

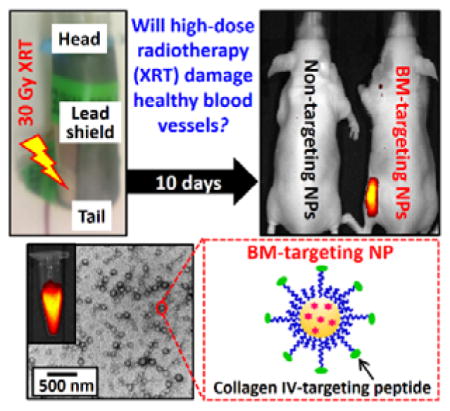
Graphical abstract
Keywords: Cancer, Radiotherapy, Cardiovascular Disease, Basement Membrane-Targeting Nanoparticles, Medical Imaging
Radiotherapy (XRT) is often utilized in the curative management of many difficult to treat cancers, such as lung, head and neck, breast and esophageal cancers.[1, 2] Over the past several decades, advances in radiation delivery have significantly reduced the side effects of XRT.[3] Despite this improvement, long-term cardiovascular side effects, such as myocardial infarction, atherosclerosis, and stroke, remains one of the most common causes of late morbidity and mortality of cancer treatment.[1, 2, 4] In one clinical study, investigators found that patients who received XRT for breast cancer are up to 4 times more likely to suffer cardiovascular events 10 to 15 years after completing their cancer treatment.[3, 5] Despite radiation-induced blood vessel damage was first reported about a century ago, the underlying mechanism remains unclear.[5] Recent studies have revealed that the late-cardiovascular events triggered by XRT may involve acute up-regulation of pro-inflammatory cytokines and adhesion molecules at the endothelium of injured blood vessel.[6] While it is widely believed that the damage of endothelium occurs shortly after XRT treatment[2], there has been no direct observation of such injury. Furthermore, the lack of understanding of this injury process prevents the development of effective therapies for the damage.
One of the characteristics of endothelium damage is the exposure of collagen IV-rich basement membrane (BM) to the circulation system.[7] A number of studies attempted to use oligo(hydroxyproline)-based collagen mimetic peptide and collagen-binding recombinant protein to target the vascular basement membrane,[8] but all these targeting ligands failed to target the collagen IV fiber that makes up of 50 % of the basement membrane. A recent study found that a synthetic peptide with an amino acid sequence of KLWVLPK binds specifically to collagen IV fiber,[9] which provides unique opportunities to develop new collagen IV-targeting molecular probe to identify early-stage blood vessel damage induced by XRT. In addition, such molecular targets afford potential novel targeted drug delivery system for preventive treatments. Biodegradable collagen IV-targeting peptide-conjugated PEGylated nanoparticles (NPs), such as poly(ethylene glycol)-poly(lactic-co-glycolic acid) (PEG-PLGA)-based di-block copolymer nanoparticles of 100 – 150 nm in diameter, are ideal for vascular basement membrane-targeting applications.[10] The active targeting PEG motif increases the circulation half-life of the NPs and allows the NPs to bind specifically to the basement membrane at the injured vessels.[10, 11] The use of biodegradable polymer can prevent the targeting NPs from irreversibly accumulating at the injury site and causing potential long-term damage.[12] In addition, techniques have been developed to encapsulate radioactive isotope and drug to di-block copolymer NPs for deep tissue imaging and preventative treatment.[13]
In this communication, we reported the first direct observation of an early-stage blood vessels injury induced by high-dose XRT via fluorescent-label BM-targeting NPs. To achieve our goal, we engineered biodegradable fluorescent-labeled collagen IV-targeting peptide-conjugated PEG-PLGA NPs. The physicochemical properties and in vitro toxicity of the BM-targeting NPs were fully investigated. Collagen IV fiber-based solid-phase binding assay and quantitative ex vivo imaging study in athymic nude (Nu) mice skin were used to elevate the binding affinity of the BM-targeting NPs to collagen IV fiber in the basement membrane. The ability of the BM-targeting NPs to identify an early-stage blood vessel injury induced by high-dose XRT was verified in a murine model via standard full-body in vivo fluorescence imaging technique.
