

Abstract
Alcohol is the most commonly abused substance among adolescents, promoting the development of substance use disorders and compromised decision-making in adulthood. We have previously demonstrated, with a preclinical model in rodents, that adolescent alcohol use results in adult risk-taking behavior that positively correlates with phasic dopamine transmission in response to risky options, but the underlying mechanisms remain unknown. Here, we show that adolescent alcohol use may produce maladaptive decision-making through a disruption in dopamine network dynamics via increased GABAergic transmission within the ventral tegmental area (VTA). Indeed, we find that increased phasic dopamine signaling after adolescent alcohol use is attributable to a midbrain circuit, including the input from the pedunculopontine tegmentum to the VTA. Moreover, we demonstrate that VTA dopamine neurons from adult rats exhibit enhanced IPSCs after adolescent alcohol exposure corresponding to decreased basal dopamine levels in adulthood that negatively correlate with risk-taking. Building on these findings, we develop a model where increased inhibitory tone on dopamine neurons leads to a persistent decrease in tonic dopamine levels and results in a potentiation of stimulus-evoked phasic dopamine release that may drive risky choice behavior. Based on this model, we take a pharmacological approach to the reversal of risk-taking behavior through normalization of this pattern in dopamine transmission. These results isolate the underlying circuitry involved in alcohol-induced maladaptive decision-making and identify a novel therapeutic target.
SIGNIFICANCE STATEMENT One of the primary problems resulting from chronic alcohol use is persistent, maladaptive decision-making that is associated with ongoing addiction vulnerability and relapse. Indeed, studies with the Iowa Gambling Task, a standard measure of risk-based decision-making, have reliably shown that alcohol-dependent individuals make riskier, more maladaptive choices than nondependent individuals, even after periods of prolonged abstinence. Using a preclinical model, in the current work, we identify a selective disruption in dopamine network dynamics that may promote maladaptive decision-making after chronic adolescent alcohol use and demonstrate its pharmacological reversal in adulthood. Together, these results highlight a novel neural mechanism underlying heightened risk-taking behavior in alcohol-dependent individuals and provide a potential therapeutic target for further investigation.
Keywords: adolescent, alcohol, decision-making, dopamine, GABA, therapeutic
Introduction
Adolescent alcohol use is increasingly recognized as a major public health concern due to findings implicating age of alcohol use onset in the development of alcohol use disorders and decision-making deficits in adulthood (Grant et al., 1997; Stout et al., 2005). Indeed, age of onset in alcohol use increases the risk for subsequent substance use disorders by 10% each year (Grant et al., 2001) and 27% of 13-year-olds report consuming alcohol (Johnston et al., 2014). Adolescence is a critical period of brain development characterized by maturation of brain regions implicated in reward and decision-making (Spear, 2000). Specifically, the mesolimbic dopamine system undergoes profound development during the adolescent time window (Marinelli and McCutcheon, 2014) and therefore may be uniquely vulnerable to environmental insults, such as alcohol exposure (Chambers et al., 2003). Dopamine signaling in this system, particularly phasic dopamine transmission in the nucleus accumbens core (NAcc), is implicated in reward learning (Flagel et al., 2011), substance abuse (Phillips et al., 2003), and decision-making under risk (St Onge and Floresco, 2009; Nasrallah et al., 2011; Sugam et al., 2012; Stopper et al., 2013; Stopper et al., 2014).
In support of the association between early-life alcohol use and perturbation to dopamine-mediated behaviors, a recent IMAGEN study demonstrated that high levels of adolescent drinking are associated with novelty-seeking behavior (Whelan et al., 2014). Similarly, data from the Iowa Gambling Task (IGT), a standard measure of risk-based decision-making, demonstrated that alcohol-dependent individuals make riskier, more maladaptive choices compared with nondependent individuals (Brevers et al., 2014). Importantly, it has been suggested that such deficits in decision-making may represent a vulnerability to addictive disorders (Redish et al., 2008). Mirroring the pattern shown with the IGT, we have demonstrated preclinically that voluntary alcohol consumption during adolescence promotes maladaptive risk-taking behavior on a probability-discounting task in adulthood (Nasrallah et al., 2011; Clark et al., 2012; Schindler et al., 2014). In addition, we have established a neural correlate of risk within the NAcc where phasic dopamine release in response to risky options is increased by alcohol exposure (Nasrallah et al., 2011). We have further demonstrated that adolescent alcohol intake potentiates stimulus-evoked phasic NAcc dopamine release and biases rats toward a dopamine-dependent incentive learning strategy (Spoelder et al., 2015). Therefore, an appealing theoretical approach is to link developmental perturbations in dopamine signaling with maladaptive decision-making and addiction vulnerability, but the underlying circuits, mechanisms, and therapeutic targets that these may engender remain poorly understood.
Here we use a multifaceted approach, including fast-scan cyclic voltammetry (FSCV), slice electrophysiology, microdialysis, and behavioral pharmacology to address these questions. We demonstrate that midbrain circuitry is compromised by early-life alcohol exposure, featuring a persistent potentiation of input to the mesolimbic dopamine system from the pedunculopontine tegmentum (PPT), a key cortical relay important to decision-making under risk (Leblond et al., 2014; Stopper et al., 2014). Further, we show that tonic dopamine levels are decreased following adolescent alcohol exposure and negatively correlate with a rat's previous risk-taking behavior. Likewise, the electrophysiological excitability of VTA dopamine neurons is decreased, and we postulate that increased inhibitory tone following adolescent alcohol intake potentially drives the increased phasic dopamine transmission in adulthood through a disinhibitory mechanism (Lobb et al., 2011a, b; Paladini and Roeper, 2014; Oster et al., 2015). These results provide evidence for a model where chronic drug/alcohol use may persistently decrease VTA dopamine neuronal activity and tonic dopamine levels in the striatum, reflecting an enduring negative affective state, while in parallel increasing phasic dopamine responses to salient drug-related stimuli that may drive relapse and continued drug abuse (Robinson and Berridge, 2003; Wanat et al., 2009; Koob, 2013). Based on these findings, we used a selective GABA(A) receptor allosteric agonist, which attenuated the increase in phasic dopamine transmission and mitigated the maladaptive risk-taking behavior seen following adolescent alcohol intake, highlighting a potential new therapeutic for further investigation.
Materials and Methods
Animals and housing.
Male Sprague Dawley rats (Charles River) aged PND 27 at the start of experiments were housed on a 12 h light/dark cycle (lights on at 06:00) individually in polycarbonate tubs. Chow and water were available ad libitum, except as noted. All procedures were approved by the University of Washington Institutional Animal Care and Use Committee. Some rats (n = 18), representing the in vivo voltammetry dataset, were used in a previous study for separate analyses (Clark et al., 2012).
Alcohol preparation, administration, and withdrawal.
Alcohol (alcohol gel) was presented to adolescent rats in a gel matrix consisting of distilled water, Knox gelatin, Polycose (10%), and ethanol (10%). Control gels had ethanol replaced with distilled water. Preparation was as previously described (Schindler et al., 2014). Rats had access to gels 24 h/d, with fresh gel-containing jars presented every day. Intake levels were monitored daily and expressed in g/kg of body weight using individual gel consumption and body weights measured daily. Experiments began with 3 d of preexposure to control gel. Subsequently, rats were split into ethanol gel and control gel groups matched by weight and baseline intake. The 20 d (PND 30–49) of 24 h/d gel exposure followed. Predetermined intake exclusion criteria included rats that failed to consume gel during the control gel preexposure period, exhibited 3 consecutive days of no consumption, or exhibited excessive (>25% of days) burying of gel in bedding once the ethanol-gel exposure began. Ethanol gel intake averaged 9.0 ± 1.2 g/kg/d. Weight gain did not differ between control and ethanol rats over the 20 d of gel exposure (two-way repeated-measures ANOVA: gel treatment × time in days: F(19,1463) = 0.2, p > 0.05). Upon completion of the 20 d exposure, gel access for all groups was discontinued, and rats were monitored daily for withdrawal symptoms for the following 20 d. No overt signs of withdrawal were observed.
