR from human full-length cDNA (cDNA clone MGC: 21106 IMAGE: 4750923). Mouse NUP93 was subcloned from mouse NUP93 full-length cDNA (cDNA clone MGC: 28230, IMAGE: 3991335). Human NUP205 subcloned from human full-length cDNA (cDNA clone MGC: 168237 IMAGE: 9020614). Human XPO5 full-length was subcloned from human full-length cDNA (cDNA clone MGC: 74566 IMAGE: 5492046). Human SMAD4 full-length was subcloned from human full-length cDNA (cDNA clone MGC: 8602 IMAGE: 2961238), human SMAD2 was subcloned from human full-length cDNA (cDNA clone MGC: 34440 IMAGE: 5221801), human SMAD5 was subcloned from human full-length cDNA (cDNA clone MGC: 8960 IMAGE:3906006). Mouse Ipo7 was subcloned from mouse full-length Importin7 (cDNA clone, MGC: 175386, IMAGE: 100014508). Expression vectors were produced using LR clonase (Invitrogen®) following the manufacturer's instruction. The following expression vectors were used in this publication: pRK5-N-Myc, pCDNA6.2-N-GFP, pCDNA6.2-C-GFP, pCS-Dest2, and pSirenRetroQ. Clones reflecting the mutations identified in individuals with SRNS were introduced in the cDNA constructed using the Quick change II XL site-directed mutagenesis kit, Agilent Technologies. This technique was also applied to generate a human SMAD4 construct lacking the canonical nuclear export signal (amino acids 142-149).45
Cell lines
Experiments shown in this publication were performed in HEK293T cells and immortalized human podocytes. HEK293T cells were purchased from the ATCC biological resource center. Human immortalized podocytes were a kind gift from Moin Saleem, University of Bristol, Bristol, UK, and were cultured as previously described.46 Cell lines were tested for mycoplasma contamination on a quarterly basis.
Coimmunoprecipitation
Coimmunoprecipitation experiments were performed as described previously.47 Briefly, cell lysates were pre-cleared with protein G or A beads. Then, cell lysates were mixed with the appropriate antibodies and incubated overnight at 4°C in lysis buffer containing the complete protease inhibitor mixture. Immune complexes were collected by binding to mixed protein G or A beads and washed four times with lysis buffer prior to immunoblotting. Coimmunoprecipitation of GFP fusion proteins was performed using Chromotek-GFP-Trap® Agarose Beads, allele bioscience. Coimmunoprecipitation experiments were performed in three independent experiments, and protein-protein interactions were confirmed reciprocally.
SMAD translocation assays
Human immortalized podocytes were transfected with scrambled or shRNA against human NUP93 (see Suppl. Table 1 for targeted sequences). For rescue experiments, a second transfection with pCDNA6.2-N-GFP-MOCK, pCDNA6.2_murine Nup93 wildtype, or pCDNA6.2_murine Nup93 mutant clones was performed after 24 hrs. 60 minutes prior to the experiment cells were exposed to 100 ng/ml recombinant BMP7 (rc-BMP7, R&D systems). The knockdown efficiency, as well as effective activation of the BMP7-SMAD1/5 pathway was confirmed by immunoblotting (Fig. 3d, Suppl. Fig. 11).
SMAD reporter gene assay
The SMAD reporter assay was performed using the Cignal SMAD Reporter (luciferase) assay (Qiagen®) following the manufacturer's protocol. In brief, HEK293 cells were transfected with the Cignal SMAD reporter construct and scrambled or shRNA against human NUP93. After 24 hours cells were transfected with murine Nup93 (WT or mutants) or pCDNA6.2-N-GFP-MOCK. Eighteen hours prior to the experiment, cells were exposed to 100 ng/ml recombinant BMP7 (rc-BMP7, R&D systems). Luciferase activities were measured using Dual-Luciferase® Reporter Assay and GloMaxTM 96 microplate luminometer (Promega) according to the manufacturer's instruction. The luciferase activities were normalized to Renilla luciferase activities and protein concentration. Data points result from three independent experiments, and are presented as mean and standard deviation. P < 0.05 was considered statistically significant. For the data shown in Suppl. Fig. 10, HEK293 cells were transfected with ON-TARGETplus SMARTpool siRNA against NUP93, NUP153, and NUP188 (GE Dharmacon) at a final concentration of 100 nM using lipofectamine® RNAiMAX (Thermo Fisher) following the manufacturer's instructions. Experiments were performed 48 hrs after transfection.