Fluorescent rhodamine B (Rhod)-labeled BM-targeting PEG-PLGA NPs were prepared via nanoprecipitation method[14] using a mixture of PEG(2K)-PLGA(15K) and collagen IV-targeting peptide-conjugated PEG(5K)-PLGA(10K) di-block copolymers as building blocks of the NPs in the presence of Rhod-labeled PLGA(30K) (Figure 1a and Experimental). During the nanoprecipitation process, the high-molecular weight hydrophobic Rhod-labeled PLGA was encapsulated within the hydrophobic PLGA core of the NPs.[15] The cysteine (C) end at the GGGC linker of the collagen IV-targeting peptide (amino acid sequence: KLWVLPKGGGC) was conjugated to maleimide-functionalized PEG(5K)-PLGA(10K) via metal-free thiol-maleimide ‘click’ reaction[16] prior to the preparation of PEG-PLGA NPs, and the structure of the peptide functionalized di-block copolymer was confirmed by 1H NMR spectroscopy (Figure S1). Non-targeting PEG-PLGA NPs have been prepared for control studies used methoxyl-functionalized PEG(5K)-PLGA(10K) instead of collagen IV-targeting peptide-conjugated PEG(5K)-PLGA(55K) as a building block of the NPs. Figure 1b shows the transmission electron microscopy (TEM) images of the BM-targeting NPs and non-targeting NPs. The number-average diameter (Dn) of the BM-targeting and non-targeting PEG-PLGA NPs were found to be 83 and 81 nm, respectively, as determined by TEM. This is consistent with the mean hydrodynamic diameter (Dh) of about 135 nm (polydispersity index, PDI ≈ 0.14, Figure S2) and mean Dn of about 110 nm (Figure 1c) determined by dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA) techniques on NP aqueous dispersions, where the long hydrophilic PEG chains were fully hydrated in aqueous environment. Based on the NTA result, it was calculated that 1 mg/mL of the BM-targeting NPs and non-targeting NPs contain approximately 9.0 ± 0.2 × 1011 NPs (i.e. 1.5 nM, Figure 1c). Aqueous electrophoresis measurements confirmed that the conjugated cationic lysine (K)-containing collagen IV-targeting peptide is located on the surface of the BM-targeting NPs with a less anionic mean zeta potential (ζ) of −21.6 mV recorded for the BM-targeting NPs (dispersed in 1 mM NaCl electrolyte) versus a more anionic mean ζ of −28.3 mV recorded for the non-targeting NPs (Figure 1d), despite both NPs having near-zero mean zeta potentials when dispersed in 0.1 M phosphate buffered saline (PBS) due to compression of electrical double-layer in the high ionic strength physiological buffer. Both PEG-PLGA NPs have negative zeta potentials at pH 7 (in 1 mM NaCl electrolyte) because of the deprotonated carboxylic acid end-group in the hydrophobic PLGA block (pKa ≈ 4).[17] Spectroscopic measurements confirmed the presence of Rhod-labeled PLGA in both nanoparticles with a characteristic Rhod B visible absorption band centered at 560 nm and a narrow emission band centered at 580 nm (Figure 1e). Further in vitro fluorescence imaging study indicated the average photon fluxes recorded at an emission wavelength of 600 nm were linearly increased with the concentration of Rhod-labeled BM-targeting NPs upon excitation at 570 nm (Figure 1f). An in vitro toxicity study indicated that relatively high concentrations (up to 15 nM) of the BM-targeting NPs and non-targeting NPs showed very low cytotoxicities against KB epithelial cells (Figure S3).
Figure 1.

