

Abstract
4-Cresol is not only a significant synthetic intermediate for production of many aromatic chemicals, but also a priority environmental pollutant because of its toxicity to higher organisms. In our previous studies, a gene cluster implicated to be involved in 4-cresol catabolism, creCDEFGHIR, was identified in Corynebacterium glutamicum and partially characterized in vivo. In this work, we report on the discovery of a novel 4-cresol biodegradation pathway that employs phosphorylated intermediates. This unique pathway initiates with the phosphorylation of the hydroxyl group of 4-cresol, which is catalyzed by a novel 4-methylbenzyl phosphate synthase, CreHI. Next, a unique class I P450 system, CreJEF, specifically recognizes phosphorylated intermediates and successively oxidizes the aromatic methyl group into carboxylic acid functionality via alcohol and aldehyde intermediates. Moreover, CreD (phosphohydrolase), CreC (alcohol dehydrogenase), and CreG (aldehyde dehydrogenase) were also found to be required for efficient oxidative transformations in this pathway. Steady-state kinetic parameters (Km and kcat) for each catabolic step were determined, and these results suggest that kinetic controls serve a key role in directing the metabolic flux to the most energy effective route.
Keywords: biodegradation, cytochrome P450, gram-positive bacteria, microbiology, phosphorylation, 4-cresol, Corynebacterium glutamicum
Introduction
The aromatic compound 4-cresol (i.e. p-cresol or p-methylphenol) is an important synthetic precursor for manufacturing a great variety of chemical products including synthetic resins, disinfectants, antioxidants, preservatives, fumigants, explosives, and others (1). However, it is also a priority environmental pollutant because of its high toxicity to many higher organisms including humans (2, 3). This compound is mainly derived from diverse industrial processes such as coal gasification and fractionation of coal tar. In nature, 4-cresol is generated by anaerobic bacteria as a byproduct during the metabolism of phenylalanine and tyrosine (4–6).
Like other aromatic compounds, 4-cresol in polluted environments is degraded by various aerobic and anaerobic microorganisms. Thus, studies on biodegradation mechanisms of 4-cresol hold significant potential for industrial application in environmental protection. In the past decades, a growing number of microorganisms capable of degrading 4-cresol have been discovered, and great efforts have been made to elucidate their biodegradation pathways (7–16).
Currently known 4-cresol catabolic pathways can be classified into one of three categories based on the initial step (Fig. 1). The microorganisms falling into the first category (Fig. 1A) begin their degradation of 4-cresol from a methyl hydroxylation step. For instance, in Gram-negative Pseudomonas species, 4-cresol is initially oxidized to 4-hydroxybenzyl alcohol, and then to 4-hydroxybenzyl aldehyde by the same enzyme 4-cresol methylhydroxylase (PCMH)3 (17–20). Subsequently, 4-hydroxybenzyl aldehyde undergoes variant modifications to form protocatechuatic acid (i.e. 3,4-dihydroxybenzoate) or benzoyl-CoA, both of which can be diverted into the central metabolism (i.e. TCA) (9, 13, 15, 19, 21).
FIGURE 1.

Known pathways for 4-cresol catabolism by variant microorganisms. Degradation of 4-cresol begins from methyl hydroxylation (A), linking fumarate to the methyl group (B), and direct aromatic ring hydroxylation (C).
In the second category, the obligate anaerobe Desulfobacterium cetonicum (12) initiates the degradation of 4-cresol by linking a fumarate moiety to the methyl group to generate 4-hydroxybenzyl succinate, which is further degraded to 4-hydroxybenzoyl-CoA via β-oxidation (Fig. 1B). Subsequent reduction to benzoyl-CoA leads the metabolic flux to the central metabolism.
The fungus Aspergillus fumigates likely adopts the third type of degradation pathway to assimilate 4-cresol (14). To form protocatechuatic acid, the hydroxylation of 4-cresol by an NADPH-dependent hydroxylase first occurs on aromatic ring to produce 4-methylcatecol, followed by a series of methyl oxidations (Fig. 1C). The intermediates and the enzyme activities for the proposed steps have been identified using cell free extracts (14). In yeast Trichosporon cutaneum (16), an ortho-fission enzyme directly transforms 4-methylcatecol into 3-methyl-cis,cis-muconic acid, which can readily enter the central metabolism via the β-ketoadipate pathway (8) (Fig. 1C).
More recently, focus has shifted toward understanding the capacity of the Gram-positive bacterium Corynebacterium glutamicum to metabolize diverse aromatic compounds including 4-cresol (22–31). This high GC content bacterium is not only an important industrial microorganism for production of amino acids and vitamins (32–37), but is also a useful model system to understand the genetics, biochemistry, and mechanisms for biodegradation, especially for assimilation of aromatic compounds (23).
In our previous studies, we employed proteomic analysis and genome mining to identify a gene cluster in C. glutamicum, creABCDEFGHIJR, which is involved in 4-cresol catabolism (22, 26). Specifically, six (creD, creE, creG, creH, creI, and creJ) of eleven cre genes were successfully knocked out from the genome of C. glutamicum RES167. Each knock-out strain lost the ability of the wild-type strain to grow in the minimal medium in which 4-cresol was the sole carbon source. For each mutant, genetic complementation with the corresponding gene restored the wild-type phenotype (22). These results clearly demonstrated that at least these six genes were required for 4-cresol biodegradation in C. glutamicum. Furthermore, subjecting the six mutants to medium containing diverse aromatic compounds as the sole carbon source, such as 4-hydroxybenzyl alcohol, 4-hydroxybenzyl aldehyde, and 4-hydroxybenzoate, suggested a catabolic pathway responsible for the degradation of 4-cresol (22). However, details of this putative catabolic pathway could not be rationalized from our previous experiments.
Although the cre pathway was genetically identified in part, the catalytic functions of the majority of enzymes were not determined. Of the remaining five genes in the pathway, creR is a putative regulatory gene and is therefore not predicted to function as a catabolic catalyst. Based on bioinformatics analysis, creA and creB are believed to reside outside of the cre gene cluster boundary. Interestingly, both Corynebacterium efficiens YS-314 and Arthrobacter sp. FB24 possess the identical cre genetic organization to that of C. glutamicum with the noted absence of homologues to creA and creB. Previously, we generated the creA and creB deletion mutant in C. glutamicum and observed that each resulting strain grew similarly to wild type on the medium supplemented with 4-cresol (22). Thus, neither creA nor creB are predicted to be involved in 4-cresol biodegradation. Finally, numerous attempts to generate creC and creF deletion mutants were unsuccessful. Therefore, the role of these gene products in 4-cresol degradation could not be anticipated.
In the current work, all biodegradation enzymes encoded by the cre gene cluster of C. glutamicum were cloned, expressed, and functionally characterized using in vitro enzymatic assays. Through these efforts, an unprecedented 4-cresol catabolic pathway was unveiled that features a novel activating mechanism and a group of enzymes possessing remarkable substrate flexibility. Of particular interest, a unique phosphorylation reaction mediated by CreHI was identified as a novel initiating step of 4-cresol biodegradation. Further, a class I cytochrome P450 (P450) system (38) consisting of CreJ (P450 enzyme), CreF (ferredoxin), and CreE (ferredoxin reductase) was elucidated during this investigation. In vitro characterization of this system demonstrated that it accepts multiple phosphorylated aromatic compounds as substrates and plays a central role in assimilation of 4-cresol.
Experimental Procedures
Strains, Plasmids, and Culture Conditions
The bacterial strains and plasmids used in this study are listed in supplemental Table S1. C. glutamicum was routinely cultivated in Luria-Bertani (LB) broth with a rotary shaker (150 rpm) at 30 °C. Escherichia coli strains were grown at 37 °C in LB broth with a rotary shaker (220 rpm) or on LB agar (2% w/v) plates. For protein expression and purification, E. coli strains were cultured in Terrific Broth medium (39). When required, kanamycin was added to a final concentration of 50 μg/ml. E. coli DH5α was used for plasmid construction, storage, and isolation. E. coli BL21 (DE3) was used for recombinant protein expression.