Nonsurvival voltammetry surgeries.
Starting 20 days after withdrawal, as previously described (Wanat et al., 2013), rats were anesthetized with 1.5 g/kg urethane (i.p.) and carbon-fiber electrodes targeting the NAc core (relative to bregma: 1.3 mm anterior, 1.3 mm lateral, 6.8–7.2 mm ventral) and an Ag/AgCl reference electrode were chronically implanted. Additional holes were drilled above the PPT (relative to bregma: 8.0 mm posterior, 2.0 mm lateral) and medial forebrain bundle (MFB) (relative to bregma: 4.6 mm anterior, 0.5 mm lateral) for stimulating electrodes.
Voltammetry recording sessions.
As previously described (Wanat et al., 2013), a triangle waveform was applied to the carbon fiber, which was ramped from −0.4 V to 1.3 V and back (vs Ag/AgCl) at a rate of 400 V/s and a frequency of 10 Hz (held at −0.4 V between scans). Dopamine release was evoked by electrical stimulation (60 pulses delivered at 60 Hz, 200 μA) via a bipolar stimulating electrode that was incrementally lowered into either the PPT or MFB. For all studies, stimulations were performed every 5 min.
Input/output experiments.
Once maximum stimulated dopamine release was achieved, stimulation current was varied from 25 to 400 μA (60 pulses, 60 Hz). Next, stimulation pulse number was varied from 60 to 3 pulses (400 μA, 60 Hz). Finally, stimulation frequency was varied from 60 to 5 Hz (200 μA, 30 pulses). Stimulations were performed every 5 min.
Pharmacology experiments.
Once maximum stimulated dopamine release was achieved, stimulations were performed every 5 min at 60 pulses, 60 Hz, and 200 μA until a stable baseline was reached (<10% deviation from the mean peak response of dopamine between 4 stimulations; i.e., 20 min of stimulations). Once a stable baseline was achieved, vehicle (35% 2-hydroxypropyl)-β-cyclodextrin (Sigma-Aldrich) or the selective GABA(A) receptor subunit 2, 3, and 5 allosteric agonist (L-838,417; 3 or 10 mg/kg, i.p.; Tocris Bioscience) was injected and stimulations continued for at least 40 min.
Behavioral experiments
Instrumental training.
Following >20 d of withdrawal, rats began food restriction to ∼90% of their free-feeding body weight. To reduce neophobia, rats were first exposed to 45 mg sucrose pellets (Bio-Serve) in their home cage. Next, rats underwent magazine training (10 sucrose pellets in 15 min) in a standard operant chamber (Med Associates), lever press training (instrumental FR1 schedule for single sucrose pellets on two separate levers; predetermined criterion of ≥24 level presses of 30 trials), and were finally autoshaped over the course of the next 4 d.
Probability-discounting task.
Following autoshaping, rats were tested on an instrumental response task as previously described (Schindler et al., 2014). Briefly, each rat was assigned one of two response levers (counterbalanced across groups) as the certain lever (certain delivery, 1.00, of two sucrose pellets) and the other as the risky lever (probabilistic delivery, 1.00, 0.75, 0.50, 0.25, or 0.00, of four sucrose pellets). Daily sessions consisted of 24 forced trials followed by 24 free choice trials. A 45 s intertrial interval was used, and failure to respond correctly within 10 s resulted in trial termination and return to the chamber intertrial interval state. All trials were cued with illumination of a light in the food tray, prompting the rat to make a nose-poke in the tray within 10 s. Following successful nose-poke, each forced trial consisted of the extension of a single lever, with a lever press resulting in illumination of the tray light and reward delivery based on the associated probability of that lever for that day (one probability per day, 5 d total). These forced choice sessions served to expose the rat to each lever's associated expected value, and the subsequent free choice trials had the same probability in effect for the uncertain lever. Each free choice trial was the same as above, but a successful nose-poke resulted in extension of both levers, allowing the rat to choose, and thus assessed the rat's preference between high/risky and low/safe reward options.
For the behavioral pharmacology experiment, vehicle or the selective GABA(A) receptor subunit 2, 3, and 5 allosteric agonist (L-838,417; 3 mg/kg, i.p.) was injected 20 min before forced trials on the 3 d of probabilistic reward on the risky lever (0.75, 0.50, and 0.25 probability). For consistency, a mock injection (i.p.) 20 min before forced trials on the 2 d of certain reward on the risky lever (1.0 and 0.00 probability) were also administered.
Slice electrophysiology.
Following 20 d of withdrawal, acute brain slices were prepared as described previously (Ting et al., 2014). Briefly, rats were anesthetized with Beuthanasia (200 mg/kg, i.p.) and were perfused with ∼40 ml of ice-cold carbogenated NMDG-ACSF containing the following (in mm): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2, 10 MgSO4, pH 7.3–7.4. The brain was removed and blocked, and 225 μm horizontal sections were made in ice-cold NMDG-ACSF. Slices were transferred to a chamber containing NMDG-ACSF at 33°C for ≤12 min and then were transferred to a second chamber containing room temperature HEPES-ACSF, composed of the following (in mm): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 2 CaCl2, 2 MgSO4. Constant bubbling of solutions with CO2/O2 was maintained throughout.
Whole-cell recordings were made using an Axopatch 700B amplifier (Molecular Devices) with filtering at 1 kHz using 4–6 m° electrodes. ACSF at 32°C containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 11 glucose, 18 NaHCO3, 2.4 CaCl2, continually bubbled with CO2/O2 and perfused over slices at a rate of ∼2 ml/min during recording. For sIPSCs, patch electrodes were filled with an internal solution containing the following (in mm): 70 KCl, 70 K-gluconate, 12 NaCl, 4 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, pH 7.2–7.4, 290 mOsm. For sEPSCs, patch electrodes were filled with an internal solution containing the following (in mm): 130 K-gluconate, 5 NaCl, 1 EGTA, 10 HEPES, 5 Mg-ATP, 0.5 Na-GTP, pH 7.2–7.4, 290 mOsm.
VTA dopamine neurons were identified by the presence of a large Ih current. Recordings were made in voltage-clamp mode at a holding potential of −60 mV. sIPSCs were recorded in the presence of kynurenic acid (2 mm), and sEPSCs were recorded in the presence of picrotoxin (100 μm). Spontaneous events were analyzed automatically and were visually inspected and confirmed using MiniAnalysis software (Synaptosoft).
Microdialysis with UPLC-tandem MS.
Following completion of the probability discounting task, control and ethanol vehicle-treated rats were anesthetized with 1.5 g/kg urethane (i.p.) and placed into a stereotaxic apparatus. A microdialysis cannula (BASi) targeting the nucleus accumbed core (relative to bregma: 1.3 mm anterior, 1.3 mm lateral, 5.25 mm ventral) was cemented in place. Next, a 2 mm in length microdialysis probe (BASi) was inserted into the cannula, flushed with artificial CSF (154.7 mm Na+, 0.82 mm Mg2+, 2.0 mm K+, 132.49 mm Cl−, 1.1 mm Ca2+, and 5.9 mm d-glucose) at 2 μl/min for 1.5 h and then at 1 μl/min for 30 min. Fractions were then collected on ice every 20 min into glass tubes (Waters) containing 0.01 N perchloric acid. An equal volume of 1.0 ng/ml 2-(3,4-dihydroxyphenyl)ethyl-1,1,2,2-d4-amine HCl (D4; as an internal standard) was added to each sample and then each fraction was rapidly frozen on dry ice and stored until further processing. Resulting fractions were analyzed on the AQUITY UPCL System (Waters), equipped with a Waters 2.1 × 100 mm HSS T3 reverse-phase. Eluting analytes were detected using a Xevo TQ-S Mass Spectrometer operating in positive mode performing dynamic multiple-reaction monitoring. The following compounds were monitored in the dialysate by the UPLC-MS method: DA and D4.