(a) Scheme summarized the preparation of BM-targeting PEG-PLGA NPs and non-targeting PEG-PLGA NPs via nanoprecipitation method. The collagen IV-targeting peptide (Col(IV)-TP) was conjugated to maleimide-functionalized PEG(5K)-PLGA(10K) prior to the prior to the preparation of BM-targeting NPs. (b) TEM images recorded for (i) BM-targeting and (ii) non-targeting NPs. (c) Number-average particle distribution curves recorded for 5 μg/mL of (i) BM-targeting NP and (ii) non-targeting NP dispersions, as determined by NTA method. It was determined that both NP dispersions contain about 4.5 × 109 particles per mL(d) Mean zeta potentials (ζ) of BM-targeting and non-targeting NPs dispersed in 1 mM NaCl electrolyte and 0.1 M PBS. (e) UV-visible absorption and fluorescence spectra recorded for BM-targeting NPs and non-targeting NPs. (f) In vitro fluorescence image recorded for (i) 0.150, (ii) 0.375, (iii) 0.750, (iv) 1.125, and (v) 1.500 nM of BM-targeting NPs. (N.B. * denotes p < 0.05, i.e. statistical significance.)
A quantitative in vitro study was performed to elevate the binding affinity of the BM-targeting NPs to collagen IV fibers. We briefly incubated 0 – 1.5 nM (0 – 1 mg/mL) of BM-targeting NPs and non-targeting NPs in a collagen IV-coated well plate, and washed with PBS and fetal bovine serum (FBS) prior to spectroscopic measurements to quantify the amount of NPs binding to collagen IV fibers. As shown in Figure 2a and Figure S4, the fluorescence intensities (at 590 nm) of collagen IV-coated wells increased with the concentration of BM-targeting NPs, suggesting that the BM-targeting NPs dose-dependently bind to collagen IV fibers. Plateau binding can be observed when the concentration of BM-targeting NPs reached about 0.74 nM. By fitting the dose-dependent fluorescence intensities to the Hill equation,[18] the Hill constant (nHill) and dissociation constant (Kd) were calculated to be 1.25 and 0.13 nM, respectively. This confirms that the BM-targeting NPs bind strongly and cooperatively to collagen IV. On the other hand, increasing the concentrations of non-targeting NPs only slightly increased the fluorescence intensities of collagen IV fibers, and the plateau fluorescent intensity is at least 10 times lower than that recorded for BM-targeting NPs. Quantitative data analysis indicated that the binding of the non-targeting NPs is non-cooperative (nHill < 1, the binding of one NP to a collagen fiber lower binding affinity for another NP[18]), and the dissociation constant of the non-targeting NPs is more than 10 times higher than that of BM-targeting NPs.
Figure 2.

(a) Concentration-dependent fluorescence intensities recorded for collagen IV-coated well plate after incubated with different concentrations of BM-targeting NPs and non-targeting NPs. The inset table summarized the binding affinities of two different NPs after fitted to Hill equation. (b) Florescence images recorded for basement membrane-exposed disease-free Nu mouse skin after incubation with (i) PBS and different concentrations of (ii–v) BM-targeting NPs and (vi–ix) non-targeting NPs. The insert cartoon shows the wide-field fluorescence imaging method. (c) Concentration-dependent fluorescence intensities at basement membrane of artificially damaged Nu mouse skin after incubation with different concentrations of BM-targeting NPs and non-targeting NPs. (d) Wide-field fluorescent images of an artificially-damaged Nu mouse skin with “U N C” letters. (N.B. * denotes p < 0.05, i.e. statistical significance. Fluorescence images were recorded using 200 ms exposure time.)