DNA Isolation and Manipulation
The genomic DNA of C. glutamicum was isolated by following the Kirby mix procedure (40). Plasmid DNA was isolated using an E.Z.N.A.TM plasmid miniprep kit (Omega Biotek, Norcross, GA). DNA fragments were purified using the Wizard SV Gel and PCR Clean-up System (Promega, Madison, WI). Ligation, transformation, agarose gel electrophoresis, and other standard techniques for E. coli were performed as previously described (41).
Construction of Expression Vectors for cre Genes
The complete open reading frames of creC, creD, creE, creF, creG, creH, creI, and creJ were PCR-amplified (35 cycles of 95 °C for 20s, 62 °C for 20s, and 72 °C for 60s) from genomic DNA of C. glutamicum using Pfu DNA polymerase (Transgen, Beijing, China). Primers used to amplify the DNA fragments of target genes are listed in supplemental Table S2. Appropriate restriction sites were introduced into the PCR primers for cloning purposes. Resulting PCR amplicons were doubly digested with a specific pair of restriction enzymes and ligated into similarly digested pET-28b (Novagen). All plasmid constructs were confirmed by DNA sequencing (Sangon Biotech, Shanghai, China). Upon sequence verification, plasmids were then transformed into E. coli BL21 (DE3) for protein expression.
Protein Expression and Purification
The E. coli BL21 (DE3) cells harboring the expression plasmids were grown at 37 °C overnight in LB broth containing 50 μg/ml kanamycin. The overnight culture was used as the seed culture to inoculate (1:100 dilution) 1 liter of Terrific Broth medium containing 5% (w/v) glycerol. The cells were grown at 37 °C with shaking until the A600 reached ∼0.6. Protein expression was initiated by adding isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.2 mm. Cultivation continued at 18 °C for an additional 20 h. Purification of His-tagged proteins was carried out as described elsewhere (42) with minor modifications. All protein purification steps were performed at 4 °C. Briefly, the cultures were harvested by centrifugation (5000 × g for 5 min). The cell pellet was resuspended in 40 ml of lysis buffer (50 mm NaH2PO4, 300 mm NaCl, 10% (w/v) glycerol, and 10 mm imidazole, pH 8.0), and the resuspended cells were lysed by sonication. The cell lysate was clarified by centrifugation at 12,000 × g for 30 min to remove cellular debris. Nickel-nitrilotriacetic acid resin (1 ml) (Qiagen) was added to each cell lysate with subsequent incubation with gentle agitation for 30 min. This slurry was loaded into an empty column and washed with ∼200 ml of wash buffer (50 mm NaH2PO4, 300 mm NaCl, 10% (w/v) glycerol, and 20 mm imidazole, pH 8.0) until no protein was detected in flow-through. Next, the nickel-bound protein was eluted from the resin with 10 ml of elution buffer (50 mm NaH2PO4, 300 mm NaCl, 10% (w/v) glycerol, and 250 mm imidazole, pH 8.0). The collected eluent was concentrated with an Amicon Ultra centrifugal filter (Merck) and exchanged into desalting buffer (50 mm NaH2PO4, 10% (w/v) glycerol, pH 7.5) using a PD-10 desalting column (GE Healthcare). The resulting purified proteins were flash frozen by liquid nitrogen and stored at −80 °C for future use.
Purity Qualification and Concentration Determination of Purified Proteins
The purity of all purified proteins was evaluated by SDS-PAGE (supplemental Fig. S1). The concentration of P450 enzyme CreJ was determined according to the method described by Omura and Sato (43). Briefly, the CO-bound reduced difference spectrum of CreJ was recorded by a UV-visible spectrophotometer DU 800 (Beckman Coulter, Fullerton, CA). The concentration of functional P450 was subsequently calculated using the extinction coefficient (ϵ450–490) of 91,000 m−1cm−1. Concentrations of non-P450 enzymes were determined by the Bradford method using BSA as the standard (44).
Enzymatic Assays
The assay employed to evaluate the activity of CreHI was developed based on the previous report by Schmeling et al. (45). The reaction mixture contained 1.0 mm of 4-cresol, 4-hydroxybenzyl alcohol, 4-hydroxybenzyl aldehyde, or 4-hydroxybenzoate as substrate, 20 mm MgCl2, 1 mm MnCl2, 2 mm ATP, and 40 μm purified CreH and CreI.
For the assay of CreD, the reaction mixture contained 1.0 mm of 4-methylbenzyl phosphate, benzylalcohol-4-phosphate, benzylaldehyde-4-phosphate, or benzoate-4-phosphate as substrate; 20 mm MgCl2; and 40 μm purified CreD.
For the assay of P450 monooxygenase activity (CreJEF), the reaction mixture contained 1.0 mm of 4-methylbenzyl phosphate, benzylalcohol-4-phosphate, or benzylaldehyde-4-phosphate as substrate, 2.0 mm NADPH, and 40 μm CreE, CreF, and CreJ (42).
The assays of CreG and CreC were performed following the previous method (22) with minor modifications. In brief, the reaction system was comprised of 1.0 mm alcohol substrate (for CreG) or aldehyde substrate (for CreC), 2.0 mm electron acceptor (NAD+ for CreG, NADP+ for CreC), and 40 μm purified CreG or CreC.
For the one-pot reaction, 200 μm 4-cresol was added, and all involved enzymes were normalized to the same concentration of 12 μm. All the above described assays were carried out in 100 mm Tris-HCl buffer (pH 8.0) at 30 °C for 120 min. Reactions were quenched by the 1:1 addition of methanol (MeOH) containing 0.2% (v/v) TFA.
Enzymatic Kinetics
The Km value of each enzyme was determined under the identical condition for its enzymatic activity assay (see above). The concentration of substrates varied from 0.1 to 15 mm, and any required cofactors were added to excess. Each enzyme was diluted to a suitable concentration to ensure that the consumption of substrate was within the linear range during the reaction. Samples from each reaction were taken at 0, 2, and 5 min. The concentrations of products or substrates were determined based on the integrations of chromatographic peak areas observed during HPLC analysis. The Km and kcat values were calculated by nonlinear regression fitting to the Michaelis-Menten equation. All reactions were carried out in triplicate, and the data were reported as the means ± S.D.
Analytical Methods
Reaction mixtures in MeOH/H2O (1:1) with protein removed by centrifugation (20,000 × g for 10 min) were analyzed on an Agilent 1260 infinity HPLC system (Agilent Technologies) equipped with an ultraviolet detector. Specifically, compounds were separated on a ZORBAX SB-C18 column (Agilent Technologies, Wilmington, DE) using a linear mobile phase gradient that ranged from 2% (v/v) MeOH in 0.1% (v/v) TFA aqueous solution to 70% (v/v) MeOH in 0.1% (v/v) TFA aqueous solution over 20 min at a flow rate of 1 ml/min. The wavelength of detection was 275 nm. Structural identification was performed by comparison of retention times of detected compounds with corresponding synthesized authentic standards. Further, eluents were collected and subjected to high resolution mass spectrometry (HRMS) examination. Spectra of electrospray ionization-MS were recorded in the negative ionization mode on an LCQ Deca XPplus ion trap mass spectrometer (Thermo-Finnigan, San Jose, CA).
Chemical Synthesis of Phosphorylated Compounds
The phosphorylated compounds were synthesized through chemical methods with reference to previous reports by McKenna et al. (46) and Kenner and Williams (47) (supplemental Fig. S2). The specific procedures are detailed in the supplemental materials. The structures of all the synthetic phosphorylated compounds were confirmed through NMR spectroscopy (supplemental Figs. S3–S8). Benzoate-4-phosphate was produced from benzylaldehyde-4-phosphate through enzymatic reaction catalyzed by the cytochrome P450 system composed of CreJ, CreE, and CreF. The proteins of this enzymatic system in crude benzoate-4-phosphate solution were removed through heat denaturation and centrifugation.