Histology.
After completion of the FSCV measurements, the carbon-fiber electrode recording site was marked by making a small electrolytic lesion at the electrode tip (∼70 μA current through the electrode for 30 s). Brains from both the FSCV and microdialysis experiments were removed, fixed in 4% PFA for 7 d, placed in 30% sucrose in PBS for 48 h, place in 10% sucrose for 48 h, flash frozen, and sectioned coronally (30 μm). Sections containing the NAcc, PPT, and MFB were mounted and stained with cresyl violet. Histology is shown in Figure 1.
Figure 1.
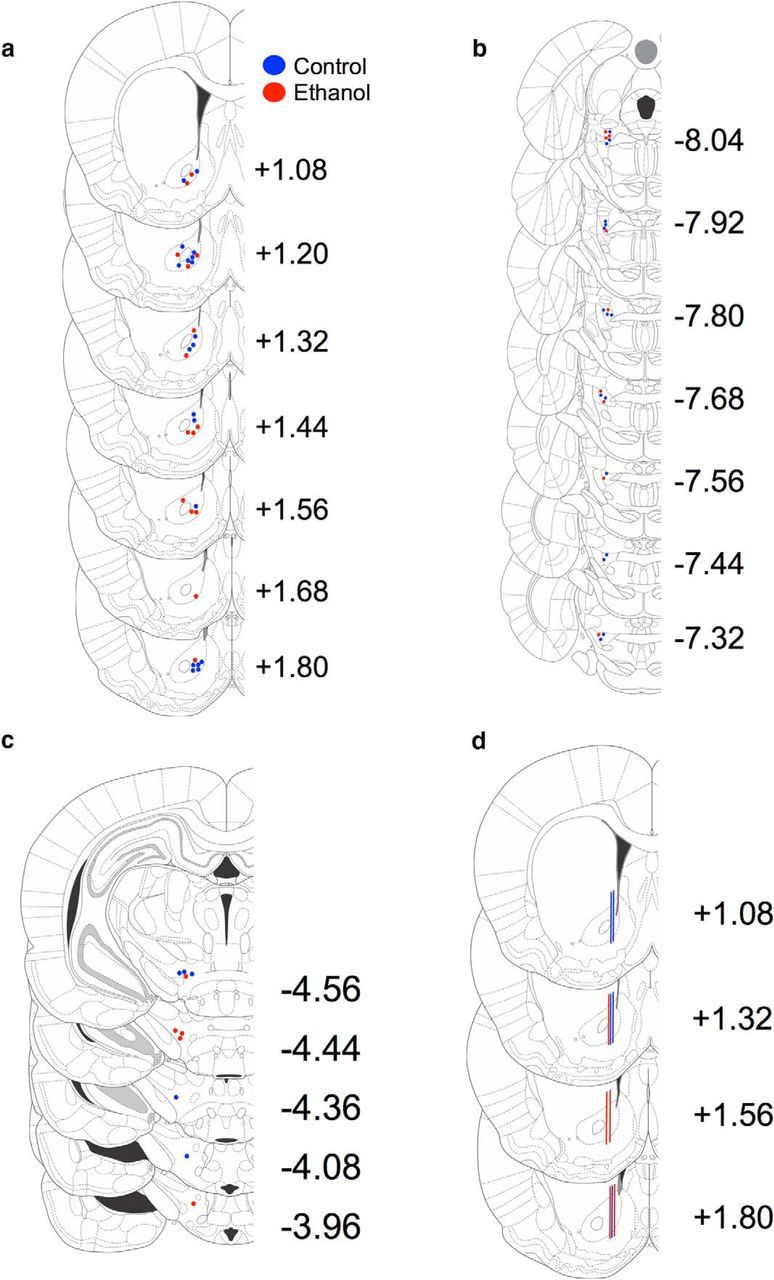
Histological verification or recording and stimulation sites. a, FSCV working electrode recording sites. b, PPT stimulating electrode sites. c, MFB stimulating electrode sites. d, Microdialysis probe sites.
Data analysis.
Chemometric analysis was used to isolate the voltammetric signal using a standard training set of stimulated dopamine release as previously described (Clark et al., 2010; Wanat et al., 2013; Heien et al., 2005). Voltammetric data analysis was performed using LabVIEW written software and was low-pass filtered at 2000 Hz. Analysis of stimulation-induced extracellular dopamine concentration was restricted to 3 s after electrical stimulation onset. For analysis of relative levels of tonic dopamine via microdialysis, each sample was normalized to its internal D4 standard. A power analysis with an α level of 0.05 and power level of 0.8, using the effect size and variance estimated from preliminary data were used to determine the number of rats used per experiment. The Kolmogorov–Smirnov test was performed on the residuals after data were fitted to a Gaussian curve with the mean and SD of each dataset to assess normality. Differences between groups were determined using repeated or nonrepeated measures one-, two-, and three-way ANOVA followed by Bonferroni post hoc. Student's unpaired, one-sample, or two-sample t tests were also used. The Pearson correlation test was used to assess the relationship between tonic dopamine and risk-taking behavior. Statistical analyses were conducted using GraphPad Prism 4.0. Rats were excluded from analysis if electrode and/or cannula placements were not correct (n = 4). Rats were also excluded if they did not meet predetermined intake and/or behavioral criteria (n = 32).
Results
Circuit-specific perturbation of phasic dopamine transmission following adolescent alcohol exposure
Voluntary consumption of ethanol during adolescence results in persistent risk-taking behavior and increased phasic dopamine transmission in response to cues signaling risky choices (Nasrallah et al., 2011). To examine the circuits underlying these findings, we used electrical stimulation of discrete brain regions coupled with FSCV in anesthetized rats. PPT stimulation increased phasic dopamine release in adult rats with and without a history of ethanol (linear regression: control; F(1,47) = 14.1, p < 0.0005, ethanol; F(1,40) = 54.3, p < 0.0001; Fig. 2a–c), consistent with previous reports (Wanat et al., 2013). However, PPT stimulation evoked significantly higher phasic dopamine release in adulthood following adolescent ethanol intake in response to increasing stimulation current (two-way repeated-measures ANOVA: adolescent treatment × current: F(6,66) = 8.8, p < 0.0001; Bonferroni post hoc analysis; Fig. 2a–d), stimulation duration (two-way repeated-measures ANOVA: adolescent treatment × duration: F(7,63) = 5.7, p < 0.0001; Bonferroni post hoc analysis; Fig. 2e,f), and stimulation frequency (two-way repeated-measures ANOVA: adolescent treatment × frequency: F(4,40) = 3.3, p < 0.02; Bonferroni post hoc analysis; Fig. 2g,h). Averaged traces for each input/output curve are shown in Figure 1d, f, and h, respectively.
Figure 2.