Next, we performed an ex vivo imaging study on freshly excised Nu mouse skin to investigate the binding affinity of BM-targeting NPs to collagen IV fibers in the basement membrane. Basement membrane was exposed to the bulk environments from the peritoneal surface prior to the ex vivo study. Figure 2b shows wide-field fluorescence images recorded for the ex vivo skin samples after incubated with different concentrations of BM-targeting NPs or non-targeting NPs. As shown in the Figure 2b(i–v), increasing the concentration of BM-targeting NPs progressively increased the fluorescence intensities at the basement membrane and allowed the identification of individual collagen fibers. Quantitative image analysis (Figure 2c) indicated the observed contrast effect is contributed by the binding of fluorescent BM-targeting NPs. Conversely, increasing the concentration of non-targeting NPs did not significantly increase the fluorescence intensities at the basement membrane (Figures 2b(vi–ix) and d). This is due to the anti-biofouling character of the high molecular weight PEG, which prevents non-specific binding to the basement membrane.[19] The fluorescence intensity of this basement membrane after incubation with 150 pM of BM-targeting NPs is about 10 times higher those incubated with non-targeting NPs. To further verify that the BM-targeting NPs bonded specifically to the basement membrane, we superficially sketched the letters “U N C” on the peritoneal surface of a piece of freshly excised mouse skin via a scalpel to expose the basement membrane before incubation with 150 pM of BM-targeting NPs. As shown in Figure 2d, “U N C” is clearly visible and strongly fluorescent, indicating specific binding of the BM-targeting NPs to the collagen IV fibers in the basement membrane that were artificially exposed before the study. Both ex vivo imaging studies confirmed that the BM-targeting NPs bonded specifically to collagen IV-rich basement membrane.
Finally, we performed in vivo fluorescence imaging study with the aim of intravenous (i.v.) administrated BM-targeting NPs to investigate early-stage blood vessel injury induced by high-dose radiotherapy in healthy Nu mice. 10 days before the imaging study, the left flank of healthy Nu mice were subjected to a single high-dose XRT (30 Gy). Full-body fluorescence images (Figure 3a and Figure S5) were recorded pre-injection and 24 after tail-vein i.v. injection of BM-targeting NPs or non-targeting NPs. As shown in Figures 3a(ii) and S5(a), the total photon flux recorded at the left flank in six out of seven Nu mice administrated with BM-targeting NPs were approximately 3.5 times higher than that recorded at the right flank (p < 0.005, Figure 3a(iii)), which indicates the accumulation of fluorescent BM-targeting NPs at the left flank. On the other hand, the total photon flux recorded at the left flank of irradiated mice administrated with non-targeting NPs was nearly identical to that recorded at the right flank (p = 0.22, Figures 3a(ii) and (iii)), and the total photon flux recorded at the right flank of mice administrated with BM-targeting NPs and non-targeting NPs was also nearly identical (p = 0.66, Figure 3a(ii), (iii) and S5). The quantitative in vivo imaging study confirmed that high-dose XRT damaged the endothelium of major blood vessels at the irradiated site, and exposed the basement membrane and nearby tissues to the circulation system, which allowed for the binding of circulating BM-targeting NPs. Further histological study was performed to investigate the contrast effect observed at the left flank of irritated mice after the administration of the BM-targeting NPs. Figure 3b shows representative optical and fluorescence images of leg histological sections collected after the in vivo imaging study. Significant vascular injuries can be observed at the tunica intima (include endothelium, basement membrane) and tunica media of the left leg histological sections that were subjected to high-dose XRT (Figure 3b), whereas the right leg blood vessels remained relatively normal. However, only the histological sections collected from Nu mice administrated with BM-targeting NPs showed strong fluorescence intensities at the injured blood arteries and nearby tissues, suggesting that the increased photon flux recorded at the left flank of Nu mice administrated with BM-targeting NP is due to the binding of BM-targeting NPs to the injured blood vessels and nearby tissues.
Figure 3.