Results
4-Cresol Is Activated via Phosphorylation by a New 4-Methylbenzyl Phosphate Synthase CreHI
Based on our previous genetic knock-out and complementation studies of cre genes and also on the identification of the metabolic intermediate 4-hydroxybenzoate (22), it was deduced that the class I P450 system (48) encoded by creJEF might be involved in the initial oxidation of 4-cresol. Similar hydroxylation of the methyl group by 4-cresol methylhydroxylase (a flavocytochrome) has been identified in Pseudomonas putida (49) and Geobacter metallireducens (50). However, we failed to detect hydroxylation of 4-cresol in reactions employing the CreJEF P450 system. Moreover, when we tested the in vitro ability of individual enzymes (CreC, CreD, CreG, CreH, or CreI), in the presence of required cofactors, to hydroxylate 4-cresol, no conversion was observed (data not shown). Taken together, these results suggest that an enzyme complex might be required for initiating the modification of 4-cresol.
Subsequent bioinformatics analysis revealed that CreH contains a phosphoenolpyruvate (PEP)-utilizing enzyme mobile domain (supplemental Fig. S9), whereas CreI contains a PEP/pyruvate binding domain (supplemental Fig. S9) (51). Their homologues Orf1 and Orf2 subunits identified in Thauera aromatica, which have 62%/45% and 63%/46% amino acid similarity/identity to CreH and CreI, respectively, were reported to be responsible for converting phenol into phenylphosphate when acting synergistically (45). In view of this report, as well as the structural similarity between 4-cresol and phenol, we hypothesized that CreH and CreI might be required in tandem to phosphorylate 4-cresol.
To test this hypothesis, recombinant CreH and CreI were produced and purified from E. coli (supplemental Fig. S1), and their combined in vitro catalytic activity against 4-cresol was evaluated. In the reaction mixture containing both CreH and CreI, as well as ATP, Mg2+, and Mn2+ (all required), 4-cresol was converted to a new product (Fig. 2A). The identity of this product was determined to be 4-methylbenzyl phosphate by HRMS (Fig. 2B) and was confirmed by comparison with the chemically synthesized authentic standard (Fig. 2A). CreHI was also able to catalyze the phosphorylation of 4-hydroxybenzyl alcohol/aldehyde (but not 4-hydroxybenzoate), into their corresponding phosphorylated products benzylalcohol-4-phosphate and benzylaldehyde-4-phosphate, respectively (Fig. 2, A, C, and D). A similar ion-dependent phenylphosphate synthase has been discovered in an anaerobic phenol degrading T. aromatica strain (45); however, the newly identified CreHI 4-methylbenzyl phosphate synthase differentiates from the T. aromatica phenylphosphate synthase by featuring a distinct substrate profile.
FIGURE 2.

CreHI catalyzed reactions. A, upper panel, CreHI reaction scheme. Lower panel, HPLC analysis (275 nm) of CreHI catalyzed reactions. s1 and s2, authentic standards; i, 4-cresol as substrate; ii, 4-hydroxybenzyl alcohol as substrate; iii, 4-hydroxybenzyl aldehyde as substrate; iv, 4-hydroxybenzoate as substrate. Traces in blue represent corresponding control reactions without the addition of enzymes. B, HRMS of 4-methylbenzyl phosphate. C, HRMS of benzylalcohol-4-phosphate. D, HRMS of benzylaldehyde-4-phosphate. Note that because of distinct extinction coefficients, the peak intensity of compounds do not necessarily reflect relative amounts.
CreJEF Represents a Unique Class I Cytochrome P450 System That Successively Oxidizes 4-Methylbenzyl Phosphate, Benzylalcohol-4-phosphate, and Benzylaldehyde-4-phosphate
Based on DNA sequence analysis, creJ putatively encodes a cytochrome P450 enzyme. The coexistence of the ferredoxin gene creF and the ferredoxin reductase gene creE in the same gene cluster strongly suggests that CreJEF should form a three-component class I cytochrome P450 system (52). CreJEF cannot directly oxidize 4-cresol; however, the discovery that CreHI produces phosphorylated products naturally led to the hypothesis that CreJEF might require phosphorylated substrate(s). As expected, CreJEF recognized 4-methylbenzyl phosphate as substrate. Interestingly, three products were observed (Fig. 3A), two of which were identified as benzylalcohol-4-phosphate and benzylaldehyde-4-phosphate (see above). The third product was determined to be benzoate-4-phosphate based on HRMS (Fig. 3B) and the proton NMR of the enzymatically prepared product (supplemental Fig. S10). Not surprisingly, CreJEF also catalyzed the oxidation of benzylalcohol-4-phosphate and benzylaldehyde-4-phosphate (Fig. 3A). Taken together, our results demonstrate that CreJEF is a class I P450 system that catalyzes the sequential oxidations from 4-methylbenzyl phosphate to benzoate-4-phosphate.
FIGURE 3.
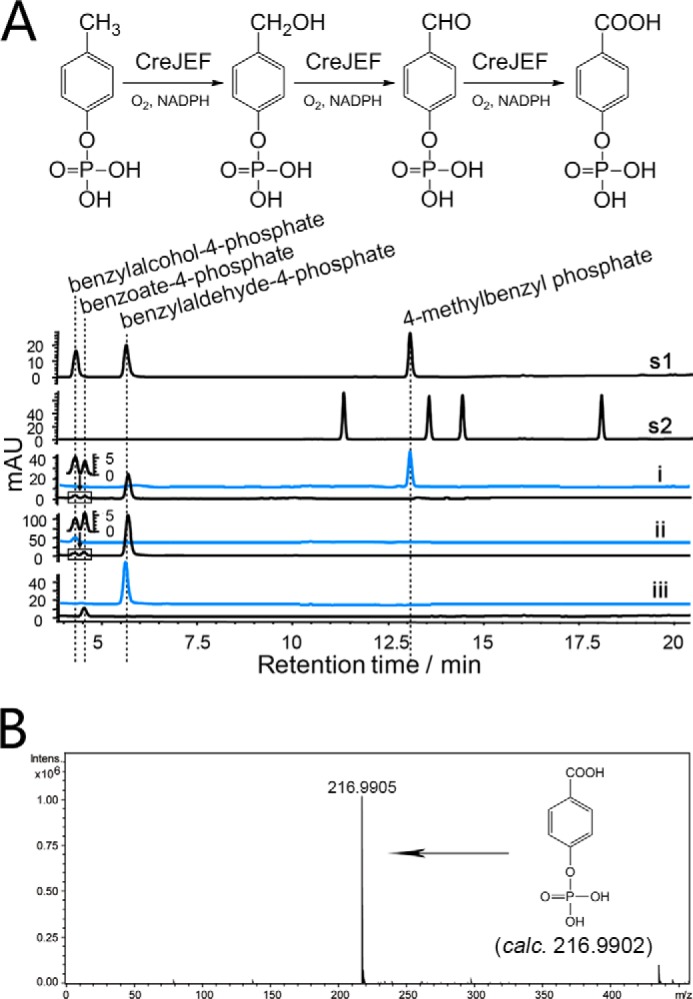
CreJEF catalyzed reactions. A, upper panel, CreJEF reactions scheme. Lower panel, HPLC analysis (275 nm) of CreJEF catalyzed reactions. s1 and s2, authentic standards; i, 4-methylbenzyl phosphate as substrate; ii, benzylalcohol-4-phosphate as substrate; iii, benzylaldehyde-4-phosphate as substrate. Traces in blue represent corresponding control reactions without addition of enzymes. B, HRMS of benzoate-4-phosphate. Note that because of distinct extinction coefficients, the peak intensity of compounds do not necessarily reflect relative amounts.
CreG Is a 4-Hydroxybenzyl Alcohol Dehydrogenase
It was previously reported that creG encodes an NAD+-dependent 4-hydroxybenzyl alcohol dehydrogenase that oxidizes 4-hydroxybenzyl alcohol into 4-hydroxybenzyl aldehyde (22). In the present work, we further demonstrated that CreG was also able to oxidize benzylalcohol-4-phosphate into benzylaldehyde-4-phosphate (Fig. 4A). Steady-state kinetic analysis indicated that CreG had higher affinity to benzylalcohol-4-phosphate (Km = 1.2 ± 0.3 mm) than to 4-hydroxybenyl alcohol (Km = 9.4 ± 0.2 mm) (Table 1). Notably, CreG used NAD+ as a preferred cofactor. The dehydrogenase activity of CreG supported by NADP+ was observed to be 1.3 × 103 times lower than that by NAD+, when 4-hydroxybenzyl alcohol was employed as substrate.