Podocyte migration assay
The Podocyte migration assay was performed as previously described.8 Briefly, real-time migration assays were performed using the xCELLigence™ system (Roche Applied Science) with CIM-plate 16 according to the manufacturer's instructions. Immortalized human podocyte with shRNA mediated knockdown of NUP93 (shRNA #1 and shRNA#3) were transfected with MOCK-GFP or mouse full-length wildtype Nup93 cDNA (Nup93_FL). 36 hrs after transfection, 4 × 104 cells were seeded in serum free medium in the upper chambers of the migration plate. The lower chambers were filled with medium containing 15% FBS as chemoattractant, or with serum-free medium as control. Changes in impedance were analyzed using the RTCA software. Results were plotted as cell index (relative podocyte migration) vs. time. Each experiment was performed in triplicates, and repeated two times independently. Results are presented as mean with standard deviation.
Podocyte proliferation assay
The proliferation assay was performed using the xCELLigence system™ with E-plates 16 (Roche Applied Science) according to the manufacturer's instructions. The experiment was performed in immortalized human podocytes with shRNA mediated knockdown of NUP93 (shRNA #1 and shRNA#3). Cells were transfected with MOCK-GFP or mouse full-length wildtype Nup93 cDNA (Nup93_FL). 36 hrs after transfection, 2.5 × 104 cells were seeded in each well of the E-plate 16. Changes in impedance were analyzed using the RTCA software. Results were plotted as cell index (relative podocyte proliferation) vs. time. Each experiment was performed in triplicates, and repeated two times independently. Results are presented as mean with standard deviation. For the data shown in Suppl. Fig. 6, immortalized human podocytes were transfected with ON-TARGETplus SMARTpool siRNA against NUP93, NUP153, and NUP188 (GE Dharmacon) at a final concentration of 100 nM using lipofectamine® RNAiMAX (Thermo Fisher) following the manufacturer's instructions. Experiments were performed 48 hrs after transfection.
In-vitro experiments to assess oxidative stress resistance
To assess oxidative stress resistance in human immortalized podocytes, H2O2 was added to cell culture medium at three different doses (100 μM, 250 μM, and 500 μM) that did not induce significant apoptosis or changes in cell morphology in control cells. After 60 minutes cell were lysed and apoptosis was assessed using an antibody against cleaved caspase-3 (cell signaling, #9661) in control (scrambled shRNA) as compared to knockdown cells. The result was shown in two independent experiments.
NUP93 depletion-addback assay in Xenopus laevis eggs
Nuclear assemblies using Xenopus egg extracts immunofluorescence on in vitro assembled nuclei, generation of affinity resins, sperm heads and floated unlabeled or DiIC18-labeled membranes were carried out as described48 and analyzed on an Olympus FV1000 confocal microscope. Nup93 was depleted as in Theerthagiri et al., 2010.31 mRNA encoding human Nup93 (GenBank accession NM_014669.4) and the corresponding mutants was prepared using the mMESSAGE mMachine kit (Life Technologies) and added to extracts at a concentration of 200 ng/μl. The experiment was repeated three times independently, and 100 nuclei were counted in each experiment. Results are shown as mean and standard deviation.
Knockdown in human podocytes
ShRNA against human NUP93 was subcloned into pSIREN RetroQ for retroviral transduction using HEK293T cells. 48 hours after transduction puromycin at a final concentration of 4 ug/ml was added to the medium for selection of transduced cells. See Suppl. Table 1 for target sequences, and Fig. 3d for knockdown efficiency.