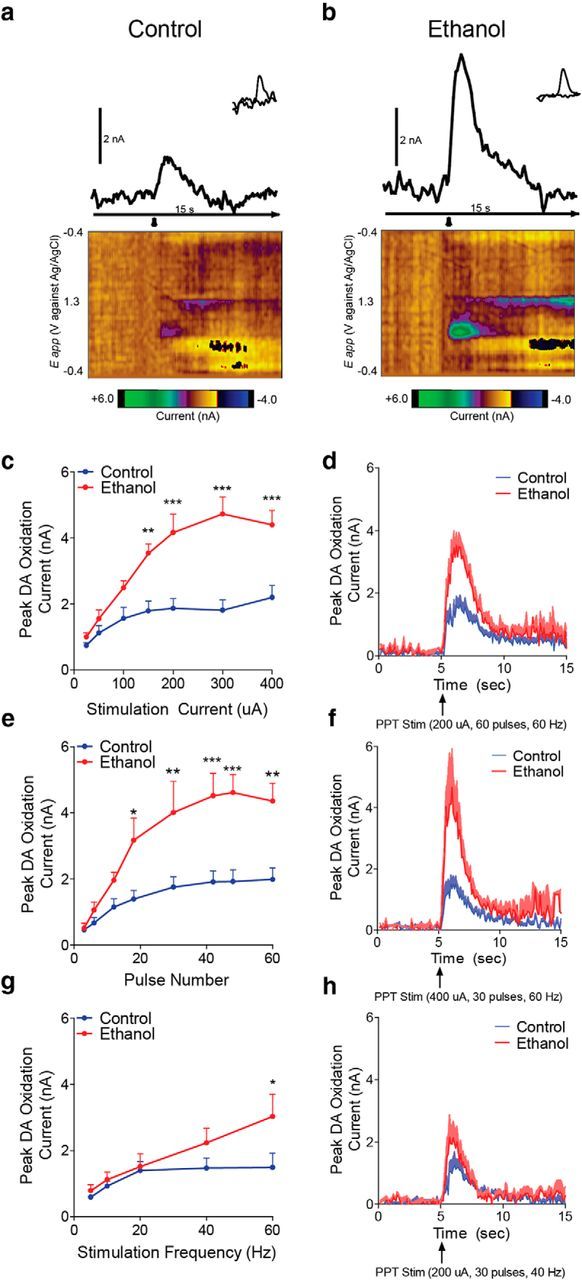
Phasic dopamine transmission is increased following adolescent alcohol use via a PPT-VTA circuit. a, b, Representative traces and corresponding background-subtracted cyclic voltammograms (inset) represent changes in dopamine oxidative current in the NAcc in response to PPT stimulation (arrowhead) in control (a) and ethanol-exposed rats (b). Color-coded observed changes in redox currents as a function of applied potential (y-axis) plotted over time (x-axis) are depicted in the pseudocolor plots. c, Following adolescent ethanol intake, PPT-evoked dopamine release is potentiated in adulthood in response to increasing stimulation current (two-way repeated-measures ANOVA interaction; Bonferroni post hoc analysis; control n = 7, ethanol n = 6). d, Average traces for stimulation current. e, Following adolescent ethanol intake, PPT-evoked dopamine release is potentiated in adulthood in response to increasing stimulation duration (two-way repeated-measures ANOVA interaction; Bonferroni post hoc analysis; control n = 6, ethanol n = 5). f, Average traces for stimulation duration. g, Following adolescent ethanol intake, PPT-evoked dopamine release is potentiated in adulthood in response to increasing stimulation frequency (two-way repeated-measures ANOVA interaction; Bonferroni post hoc analysis; control n = 6, ethanol n = 6), but only at the highest frequency. h, Average traces for stimulation frequency. Error bars indicate mean ± SEM. *p < 0.05. **p < 0.01. ***p < 0.001.
A potentiation in PPT-evoked phasic dopamine release could be attributable to changes in either the cells bodies located in the VTA or the presynaptic terminals located in the NAcc. To differentiate between these two alternatives, we next stimulated the MFB to bypass the VTA and specifically isolate terminals in the NAcc. MFB stimulation evoked robust dopamine release in both groups (Fig. 3a,b). However, unlike the PPT, MFB-evoked dopamine release did not differ between groups in response to increasing stimulation current (two-way repeated-measures ANOVA: adolescent treatment × current: F(6,48) = 0.3, p = 0.90; Fig. 3a–d), stimulation duration (two-way repeated-measures ANOVA: adolescent treatment × duration: F(7,42) = 0.5, p = 0.79; Fig. 3e,f), or stimulation frequency (two-way repeated-measures ANOVA: adolescent treatment × frequency: F(4,24) = 0.8, p = 0.51; Fig. 3g,h). Averaged traces for each input/output curve are shown in Figure 3d, f, and h, respectively.
Figure 3.
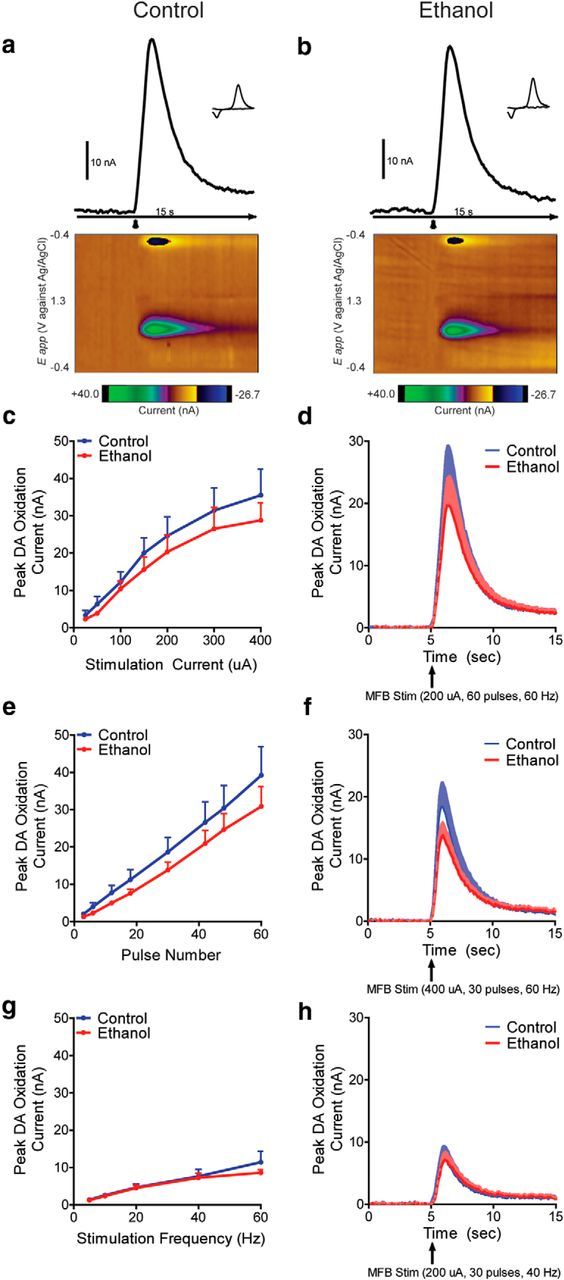
Adolescent alcohol does not increase MFB-stimulated NAcc phasic dopamine transmission in adulthood. a, b, Representative traces and corresponding background-subtracted cyclic voltammograms (inset) represent changes in dopamine oxidative current in the NAcc in response to MFB electrical stimulation (arrowhead) in control (a) and ethanol (b) exposed rats. Color-coded observed changes in redox currents as a function of applied potential (y-axis) plotted over time (x-axis) are depicted in the pseudocolor plots. c, MFB stimulation did not change NAcc phasic dopamine release in adulthood following adolescent alcohol intake in response to increasing stimulation current (two-way repeated-measures ANOVA interaction; control n = 5, ethanol n = 5). d, Average traces for stimulation current. e, MFB stimulation did not change NAcc phasic dopamine release in adulthood following adolescent alcohol intake in response to increasing stimulation duration (two-way repeated-measures ANOVA interaction; control n = 4, ethanol n = 4). f, Average traces for stimulation duration. g, MFB stimulation did not change NAcc phasic dopamine release in adulthood following adolescent alcohol intake in response to increasing stimulation frequency (two-way repeated-measures ANOVA interaction; control n = 4; ethanol n = 4). h, Average traces for stimulation frequency. Error bars indicate mean ± SEM.
Inhibitory tone on VTA dopamine neurons is increased, and tonic NAcc dopamine decreased, following adolescent alcohol exposure
We next used slice electrophysiology to measure excitatory and inhibitory inputs onto VTA dopamine neurons from rats with and without a history of adolescent alcohol consumption. Spontaneous EPSCs onto VTA dopamine neurons did not significantly differ in adult rats following adolescent ethanol intake (Fig. 4a) in either amplitude (two-way ANOVA: adolescent treatment × event amplitude: F(40,1148) = 0.4, p = 1.0; Fig. 4b) or frequency (two-way ANOVA: adolescent treatment × interevent interval: F(70,1142) = 0.2, p = 1.0; Fig. 4c). However, spontaneous IPSCs onto VTA dopamine neurons were significantly increased following ethanol intake (Fig. 4d) for amplitude (two-way ANOVA: adolescent treatment × event amplitude: F(50,867) = 2.0, p < 0.0001; Fig. 4e) and frequency (two-way ANOVA: adolescent treatment × interevent interval: F(60,1037) = 2.0, p < 0.0001; Fig. 4f).