FIGURE 4.

Reactions catalyzed by CreG and CreC. A, upper panel, CreG reactions scheme. Lower panel, HPLC analysis (275 nm) of CreG catalyzed reactions. s1 and s2, authentic standards; i, 4-hydroxybenzyl alcohol as substrate; ii, benzylalcohol-4-phosphate as substrate. B, upper panel, CreC reactions scheme. Lower panel, HPLC analysis of CreC catalyzed reactions. s1 and s2, authentic standards; i, 4-hydroxybenzyl aldehyde as substrate; ii, benzylaldehyde-4-phosphate as substrate. Traces in blue represent corresponding control reactions without addition of enzymes. Note that because of distinct extinction coefficients, the peak intensity of compounds do not necessarily reflect relative amounts.
TABLE 1.
Steady-state kinetics of enzymes encoded by the cre cluster
The values were calculated by nonlinear regression fitting to the Michaelis-Menten equation. All reactions were carried out in triplicate, and the data are reported as means ± S.D.
| Enzyme | Substrate | Km | kcat | kcat/Km |
|---|---|---|---|---|
| mm | min−1 | mm−1 min−1 | ||
| CreHI | 4-Cresol | 0.61 ± 0.06 | 60.4 ± 1.8 | 99 |
| CreHI | 4-Hydroxybenzyl alcohol | 0.10 ± 0.03 | 36.3 ± 2.7 | 3.7 × 102 |
| CreHI | 4-Hydroxybenzyl aldehyde | 0.21 ± 0.02 | 14.9 ± 2.7 | 71 |
| CreJEF | 4-Methylbenzyl phosphate | 0.34 ± 0.06 | 44.5 ± 2.3 | 1.3 × 102 |
| CreJEF | Benzylalcohol-4-phosphate | 0.38 ± 0.09 | 5.21 ± 0.33 | 14 |
| CreJEF | Benzylaldehyde-4-phosphate | 0.90 ± 0.22 | 6.61 ± 0.59 | 7.4 |
| CreD | 4-Methylbenzyl phosphate | 0.62 ± 0.09 | 40.2 ± 1.9 | 65 |
| CreD | Benzylalcohol-4-phosphate | 0.52 ± 0.06 | 11.3 ± 1.5 | 22 |
| CreD | Benzylaldehyde-4-phosphate | 0.32 ± 0.02 | 23.3 ± 0.7 | 72 |
| CreC | 4-Hydroxybenzyl aldehyde | 0.058 ± 0.006 | 4.43 ± 0.20 | 77 |
| CreC | Benzylaldehyde-4-phosphate | 0.12 ± 0.04 | (1.41 ± 0.10) ×103 | 1.2 × 104 |
| CreG | 4-Hydroxybenzyl alcohol | 9.4 ± 0.2 | 24.8 ± 0.4 | 2.6 |
| CreG | Benzylalcohol-4-phosphate | 1.2 ± 0.3 | 1.74 ± 0.20 | 1.5 |
CreC Is a Benzaldehyde-4-phosphate Dehydrogenase
Conserved domain searches suggested that CreC belongs to the aldehyde dehydrogenase superfamily containing an aldehyde dehydrogenase domain and an NAD(P)+ cofactor-binding domain. Based on protein sequence alignment, CreC displays 34% amino acid identity to PcuC, a known 4-hydroxybenzyl aldehyde dehydrogenase from Pseudomonas species (7, 8). Catalytically, CreC was able to oxidize both 4-hydroxybenzyl aldehyde and benzylaldehyde-4-phosphate to corresponding carboxylic acids (Fig. 4B), respectively. Evaluation of the catalytic efficiency with both substrates revealed that CreC was much more efficient in its ability to oxidize benzylaldehyde-4-phosphate (kcat/Km = 1.2 × 104 mm−1min−1) than in oxidizing 4-hydroxybenzyl aldehyde (kcat/Km = 77 mm−1 min−1) (Table 1). Unlike CreG, CreC can only employ NADP+ as a cofactor, because no catalytic activity was detected in the presence of NAD+. This feature is distinct to other benzylaldehyde dehydrogenases, which have been reported to have the capacity to utilize both NAD+ and NADP+ as cofactors (53–55).
CreD Is a Universal Phosphohydrolase
Our previous study demonstrated that CreD displayed phosphohydrolase activity against the unnatural substrate 4-nitrophenylphosphate (22). However, its physiological function remained elusive because there were no substrate candidates available for testing at that time. In this work, the discovery that CreHI catalysis produced phosphorylated products inspired us to examine the activity of CreD against these phosphorylated compounds. As expected, CreD hydrolyzed 4-methylbenzyl phosphate, benzylalcohol-4-phosphate, benzylaldehyde-4-phosphate, and benzoyl-4-phosphate into 4-cresol, 4-hydroxybenzyl alcohol, 4-hydroxybenzyl aldehyde, and 4-hydroxybenzoate, respectively (Fig. 5).
FIGURE 5.
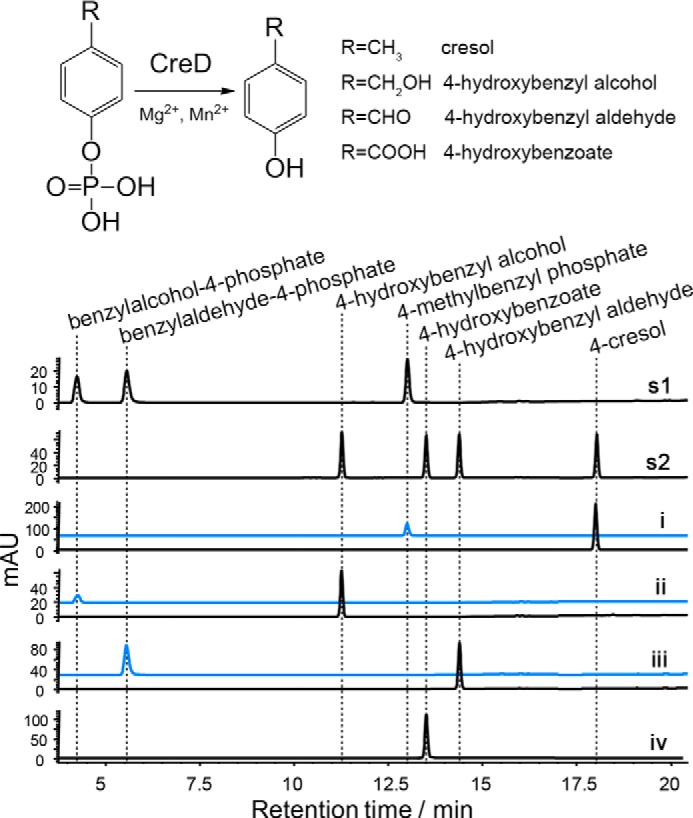
Reactions catalyzed by CreD. Upper panel, CreD reactions scheme. Lower panel, HPLC analysis (275 nm) of CreD catalyzed reactions. s1 and s2, authentic standards; i, 4-methylbenzyl phosphate as substrate; ii, benzylalcohol-4-phosphate as substrate; iii, benzylaldehyde-4-phosphate as substrate; iv, benzoate-4-phosphate as substrate. Traces in blue represent corresponding control reactions without addition of enzymes. Note that because of distinct extinction coefficients, the peak intensity of compounds do not necessarily reflect relative amounts.
In Vitro Assembly of the 4-Cresol Catabolic Pathway
Elucidation of the catalytic activities of CreHI, CreJEF, CreG, CreC, and CreD has led to the proposal of a unique 4-cresol biodegradation pathway in C. glutamicum (Fig. 6). This pathway has two unusual features. First, all involved biocatalysts can accept multiple metabolic intermediates as substrates, which results in two alternative but interactive routes. The first route of 4-cresol degradation proceeds through phosphorylated intermediates (steps 1 → 3 → 4 → 10 → 14), whereas the second route proceeds through nonphosphorylated compounds (steps 1 → 3 → 7 → 8 → 13). Second, CreHI and CreD catalyze reverse reactions. This unique paradigm appears to be energy wasteful because the net outcome of ATP consuming phosphorylation reaction catalyzed by CreHI plus phosphohydrolysis by CreD is equivalent to the uneconomical consumption of ATP.