(a) Representative in vivo optical and fluorescence overlaid images of high-dose X-ray irradiated healthy Nu mice recorded (i) pre-injection and (ii) 24 h after tail-vein i.v. injection of BM-targeting NPs (BM-NPs) or non-targeting NPs (NT-NPs). (See Figure S5 for the overlaid images of the remaining 6 Nu mice recorded 24 h after administration of BM-targeting NPs and non-targeting NPs.) 6 out of 7 of the X-ray irradiated mice showed increased photon flux (shown in yellow-red) at left flank (irradiated site) after the administration of the BM-targeting NPs. (iii) The total photon fluxes at the left and right flanks of the irradiated mice recorded 24 h after i.v. administration of BM-targeting NPs or non-targeting NPs, as quantified from the corresponding in vivo fluorescence images. The inset cartoon and digital image show the setup for site-specific XRT. (b) Representative low- and high-magnification optical and fluorescent histological images of (i) left and (ii) right leg histological sections that collected after the in vivo imaging study. The blue arrows labeled the vascular injury region. Fluorescence basement membrane can be easily identified from the high magnification fluorescence image of left leg section collected from mouse administrated with BM-targeting NPs. (N.B. n.s. denotes statistical insignificant; * denotes p < 0.05, i.e. statistical significance. L denotes lumen; TI denotes tunica intima which compose of endothelium (ED) and basement membrane (BM); TM denotes tunica media. Low magnification fluorescence images were recorded using 200 ms exposure time, high magnification images were recorded using 100 ms exposure time.)
In summary, we successfully engineered new fluorescent-labeled collagen IV-targeting peptide-conjugated BM-targeting PEG-PLGA NPs for the first direct observation of an early-stage vascular injury-induced by high-dose XRT. Quantitative in vitro study in collagen IV-based solid-phase binding assay and quantitative ex vivo imaging studies in Nu mice skin demonstrated that BM-targeting NPs bind strongly and specifically to collagen IV fibers in the basement membrane. Intravenous administration of fluorescent BM-targeting NPs allowed us to observe an early-stage blood vessels injury-induced by high-dose XRT in a murine model via standard full body in vivo fluorescence imaging technique due to specific binding of BM-targeting NPs to the basement membrane of damaged blood vessels and surrounding tissues, which was confirmed by further histological study. The successful identification of an early-stage high-dose XRT-induced blood vessel injury should facilitates the developments of new preventative treatments for high-dose XRT-induced vascular diseases. In addition, the successful fabrication of BM-targeting PEG-PLGA NPs should facilitate the development of a new basement membrane-targeting drug delivery system to treat basement membrane-related complications, such as surgical adhesion, that have posed a major challenge in medicine for many years.
Experimental Section
Materials and Experimental Setup are provided in the Supporting Information.
Supplementary Material
Figure S1. Assigned 1H NMR(500 MHz, 2,048 scans) spectra of (i) maleimide-functionalized PEG(5K)-PLGA(55K) and (ii) collagen IV-targeting peptide conjugated PEG(5K)-PLGA(55K). The disappearance of sharp maleimide protons (Ha and Hb) at 6.7 ppm and the appearance of new broad protons bands at 0.5, 0.8, 1.25, 2.9 – 3.0 and 8.0 – 8.25 ppm (all contributed from collagen IV-targeting peptide, labeled as *) confirmed the complete conjugation of collagen IV-targeting peptide to the di-block copolymer. (N.B. The collagen IV-targeting peptide-conjugated PEG-PLGA was dried under nitrogen for 3 days to vaporize the DMF (solvent) and trimethylamine (catalyst) before the NMR study.)
Figure S2. Intensity-average diameter (Dh) distribution curves recorded for (i) BM-targeting NPs and (ii) non-targeting NPs dispersed in 0.1 M PBS, as determined by DLS.
Figure S3. In vitro toxicity of 0.02 – 15.00 nM of non-fluorescent BM-targeting NPs and non-targeting NPs against KB epithelial cells. The toxicity study indicate both PEG-PLGA NPs are non-toxic (cell viability ≥ 90 %). All toxicity study were performed in triplicate.