Figure 4.
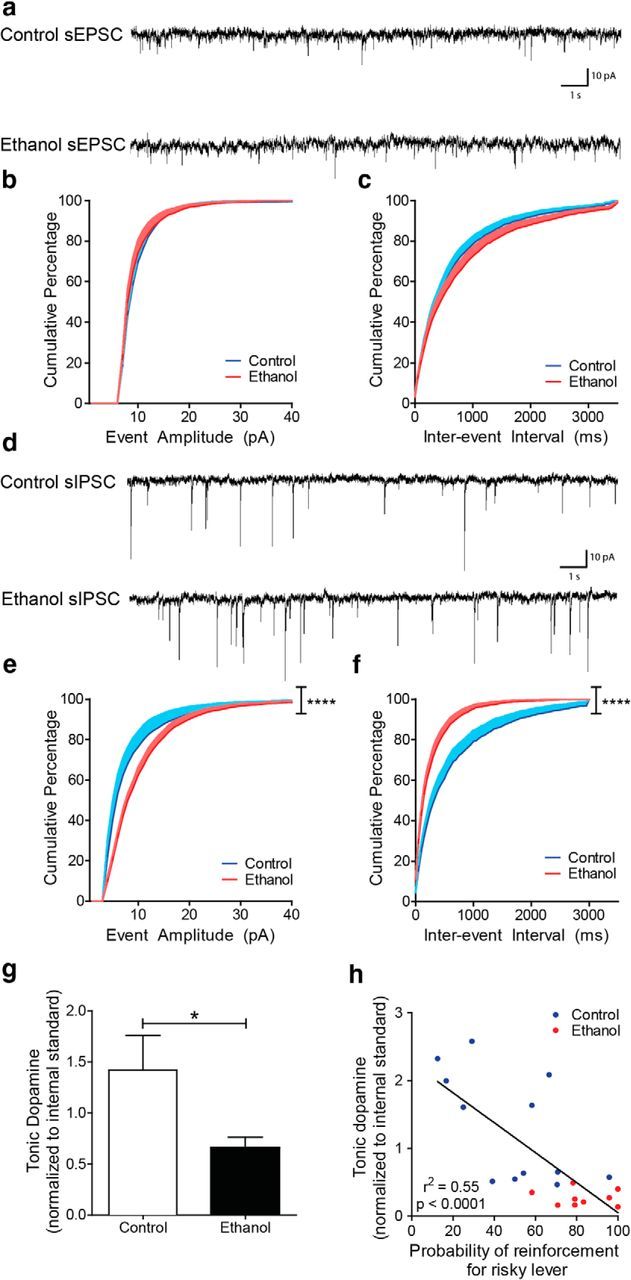
IPSCs onto VTA dopamine neurons are increased, and tonic NAcc dopamine is decreased following adolescent alcohol intake. a, Example traces of spontaneous EPSCs. b, The cumulative distribution of spontaneous EPSC amplitudes does not change following adolescent ethanol intake (two-way repeated-measures ANOVA interaction; control n = 15 cells, ethanol n = 15 cells). c, The cumulative distribution of spontaneous EPSC interevent intervals does not change following adolescent ethanol intake (two-way repeated-measures ANOVA interaction; control n = 15, ethanol n = 15 cells). d, Example traces of spontaneous IPSCs. e, The cumulative distribution of spontaneous IPSC amplitudes shifts toward higher amplitude events in VTA dopamine neurons following adolescent ethanol intake (two-way ANOVA interaction; control n = 9 cells, ethanol n = 10 cells). f, The cumulative distribution of spontaneous IPSC interevent intervals shifts toward shorter intervals in VTA dopamine neurons following adolescent ethanol intake (two-way ANOVA interaction; control n = 9 cells, ethanol n = 10 cells). g, Tonic NAcc dopamine is decreased relative to controls following ethanol intake (Student's t test; control n = 5, ethanol n = 5). h, Tonic NAcc dopamine level is negatively correlated with risk preference (Pearson correlation test; control n = 12, ethanol n = 9). Error bars indicate mean ± SEM. *p < 0.05. ****p < 0.0001.
Changes to VTA dopamine neuron activity have previously been shown to coincide with changes in tonic NAc dopamine levels (Diana et al., 1993; Floresco et al., 2003), and tonic dopamine levels have also been previously linked to risk-taking behavior (St. Onge et al., 2012). Using microdialysis and UPLC-MS, we found that basal NAcc dopamine levels were lower in adulthood in adolescent ethanol-exposed rats relative to controls (Student's unpaired t test, t(8) = 2.2, p < 0.05; Fig. 4g). Furthermore, we found a significant negative correlation between a rat's previous risk-taking behavior and tonic dopamine levels (Pearson correlation, r = −0.7, F(1,19) = 22.9, p < 0.0001; Fig. 4h), potentially linking lower tonic dopamine levels to increased risk-taking behavior.
Pharmacological reversal of alcohol-induced changes in dopamine transmission and risk-taking behavior in adulthood
Based on the electrophysiological and microdialysis data, we next used a GABA(A) allosteric agonist to explore pharmacological mitigation of the behavioral and neurochemical consequences of adolescent ethanol consumption. The GABA(A) α2, α3, α4, but not α1, subunit mRNAs are found in VTA dopamine neurons (Okada et al., 2004). Therefore, we exploited this heterogeneity and the increased GABAergic transmission within the VTA that the electrophysiological results suggest to specifically target VTA dopamine neurons with the GABA(A) α2, α3, and α5 subunit-selective allosteric agonist L-838,417. Systemic administration of this drug (i.p.) in adult anesthetized control rats produced a dose-dependent decrease in PPT-evoked dopamine release (one-way ANOVA: F(2,103) = 54.3, p < 0.0001; Bonferroni post hoc analysis; Fig. 5a). Consistent with increased inhibitory tone in the VTA, rats that consumed ethanol during adolescence showed an L-838,417-induced decrease in PPT-evoked dopamine release in adulthood (two-way ANOVA: main effect of adolescent treatment: F(1,164) = 4.872, p = 0.03, main effect of drug: F(1,164) = 98.54, p < 0.0001; Bonferroni post hoc analysis; Fig. 5b), where dopamine returned to control baseline levels when the data are visualized in raw form (two-way ANOVA: main effect of adolescent treatment: F(1,164) = 86.07, p < 0.0001, main effect of drug: F(1,164) = 63.82, p < 0.0001; Bonferroni post hoc analysis; Fig. 5c).
Figure 5.