FIGURE 6.

The catabolic pathway for 4-cresol from C. glutamicum. Bold arrows indicate the major phosphorylated pathway comprised of enzymatic steps with greater kcat/Km values. Each number in bracket denotes an individual enzymatic step.
To understand this seemingly unreasonable network with enzyme functional redundancies (i.e. a common reaction catalyzed by different enzymes) and unfavorable energy economy, we carried out a one-pot enzymatic reaction to evaluate the efficiency of ATP utilization. In this one-pot pathway reconstitution experiment, all enzymes were normalized to the same concentrations (12.0 μm) in the reaction mixture containing 200 μm of 4-cresol and all required cofactors. This experimental design is based on the assumption that all involved enzymes are physiologically expressed simultaneously. This assumption was supported by a series of RT-PCR experiments. To determine whether all cre genes were expressed coordinately, we attempted to amplify all intergenic regions between two adjacent genes within the cre cluster using a common cDNA sample as template. The RT-PCR results (supplemental Fig. S11) indicate that creC-R were transcribed as a single operon and likely expressed coordinately. However, the cellular enzyme levels could vary dynamically because of differential affinity of ribosome binding sites across the polycistronic mRNA or diverse epigenetic effects. Thus, the one-pot reaction containing the same levels of enzymes represented a simplified model system for understanding the kinetic behavior of mixed 4-cresol degrading enzymes.
To initiate the multienzymatic catalysis, varying ATP doses (200, 400, and 600 μm, corresponding to molar ratios of 1:1. 1:2, and 1:3 for -cresol:ATP, respectively) were added into the reaction mixtures. Results showed that 40.8% and 78.8% of 4-cresol were consumed when the substrate:ATP ratios were 1:1 and 1:2, respectively. 4-Cresol was completely consumed when the 4-cresol:ATP ratio was 1:3 (Fig. 7A). These quantitative results indicate that 4-cresol can be efficiently converted into downstream metabolic intermediates; meanwhile the futile ATP consumption indeed occurred to some extent.
FIGURE 7.
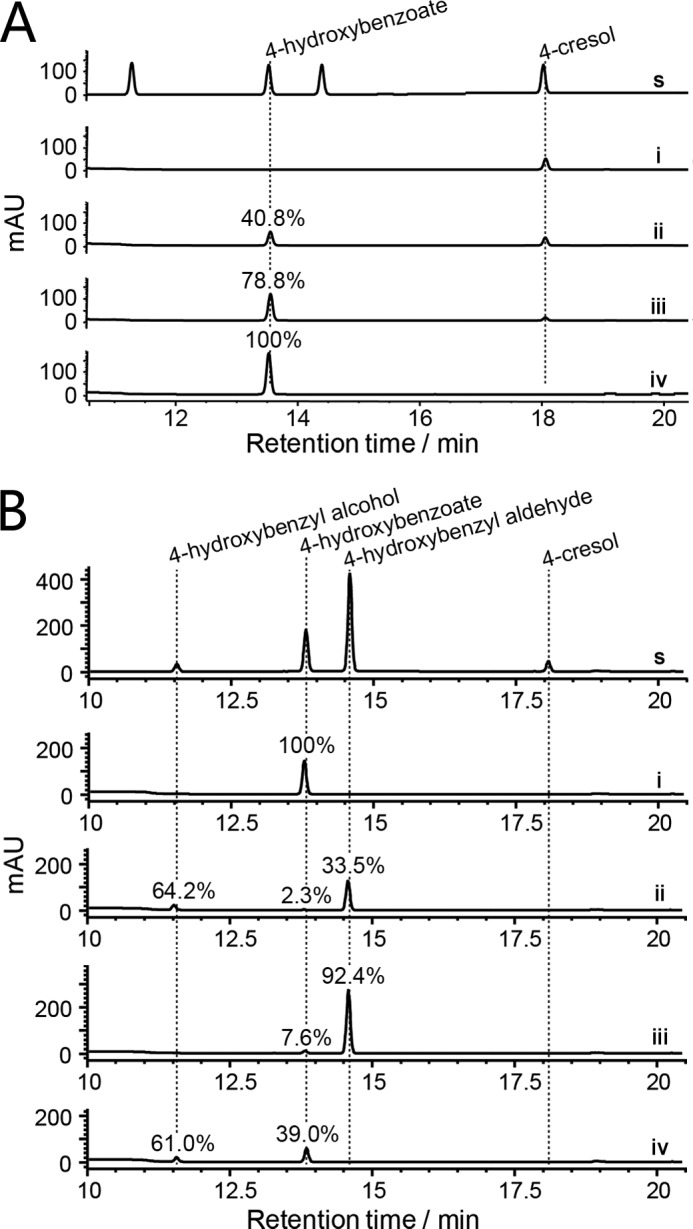
One-pot reaction analyses. A, HPLC analysis (275 nm) of the one-pot reaction containing all enzymes (CreHI, CreJEF, CreD, CreG, and CreC) under different molar ratios of 4-cresol:ATP. s, authentic standards; i–iv, ATP:4-cresol = 0, 1, 2, 3. B, HPLC analysis (275 nm) of the one-pot reaction with different enzyme combinations. s, authentic standards; i, all enzymes (CreHI, CreJEF, CreD, CreG, and CreC) in one pot; ii, all enzymes except CreC and CreG; iii, all enzymes except CreC; iv, all enzymes except CreG. All involved enzymes were normalized to the same concentration of 12.0 μm. All assays used 200 μm 4-cresol as substrate and were carried out in 100 mm Tris-HCl buffer (pH 8.0) at 30 °C for 120 min. In assays of B, the initial concentration of ATP was 600 μm, and all conversions of 4-cresol were complete. Note that each percentage indicates the conversion ratio from 4-cresol to the specific product. Because of distinct extinction coefficients, the peak intensity of compounds do not necessarily reflect relative amounts shown in percentage.
Enzyme Kinetics Regulates the Metabolic Flux in the 4-Cresol Biodegradation Pathway
To further understand the metabolic flux in the complex 4-cresol catabolic pathway, the steady-state enzymatic kinetic parameters of each step were determined (Table 1 and supplemental Figs. S12–S24). The phosphorylation of 4-cresol by CreHI had an apparent Km value of 0.61 ± 0.06 mm and a kcat value of 60.4 ± 1.8 min−1, resulting in a calculated catalytic efficiency (kcat/Km) of 99 mm−1min−1 for this initial step. The catalytic efficiency of CreHI was slightly higher than the phosphohydrolysis of 4-methylbenzyl phosphate (kcat/Km = 65 mm−1min−1) by CreD. Interestingly, the oxidation of 4-methylbenzyl phosphate by CreJEF was more efficient than the phosphohydrolysis by CreD, because CreJEF had lower Km and higher kcat (∼2-fold more efficient than CreD for 4-methylbenzyl phosphate). The competition experiment between CreJEF and CreD showed that the final ratio between oxidation and hydrolysis products was 59:41 (supplemental Fig. S25), which is consistent with the kinetic data.
Next, additional one-pot reactions were performed to evaluate the contribution of CreC and CreG in this catabolic pathway (Fig. 7B). Again, 4-cresol was completely converted to 4-hydroxybenoate in the one-pot reaction containing all enzymes. When both CreC and CreG were absent, the main products turned out to be 4-hydroxybenzyl alcohol (64.2%) and 4-hydroxybenzyl aldehyde (33.5%), whereas the terminal-product 4-hydroxybenzoate only accounted for 2.3% of all products. This result is consistent with the enzyme kinetic studies, where the kcat/Km values of step 4 (14 mm−1min−1) and step 9 (7.4 mm−1min−1) were lower than those of step 7 (22 mm−1min−1) and step 12 (72 mm−1min−1), respectively. When only CreC was omitted, 4-hydroxybenzyl aldehyde was the predominant product (92.4%), 4-hydroxybenzoate was the minor product (7.6%), whereas 4-hydroxybenzyl alcohol was not detected. If only CreG was omitted, 4-hydroxybenzyl alcohol and 4-hydroxybenzoate accounted for 61.0 and 39.0% of products, respectively. In sum, the involvement of CreC and CreG in the reaction pathway serves to make these oxidative conversions more efficient and energy-effective.