Antibodies
For immunofluorescence experiments the following primary antibodies were used: mouse anti-NUP93 (F2), sc-374400, Santa Cruz Biotechnology; mouse anti-SMAD4 (B-8), sc-7966, Santa Cruz Biotechnology; and goat anti-Importin7, NB100-1081, Novus bioscience; and anti-TRP, mAB414, Covance. Antibodies against Xenopus Nup20531 and Nup9349 have been described. Donkey anti-goat Alexa 488 and Alexa-594 conjugated secondary antibodies, and DAPI (4′,6-Diamidino-2-Phenylindole, Dihydrochloride) were obtained from Invitrogen. For immunoblotting the following primary antibodies were used: mouse anti-NUP93 (F2), sc-374400, Santa Cruz Biotechnology; mouse anti-SMAD4 (B-8), sc-7966, Santa Cruz Biotechnology; goat anti-Importin7, NB100-1081, Novus bioscience; rabbit anti-NUP205, HPA024574, Atlas antibodies, Sigma; rabbit anti-phospho-Smad1/5, #9526, cell signaling. HRP labeled secondary antibodies were purchased from Santa Cruz. See Supplementary Fig. 12 for characterization of antibodies against human NUP93 and NUP205.
Immunofluorescence and confocal microscopy in cell lines
For immunostaining human immortalized podocytes were seeded on coverglasses, and grown at permissive temperature. For overexpression studies human podocytes were transiently transfected using lipofectamine 2000® following the manufacturer's instructions. Experiments were performed 24-48 hrs after transfection. Cells were fixed and permeabilized for 10 min using 4% paraformaldehyde and 0.25%Triton-X100. After blocking, cells were incubated with primary antibody over-night at 4°C. The cells were incubated in secondary antibodies for 90 min at room-temperature, followed by 5 min. staining with 1 × DAPI/PBS. Confocal imaging was performed using Leica SP5X system with an upright DM6000 microscope and images were processed with the Leica AF software suite. Immunofluorescence experiments were repeated at least twice in independent experiments.
Immunofluorescence and confocal microscopy on rat tissues
Frozen tissue sections were permeabilized in 0.25%Triton-X100, blocked in 10% donkey serum for an hour at room temperature, and incubated in primary antibody overnight. The cells were incubated in secondary antibodies for 90 min at room-temperature, followed by 5 min. staining with 1 × DAPI/PBS. Confocal imaging was performed using Leica SP5X system with an upright DM6000 microscope and images were processed with the Leica AF software suite.
Statistical analysis
Results in Fig. 5 are presented as means ± SD for the indicated number of experiments. Statistical analysis was performed with a 2-tailed Student t test using GraphPad Prism®. P < 0.05 was considered statistically significant.
Bioinformatics
Genetic location is according to the February 2009 Human Genome Browser data, hg19 assembly.
Supplementary Material
Acknowledgments
We are grateful to the families and study individuals for their contribution.
We thank the Yale Center for Mendelian Genomics for whole exome sequencing analysis and Michael Mihatsch and H. Hopfer (Basel, Switzerland) for providing histology data.
This research was supported by grants from the National Institutes of Health to F.H. (DK076683) and by core funding of the Max Planck Society to W.A.
H.Y.G. is supported by the NephCure/ASN Foundation for Kidney Research.
W.T. is supported by the ASN Foundation for Kidney Research.
F.O. is supported by the European Community's Seventh Framework Programme (FP7/2007-2013) (EURenOmics; grant 2012-305608). The Nephrogenetics Laboratory at Hacettepe University was established by the Hacettepe University Infrastructure Project (grant number 06A101008)
F.H. is an Investigator of the Howard Hughes Medical Institute, a Doris Duke Distinguished Clinical Scientist, and a Warren E. Grupe Professor.
Footnotes
Author Contributions: D.A.B., C.E.S., S.K., S.L., W.L.P., S.A., H.Y.G., S.S., M.A., W.T., J.R., D.S., W.I.C., T.H., R.P.L. and F.H. generated total genome linkage data, performed exome capture with massively parallel sequencing, and performed whole exome evaluation and mutation analysis.
D.A.B. performed luciferase reporter gene assay, podocyte in-vitro experiments, and co-immunoprecipitation experiments.
D.A.B., S.K., and J.A.L. performed immunofluorescence and subcellular localization studies in tissues and cell lines by confocal microscopy.
W.A. and S.A.A. performed depletion-addback assay, and co-immunoprecipitation experiments analyzing Nup93-Nup205 interaction.
M.K., M.P., F.O., M. Ko., R.B., R.Bu., E.S, U.H., D.A.B., C.E.S., S.K., S.L., W.L.P., S.A., H.Y.G., S.S., and F.H. recruited patients and gathered detailed clinical information for the study.
All authors critically reviewed the paper.
F.H. conceived of and directed the project and wrote the paper.
Competing interests statement: The authors declare that they have no competing financial interests.