Increased phasic dopamine and maladaptive risk-taking behavior following adolescent alcohol intake are reversed by a GABA(A) allosteric agonist. a, L-838,417 dose-dependently decreases PPT-evoked phasic dopamine release in control rats (one-way ANOVA; Bonferroni post hoc analysis; control vehicle n = 5, control 3 mg/kg n = 4, control 10 mg/kg n = 4). b, L-838,417 (3 mg/kg, i.p.) decreases PPT-evoked phasic dopamine release in adulthood in both groups (two-way ANOVA main effect of adolescent treatment, two-way ANOVA main effect of drug; Bonferroni post hoc analysis; control vehicle n = 5, control 3 mg/kg n = 4, ethanol vehicle n = 6; ethanol 3 mg/kg n = 6). c, L-838,417 (3 mg/kg, i.p.) returns PPT-evoked dopamine release following adolescent ethanol intake to baseline control levels (two-way ANOVA main effect of adolescent treatment and main effect of drug; Bonferroni post hoc analysis; control vehicle n = 5, control 3 mg/kg n = 4, ethanol vehicle n = 6; alcohol 3 mg/kg n = 6). d, L-838,417 (3 mg/kg, i.p.) administered before behavioral sessions reverses maladaptive risk-taking behavior seen following adolescent ethanol intake (two-way repeated-measures ANOVA interaction; ethanol vehicle n = 7, ethanol 3 mg/kg n = 8) but has no effect on control rats (two-way repeated-measures ANOVA interaction; control vehicle n = 11, control 3 mg/kg n = 6). e, L-838,417 (3 mg/kg, i.p.) administered before behavioral sessions also reverses the risk preference score seen following adolescent ethanol intake (one-sample t test from a theoretical mean = 50.0: ethanol/vehicle-treated, n = 21, ethanol/L-838–417-treated, n = 24). f, L-838,417 (3 mg/kg, i.p.) administered before behavioral sessions reverses the increase in win-stay behavior following adolescent ethanol intake (three-way ANOVA interaction followed by separate two-way ANOVAs for win-stay and lose shift; Bonferroni post hoc analysis; control vehicle n = 11, control 3 mg/kg n = 6, ethanol vehicle n = 7, ethanol 3 mg/kg n = 7). Error bars indicate mean ± SEM. *p < 0.05. **p < 0.01. ***p < 0.001. n.s., Not significant.
We next investigated the potential for this drug to act as a therapeutic agent during decision-making. When administered systemically 20 min before the session, L-838,417 (3 mg/kg, i.p.) was able to decrease risk-taking behavior in adulthood following adolescent ethanol intake (two-way repeated-measures ANOVA: drug × probability: F(4,52) = 2.7, p = 0.04; Fig. 5d), with no effect in control rats (two-way repeated-measures ANOVA: drug × probability: F(4,60) = 0.07, p = 0.99; Fig. 5d). Importantly, choice behavior during conditions where risk was not present (i.e., at 1.00 and 0.00) was unaffected by L-838,417 administration, suggesting that the drug did not influence reward processing nonspecifically (Fig. 5d). To generate an overall risk preference score (Nasrallah et al., 2011; Schindler et al., 2014), choice behavior for the uncertain reward option over the probabilistic test conditions (0.75, 0.50, and 0.25) was collapsed, as the expected value for the options are equal when the conditions are combined. In line with our previous results (Schindler et al., 2014), rats that consumed ethanol during adolescence demonstrated a risk preference score that was significantly elevated above risk neutrality in adulthood (50% choice for the risky option; one-sample t test, t(20) = 4.5, p < 0.0002; Fig. 5e). However, ethanol-exposed rats that received L-838,417 (3 mg/kg, i.p.) returned to risk neutrality (one-sample t test, t(23) = 0.5, p = 0.61; Fig. 5e). Importantly, the drug did not change the risk neutrality of control rats when administered at this dose (one-sample t test, t(17) = 0.7, p = 0.52; Fig. 5e). L-838,417 did not alter food consumption (two-way ANOVA: adolescent treatment × drug: F(1,92) = 2.1, p = 0.15; data not shown), the number of omitted forced trials (two-way ANOVA: adolescent treatment × drug: F(1,92) = 2.6, p = 0.11; data not shown), the number of omitted choice trials (two-way ANOVA: adolescent treatment × drug: F(1,89) = 1.6, p = 0.20; data not shown), or latency to respond (two-way ANOVA: adolescent treatment × drug: F(1,92) = 1.8, p = 0.18; data not shown).
Finally, we also analyzed “win-stay” and “lose-shift” choice patterns to determine the effects of adolescent ethanol exposure and L-838,417 on positive and negative feedback sensitivity, respectively (Stopper et al., 2014). Ethanol intake during adolescence selectively increased win-stay behavior in adulthood, and pretreatment with L-838,417 (3 mg/kg, i.p.) was able to reverse this behavior without affecting the behavior of control rats (three-way ANOVA: adolescent treatment × drug × win-stay/lose-shift: F(1,54) = 6.41, p = 0.01; win-stay two-way ANOVA: adolescent treatment × drug treatment: F(1,27) = 9.3, p < 0.01; Bonferroni post hoc analysis and lost-shift two-way ANOVA: adolescent treatment × drug treatment: F(1,27) = 0.2 p > 0.05; Fig. 5f).
Discussion
Recent reports evaluating the impact of drug abuse on society conclude that alcohol ranks as the most harmful of all abused substances (Nutt et al., 2010; World Health Organization, 2014). In addition, despite continuing efforts to curb its use, alcohol remains the most commonly abused substance among adolescents (Witt, 2010). That such experience can be antecedent to problem drinking has been recognized for some time; that such experience may also have long-lasting effects on decision-making processes is a relatively recent consideration (Stout et al., 2005; Johnson et al., 2008). In the current work, we identify a specific alcohol-induced perturbation in dopamine network dynamics where a potentiation of PPT-evoked dopamine release may drive maladaptive decision-making following adolescent alcohol intake. Emerging evidence indicates that there are multiple excitatory inputs to the VTA, including the PPT, and that these circuits may differ in the information that is conveyed and the behaviors that are supported (Geisler et al., 2007; Watabe-Uchida et al., 2012). The PPT in particular has been linked to the assignment of positive incentive value (Lisman and Grace, 2005), signaling of dopamine-dependent positive reward prediction errors (Pan and Hyland, 2005), and the activation/inactivation of this structure has been shown to alter choice behavior under risk (Leblond et al., 2014; Stopper et al., 2014). Consistent with these results and the proposed role of the PPT, we have shown that positive reward prediction errors are altered by adolescent alcohol exposure (Spoelder et al., 2015) as well as choice behavior under risk. However, the PPT sends both glutamatergic and cholinergic projections to the VTA, and the approach taken here cannot resolve a separable contribution of each. Thus, future studies will be required to determine whether glutamatergic or cholinergic input (or both) regulates the effect of adolescent alcohol seen in the present study.
We next examined the excitability of VTA dopamine neurons and found an increase in inhibitory tone following adolescent alcohol intake. In line with these results, previous reports have demonstrated that a single injection of ethanol can induce a long-lasting potentiation of GABAergic synapses in VTA dopamine neurons (Melis et al., 2002); and following cessation of chronic ethanol administration, basal VTA dopamine neuronal population activity is reduced (Shen et al., 2007). Increased phasic dopamine release in conjunction with increased inhibitory tone onto VTA dopamine neurons shown here provides novel in vivo support for previous modeling work implicating a role for GABAergic signaling in the midbrain for disinhibition-induced burst firing of dopamine neurons (Lobb et al., 2011a, b; Paladini and Roeper, 2014; Oster et al., 2015). In this model, bursts in dopamine neuronal activity can be induced by disinhibition (i.e., phasic removal of inhibitory GABA(A) receptor conductance), and importantly bursting is enhanced by increasing the degree of this disinhibition. We find increased spontaneous IPSCs following adolescent alcohol intake; therefore, the enhanced phasic dopamine release we find may be the result of a larger degree of disinhibition (mediated via increased IPSCs) in response to salient stimuli or PPT stimulation. Additional studies will be required to determine whether disinhibition is responsible for the enhanced phasic dopamine release (evoked by salient cues and/or drug related stimuli) seen following adolescent alcohol intake. Thus, future work will focus on the source of inhibition (VTA interneurons, RMTg, striatum) and the mechanism of disinhibition that may result from excitatory input from the PPT.