Discussion
Two unusual enzymatic systems represent the most important discoveries of the current study: 1) CreHI initiates the pathway by phosphorylating 4-cresol to give 4-methylbenzyl phosphate, and 2) CreJEF catalyzes the three-step sequential oxidation of 4-methylbenzyl phosphate to benzylalcohol-4-phosphate, benzylaldehyde-4-phosphate, and benzoate-4-phosphate.
Conserved domain searches revealed that CreH has a PEP-utilizing enzyme mobile domain, whereas CreI has a PEP/pyruvate binding domain (51). Interestingly, both CreH and CreI possess respective conserved domains with strong similarity to different parts of the PEP synthase of E. coli (supplemental Fig. S9), suggesting a possible evolutionary relationship between CreH, CreI, and PEP synthase. The Orf1 and Orf2 subunits from the anaerobic T. aromatica, which show 45 and 46% amino acid identity to CreH and CreI, respectively, are responsible for converting phenol into phenylphosphate (45, 56, 57). The creHI homologous genes are also found in many other microbial genomes such as C. efficiens, Cladosporium halotolerans, Brevibacterium flavum, Arthrobacter species, Kocuria palustris, Bradyrhizobium species, and Runella slithyformis. Taken together, these gene products may form a new enzyme family capable of phosphorylating phenolic compounds. Moreover, the genomes of C. efficiens, C. halotolerans, B. flavum, Arthrobacter species, and K. palustris contain gene clusters with strong homology to the cre gene cluster of C. glutamicum, suggesting that these bacteria are likely able to degrade 4-cresol using the same mechanism employed by C. glutamicum.
CreJEF is able to catalyze the successive oxidations of phosphorylated intermediates. It is possible that the phosphate group of substrates could act as an anchoring group that delivers the substrate to the correct position within the active site of CreJ, as done by the deoxysugar that is attached to substrates of the PikC P450 enzyme (58, 59). To the best of our knowledge, this is the first reported example of a P450 enzyme that recognizes a spectrum of substrates via a phosphate group. This discovery may have great biotechnological potential for selective oxidation of unactivated C–H bonds.
Some known P450 enzymes are capable of catalyzing the complete oxidation of a C–H bond into a carboxylic group. One important example is the multifunctional ent-kaurene oxidase encoded by the P450–4 gene in the gibberellin biosynthetic pathway of Gibberella fujikuroi. This P450 enzyme catalyzes three successive oxidation steps between ent-kaurene and ent-kaurenoic acid (60). Another example is CYP71AV1 from Artemisia annua, which performs a three-step oxidation of amorpha-4,11-diene to form artemisinic acid (61). To complete this challenging six-electron oxidation, CYP71AV1 recruits an artemisinic aldehyde dehydrogenase and an alcohol dehydrogenase to achieve the optimal transformation (62). This is similar to what we have observed with CreJEF, CreC, and CreG. Likewise, in the tirandamycin biosynthetic pathway, TamI P450 enzyme employs the FAD-containing TamL oxidase to overcome the conversion from an alcohol to a keto group (63). These examples suggest that the successive oxidations are likely a heavy burden for P450 enzymes to accomplish on their own; therefore, assistance is required from other types of oxidases.
It is obvious that the initial oxidation of 4-cresol requiring a prephosphorylation step in C. glutamicum is more complicated than the direct oxidation catalyzed by a 4-cresol methylhydroxylase (PCMH). Kinetically, the latter flavocytochrome is much more efficient. For example, the PCMH from a denitrifying Achromobacter sp. exhibited Km and kcat values for 4-cresol of 21 μm and 112 s−1, respectively (15). The kcat/Km value of another PCMH from P. putida NCIMB 9869 was calculated to be 2.9 × 105 mm−1 min−1 (17). Both enzymes are >2000 times more efficient than CreJEF using 4-methylbenzyl phosphate as substrate (Km = 0.34 mm, kcat = 44.5 min−1; Table 1). This is not surprising because the kcat values of many P450 enzymes are within 1–300 min−1, which can mainly be attributed to the slow electron transfer step (64). Considering that the additional enzymes in the pathway (except for CreC against benzylaldehyde-4-phosphate) also displayed relatively high Km and low kcat values (Table 1), C. glutamicum appears not to be a good 4-cresol assimilating microorganism. However, without a PCMH gene on its genome (data not shown), it is intriguing to speculate that C. glutamicum was forced to evolve a novel, albeit less efficient, pathway for 4-cresol degradation.
Based on the kinetic analyses and competition experiments, we propose that the primary degradation pathway for 4-cresol in C. glutamicum is the phosphorylated route (Fig. 6): 4-cresol is first activated by CreHI via conversion into 4-methylbenzyl phosphate, and subsequent oxidations of phosphorylated intermediates are co-mediated by CreJEF, CreG, and CreC. This proposed pathway sequence is supported by the fact that the conversion from benzoate-4-phosphate to 4-hydroxybenzoate is a unidirectional reaction (Fig. 6), because CreHI does not take 4-hydroxybenoate as substrate.
However, it remains yet premature to suggest that the primary route consisting of steps 1 → 3 → 4 → 10 → 14 represents the physiological pathway for 4-cresol degradation in C. glutamicum because of details lacking with respect to the cellular concentration and localization of enzymes, availability of cofactors, and other intracellular environmental factors. It is noteworthy that there is both agreement as well as inconsistency between the proposed pathway in this study and the previous in vivo results (22). For example, according to the pathways shown in Fig. 6, the inactivation of CreJEF, CreHI, or CreD would block the major catabolic pathway, thus abolishing the ability of knock-out strains to grow on 4-cresol as the sole carbon source. This observation is consistent with the previous observations (22). In contrast, based on the results obtained from the one-pot reactions (Fig. 7B), CreG and CreC appear to be unnecessary in the major route from 4-cresol to 4-hydroxybenzoate. This observation, however, contradicts our earlier finding that the creG knock-out strain was unable to grow using 4-cresol as the sole carbon source. We reason that this inconsistency might be derived from a yet unknown function of CreG or the result of the complexity within the in vivo environment such as balances of energy and cofactors or potential toxicity of produced intermediates that could adversely affect cell growth.
Another interesting aspect of the 4-cresol catabolic pathway is the differential usage of cofactors by CreJEF (NADPH), CreG (NAD+), and CreC (NADP+). This may be of physiological importance, because these enzymes may rely on a cofactor balancing system during degradation of 4-cresol. For instance, CreJEF and CreC may form an NADPH/NADP+ recycling system in vivo to avoid too much loss of reducing power during multiple P450 oxidations.
It is intriguing to speculate that the substrate flexibility of the catabolic enzymes involved in 4-cresol degradation may have physiological significance because this pathway could also assimilate other phenolic compounds. To further assess this possibility, we are currently pursuing more detailed biochemical studies in our laboratories. Enzyme function redundancy may be useful when microorganisms face harsh polluted environments because multiple enzymes with a common functionality could maximally release the metabolic potential by better taking advantage of substrates, cofactors, and energy stored in different forms.
Author Contributions
L. D., X. W., S.-J. L., and S. L. conceived this study, analyzed the results, and wrote the manuscript. L. D., L. M., F. Q., X. Z., C. J., and A. L. conducted experiments. All authors read and approved the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Shaohua Huang, Dr. Ying Yang, and Fali Bai (Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences) for assistance during NMR and LC-MS data collections. We are grateful to Dr. Jeffrey D. Kittendorf (Alluvium Biosciences Inc.) for proofreading the manuscript.
This work was supported by National Natural Science Foundation of China under Grant NSFC 31422002 and Shandong Provincial Natural Science Foundation Grant JQ201407, and the funding from Recruitment Program of Global Experts (to S. L.) and 973 Project from Ministry of Science and Technology Grant 2012CB7211-04 (to S.-J. L.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1 and S2 and Figs. S1–S25.
- PCMH
- 4-cresol methylhydroxylase
- P450
- cytochrome P450 enzyme
- HRMS
- high resolution mass spectrometry
- PEP
- phosphoenolpyruvate.