Figure S4. Concentration-dependent fluorescence intensities recorded for 0.0015 – 1.5 nM (0.00005 to 1 mg/mL) of (i) BM-targeting NPs and (ii) non-targeting NPs via a 96-well plate reader (Infinite® 200 Pro, Tecan i-control), using an excitation wavelength (λex) of 535 nm and an emission wavelength (λem) of 595 nm.
Figure S5. Representative in vivo optical and fluorescence overlaid images of high-dose X-ray irradiated healthy Nu mice recorded 24 h after i.v. administration of (a) BM-targeting NPs or (b) non-targeting NPs.
Acknowledgments
We thank Microscopy Service Laboratory Core, Animal Study Core, Small Animal Imaging Facility, and Animal Histopathology Core Facility in the School of Medicine at the University of North Carolina at Chapel Hill for their assistance with procedures in this manuscript. Peptide synthesis was performed in the High-Throughput Peptide Synthesis and Arrays Core Facility at University of North Carolina in Chapel Hill. North Carolina Biotechnology Center Institutional Development Grant provided funding to develop this facility. This work was supported by the University Cancer Research Fund from the University of North Carolina and R01CA178748 from the National Institutes of Health/National Cancer Institute. AZW was also supported by the National Institutes of Health Center for Nanotechnology Excellence Grant U54-CA151652.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Kin Man Au, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Sayed Nabeel Hyder, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Kyle Wagner, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Dr. Caihong Shi, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Shenyang Pharmaceutical University, 103 Wenhua Road, Shenhe, Shenyang, Liaoning, 110016, China
Dr. Young Seok Kim, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Asan Medical Center, College of Medicine, University of Ulsan, Seoul 138-736, Korea
Dr. Joseph M Caster, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Dr. Xi Tian, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Dr. Yuanzeng Min, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Dr. Andrew Z. Wang, Email: zawang@med.unc.edu, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
References
- 1.Russell NS, Hoving S, Heeneman S, Hage JJ, Woerdeman LA, de Bree R, Lohuis PJ, Smeele L, Cleutjens J, Valenkamp A, Dorresteijn LD, Dalesio O, Daemen MJ, Stewart FA. Radiother Oncol. 2009;92:477. doi: 10.1016/j.radonc.2009.05.021. [DOI] [PubMed] [Google Scholar]; Lancet. 2000;355:1757. [Google Scholar]; Adams MJ, Lipsitz SR, Colan SD, Tarbell NJ, Treves ST, Diller L, Greenbaum N, Mauch P, Lipshultz SE. J Clin Oncol. 2004;22:3139. doi: 10.1200/JCO.2004.09.109. [DOI] [PubMed] [Google Scholar]; Aleman BM, van den Belt-Dusebout AW, Klokman WJ, Van’t Veer MB, Bartelink H, van Leeuwen FE. J Clin Oncol. 2003;21:3431. doi: 10.1200/JCO.2003.07.131. [DOI] [PubMed] [Google Scholar]; Aleman BM, van den Belt-Dusebout AW, De Bruin ML, van’t Veer MB, Baaijens MH, de Boer JP, Hart AA, Klokman WJ, Kuenen MA, Ouwens GM, Bartelink H, van Leeuwen FE. Blood. 2007;109:1878. doi: 10.1182/blood-2006-07-034405. [DOI] [PubMed] [Google Scholar]; Bowers DC, McNeil DE, Liu Y, Yasui Y, Stovall M, Gurney JG, Hudson MM, Donaldson SS, Packer RJ, Mitby PA, Kasper CE, Robison LL, Oeffinger KC. J Clin Oncol. 