We next used microdialysis and UPLC-MS to demonstrate a decrease in basal NAcc dopamine levels relative to controls following adolescent alcohol intake, which was negatively correlated with risk-taking behavior. Previous reports have demonstrated a reduction in tonic NAcc dopamine levels following cessation of chronic ethanol exposure (Rossetti et al., 1992; Diana et al., 1993). Likewise, withdrawal from a variety of drugs of abuse results in decreased NAcc tonic dopamine levels and/or attenuated tonic VTA dopamine neuron firing (Parsons et al., 1991; Robertson et al., 1991; Rossetti et al., 1992; Liu and Jin, 2004). Additionally, previous work has demonstrated that modulation of VTA dopamine cell activity coincides with changes in NAc tonic dopamine levels (Diana et al., 1993; Floresco et al., 2003), as demonstrated in the present study. This body of work suggests a complex interplay between tonic and phasic dopamine where chronic substance use may persistently decrease tonic dopamine levels in the striatum while in parallel increasing phasic dopamine responses to salient stimuli, thereby increasing the “signal-to-noise” ratio of phasic to tonic dopamine in the NAcc (Wanat et al., 2009). Notably, the findings reported here support theoretical work positing that chronic alcohol exposure may provoke a persistent negative affective state and subsequently motivate maladaptive drug-seeking behavior via alcohol-induced deficits in NAcc baseline dopamine levels, leading to a much greater percentage increase in phasic dopamine release upon continued drug self-administration (Koob, 2013).
Finally, based on the overall pattern of electrochemical and electrophysiological findings, we hypothesized that ethanol exposure enhances GABA transmission to reduce tonic dopamine but may be insufficient to reduce phasic dopamine, thus generating an imbalance in the tonic to phasic signaling ratio. If correct, then additional modulation of GABA input may be expected to increase this inhibition further and be sufficient to reduce phasic dopamine, restore the balance, and mitigate maladaptive behavior. Consistent with our electrophysiological data showing increased GABA-mediated inhibitory tone onto VTA dopamine neurons, we found that the selective GABA(A) allosteric agonist L-838,417 was able to return PPT-evoked dopamine release to control baseline levels in rats that previously consumed alcohol in adolescence. We then applied these findings in our behavioral pharmacology study and, using the 3 mg/kg dose of L-838,417, we were able to reverse maladaptive decision-making in rats that previously consumed alcohol during adolescence, without affecting the behavior of control rats. Our results are in line with previous reports demonstrating that choice behavior on a risk/reward probability discounting task is highly sensitive to pharmacological manipulations of the mesolimbic dopamine system (St Onge and Floresco, 2009; Stopper et al., 2013), although additional studies will be required to specifically investigate the effect of L-838,417 on phasic dopamine release while the rats are performing the risky decision-making task.
Although risk attitude is often attributed to loss sensitivity (Tom et al., 2007), our work has suggested that a change in the processing of gains is critical to generating risk preference after chronic alcohol use (Clark et al., 2012; Spoelder et al., 2015). Specifically, we have previously demonstrated that adult rats that have previously consumed alcohol as adolescents show increased dopamine-dependent incentive (gain) learning strategies and potentiated stimulus-evoked NAcc phasic dopamine release (Clark et al., 2012; Spoelder et al., 2015). Indeed, a recent report with the IGT demonstrates that alcohol-dependent individuals are more prone to risky choices and display between-group differences in gain but not loss sensitivity (Brevers et al., 2014). Consistent with data in human alcoholics, we show here that adolescent alcohol use selectively increases win-stay (gains) but not lose-shift (losses) choice patterns. Importantly, we show that the reduction in risk preference by L-838,417 is attributable to its effect on win-stay behavior selectively. This result is consistent with a recent report showing that electrical stimulation of the lateral habenula, which suppresses phasic dopamine signaling, is sufficient to decrease risk-taking behavior and win-stay tendencies (Stopper et al., 2014).
In conclusion, this work highlights a potential neural mechanism that promotes maladaptive decision-making after adolescent alcohol use and demonstrates its pharmacological reversal in adulthood. Indeed, we present data from multiple experimental approaches supporting a model where adolescent alcohol use produces increased inhibitory tone on dopamine neurons in parallel with a persistent decrease in basal dopamine levels and a potentiation in stimulus-evoked phasic dopamine release. These results provide unique insight into the dynamic interplay between different modes of dopamine transmission within the mesolimbic network, the underlying mechanisms involved in drug-induced maladaptive risk-taking behavior, and highlight a potential new therapeutic target for further investigation.
Footnotes
This work was supported by the National Institutes of Health Grants R01AA021121 and U01AA024599 to J.J.C., Grant T32AA07455 to A.G.S., and Grant R01MH094536 to M.E.S. and L.S.Z. We thank Scott Ng-Evans and Dale Whittington for considerable technical assistance; and Paul Phillips for helpful comments.
The authors declare no competing financial interests.
References
- Brevers D, Bechara A, Cleeremans A, Kornreich C, Verbanck P, Noël X. Impaired decision-making under risk in individuals with alcohol dependence. Alcohol Clin Exp Res. 2014;38:1924–1931. doi: 10.1111/acer.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Taylor JR, Potenza MN. Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. Am J Psychiatry. 2003;160:1041–1052. doi: 10.1176/appi.ajp.160.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JJ, Sandberg SG, Wanat MJ, Gan JO, Horne EA, Hart AS, Akers CA, Parker JG, Willuhn I, Martinez V, Evans SB, Stella N, Phillips PE. Chronic microsensors for longitudinal, subsecond dopamine detection in behaving animals. Nat Methods. 2010;7:126–129. doi: 10.1038/nmeth.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JJ, Nasrallah NA, Hart AS, Collins AL, Bernstein IL, Phillips PE. Altered risk-based decision-making following adolescent alcohol use results from an imbalance in reinforcement learning in rats. PLoS One. 2012;7:e37357. doi: 10.1371/journal.pone.0037357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana M, Pistis M, Carboni S, Gessa GL, Rossetti ZL. Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: electrophysiological and biochemical evidence. Proc Natl Acad Sci U S A. 1993;90:7966–7969. doi: 10.1073/pnas.90.17.7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagel SB, Clark JJ, Robinson TE, Mayo L, Czuj A, Willuhn I, Akers CA, Clinton SM, Phillips PE, Akil H. A selective role for dopamine in stimulus-reward learning. Nature. 2011;469:53–57. doi: 10.1038/nature09588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Geisler S, Derst C, Veh RW, Zahm DS. Glutamatergic afferents of the ventral tegmental area in the rat. J Neurosci. 2007;27:5730–5743. doi: 10.1523/JNEUROSCI.0012-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Dawson DA. Age at onset of alcohol use and its association with DSM-IV alcohol abuse and dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. J Subst Abuse. 1997;9:103–110. doi: 10.1016/S0899-3289(97)90009-2. [DOI] [PubMed] [Google Scholar]
- Grant BF, Stinson FS, Harford TC. Age at onset of alcohol use and DSM-IV alcohol abuse and dependence: a 12-year follow-up. J Subst Abuse. 2001;13:493–504. doi: 10.1016/S0899-3289(01)00096-7. [DOI] [PubMed] [Google Scholar]