References
- 1. Fiege H. (2000) Cresols and Xylenols. in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Inc., New York [Google Scholar]
- 2. (2008) Toxicological Profile for Cresols, Agency for Toxic Substances and Disease Registry, U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA: [PubMed] [Google Scholar]
- 3. Back K. C., Thomas A. A., and MacEwen J. D. (1972) Reclassification of Materials Listed as Transportation Health Hazards, 6570th Aerospace Medical Research Laboratory, Wright-Patterson Air Force Base, Fairborn, OH [Google Scholar]
- 4. Curtius H. C., Mettler M., and Ettlinger L. (1976) Study of the intestinal tyrosine metabolism using stable isotopes and gas chromatography-mass spectrometry. J. Chromatogr. 126, 569–580 [DOI] [PubMed] [Google Scholar]
- 5. Yu L., Blaser M., Andrei P. I., Pierik A. J., and Selmer T. (2006) 4-hydroxyphenylacetate decarboxylases: properties of a novel subclass of glycyl radical enzyme systems. Biochemistry 45, 9584–9592 [DOI] [PubMed] [Google Scholar]
- 6. Yokoyama M. T., and Carlson J. R. (1981) Production of skatole and para-cresol by a rumen Lactobacillus sp. Appl. Environ. Microbiol. 41, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cho A. R., Lim E. J., Veeranagouda Y., and Lee K. (2011) Identification of a p-cresol degradation pathway by a GFP-based transposon in Pseudomonas and its dominant expression in colonies. J. Microbiol. Biotechnol. 21, 1179–1183 [DOI] [PubMed] [Google Scholar]
- 8. Jõesaar M., Heinaru E., Viggor S., Vedler E., and Heinaru A. (2010) Diversity of the transcriptional regulation of the pch gene cluster in two indigenous p-cresol-degradative strains of Pseudomonas fluorescens. FEMS Microbiol. Ecol. 72, 464–475 [DOI] [PubMed] [Google Scholar]
- 9. Peters F., Heintz D., Johannes J., van Dorsselaer A., and Boll M. (2007) Genes, enzymes, and regulation of para-cresol metabolism in Geobacter metallireducens. J. Bacteriol. 189, 4729–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butler J. E., He Q., Nevin K. P., He Z., Zhou J., and Lovley D. R. (2007) Genomic and microarray analysis of aromatics degradation in Geobacter metallireducens and comparison to a Geobacter isolate from a contaminated field site. BMC Genomics 8, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tallur P. N., Megadi V. B., Kamanavalli C. M., and Ninnekar H. Z. (2006) Biodegradation of p-cresol by Bacillus sp strain PHN 1. Curr. Microbiol. 53, 529–533 [DOI] [PubMed] [Google Scholar]
- 12. Müller J. A., Galushko A. S., Kappler A., and Schink B. (2001) Initiation of anaerobic degradation of p-cresol by formation of 4-hydroxybenzylsuccinate in Desulfobacterium cetonicum. J. Bacteriol. 183, 752–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Londry K. L., Suflita J. M., and Tanner R. S. (1999) Cresol metabolism by the sulfate-reducing bacterium Desulfotomaculum sp. strain Groll. Can. J. Microbiol. 45, 458–463 [PubMed] [Google Scholar]
- 14. Jones K. H., Trudgill P. W., and Hopper D. J. (1993) Metabolism of p-cresol by the fungus Aspergillus fumigatus. Appl. Environ. Microbiol. 59, 1125–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hopper D. J., Bossert I. D., and Rhodes-Roberts M. E. (1991) p-Cresol methylhydroxylase from a denitrifying bacterium involved in anaerobic degradation of p-cresol. J. Bacteriol. 173, 1298–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Powlowski J. B., and Dagley S. (1985) β-Ketoadipate pathway in Trichosporon cutaneum modified for methyl-substituted metabolites. J. Bacteriol. 163, 1126–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Efimov I., Cronin C. N., Bergmann D. J., Kuusk V., and McIntire W. S. (2004) Insight into covalent flavinylation and catalysis from redox, spectral, and kinetic analyses of the R474K mutant of the flavoprotein subunit of p-cresol methylhydroxylase. Biochemistry 43, 6138–6148 [DOI] [PubMed] [Google Scholar]
- 18. Cunane L. M., Chen Z. W., Shamala N., Mathews F. S., Cronin C. N., and McIntire W. S. (2000) Structures of the flavocytochrome p-cresol methylhydroxylase and its enzyme-substrate complex: gated substrate entry and proton relays support the proposed catalytic mechanism. J. Mol. Biol. 295, 357–374 [DOI] [PubMed] [Google Scholar]
- 19. Kim J., Fuller J. H., Cecchini G., and McIntire W. S. (1994) Cloning, sequencing, and expression of the structural genes for the cytochrome and flavoprotein subunits of p-cresol methylhydroxylase from two strains of Pseudomonas putida. J. Bacteriol. 176, 6349–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mathews F. S., Chen Z. W., Bellamy H. D., and McIntire W. S. (1991) 3-Dimensional structure of para-cresol methylhydroxylase (flavocytochrome-C) from Pseudomonas putida at 3.0-Å resolution. Biochemistry 30, 238–247 [DOI] [PubMed] [Google Scholar]
- 21. Tallur P. N., Megadi V. B., and Ninnekar H. Z. (2009) Biodegradation of p-cresol by immobilized cells of Bacillus sp. strain PHN 1. Biodegradation 20, 79–83 [DOI] [PubMed] [Google Scholar]
- 22. Li T., Chen X., Chaudhry M. T., Zhang B., Jiang C. Y., and Liu S. J. (2014) Genetic characterization of 4-cresol catabolism in Corynebacterium glutamicum. J. Biotechnol. 192, 355–365 [DOI] [PubMed] [Google Scholar]
- 23. Shen X. H., Zhou N. Y., and Liu S. J. (2012) Degradation and assimilation of aromatic compounds by Corynebacterium glutamicum: another potential for applications for this bacterium? Appl. Microbiol. Biotechnol. 95, 77–89 [DOI] [PubMed] [Google Scholar]
- 24. Zhao K. X., Huang Y., Chen X., Wang N. X., and Liu S. J. (2010) PcaO positively regulates pcaHG of the β-ketoadipate pathway in Corynebacterium glutamicum. J. Bacteriol. 192, 1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang Y., Zhao K. X., Shen X. H., Jiang C. Y., and Liu S. J. (2008) Genetic and biochemical characterization of a 4-hydroxybenzoate hydroxylase from Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 78, 75–83 [DOI] [PubMed] [Google Scholar]
- 26. Qi S. W., Chaudhry M. T., Zhang Y., Meng B., Huang Y., Zhao K. X., Poetsch A., Jiang C. Y., Liu S., and Liu S. J. (2007) Comparative proteomes of Corynebacterium glutamicum grown on aromatic compounds revealed novel proteins involved in aromatic degradation and a clear link between aromatic catabolism and gluconeogenesis via fructose-1,6-bisphosphatase. Proteomics 7, 3775–3787 [DOI] [PubMed] [Google Scholar]
- 27. Chaudhry M. T., Huang Y., Shen X. H., Poetsch A., Jiang C. Y., and Liu S. J. (2007) Genome-wide investigation of aromatic acid transporters in Corynebacterium glutamicum. Microbiology 153, 857–865 [DOI] [PubMed] [Google Scholar]
- 28. Huang Y., Zhao K. X., Shen X. H., Chaudhry M. T., Jiang C. Y., and Liu S. J. (2006) Genetic characterization of the resorcinol catabolic pathway in Corynebacterium glutamicum. Appl. Environ. Microbiol. 72, 7238–7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brinkrolf K., Brune I., and Tauch A. (2006) Transcriptional regulation of catabolic pathways for aromatic compounds in Corynebacterium glutamicum. Genet. Mol. Res. 5, 773–789 [PubMed] [Google Scholar]
- 30. Shen X. H., Jiang C. Y., Huang Y., Liu Z. P., and Liu S. J. (2005) Functional identification of novel genes involved in the glutathione-independent gentisate pathway in Corynebacterium glutamicum. Appl. Environ. Microbiol. 71, 3442–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen X. H., Huang Y., and Liu S. J. (2005) Genomic analysis and identification of catabolic pathways for aromatic compounds in Corynebacterium glutamicum. Microbes Environ. 20, 160–167 [Google Scholar]