References
- 1.Grossman E, Medalia O, Zwerger M. Functional architecture of the nuclear pore complex. Annual review of biophysics. 2012;41:557–584. doi: 10.1146/annurev-biophys-050511-102328. [DOI] [PubMed] [Google Scholar]
- 2.Basel-Vanagaite L, et al. Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Annals of neurology. 2006;60:214–222. doi: 10.1002/ana.20902. [DOI] [PubMed] [Google Scholar]
- 3.Tullio-Pelet A, et al. Mutant WD-repeat protein in triple-A syndrome. Nature genetics. 2000;26:332–335. doi: 10.1038/81642. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008;135:1017–1027. doi: 10.1016/j.cell.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 5.Machuca E, Benoit G, Antignac C. Genetics of nephrotic syndrome: connecting molecular genetics to podocyte physiology. Human molecular genetics. 2009;18:R185–194. doi: 10.1093/hmg/ddp328. [DOI] [PubMed] [Google Scholar]
- 6.Smith JM, Stablein DM, Munoz R, Hebert D, McDonald RA. Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Pediatr Transplant. 2007;11:366–373. doi: 10.1111/j.1399-3046.2007.00704.x. [DOI] [PubMed] [Google Scholar]
- 7.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney international. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 8.Gee HY, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. The Journal of clinical investigation. 2013;123:3243–3253. doi: 10.1172/JCI69134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gee HY, et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. The Journal of clinical investigation. 2015;125:2375–2384. doi: 10.1172/JCI79504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashraf S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. The Journal of clinical investigation. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadowski CE, et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. Journal of the American Society of Nephrology : JASN. 2014;26:1279–1289. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hildebrandt F, et al. A Systematic Approach to Mapping Recessive Disease Genes in Individuals from Outbred Populations. PloS Genetics. 2009;5:31000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otto EA, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nature genetics. 2010;42:840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halbritter J, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. Journal of medical genetics. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 15.Halbritter J, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Human genetics. 2013;132:865–884. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grandi P, Doye V, Hurt EC. Purification of NSP1 reveals complex formation with ‘GLFG’ nucleoporins and a novel nuclear pore protein NIC96. The EMBO journal. 1993;12:3061–3071. doi: 10.1002/j.1460-2075.1993.tb05975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vollmer B, Antonin W. The diverse roles of the Nup93/Nic96 complex proteins - structural scaffolds of the nuclear pore complex with additional cellular functions. Biological chemistry. 2014;395:515–528. doi: 10.1515/hsz-2013-0285. [DOI] [PubMed] [Google Scholar]
- 18.Allende ML, et al. Insertional mutagenesis in zebrafish identifies two novel genes, pescadillo and dead eye, essential for embryonic development. Genes & development. 1996;10:3141–3155. doi: 10.1101/gad.10.24.3141. [DOI] [PubMed] [Google Scholar]
- 19.Hildebrandt F, Heeringa SF. Specific podocin mutations determine age of onset of nephrotic syndrome all the way into adult life. Kidney international. 2009;75:669–671. doi: 10.1038/ki.2008.693. [DOI] [PubMed] [Google Scholar]
- 20.Ruf RG, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. Journal of the American Society of Nephrology : JASN. 2004;15:722–732. doi: 10.1097/01.asn.0000113552.59155.72. [DOI] [PubMed] [Google Scholar]
- 21.Amlacher S, et al. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell. 2011;146:277–289. doi: 10.1016/j.cell.2011.06.039. [DOI] [PubMed] [Google Scholar]
- 22.Brownawell AM, Macara IG. Exportin-5, a novel karyopherin, mediates nuclear export of double-stranded RNA binding proteins. The Journal of cell biology. 2002;156:53–64. doi: 10.1083/jcb.200110082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohnsack MT, et al. Exp5 exports eEF1A via tRNA from nuclei and synergizes with other transport pathways to confine translation to the cytoplasm. The EMBO journal. 2002;21:6205–6215. doi: 10.1093/emboj/cdf613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 25.Wente SR, Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol. 2010;2:a000562. doi: 10.1101/cshperspect.a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ebarasi L, et al. Defects of CRB2 Cause Steroid-Resistant Nephrotic Syndrome. American journal of human genetics. 2014;96:153–161. doi: 10.1016/j.ajhg.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gee HY, et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. American journal of human genetics. 2014;94:884–890. doi: 10.1016/j.ajhg.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gant TM, Wilson KL. Nuclear assembly. Annu Rev Cell Dev Biol. 1997;13:669–695. doi: 10.1146/annurev.cellbio.13.1.669. [DOI] [PubMed] [Google Scholar]