2005;23:6508. doi: 10.1200/JCO.2005.15.107. [DOI] [PubMed] [Google Scholar]; Bowers DC, Liu Y, Leisenring W, McNeil E, Stovall M, Gurney JG, Robison LL, Packer RJ, Oeffinger KC. J Clin Oncol. 2006;24:5277. doi: 10.1200/JCO.2006.07.2884. [DOI] [PubMed] [Google Scholar]; Clarke M, Collins R, Darby S, Davies C, Elphinstone P, Evans E, Godwin J, Gray R, Hicks C, James S, MacKinnon E, McGale P, McHugh T, Peto R, Taylor C, Wang Y EB CT CG (EBCTCG) Lancet. 2005;366:2087. doi: 10.1016/S0140-6736(05)67887-7. [DOI] [PubMed] [Google Scholar]; Darby S, McGale P, Peto R, Granath F, Hall P, Ekbom A. BMJ. 2003;326:256. doi: 10.1136/bmj.326.7383.256. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dorresteijn LD, Kappelle AC, Boogerd W, Klokman WJ, Balm AJ, Keus RB, van Leeuwen FE, Bartelink H. J Clin Oncol. 2002;20:282. doi: 10.1200/JCO.2002.20.1.282. [DOI] [PubMed] [Google Scholar]; Dorresteijn LD, Kappelle AC, Scholz NM, Munneke M, Scholma JT, Balm AJ, Bartelink H, Boogerd W. Eur J Cancer. 2005;41:1026. doi: 10.1016/j.ejca.2005.01.020. [DOI] [PubMed] [Google Scholar]; Hooning MJ, Aleman BM, van Rosmalen AJ, Kuenen MA, Klijn JG, van Leeuwen FE. Int J Radiat Oncol Biol Phys. 2006;64:1081. doi: 10.1016/j.ijrobp.2005.10.022. [DOI] [PubMed] [Google Scholar]; Hooning MJ, Botma A, Aleman BM, Baaijens MH, Bartelink H, Klijn JG, Taylor CW, van Leeuwen FE. J Natl Cancer Inst. 2007;99:365. doi: 10.1093/jnci/djk064. [DOI] [PubMed] [Google Scholar]; Jagsi R, Griffith KA, Koelling T, Roberts R, Pierce LJ. J Clin Oncol. 2006;24:2779. doi: 10.1200/JCO.2005.04.0014. [DOI] [PubMed] [Google Scholar]; Ural AU, Avcu F, Baran Y. Cancer. 2007;110:469. doi: 10.1002/cncr.22804. [DOI] [PubMed] [Google Scholar]; Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Biomaterials. 2007;28:869. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weintraub NL, Jones WK, Manka D. J Am Coll Cardiol. 2010;55:1237. doi: 10.1016/j.jacc.2009.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baskar R, Lee KA, Yeo R, Yeoh KW. J Med Sci Int. 2012;9:193. doi: 10.7150/ijms.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bhide SA, Nutting CM. BMC Med. 2010;8:25. doi: 10.1186/1741-7015-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fajardo LF. Cardiovasc Radiat Med. 1999;1:108. doi: 10.1016/s1522-1865(98)00012-2. [DOI] [PubMed] [Google Scholar]
- 5.Fajardo LF, Berthrong M. Pathol Annu. 1988;23(Pt 1):297. [PubMed] [Google Scholar]; Jurado JA, Bashir R, Burket MW. Catheter Cardiovasc Interv. 2008;72:563. doi: 10.1002/ccd.21681. [DOI] [PubMed] [Google Scholar]
- 6.Halle M, Gabrielsen A, Paulsson-Berne G, Gahm C, Agardh HE, Farnebo F, Tornvall P. J Am Coll Cardiol. 2010;55:1227. doi: 10.1016/j.jacc.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 7.Hudson BG, Reeders ST, Tryggvason K. J Biol Chem. 1993;268:26033. [PubMed] [Google Scholar]; Kalluri R. Nat Rev Cancer. 2003;3:422. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]; Blann AD. Pathophysiol Haemost Thromb. 2003;33:445. doi: 10.1159/000083843. [DOI] [PubMed] [Google Scholar]