- 32. Ault A. (2004) The monosodium glutamate story: the commercial production of MSG and other amino acids. J. Chem. Educ. 81, 347–355 [Google Scholar]
- 33. Becker J., Zelder O., Häfner S., Schröder H., and Wittmann C. (2011) From zero to hero-Design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metab. Eng. 13, 159–168 [DOI] [PubMed] [Google Scholar]
- 34. Ikeda M., Ohnishi J., Hayashi M., and Mitsuhashi S. (2006) A genome-based approach to create a minimally mutated, Corynebacterium glutamicum strain for efficient l-lysine production. J. Ind. Microbiol. Biotechnol. 33, 610–615 [DOI] [PubMed] [Google Scholar]
- 35. Jojima T., Fujii M., Mori E., Inui M., and Yukawa H. (2010) Engineering of sugar metabolism of Corynebacterium glutamicum for production of amino acid l-alanine under oxygen deprivation. Appl. Microbiol. Biotechnol. 87, 159–165 [DOI] [PubMed] [Google Scholar]
- 36. Peters-Wendisch P., Stolz M., Etterich H., Kennerknecht N., Sahm H., and Eggeling L. (2005) Metabolic engineering of Corynebacterium glutamicum for l-serine production. Appl. Environ. Microbiol. 71, 7139–7144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stäbler N., Oikawa T., Bott M., and Eggeling L. (2011) Corynebacterium glutamicum as a host for synthesis and export of d-amino acids. J. Bacteriol. 193, 1702–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hannemann F., Bichet A., Ewen K. M., and Bernhardt R. (2007) Cytochrome P450 systems: biological variations of electron transport chains. Biochim. Biophys. Acta 1770, 330–344 [DOI] [PubMed] [Google Scholar]
- 39. Anzai Y., Li S., Chaulagain M. R., Kinoshita K., Kato F., Montgomery J., and Sherman D. H. (2008) Functional analysis of MycCI and MycG, cytochrome P450 enzymes involved in biosynthesis of mycinamicin macrolide antibiotics. Chem. Biol. 15, 950–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kieser T., Bibb M. J., Buttner M. J., Chater K. F., and Hopwood D. A. (2000) Practical Streptomyces Genetics, John Innes Foundation, Norwich, UK [Google Scholar]
- 41. Green M. R., and Sambrook J. (2012) Molecular Cloning: A Laboratory Manual, 4th Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, New York [Google Scholar]
- 42. Liu Y., Wang C., Yan J. Y., Zhang W., Guan W. N., Lu X. F., and Li S. Y. (2014) Hydrogen peroxide-independent production of α-alkenes by OleTJE P450 fatty acid decarboxylase. Biotechnol. Biofuels 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Omura T., and Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes: II. solubilization, purification, and properties. J. Biol. Chem. 239, 2379–2385 [PubMed] [Google Scholar]
- 44. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 45. Schmeling S., Narmandakh A., Schmitt O., Gad'on N., Schühle K., and Fuchs G. (2004) Phenylphosphate synthase: a new phosphotransferase catalyzing the first step in anaerobic phenol metabolism in Thauera aromatica. J. Bacteriol. 186, 8044–8057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McKenna C. E., Higa M. T., Cheung N. H., and McKenna M.-C. (1977) The facile dealkylation of phosphonic acid dialkyl esters by bromotrimethylsilane. Tetrahedron Lett. 18, 155–158 [Google Scholar]
- 47. Kenner G. W., and Williams N. R. (1955) A method of reducing phenols to aromatic hydrocarbons. J. Chem. Soc. 522–525 [Google Scholar]
- 48. Werck-Reichhart D., and Feyereisen R. (2000) Cytochromes P450: a success story. Genome Biol. 1, reviews3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Keat M. J., and Hopper D. J. (1978) para-Cresol and 3,5-xylenol methylhydroxylases in Pseudomonas putida Ncib-9869. Biochem. J. 175, 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Johannes J., Bluschke A., Jehmlich N., von Bergen M., and Boll M. (2008) Purification and characterization of active-site components of the putative p-cresol methylhydroxylase membrane complex from Geobacter metallireducens. J. Bacteriol. 190, 6493–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herzberg O., Chen C. C., Kapadia G., McGuire M., Carroll L. J., Noh S. J., and Dunaway-Mariano D. (1996) Swiveling-domain mechanism for enzymatic phosphotransfer between remote reaction sites. Proc. Natl. Acad. Sci. U.S.A. 93, 2652–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hanukoglu I. (1996) Electron transfer proteins of cytochrome P450 systems. in Advances in Molecular and Cell Biology, JAI Press Inc., Stamford, CT [Google Scholar]
- 53. Ding W., Si M. R., Zhang W. P., Zhang Y. L., Chen C., Zhang L., Lu Z. Q., Chen S. L., and Shen X. H. (2015) Functional characterization of a vanillin dehydrogenase in Corynebacterium glutamicum. Sci. Rep. 5, 8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Y. F., Chao H., and Zhou N. Y. (2014) The catabolism of 2,4-xylenol and p-cresol share the enzymes for the oxidation of para-methyl group in Pseudomonas putida NCIMB 9866. Appl. Microbiol. Biotechnol. 98, 1349–1356 [DOI] [PubMed] [Google Scholar]
- 55. Di Gioia D., Luziatelli F., Negroni A., Ficca A. G., Fava F., and Ruzzi M. (2010) Metabolic engineering of Pseudomonas fluorescens for the production of vanillin from ferulic acid. J. Biotechnol. 156, 309–316 [DOI] [PubMed] [Google Scholar]
- 56. Breinig S., Schiltz E., and Fuchs G. (2000) Genes involved in anaerobic metabolism of phenol in the bacterium Thauera aromatica. J. Bacteriol. 182, 5849–5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schühle K., and Fuchs G. (2004) Phenylphosphate carboxylase: a new C-C lyase involved in anaerobic in phenol metabolism in Thauera aromatica. J. Bacteriol. 186, 4556–4567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li S., Chaulagain M. R., Knauff A. R., Podust L. M., Montgomery J., and Sherman D. H. (2009) Selective oxidation of carbolide C-H bonds by an engineered macrolide P450 mono-oxygenase. Proc. Natl. Acad. Sci. U.S.A. 106, 18463–18468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sherman D. H., Li S., Yermalitskaya L. V., Kim Y., Smith J. A., Waterman M. R., and Podust L. M. (2006) The structural basis for substrate anchoring, active site selectivity, and product formation by P450 PikC from Streptomyces venezuelae. J. Biol. Chem. 281, 26289–26297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tudzynski B., Hedden P., Carrera E., and Gaskin P. (2001) The P450–4 gene of Gibberella fujikuroi encodes ent-kaurene oxidase in the gibberellin biosynthesis pathway. Appl. Environ. Microbiol. 67, 3514–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ro D. K., Paradise E. M., Ouellet M., Fisher K. J., Newman K. L., Ndungu J. M., Ho K. A., Eachus R. A., Ham T. S., Kirby J., Chang M. C., Withers S. T., Shiba Y., Sarpong R., and Keasling J. D. (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940–943 [DOI] [PubMed] [Google Scholar]
- 62. Paddon C. J., Westfall P. J., Pitera D. J., Benjamin K., Fisher K., McPhee D., Leavell M. D., Tai A., Main A., Eng D., Polichuk D. R., Teoh K. H., Reed D. W., Treynor T., Lenihan J., et al. (2013) High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496, 528–532 [DOI] [PubMed] [Google Scholar]
- 63. Carlson J. C., Li S., Gunatilleke S. S., Anzai Y., Burr D. A., Podust L. M., and Sherman D. H. (2011) Tirandamycin biosynthesis is mediated by co-dependent oxidative enzymes. Nat. Chem. 3, 628–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bernhardt R., and Urlacher V. B. (2014) Cytochromes P450 as promising catalysts for biotechnological application: chances and limitations. Appl. Microbiol. Biotechnol. 98, 6185–6203 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.