- 29.Sachdev R, Sieverding C, Flotenmeyer M, Antonin W. The C-terminal domain of Nup93 is essential for assembly of the structural backbone of nuclear pore complexes. Mol Biol Cell. 2012;23:740–749. doi: 10.1091/mbc.E11-09-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grandi P, et al. Nup93, a vertebrate homologue of yeast Nic96p, forms a complex with a novel 205-kDa protein and is required for correct nuclear pore assembly. Mol Biol Cell. 1997;8:2017–2038. doi: 10.1091/mbc.8.10.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Theerthagiri G, Eisenhardt N, Schwarz H, Antonin W. The nucleoporin Nup188 controls passage of membrane proteins across the nuclear pore complex. J Cell Biol. 2010;189:1129–1142. doi: 10.1083/jcb.200912045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, Xu L. Specific nucleoporin requirement for Smad nuclear translocation. Molecular and cellular biology. 2010;30:4022–4034. doi: 10.1128/MCB.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao X, Chen X, Cottonham C, Xu L. Preferential utilization of Imp7/8 in nuclear import of Smads. The Journal of biological chemistry. 2008;283:22867–22874. doi: 10.1074/jbc.M801320200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dudley AT, Lyons KM, Robertson EJ. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes & development. 1995;9:2795–2807. doi: 10.1101/gad.9.22.2795. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura J, Yanagita M. Bmp modulators in kidney disease. Discovery medicine. 2012;13:57–63. [PubMed] [Google Scholar]
- 36.Meng XM, et al. Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney international. 2012;81:266–279. doi: 10.1038/ki.2011.327. [DOI] [PubMed] [Google Scholar]
- 37.Mitu GM, Wang S, Hirschberg R. BMP7 is a podocyte survival factor and rescues podocytes from diabetic injury. American journal of physiology. Renal physiology. 2007;293:F1641–1648. doi: 10.1152/ajprenal.00179.2007. [DOI] [PubMed] [Google Scholar]
- 38.Leeuwis JW, et al. Direct visualization of Smad1/5/8-mediated transcriptional activity identifies podocytes and collecting ducts as major targets of BMP signalling in healthy and diseased kidneys. The Journal of pathology. 2011;224:121–132. doi: 10.1002/path.2844. [DOI] [PubMed] [Google Scholar]
- 39.Klaassen I, et al. Response to cyclosporine in steroid-resistant nephrotic syndrome: discontinuation is possible. Pediatric nephrology. 2015 doi: 10.1007/s00467-015-3109-3. [DOI] [PubMed] [Google Scholar]
- 40.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. American journal of human genetics. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 41.Strauch K, et al. Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. American journal of human genetics. 2000;66:1945–1957. doi: 10.1086/302911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nature genetics. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 43.Sayer JA, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nature genetics. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 44.Boyden LM, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe M, Masuyama N, Fukuda M, Nishida E. Regulation of intracellular dynamics of Smad4 by its leucine-rich nuclear export signal. EMBO reports. 2000;1:176–182. doi: 10.1093/embo-reports/kvd029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saleem MA, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. Journal of the American Society of Nephrology : JASN. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 47.Zariwala, Maimoona A, et al. ZMYND10 Is Mutated in Primary Ciliary Dyskinesia and Interacts with LRRC6. The American Journal of Human Genetics. 2013;93:336–345. doi: 10.1016/j.ajhg.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisenhardt N, Schooley A, Antonin W. Xenopus in vitro assays to analyze the function of transmembrane nucleoporins and targeting of inner nuclear membrane proteins. Methods in cell biology. 2014;122:193–218. doi: 10.1016/B978-0-12-417160-2.00009-6. [DOI] [PubMed] [Google Scholar]
- 49.Franz C, et al. MEL-28/ELYS is required for the recruitment of nucleoporins to chromatin and postmitotic nuclear pore complex assembly. EMBO reports. 2007;8:165–172. doi: 10.1038/sj.embor.7400889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.