- 8.Santos JL, Li Y, Culver HR, Yu MS, Herrera-Alonso M. Chem Commun (Camb) 2014;50:15045. doi: 10.1039/c4cc06056c. [DOI] [PMC free article] [PubMed] [Google Scholar]; Danila D, Johnson E, Kee P. Nanomedicine. 2013;9:1067. doi: 10.1016/j.nano.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Chan JM, Rhee JW, Drum CL, Bronson RT, Golomb G, Langer R, Farokhzad OC. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19347. doi: 10.1073/pnas.1115945108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Mol Pharm. 2008;5:505. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Adv Drug Deliv Rev. 2014;66:2. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. J Control Release. 2012;161:505. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 13.Werner ME, Karve S, Sukumar R, Cummings ND, Copp JA, Chen RC, Zhang T, Wang AZ. Biomaterials. 2011;32:8548. doi: 10.1016/j.biomaterials.2011.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quintanar-Guerrero D, Allémann E, Fessi H, Doelker E. Drug Dev Ind Pharm. 1998;24:1113. doi: 10.3109/03639049809108571. [DOI] [PubMed] [Google Scholar]
- 15.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Proc Natl Acad Sci U S A. 2008;105:17356. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pounder RJ, Stanford MJ, Brooks P, Richards SP, Dove AP. Chem Commun (Camb) 2008:5158. doi: 10.1039/b809167f. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Pei Y, Zhang X, Gu Z, Zhou Z, Yuan W, Zhou J, Zhu J, Gao X. J Control Release. 2001;71:203. doi: 10.1016/s0168-3659(01)00218-8. [DOI] [PubMed] [Google Scholar]
- 18.Weiss JN. FASEB J. 1997;11:835. [PubMed] [Google Scholar]
- 19.Knop K, Hoogenboom R, Fischer D, Schubert US. Angew Chem Int Ed Engl. 2010;49:6288. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Assigned 1H NMR(500 MHz, 2,048 scans) spectra of (i) maleimide-functionalized PEG(5K)-PLGA(55K) and (ii) collagen IV-targeting peptide conjugated PEG(5K)-PLGA(55K). The disappearance of sharp maleimide protons (Ha and Hb) at 6.7 ppm and the appearance of new broad protons bands at 0.5, 0.8, 1.25, 2.9 – 3.0 and 8.0 – 8.25 ppm (all contributed from collagen IV-targeting peptide, labeled as *) confirmed the complete conjugation of collagen IV-targeting peptide to the di-block copolymer. (N.B. The collagen IV-targeting peptide-conjugated PEG-PLGA was dried under nitrogen for 3 days to vaporize the DMF (solvent) and trimethylamine (catalyst) before the NMR study.)
Figure S2. Intensity-average diameter (Dh) distribution curves recorded for (i) BM-targeting NPs and (ii) non-targeting NPs dispersed in 0.1 M PBS, as determined by DLS.
Figure S3. In vitro toxicity of 0.02 – 15.00 nM of non-fluorescent BM-targeting NPs and non-targeting NPs against KB epithelial cells. The toxicity study indicate both PEG-PLGA NPs are non-toxic (cell viability ≥ 90 %). All toxicity study were performed in triplicate.
Figure S4. Concentration-dependent fluorescence intensities recorded for 0.0015 – 1.5 nM (0.00005 to 1 mg/mL) of (i) BM-targeting NPs and (ii) non-targeting NPs via a 96-well plate reader (Infinite® 200 Pro, Tecan i-control), using an excitation wavelength (λex) of 535 nm and an emission wavelength (λem) of 595 nm.
Figure S5. Representative in vivo optical and fluorescence overlaid images of high-dose X-ray irradiated healthy Nu mice recorded 24 h after i.v. administration of (a) BM-targeting NPs or (b) non-targeting NPs.