- Heien ML, Khan AS, Ariansen JL, Cheer JF, Phillips PE, Wassum KM, Wightman RM. Real-time measurement of dopamine fluctuations after cocaine in the brain of behaving rats. Proc Natl Acad Sci U S A. 2005;102:10023–10028. doi: 10.1073/pnas.0504657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CA, Xiao L, Palmer P, Sun P, Wang Q, Wei Y, Jia Y, Grenard JL, Stacy AW, Bechara A. Affective decision-making deficits, linked to a dysfunctional ventromedial prefrontal cortex, revealed in 10th grade Chinese adolescent binge drinkers. Neuropsychologia. 2008;46:714–726. doi: 10.1016/j.neuropsychologia.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LD, O'Malley PM, Bachman JG, Schulenberg JE, Miech RA. Monitoring the future national survey results on drug use, 1975–2013: Volume I, Secondary school students. Ann Arbor, MI: Institute for Social Research, University of Michigan; 2014. [Google Scholar]
- Koob GF. Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci. 2013;13:3–30. doi: 10.1007/7854_2011_129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond M, Sukharnikova T, Yu C, Rossi MA, Yin HH. The role of pedunculopontine nucleus in choice behavior under risk. Eur J Neurosci. 2014;39:1664–1670. doi: 10.1111/ejn.12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into longterm memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Liu ZH, Jin WQ. Decrease of ventral tegmental area dopamine neuronal activity in nicotine withdrawal rats. Neuroreport. 2004;15:1479–1481. doi: 10.1097/01.wnr.0000126218.25235.b6. [DOI] [PubMed] [Google Scholar]
- Lobb CJ, Troyer TW, Wilson CJ, Paladini CA. Disinhibition bursting of dopaminergic neurons. Front Syst Neurosci. 2011a;5:25. doi: 10.3389/fnsys.2011.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb CJ, Wilson CJ, Paladini CA. High-frequency, short-latency disinhibition bursting of midbrain dopaminergic neurons. J Neurophysiol. 2011b;105:2501–2511. doi: 10.1152/jn.01076.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli M, McCutcheon JE. Heterogeneity of dopamine neuron activity across traits and states. Neuroscience. 2014;282:176–197. doi: 10.1016/j.neuroscience.2014.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah NA, Clark JJ, Collins AL, Akers CA, Phillips PE, Bernstein IL. Risk preference following adolescent alcohol use is associated with corrupted encoding of costs but not rewards by mesolimbic dopamine. Proc Natl Acad Sci U S A. 2011;108:5466–5471. doi: 10.1073/pnas.1017732108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt DJ, King LA, Phillips LD. Drug harms in the UK: a multicriteria decision analysis. Lancet. 2010;376:1558–1565. doi: 10.1016/S0140-6736(10)61462-6. [DOI] [PubMed] [Google Scholar]
- Okada H, Matsushita N, Kobayashi K, Kobayashi K. Identification of GABAA receptor subunit variants in midbrain dopaminergic neurons. J Neurochem. 2004;89:7–14. doi: 10.1111/j.1471-4159.2004.02271.x. [DOI] [PubMed] [Google Scholar]
- Oster A, Faure P, Gutkin BS. Mechanisms for multiple activity modes of VTA dopamine neurons. Front Comput Neurosci. 2015;9:95. doi: 10.3389/fncom.2015.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini CA, Roeper J. Generating bursts (and pauses) in the dopamine midbrain neurons. Neurosci. 2014;282C:109–121. doi: 10.1016/j.neuroscience.2014.07.032. [DOI] [PubMed] [Google Scholar]
- Pan WX, Hyland BI. Pedunculopontine tegmental nucleus controls conditioned responses of midbrain dopamine neurons in behaving rats. J Neurosci. 2005;25:4725–4732. doi: 10.1523/JNEUROSCI.0277-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons LH, Smith AD, Justice JB., Jr Basal extracellular dopamine is decreased in the rat nucleus accumbens during abstinence from chronic cocaine. Synapse. 1991;9:60–65. doi: 10.1002/syn.890090109. [DOI] [PubMed] [Google Scholar]
- Phillips PE, Stuber GD, Heien M, Wightman RM, Carelli RM. Subsecond dopamine release promotes cocaine seeking. Nature. 2003;422:614–618. doi: 10.1038/nature01476. [DOI] [PubMed] [Google Scholar]
- Redish AD, Jensen S, Johnson A. Addiction as vulnerabilities in the decision process. Behav Brain Sci. 2008;31:448–461. doi: 10.1017/S0140525X0800472X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson MW, Leslie CA, Bennett JP., Jr Apparent synaptic dopamine deficiency induced by withdrawal from chronic cocaine treatment. Brain Res. 1991;538:337–339. doi: 10.1016/0006-8993(91)90451-Z. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Addiction. Annu Rev Psychol. 2003;54:25–53. doi: 10.1146/annurev.psych.54.101601.145237. [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Hmaidan Y, Gessa GL. Marked inhibition of mesolimbic dopamine release: a common feature of ethanol, morphine, cocaine and amphetamine abstinence in rats. Eur J Pharmacol. 1992;221:227–234. doi: 10.1016/0014-2999(92)90706-A. [DOI] [PubMed] [Google Scholar]
- Schindler AG, Tsutsui KT, Clark JJ. Chronic alcohol intake during adolescence, but not adulthood, promotes persistent deficits in risk-based decision-making. Alcohol Clin Exp Res. 2014;38:1622–1629. doi: 10.1111/acer.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen RY, Choong KC, Thompson AC. Long-term reduction in ventral tegmental area dopamine neuron population activity following repeated stimulant or ethanol treatment. Biol Psychiatry. 2007;61:93–100. doi: 10.1016/j.biopsych.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobeh Rev. 2000;24:417–463. doi: 10.1016/S0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Spoelder M, Tsutsui KT, Lesscher HM, Vanderschuren LJ, Clark JJ. Adolescent alcohol exposure amplifies the incentive value of reward-predictive cues through potentiation of phasic dopamine signaling. Neuropsychopharmacology. 2015;40:2873–2885. doi: 10.1038/npp.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Onge JR, Floresco SB. Dopaminergic modulation of risk-based decision-making. Neuropsychopharmacology. 2009;34:681–697. doi: 10.1038/npp.2008.121. [DOI] [PubMed] [Google Scholar]
- St Onge JR, Ahn S, Phillips AG, Floresco SB. Dynamic fluctuations in dopamine efflux in the prefrontal cortex and nucleus accumbens during risk-based decision-making. J Neurosci. 2012;32:16880–16891. doi: 10.1523/JNEUROSCI.3807-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopper CM, Khayambashi S, Floresco SB. Receptor-specific modulation of risk-based decision-making by nucleus accumbens dopamine. Neuropsychopharmacology. 2013;38:715–728. doi: 10.1038/npp.2012.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopper CM, Tse MT, Montes DR, Wiedman CR, Floresco SB. Overriding phasic dopamine signals redirects action selection during risk/reward decision-making. Neuron. 2014;84:177–189. doi: 10.1016/j.neuron.2014.08.033. [DOI] [PubMed] [Google Scholar]
- Stout JC, Rock SL, Campbell MC, Busemeyer JR, Finn PR. Psychological processes underlying risky decisions in drug abusers. Psychol Addictive Behav. 2005;19:148–157. doi: 10.1037/0893-164X.19.2.148. [DOI] [PubMed] [Google Scholar]
- Sugam JA, Day JJ, Wightman RM, Carelli RM. Phasic nucleus accumbens dopamine encodes risk-based decision-making behavior. Biol Psychiatry. 2012;71:199–205. doi: 10.1016/j.biopsych.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol. 2014;1183:221–242. doi: 10.1007/978-1-4939-1096-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom SM, Fox CR, Trepel C, Poldrack RA. The neural basis of loss aversion in decision-making under risk. Science. 2007;315:515–518. doi: 10.1126/science.1134239. [DOI] [PubMed] [Google Scholar]
- Wanat MJ, Willuhn I, Clark JJ, Phillips PE. Phasic dopamine release in appetitive behaviors and drug addiction. Curr Drug Abuse Rev. 2009;2:195–213. doi: 10.2174/1874473710902020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Bonci A, Phillips PE. CRF acts in the midbrain to attenuate accumbens dopmaine release to rewards but not their predictors. Nat Neurosci. 2013;16:383–385. doi: 10.1038/nn.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron. 2012;74:858–873. doi: 10.1016/j.neuron.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Whelan R, Watts R, Orr CA, Althoff RR, Artiges E, Banaschewski T, Barker GJ, Bokde AL, Büchel C, Carvalho FM, Conrod PJ, Flor H, Fauth-Bühler M, Frouin V, Gallinat J, Gan G, Gowland P, Heinz A, Ittermann B, Lawrence C, et al. Neuropsychosocial profiles of current and future adolescent alcohol misusers. Nature. 2014;512:185–189. doi: 10.1038/nature13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Global Status Report on Alcohol and Health 2014. Geneva: World Health Organization; 2014. [Google Scholar]
- Witt ED. Research on alcohol and adolescent brain development: opportunities and future directions. Alcohol. 2010;44:119–124. doi: 10.1016/j.alcohol.2009.08.011. [DOI] [PubMed] [Google Scholar]